Simple Summary
Studies conducted in a recent decade revealed that acute lymphoblastic leukemia cells exploit various mechanisms to avoid immune recognition and destruction by the immune system. As a consequence, leukemia cells become more resistant to treatment, which results in poor patient outcomes. In this review, we describe the interactions between acute lymphoblastic leukemia (ALL) cells and selected populations of immune cells within the bone marrow microenvironment that contribute to leukemia development, promotion, and relapse. We also discuss how various elements of the bone marrow niche affect ALL chemo- and immunotherapy and present recently discovered treatment strategies that restore or stimulate the immune response to ALL.
Abstract
Acute lymphoblastic leukemia (ALL) results from a clonal expansion of abnormal lymphoid progenitors of B cell (BCP-ALL) or T cell (T-ALL) origin that invade bone marrow, peripheral blood, and extramedullary sites. Leukemic cells, apart from their oncogene-driven ability to proliferate and avoid differentiation, also change the phenotype and function of innate and adaptive immune cells, leading to escape from the immune surveillance. In this review, we provide an overview of the genetic heterogeneity and treatment of BCP- and T-ALL. We outline the interactions of leukemic cells in the bone marrow microenvironment, mainly with mesenchymal stem cells and immune cells. We describe the mechanisms by which ALL cells escape from immune recognition and elimination by the immune system. We focus on the alterations in ALL cells, such as overexpression of ligands for various inhibitory receptors, including anti-phagocytic receptors on macrophages, NK cell inhibitory receptors, as well as T cell immune checkpoints. In addition, we describe how developing leukemia shapes the bone marrow microenvironment and alters the function of immune cells. Finally, we emphasize that an immunosuppressive microenvironment can reduce the efficacy of chemo- and immunotherapy and provide examples of preclinical studies showing strategies for improving ALL treatment by targeting these immunosuppressive interactions.
1. Introduction
Acute lymphoblastic leukemia (ALL) results from a clonal expansion of abnormal, immature lymphoid progenitors of B cell or T cell origin that invade bone marrow (BM), peripheral blood, and extramedullary sites [1]. The incidence of ALL is highest among children aged 1–4 years and declines with age rising slightly after the age of 50. Several genetic alterations have been already found as drivers of ALL, uncovering the genetic heterogeneity of the disease. Age at diagnosis of ALL considerably affects the 5-year overall survival (OS), reaching 90% in pediatric patients and only 30–40% in adults older than 40 years [2]. High frequency of aggressive molecular ALL subtypes and an increased risk of treatment-induced toxicities contribute to poor prognosis in adult patients. ALL patients who do not respond to conventional chemotherapy are now treated with various types of immunotherapy. However, the efficacy of the immunotherapy is greatly affected by the interactions of leukemic cells with the microenvironment within the BM niche.
As other cancer cells, ALL cells, due to genetic alterations and dysregulated post-translational modifications, express neoantigens and can induce tumor-specific T cell responses. Indeed, though ALL is a malignancy with relatively low mutational burden, recent reports show that leukemic, antigen-specific T cells are present in the BM microenvironment [3]. There is also growing evidence that natural killer (NK) cells play a role in the immunosurveillance of ALL [4]. However, developing leukemia impairs the key components of the immune system responsible for mounting an anticancer response, particularly in patients poorly responding to treatment or at the relapse stage [5,6,7]. Cancer cells can avoid recognition and elimination by the immune system by various, cancer type-specific mechanisms, which are already well documented in solid tumors and are just being discovered in ALL. Downregulation or loss of human leukocyte antigen (HLA) class I molecules, which leads to impaired recognition of leukemic cells by cytotoxic T lymphocytes, is a rare but functionally significant mechanism of immune evasion in ALL [8]. Moreover, recent data uncovers the mechanism of T cell exhaustion operating in ALL [5,9,10], opening the possibility to employ appropriate immune checkpoint inhibitors to ALL treatment protocols. Finally, novel, high-throughput methods, in particular single cell RNA sequencing, reveal the important role of suppressive myeloid cell populations in promoting leukemia progression and hampering treatment efficacy [11].
In this review, we describe the crosstalk between ALL cells and the BM microenvironment that contributes to leukemia development, promotion, and relapse. We summarize recent findings on how ALL avoids destruction by the immune system and describe the interactions between ALL cells and selected populations of immune cells. The overview of the mutual interactions between ALL cells and the key cells of the BM microenvironment is presented in Figure 1.
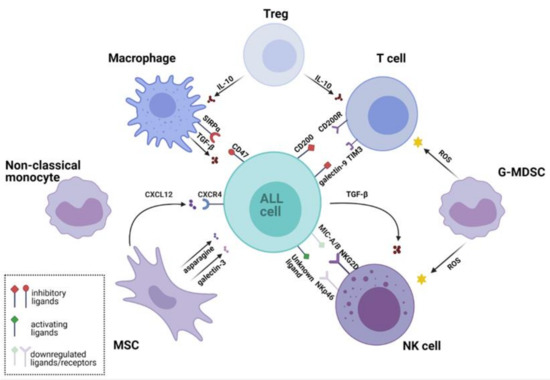
Figure 1.
Leukemic microenvironment supports survival of acute lymphoblastic leukemia (ALL) cells and their immune evasion through multiple interactions. Various cell populations shape the leukemic microenvironment. Regulatory T cells (Tregs) secrete inhibitory cytokines that suppress the cytotoxic activity of T cells and reduce macrophage phagocytosis. Granulocytic Monocyte Derived Suppressor Cells (G-MDSC) produce reactive oxygen species (ROS) that inhibit activity of T cells and natural killer (NK) cells. NK cells express low levels of natural cytotoxicity triggering receptor p46 (NKp46) activating receptor, while ALL cells downregulate major histocompatibility complex class I-related chains A/B (MIC-A/B)-a ligand for natural killer group 2 member D (NKG2D) activating receptor. ALL also drives NK cell dysfunction by secreting immunosuppressive transforming growth factor beta (TGF-β). Mesenchymal stem cells (MSCs) secrete chemokines, e.g., C-X-C chemokine ligand 12 (CXCL12), which binds C-X-C chemokine receptor type 4 (CXCR4) and promotes ALL engraftment into the vascular niche. Furthermore, MSCs protect ALL cells against the treatment by secretion of galectin-3, which activates the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathway. MSCs also secrete metabolites, such as asparagine, which reduces the cytotoxicity of L-asparaginase. Non-classical (CD16+) monocytes infiltrate the leukemic microenvironment and are thought to be involved in ALL cells protection. Macrophages from the leukemic niche acquire immunosuppressive properties and secrete tumor-promoting cytokine TGF-β. Their phagocytic activity is reduced by the interaction of signal regulatory protein α (SIRPα) with cluster of differentiation 47 (CD47)—a “do not eat me signal” expressed by leukemic cells. The figure was created in BioRender (https://biorender.com/; accessed on 27 February 2021). Other abbreviations: CD200, cluster of differentiation 200; CD200R, CD 200 receptor; G-MDSC, Granulocytic Monocyte Derived Suppressor Cells; IL-10, Interleukin 10; TIM3, T-cell immunoglobulin domain and mucin domain 3.
We also discuss how various elements of the BM niche affect ALL chemo- and immunotherapy and present recently discovered treatment strategies that restore or stimulate the immune response to ALL.
2. Immunophenotype and Genotype of Acute Lymphoblastic Leukemia
Depending on the cell of origin, ALL is classified as a B cell precursor ALL (BCP-ALL) and T cell ALL (T-ALL). BCP-ALL accounts for 80–85% of ALL [2]. According to the Associazone Italiana Ematologia Oncologia Pediatrica and the Berlin-Frankfurt-Münster (AIEOP-BFM) Consensus Guidelines for Flow Cytometric Immunophenotyping, BCP-ALL is diagnosed when dominant leukemic clone in the BM shows strong expression of at least two of the following antigens: CD19, CD10, i(intracellular)CD22, or iCD79a [12]. Further classification of the blasts’ immunophenotype dissects four subtypes of BCP-ALL (BI: pro-B; BII: common; BIII: pre-B; and BIV: mature B), based on the differences in the expression of CD10, iIgM, λ, and κ chains [12]. BCP-ALL is also classified into several molecular subtypes according to the major (initiating) chromosomal abnormality and specific secondary lesions, which both result in a unique gene expression profile and chemosensitivity of leukemic cells [1]. This heterogeneous mutational landscape of BCP-ALL is reflected in the revised version of World Health Organization (WHO) classification of myeloid neoplasms and acute leukemias, which distinguishes eleven subtypes of ALL, based on the presence of somatic molecular lesions [13]. Genetic abnormalities that confer to BCP-ALL development may associate with the expression of the specific antigens in leukemic cells and serve as prognostic factors [12] (Table 1). Selected molecular lesions are currently used in the clinical practice to predict the risk for BCP-ALL recurrence in the individual patient and then adapt the intensity of the therapy accordingly. Moreover, dissection of genomically defined ALL subtypes led to the development of relevant molecularly targeted therapies that contributed to further significant improvement in patients’ survival. The examples are inhibitors targeting ABL1/ABL2, which increased the response rate among ALL patients with translocation t(9;22) (q34;q11), resulting in breakpoint cluster region–Abelson murine leukemia viral oncogene homolog 1 (BCR–ABL1) gene fusion (Philadelphia chromosome), or inhibitors of Janus kinase 2 (JAK2), which improved treatment outcome of individuals with Philadelphia chromosome-like ALL (Ph-like ALL) [14].

Table 1.
Molecular subtypes of B cell precursor acute lymphoblastic leukemia [1,2].
T-ALL represents approximately 12–15% and 20% of all ALL cases in children and adults, respectively [2]. It clearly shows a distinct biology, clinical features (hyperleukocytosis with extramedullary involvement of lymph nodes, central nervous system infiltration, and the presence of a mediastinal mass, arising from the thymus) and response to particular elements of therapy, as compared to BCP-ALL. The immunophenotype of T-ALL blast cells is distinguished by the presence of iCD3, iCD7, and weak expression or lack of iMPO [12]. There are four subtypes of T-ALL according to the European Group for the Immunological Classification of Leukemia (EGIL): pro-T EGIL T-I (iCD3+, CD7+), pre-T EGIL T-II (iCD3+, CD7+ and CD5/CD2+), cortical T EGIL T-III (iCD3+, Cd1a+, sCD3+/−), and mature-T EGIL T-IV (iCD3+, sCD3+, CD1a−) [25]. Early T cell Precursor (ETP) ALL represents an additional subtype, showing a unique immunophenotype (lack of expression of CD1a and CD8, weak CD5 expression, and expression of 1 or more myeloid or stem cell markers) and a specific genomic profile [26,27]. The genetic landscape of T-ALL varies according to the maturation stage of the dominant leukemic clone and includes chromosomal rearrangements resulting in gene fusions, point mutations, and copy number alterations (Table 2). Although these aberrations significantly contribute to malignant lymphoproliferation of T-ALL and several are even related to treatment outcome, none of them is presently used for risk stratification [28,29].
Table 2.
Molecular subtypes in T cell acute lymphoblastic leukemia, based on [28,29].
3. Treatment and Prognosis of Acute Lymphoblastic Leukemia
The first-line treatment of ALL usually consists of four phases: induction, consolidation, intensification, long-term maintenance, and directed therapy preventing central nervous system relapse of leukemia. In each phase, patients receive courses of different combinations of drugs, including glucocorticoids, vinca alkaloids (vincristine), anthracyclines (doxorubicin), antimetabolites (cytarabine, mercaptopurine, methotrexate), alkylating agents (cyclophosphamide), and L-asparaginase. While this intensive therapeutic approach leads to a 5-year OS of 90% in childhood ALL, adults achieve a 5-year OS of less than 45% [2]. Several studies have demonstrated improved long-term outcomes in adolescents and young adults (AYA) treated with pediatric-intensive chemotherapy [30]. Additionally, in case of chemotherapy failure or insufficiency, ALL patients may be treated with various forms of immunotherapy (Table 3), including the oldest one-hematopoietic stem cell transplantation (HSCT). The curative effect of HSCT results from direct cytotoxicity from the chemo-radiotherapy administered in the conditioning regimen, along with an immune effect termed graft-versus-leukemia (GVL) [31].
HSCT is reserved for patients with high risk ALL who show specific genetic abnormalities (e.g., low hypodiploidy) and/or persistent minimal residual disease (MRD), are fit, and have an available donor [2]. Whereas HSCT is still perceived as a standard consolidative therapy preventing relapse in many adult patients with Ph-negative ALL [32], the role of HSCT in childhood ALL is continuously redefined as advances in immunotherapy and targeted treatment are made. A detailed discussion of the indications of HSCT in childhood ALL, as depending on the molecular aberrations and response to therapy, was presented by Merli et al. [33].
Bone marrow transplantation has severe limitations, e.g., graft versus host disease (GVHD), increased infection rates due to delayed immune reconstitution, and difficulties in finding suitable, HLA-matched donors. For BCP-ALL patients, other forms of more selective immunotherapy targeting B cell-specific antigens, CD19, CD20, and CD22, are already available [2,34]. The first group includes therapeutic monoclonal antibodies (mAbs) and their derivatives, such as rituximab (anti-CD20 “naked” mAb), inotuzumab ozogamycin (anti-CD22 mAb conjugated with a drug calicheamicin), and blinatumomab (anti-CD19/anti-CD3 bispecific T cell engager) [35,36,37]. Since 2017, cellular immunotherapy using patient-derived T cells, genetically modified with chimeric antigen receptors (CAR-T cells), has been approved. CARs are synthetic constructs that consist of an extracellular antibody-derived domain that is responsible for antigen recognition, intracellular activating domain derived from T cell receptor (CD3ζ chain), and different co-stimulatory domains (CD28, 4-1BB, ICOS, OX40). Upon introduction into patient’s T cells, CARs reprogram the cells to recognize antigens present on leukemic cells, which results in T cell activation and leukemic cell death [38]. Tisagenlecleucel (autologous, anti-CD19 CAR-T cells) is approved by the FDA (U.S. Food and Drug Administration) for the treatment of relapsed or refractory pediatric and adult BCP-ALL. Despite a spectacular initial response, the long-term outcome of refractory/relapsed BCP-ALL patients treated with CAR-T cells is limited, with overall survival reaching only 12.9 months [39]. The development of CAR-T cells against other B cell antigens is ongoing. Various types of immunotherapy, already approved and tested in ALL patients, are presented in Table 3.
Table 3.
Overview of immunotherapy used against acute lymphoblastic leukemia.
Table 3.
Overview of immunotherapy used against acute lymphoblastic leukemia.
| Applied in clinics (BCP-ALL) | |||
|---|---|---|---|
| Type of Immunotherapy | Drug/Therapy Name | Mechanism of Action | References/ Clinical Trial No. |
| HSCT | Infusion of hematopoietic stem/progenitor cells | [40] | |
| mAbs | Blinatumomab | Anti-CD19/CD3 bi-specific T-cell-mAb, which binds simultaneously to CD3-positive cytotoxic T cells and to CD19-positive B cells; endogenous T cells recognize and eliminate CD19-positive ALL blasts | [41] NCT02013167 NCT03628053 |
| Rituximab | Humanized murine mAb targeting CD20 on BCP-ALL cells | [42] | |
| Ofatumumab | Second-generation anti-CD20 mAb. Binds to a different epitope on the CD20 than Rituximab. | [43] | |
| ADC (antibody-drug conjugates) | Inotuzumab ozogamycin | Humanized anti-CD22 antibody conjugated to a calicheamicin (cytotoxic drug); the CD22–conjugated complex is rapidly internalized and calicheamicin is released, which induces DNA double strand breaks | [44] |
| CAR-T cells | Tisagenlecleucel | Chimeric antigen receptor (CAR) T-cells targeting CD19 antigen and containing 4-1BB zeta co-stimulatory domain | [45] NCT03123939 |
| Tested in clinical trials (BCP-ALL) | |||
| mAbs | Epratuzumab | Humanized anti-CD22 mAb (IgG1) | [46] NCT02844530 NCT01354457 NCT01802814 |
| Alemtuzumab | Fully humanized anti-CD52 mAb | [47,48] | |
| Blinatumomab + Nivolumab +/− Ipilimumab | Anti-CD19/CD3 bi-specific T-cell-mAb + Humanized anti-PD-1 mAb +/− Humanized anti-CTLA4 mAb | NCT02879695 | |
| Blinatumomab +Nivolumab | Anti-CD19/CD3 bi-specific T-cell-mAb + Humanized anti-PD-1 mAb | NCT04546399 | |
| Blinatumomab + Pembrolizumab | Humanized anti-PD-1 mAb | NCT03160079 | |
| TTI-621 + Rituximab/Nivolumab | TTI-621 (SIRPαFc) is a soluble recombinant fusion protein composed of the N-terminal CD47 binding domain of human SIRPα and the Fc domain of human immunoglobulin (IgG1); TTI-621 binds to CD47 and prevents “do not eat” (anti- phagocytic) signaling | NCT02663518 | |
| ADC | Denintuzumab | Humanized anti-CD19 antibody conjugated to a microtubule-disrupting agent monomethyl auristatin F (MMAF) | [49] |
| Coltuximab ravtansine (SAR3419) | Anti-CD19 monoclonal antibody conjugated to potent inhibitor of tubulin polymerization and microtubule assembly, maytansinoid, DM4 | [50] | |
| CAR-T cells | CD19-CD22 CAR-T | Modified autologous T cells expressing anti-CD19 and anti-CD22 CARs | [39] NCT04626765 |
| CD22 CAR-T | Modified autologous T cells expressing anti-CD22 CARs | [39] NCT04626765 | |
| CD19–28z CAR-T | Modified autologous T cells expressing anti-CD19 CARs with CD28 co-stimulatory domain | [39] | |
| NK cells | Allogenic activated NK cells | Infusion of IL-15/IL-21-activated NK cells after HLA-mismatched HSCT | [51] |
| Allogenic activated NK cells | Activated and expanded natural killer cells (NKAEs) from haploidentical donor infused to patients | NCT02074657 | |
| Autologous NK cells | Enriched and expanded autologous NK cells | NCT02185781 | |
| Cord blood NK cells | Personalized cord blood (CB)-derived NK cells for HLA-C2/C2 patients after chemotherapy | NCT02727803 | |
| Cord blood NK cells | CD19-CD28-zeta-2A-iCasp9-IL15-transduced cord blood natural killer (CB-NK) cells recognizing CD19+ tumor cells | NCT03056339 | |
| Tested in clinical trials (T-ALL) | |||
| mAbs | Isatuximab | Anti-CD38 mAb | NCT02999633 NCT03860844 |
| Daratumumab | Anti-CD38 mAb | NCT03384654 | |
| Alemtuzumab | Anti-CD52 mAb | NCT00199030 NCT00061048 NCT00061945 | |
| CAR-T cells | CD4 CAR-T | Modified T cells expressing anti-CD4 CARs | NCT03829540 NCT04162340 |
| CD5 CAR-T | Modified T cells expressing anti-CD5 CARs | NCT03081910 | |
| CD7 CAR-T | Modified T cells expressing anti-CD7 CARs | NCT04004637 NCT04264078 NCT03690011 NCT04033302 NCT04480788 | |
| NK cells | CD7 CAR NK cells | Modified NK cells expressing anti-CD7 CARs | NCT02742727 |
Abbreviations: ADC, antibody-drug conjugates; Casp9, caspase 9; CTLA-4, cytotoxic T-lymphocyte-associated protein 4; HSCT, hematopoietic stem-cell transplantation; mAbs, monoclonal antibodies; PD-1, programmed death receptor 1; SIRPαFc, signal regulatory protein α fragment crystallizable.
In contrast to the wide range of immune targets in BCP-ALL, treatments targeting T-ALL-specific antigens are less available in clinical practice. Nevertheless, Hening et al. has recently shown an effective eradication of residual disease and prolonged disease-free survival in patients with advanced relapse of T-ALL who obtained recombinant monoclonal anti-CD38 antibody (daratumumab) [52]. In addition, mimicking negative selection of T lymphocytes in the thymus using anti-CD3 antibody showed promise in preclinical studies in CD3/TCR-positive T-ALL [53].
There is a wide range of biological and clinical factors predicting the outcome of ALL, including age, sex, race, blood count at diagnosis, immunophenotype, and genetic alterations, but the key prognostic determinant is the response to treatment measured through serial minimal residual disease (MRD) monitoring [2]. Chemosensitivity of ALL, as well as response to immunotherapies, depend on intrinsic biological features of cancer cells but are also modified by diverse interactions between leukemic cells and the BM microenvironment. The long-term response to immune-based therapy might be limited as a result of clonal evolution of leukemia. This process promotes selection of ALL subclones exhibiting treatment-refractory phenotype through progressive and diverse accumulation of genetic alterations by neoplastic cells under therapeutic pressure [54,55]. There are several examples proving that clonal evolution and intratumor heterogeneity affect the efficacy of targeted immunotherapy [56,57]. One of the most illustrative in this context is successful elimination of CD19+ cells by patients with KMT2A-rearranged B-ALL receiving CD19 CAR-T [54].
4. Bone Marrow Niche as a Sanctuary Site Supporting Acute Lymphoblastic Leukemia Development
Physiologically, BM niche is a specialized area of interactions between cells, matrix, biophysical forces, and metabolites. These factors provide the appropriate conditions for hematopoietic stem cells (HSCs) quiescence, self-renewal, proliferation, multi-lineage differentiation capacity, and localization [58]. There are two functionally distinct BM niches: osteoblastic (endosteal) and vascular. The osteoblastic niche is located at the endosteum and contains diverse non-hematopoietic cell types, including osteoblasts, osteoclasts, glial non-myelinating Schwann cells, and regulatory T cells (Tregs). The vascular niche is enriched at the sinusoidal walls and comprises mesenchymal stem cells (MSCs), reticular cells, leptin receptor-positive perivascular stromal cells, macrophages, arteriolar and sinusoidal endothelial cells, as well as other immune cells. All these diverse cell types provide juxtacrine and paracrine signals (growth factors, chemokines, cytokines, morphogens, and adhesion molecules) to either normal hematopoietic or leukemic cells [59,60]. Osteoblastic niche supports HSCs quiescence while vascular niche stimulates cell proliferation, differentiation, and mobilization [61]. Additionally, multi-lineage differentiation of HSCs, as well as progression of leukemia within the niche, are regulated by extracellular matrix proteins (heparin sulfate, tenascin-C, and osteopontin) and by the extrinsic factors (calcium ions, the oxygen tension, the local concentration of reactive oxygen species (ROS), nitric oxide (NO), and pH) [59].
4.1. Interactions of Leukemic Cells with the Bone Marrow Microenvironment
Leukemogenesis can no longer be perceived as a process that exclusively affects HSC or more differentiated lineage precursors. Some genetic alterations, epigenetic changes, and transcriptomic variations are already present within stromal compartment at cancer diagnosis, but their incidence in patients with ALL has not been precisely defined [62,63,64]. Moreover, the possible role of the microenvironment in promoting hematopoietic dysregulation is not restricted to the small percentage of patients harboring germline predisposition to leukemia. At least, in the case of myeloid malignancies, genomic and transcriptomic variations in stromal compartment have been also observed outside of constitutional defects [65]. Moreover, leukemic cells modify the BM microenvironment in a way that allows maintenance of cancer cells survival and the protection against chemotherapy. Blasts may affect the osteoblastic niche, leading to the loss of osteoblastic cells and an impaired function of normal HSCs [60]. They contribute to the remodeling of the extracellular matrix (ECM) through secretion of matrix metalloproteinase-9 (MMP-9) [66]. Interestingly, overexpression of C-X-C chemokine receptor type 4 (CXCR4) on leukemic cells is associated with extra-medullary organs involvement and poor patients’ outcome [67]. Due to the strong effect of CXCR4-mediated interaction between the BM microenvironment and leukemic cells, several CXCR4 antagonists have been successfully tested in preclinical models. They decreased the number of leukemic cells in peripheral blood and limited the dissemination of ALL cells to extra-medullary sites [68]. However, these promising biological effects did not fully translate into clinical practice. Plerixafor, a reversible CXCR4 antagonist, showed only a modest therapeutical effect among heavily pretreated pediatric patients diagnosed with refractory/relapsed acute leukemias [69].
An interaction between leukemic cells and the BM microenvironment may also be directly associated with genetic alterations of leukemic cells and might be targeted to augment the existing treatment strategies. This can be illustrated by genetic abnormalities within the IKZF1 gene in kinase-driven (Ph-positive and Ph-like) ALL, which result in acquisition of a hematopoietic stem cell-like phenotype and elevated cell surface expression of adhesion molecules. The acquisition of this phenotype causes extravascular invasion, residence in the niche, and activation of integrin signaling pathways. This leads to peculiar BM stromal cell adhesion and mislocalization of the IKZF1-mutated ALL cells and results in decreased chemosensitivity. Interestingly, retinoids and focal adhesion kinase (FAK) inhibitors restore sensitivity to chemotherapy [70].
4.2. Growth-Supporting and Protective Role of Mesenchymal Stem Cells against Leukemic Cells
MSCs are essential components of the hematopoietic microenvironment. They are mainly located close to the vascular BM niche, where they give rise to different cell lineages, including osteoblasts, chondrocytes, adipocytes, and myocytes. MSCs produce HSC-supporting substances, including chemokines (stromal cell-derived factor 1(SDF1/CXCL12)), cytokines, angiopoietin, stem cell factor (SCF/Kit ligand), galectin-3, extracellular matrix (ECM) proteins, and metabolites. Examples of the effects of these factors on leukemia development, progression, and modulation of response to therapy are discussed below [60,61]. Importantly, human MSCs co-cultured ex vivo with primary ALL cells support ALL cells’ survival and enable in vitro drug testing that allows to better predict in vivo response [69,71,72].
ALL cells tend to engraft in a vascular niche that is particularly enriched in E-selectin and stromal-derived factor 1 (SDF-1/CXCL12). Together with IL-7, CXCL12 stimulates proliferation of both T- and BCP-ALL [73,74]. Other cytokines (IL-1, IFN-γ, and TNF-α) and the soluble HLA-G in the bone marrow microenvironment may also contribute to T-ALL recurrence by inducing immunotolerance [66,75]. Recently, tunneling nanotubes were identified as a mechanism of the crosstalk between ALL cells and MSCs within the niche. This intercellular communication leads to the secretion of prosurvival cytokines (IL-8) and chemoattractants interferon gamma-induced protein/ CXC chemokine ligand 10 (IP10/CXCL10), monocyte chemotactic protein-1 (MCP-1)/CCL2) enhancing the resistance of leukemic cells to steroids [76]. In addition, via the expression of heme oxygenase-1 and the release of vascular endothelial growth factor (VEGF), MSCs promote resistance to vincristine [77]. MSCs can also protect ALL cells against the treatment by secretion of soluble or stroma-cell-bound galectin-3, which stimulates expression of endogenous LGALS3 mRNA and activates the tonic NF-κB pathway in leukemic cells [78]. The proliferation and dissemination of ALL cells in the niche is also stimulated by MSC-derived ECM proteins, such as periostin and osteopontin. Periostin induces the expression of C-C Motif Chemokine Ligand 2 (CCL2) in BCP-ALL cells [79]. Osteopontin controls ALL cell dormancy in the niche and its neutralization sensitizes ALL cells to chemotherapy with cytarabine [80]. Finally, MSCs also secrete metabolites, such as asparagine, that reduces the cytotoxicity of L-asparaginase [81].
4.3. Leukemia-Induced Loss of Immunosurveillance within the Bone Marrow Niche
Bone marrow provides a microenvironmental framework supporting development and function of all immune cells and acts as a host for various mature cell types, including B and T cells, plasma cells, dendritic cells, neutrophils, and macrophages that reside in the BM niche [61]. Therefore, the niche becomes the main and essential place of mutual interactions of cancer and immune cells and, as a result of these interactions, leukemic cells should be recognized and eliminated. However, during leukemogenesis, proliferating blasts affect the differentiation and function of immune cells, leading to loss of immunosurveillance and cancer progression. Below we provide examples of how developing leukemia affects key cells of the immune system involved in the immunosurveillance, such as myeloid cells, NK cells, and various subpopulations of T cells.
5. Myeloid Cells
Cells of myeloid origin at various stages of differentiation and maturation contribute to immune evasion in ALL. The immature myeloid populations involved in immunoregulation are collectively referred to as myeloid-derived suppressor cells (MDSCs). The mature myeloid populations include granulocytes, monocytes, macrophages, and dendritic cells (DCs). All myeloid cell types have distinct immunological functions and can be identified by their immunophenotype as described in [82,83,84,85].
The development of myeloid lineage begins in the bone marrow from granulocyte-monocyte progenitors (GMPs) or monocyte-dendritic cell progenitors (MDPs) [86]. Some of them do not undergo a full differentiation into monocytes and remain in an immature state to become MDSC - immunoregulatory cells, best known for their role in promotion of tumor development, also in hematological malignancies [87,88]. Two distinct populations of MDSCs, granulocytic (G) and monocytic (M), can be distinguished depending on the phenotypic differences, as reviewed in [89,90]. This classification also implies functional differences: G-MDSCs induce immunosuppression mostly by reactive oxygen species (ROS)-dependent mechanisms, while M-MDSCs act rather by secretion of immunosuppressive cytokines (IL-10, TGFβ), arginase, and NO.
Unlike MDSCs, most myeloid progenitors continue their development to become granulocytes or monocytes. The latter ones can be divided into three main subsets depending on the expression of CD14 and CD16 surface markers. Around 90% of monocytes are characterized by high expression of CD14 and are known as classical (CD14++CD16−) monocytes. The other 10% are non-classical (CD14+CD16++) and intermediate (CD14+CD16+) monocytes [91]. Monocytes act as early responders to inflammation or infection. Additionally, non-classical monocytes are involved in tissue repair and healing processes [92].
After several days of circulation in the blood, monocytes tend to migrate to other tissues and transform into macrophages or dendritic cells. Both populations are involved in phagocytosis and antigen presentation. Macrophages are traditionally divided into two subtypes: classically activated, antitumor M1 macrophages, and alternatively activated, tumor-supporting M2 macrophages [93]. This paradigm is now viewed as a basis for a spectrum of numerous macrophage phenotypes located between those two extremes [94,95,96]. Macrophages display great phenotypic flexibility, depending on microenvironmental stimuli. The studies on different macrophage subtypes revealed that in the tumor microenvironment, some of the macrophages acquire immunosuppressive properties and become tumor associated macrophages (TAM), or leukemia associated macrophages (LAM). Such macrophages promote tumor cells proliferation, migration, and angiogenesis [97]. Most LAMs are of the M2-like phenotype. Similarly, the dendritic cells are also divided into several subpopulations with distinct immunological features [98].
5.1. Myeloid-Derived Suppressor Cells
Though the pro-tumorigenic function of MDSCs is well established in solid tumors and some hematological neoplasms, their role in the development and progression of ALL is still poorly understood. Several studies have reported that the numbers of MDSCs are higher in the blood and the BM of patients diagnosed with BCP-ALL in comparison with healthy donors [10,99]. It was also shown that some chemotherapeutic drugs used in the treatment of ALL, as cyclophosphamide and doxorubicin, increase the already elevated number of circulating MDSCs [99,100].
Substantial evidence of G-MDSCs (but not M-MDSCs) being involved in evading immune surveillance in BCP-ALL was documented in the study of Liu et al. [101]. The authors compared the numbers of G-MDSCs in peripheral blood and BM of BCP-ALL patients and healthy donors and discovered a significant elevation of these suppressive cells in patients’ samples. Moreover, the G-MDSCs numbers positively correlated with blast clearance and a worse therapeutic response. Ex vivo tests confirmed that G-MDSCs suppress T cell and NK cell responses via ROS [101].
5.2. Non-Classical Monocytes
The accumulation of non-classical monocytes was the major immune cell alteration observed recently in BCP-ALL patients’ BM [11]. To establish the main differences between a healthy and leukemic BM microenvironment, the authors employed a single-cell RNA sequencing-based approach. They found that the myeloid cell compartment was most significantly changed in BCP-ALL in patients’ BM and that leukemic cells promoted non-classical monocyte differentiation, likely in response to vascular damage. Accordingly, the pan-myeloid depletion via colony stimulating factor 1 receptor (CSF1R) blockade increased sensitivity to nilotinib in an in vivo mouse model of BCP-ALL, indirectly confirming the role of non-classical monocytes in the protection of leukemic cells [11].
5.3. Macrophages and Dendritic Cells
Elevated levels of macrophages expressing M2 markers, including CD68, CD163, and CD206, were found in the BM of BCP-ALL patients [10,102]. Valencia et al. reported that culturing of macrophages in ALL-conditioned medium resulted in the upregulation of tumor-promoting cytokines (IL-10, TGF-β), enzymes (IDO1, MMP9), and VEGF. Similarly, human dendritic cells have also acquired the same immunosuppressive features after analogous culturing [103].
M2 macrophages were also shown to provide pro-survival signals to T-ALL cells. Human macrophages cultured in vitro secreted insulin-like growth factor IGF1, which contributed to increased survival of T-ALL cells expressing IGF1-R (insulin-like growth factor 1 receptor) [104]. Accordingly, depletion of myeloid cells led to reduced leukemic burden and improved survival of mice in the LN3 transgenic model of T-ALL [104].
Apart from promoting M2 differentiation, ALL cells can also overcome immune surveillance by escaping the effector mechanisms of functional macrophages. This can be done by overexpression of CD47 - a surface protein that acts as a “do not eat me signal” for macrophages, by triggering its inhibitory anti-phagocytic pathway mediated by the SIRPα receptor. Chao et al. have confirmed the presence of CD47 on T-ALL and BCP-ALL cells and used the humanized anti-CD47 antibody to promote ALL cells’ phagocytosis in vitro and inhibit ALL engraftment in NSG mice [105]. Accordingly, a fully humanized anti-CD47 antibody induced an efficient phagocytosis of human T-ALL cell line CCRF in vitro, as well as inhibited engraftment of this cell line in immunodeficient BALB/c nude mice. A strategy aiming to prevent this inhibitory pathway, employing recombinant fusion protein composed of a domain of SIRPα and Fc of human IgG1, is currently being tested in a clinical trial (NCT02663518).
5.4. Use of Myeloid Cells in Acute Lymphoblastic Leukemia Therapy
An interesting approach combining monocyte-based adoptive immunotherapy and gene therapy for BCP-ALL treatment was presented by Escobar et al. [106]. The high infiltration of BM microenvironment by monocytes was utilized to counteract immune evasion. An innovative gene therapy was designed to deliver IFNα into the bone marrow niche by using genetically engineered monocyte progenitors. Such an approach, tested in a mouse model of BCP-ALL, resulted in the promotion of T cell activity and synergized with tumor-specific CAR-T cells treatment, successfully overcoming inhibitory signals from the leukemic microenvironment [106].
6. Natural Killer Cells
Natural killer (NK) cells are innate lymphoid cells that recognize and kill virus-infected or malignant cells (target cells). They utilize several pathways to kill target cells, which are extensively summarized in [107,108]. The ability of NK cells to kill ALL blasts depends on the balance between the activating and inhibitory receptors on NK cells as well as by the presence of their corresponding ligands on ALL cells [109]. The inhibitory receptors, among others, include killer immunoglobulin-like receptors (KIR), such as killer cell immunoglobulin-like receptor 2DL (KIR2DL), killer cell immunoglobulin-like receptor 3DL (KIR3DL), and natural killer group 2A (NKG2A). These receptors recognize classical and non-classical HLA class I molecules. The activating receptors include killer cell immunoglobulin-like receptor 2DS (KIR2DS) and killer cell immunoglobulin-like receptor 3DS (KIR3DS); natural killer group 2D (NKG2D); DNAX accessory molecule 1 (DNAM-1); and Natural Cytotoxicity Receptors (NCRs), e.g., NKp46. The NKG2D receptor recognizes the stress inducible molecules, MHC class I chain-related proteins A and B (MIC A/B), and UL16-binding proteins (ULBP), whereas DNAM-1 binds poliovirus receptor (PVR) and Nectin-2. The ligands for the other activating receptors are not well defined. The occurrence of HLA molecules on normal cells protects them from killing by NK cells [107,110,111,112]. As the genes encoding HLA molecules are polygenic and polymorphic, it was shown that specific alleles may promote leukemia development, highlighting the role for NK cells in ALL immunosurveillance. The occurrence of the C2 variant of HLA-C, which binds the inhibitory KIR2DL1 receptor with high affinity [113], correlated with leukemia susceptibility and risk of late relapse [114].
6.1. Expression of Ligands for NK Receptors on Leukemic Cells
Several studies reported that ALL blasts are not effectively killed by NK cells in vitro [115,116,117,118]. Although the reasons are not yet precisely understood, it is known that developing leukemia modifies NK cells’ education and maturation. Moreover, leukemic blasts avoid NK cell-mediated killing by the expression of selected ligands for NK cell receptors. Romanski et al. showed that BCP-ALL cell lines and primary cells, but not T-ALL cells, are resistant to NK cell (NK-92)-mediated lysis due to low levels of MICA/B ligands for activating receptor NKG2D [115,118]. Interestingly, adult and pediatric ALL blasts showed different expression of the ligands for the NKG2D and DNAM-1 receptors [119]. Higher levels of Nec-2 (ligand of DNAM-1), ULBP1, and ULBP3 (ligands of NKG2D) were observed in pediatric patients as compared to adults. Moreover, differences in the expression of the ligands for NK cell receptors were observed between genetically defined BCP-ALL subgroups. Blasts from patients carrying the BCR-ABL1 gene fusion had significantly higher levels of NKG2D and DNAM-1 ligands and were more sensitive to NK cell-mediated lysis than blasts without this genetic aberration [119]. Although lower levels of HLA-C and HLA-E, the ligands for NK cell inhibitory receptors, were also observed in BCP-ALL primary cells collected at diagnosis, the activation of NK cells due to “missing-self” was not observed [116]. All the above results suggest that ALL blasts escape from NK cell lysis predominantly by downregulation of the ligands for NK cell-activating receptors.
6.2. Alterations in the Numbers and Activity of NK Cells
Patients with ALL show differences in the number, phenotype, and activity of NK cells. Early studies showed that patients suffering from ALL have abnormal percentages and absolute numbers of NK cells [120]. Moreover, Valenzuela-Vazquez et al. confirmed these observations in Hispanic ALL patients, especially in those diagnosed with T-ALL [121]. Already in 1989, Gabrilovac et al. observed impairment of NK cell function in ALL [122]. More recent studies confirmed alterations in the phenotype and cytotoxicity of NK cells derived from ALL patients [118,121]. As compared to healthy donors, NK cells from pediatric ALL patients had lower levels of NKp46 activating receptors and higher levels of inhibitory NKG2A receptors [118]. Moreover, factors limiting NK cell activity (e.g., low expression of activating receptor NKp46) correlated with MRD positivity [123]. Rouce et al. observed that the dysfunction of NK cells from ALL patients results from the release of TGF1 by BCP-ALL cells, which leads to the activation of the TGF/SMAD pathway and in consequence to NK cell dysfunction [118].
6.3. NK Cells in Acute Lymphoblastic Leukemia Immunotherapy
NK cells also greatly affect the efficacy of ALL therapy. Patients’ NK cells are the major cytotoxic effector cells that mediate the therapeutic antibody-dependent cellular cytotoxicity (ADCC) effect. The NK cell receptor responsible for ADCC is CD16, which recognizes the Fc fragment of immunoglobulin G (FcγRIII). NK cells are particularly important for the efficacy of “naked” therapeutic monoclonal antibodies, such as rituximab. Bispecific antibodies triggering NK cells, named bispecific killer cell engagers (BiKE), with analogy to BiTE, were effective against primary ALL cells in preclinical tests [124]. It is worth emphasizing, however, that the effectiveness of such therapeutic monoclonal antibodies, both mono and multi-specific, is determined by the condition and cytotoxic capability of the patient’s NK cells, which is compromised. In this context, immunotherapy using NK cells derived from healthy donors may be a more effective therapeutic option.
Extensive work is underway to optimize in vitro NK cell expansion protocols. The activation of healthy donor-derived NK cells in vitro by interleukin 2 (IL-2), interleukin 12 (IL-12), interleukin 15 (IL-15), and interleukin 18 (IL-18) results in more effective lysis of ALL blasts [125,126,127]. Recently, Liu et al. established a novel platform for selective expansion of natural killer group 2 C (NKG2C)+ NK cells that express a single KIR receptor (KIR2DL3 or KIR2DL1) and specifically kill allogeneic ALL cells lacking the cognate HLA ligand [127]. To enhance the killing potential and increase cancer specific recognition, NK cells may be genetically modified to express CAR molecules. A CD19-specific CAR-modified NK cells reveal high cytotoxic activity against malignancies derived from mature B cells (chronic lymphocytic leukemia and non-Hodgkin lymphoma), without causing toxic effects [128]. CD19-CAR-NK cells also presented preliminary efficacy against BCP-ALL cell lines in in vitro assays [129]. NK cell-based immunotherapies tested against ALL are presented in Table 3.
Finally, NK cells also play a central role in HSCT. The HSCT from HLA-haploidentical donors has been claimed to benefit from the graft-vs-leukemia (GVL) effect, which is achieved by alloreactive NK cells from the donor [130,131,132]. The molecular basis for the NK cells’ alloreactivity is the recognition of specific ligands for NK cell activating receptors and lack of recognition by the inhibitory receptors (“missing-self” reactivity) [64]. Research from the last decade provides information how HLA and KIR genotypes of the donors shape NK cell repertoire and alloreactivity of NK cells [117]. Pende et al. presented that the activating receptor KIR2DS1, which recognizes HLA-C2 alleles on ALL cells, plays a major role in the lysis of leukemia cells by donor NK cells [130]. These results may guide optimal selection of HSCT donors for a more effective treatment of ALL.
7. T Cells
T cells possess a wide range of effector functions and play a substantial role in adaptive immune responses. Based on the expression of either CD4 or CD8 molecules, T cells are respectively classified as T helper (Th) or T cytotoxic cells (Tc). Physiological function of Tc cells is to eliminate host cells displaying foreign antigens. Several subsets of Th cells on the other hand, are responsible for the regulation of cell-mediated and humoral immune responses. A specific subset of CD4+ T cells expressing CD25 antigen and intracellular transcription factor Forkhead box P3 (FOXP3), known as regulatory T cells (Tregs), inhibits adaptive immune responses mainly through interaction with other subpopulations of T cells and antigen-presenting cells [133].
It is currently well established that T cells are capable of recognizing and eradicating tumor cells. Accordingly, exploring the landscape of tumor-specific and tumor-associated antigens and their implication for immunotherapies in ALL has been a goal of several preclinical studies [134,135,136,137]. Deeper insight into the occurrence of ALL antigens-specific CD8+ T cells was provided in a recent study by Zamora et al. [3]. Employing high-throughput approaches, the authors identified cancer specific antigens, which elicited T cells expansion and efficient responses to majority of tested neoantigens, including ETV6-RUNX1 fusion protein. Nevertheless, the sole occurrence and presentation of antigens with a proven immunogenic potential is insufficient to persistently activate T cells and provide immune responses that ensure tumor clearance. Two main mechanisms that attenuate the T cell-mediated antitumor immune response are well recognized: the expression of inhibitory checkpoint receptors on conventional T cells, and the accumulation and augmented suppressive function of Tregs. Moreover, in the case of T-ALL, a T cell-derived malignancy, even more challenges for potent T cell-mediated immune responses/immunotherapies are met [138]. This is partially due to the target antigens overlapping between leukemic and normal T cells, which can lead to various off-target effects.
7.1. Immune Checkpoints and Their Ligands
Among others, inhibitory receptors present on T cells include cytotoxic T cell antigen 4 (CTLA-4), programmed cell death protein 1 (PD-1), T cell immunoglobulin and mucin domain-containing protein 3 (TIM-3), and lymphocyte-activation gene 3 (LAG-3). The detailed characteristics of the various inhibitory pathways and their involvement in T cell exhaustion (defects in T cell effector functions) are extensively reviewed in [139,140,141,142]. Therefore, to ensure easier interpretation of results discussed in this review, we only briefly summarized several components of the immune checkpoint pathways, and their main mechanisms of action (Table 4). Interactions between inhibitory molecules on T cells and their respective ligands on the leukemic cells/antigen-presenting cells investigated so far in ALL patients and in ALL murine models, include CTLA-4/CD80 or CD86, PD-1/PD-L1, TIM-3/galectin-9, TIGIT/CD155, and CD200/CD200R.
Table 4.
Overview of the inhibitory immune checkpoint interactions.
CTLA-4 is the first immune checkpoint molecule described and the first clinical target for immune checkpoint inhibition [143,144]. Using multiplexed immunohistochemistry, Hohtari et al. [10] compared healthy and leukemic bone marrow derived from adult BCP-ALL patients and revealed high CTLA-4 expression in CD4+ and CD8+ T cells in patients’ bone marrow. Increased CTLA-4 expression was also confirmed in various subsets of peripheral T cells in pediatric BCP-ALL. Interestingly, the levels of this checkpoint molecule were more pronounced in high risk ALL subtypes and correlated with decreased relapse-free survival [7].
Other inhibitory interactions involving leukemic T cells, such as PD-1/PD-L1, TIM-3/galectin-9, TIGIT/CD155, and their effects on the immune anti-leukemic responses were recently described [3,5,158]. For instance, in a syngeneic murine BCP-ALL leukemia model, the PD-1 level was increased in CD4+ and CD8+ T cells, and, to some extent, ALL-induced PD-1 expression was independent of TCR activation [159]. Interestingly, PD-1 expression alone was not responsible for T cell exhaustion in this BCP-ALL murine model as the immune checkpoint inhibition had no impact on disease progression. In addition, the authors presented that introducing murine CD19 CARs into T cells isolated from leukemia-bearing mice did not fully improve their effector functions, indicating significant exhaustion of the leukemic T cells. In contrast to this finding, only small amounts of neoantigen-specific CD8+ T cells expressed PD-1 in human ALL [3]. Hence, it seems that despite ambiguous PD-1 expression, the PD-1/PD-L1 pathway has little impact on T cell responses in ALL, and that other inhibitory pathways may be involved in this process. Of note, Anand et al. [9] presented the involvement of TIM-3 and its ligand, galectin-9, in the inhibition of CD8+ T cell responses in early T-cell precursor ALL. Moreover, higher numbers of TIM-3+/PD-1+ CD4+ T cells predicted poor treatment outcome in adult BCP-ALL [10]. In line with this data, Blaeschke et al. [5] presented that TIM-3+ CD4+ T cells were enriched in BCP-ALL pediatric patients and were strongly associated with increased risk of relapse. Genetically modified, TIM-3 overexpressing T cells co-cultured in vitro with ALL cells possessed lower activation and proliferation rates than observed in wild-type T cells. Interestingly, the authors discovered that ALL-mediated induction of TIM-3 on T cells is mediated by CD200, yet another immune checkpoint molecule; however, the precise mechanism how CD200 augments TIM-3 expression needs further evaluation.
Inhibition of T cell responses elicited by interaction of CD200 with its cognate receptor, CD200R, was confirmed in various hematological cancers [160,161], showing potential for development of novel checkpoint inhibitors. Importantly, CD200 expression was widely detected in BCP-ALL; however, the expression pattern is variable and could be subtype specific, as shown in several studies [5,162,163]. Thus far, not much data on the CD200 levels are available for T-ALL.
7.2. Regulatory T Cells
Although the major function of regulatory T cells (Tregs) is maintaining self-tolerance [164], the accumulation and altered function of these cells has also been implicated in cancer progression [165]. In several tumor types, including hematological cancers, Tregs accumulation is associated with increased disease progression and suppression of anti-tumor responses [166,167,168,169]. The prevalence of the Tregs population in pediatric and adult ALL has been recently summarized by Niedzwiecki et al. [170]. In all the reviewed literature on ALL, the authors found increased frequency of Tregs in either peripheral blood or the BM of leukemic patients. Notably, rare studies provide a correlation of the Tregs number with disease progression or a detailed explanation of how they elicit potent suppressive mechanisms; thus, more functional studies in this context are of great need. Furthermore, in recent studies, an increased circulating Tregs frequency has been linked with diminished potency of selected immunotherapies against BCP-ALL, such as blinatumomab [6] or CAR-T cells [171]. A more detailed explanation of immunosuppression mediated by blinatumomab-stimulated Tregs in BCP-ALL involves not only IL-10 production but also direct cell-to-cell contacts with other T cell subtypes. Collectively, these results show that an elevated Tregs number is an important prognostic factor for poor response to some immunotherapies. Hence, strategies focused on Tregs ablation or selective inactivation as a component of combination therapies against ALL should be further exploited.
8. Conclusions
Studies conducted in the last decade revealed that ALL cells exploit various mechanisms to avoid immune recognition and destruction by the immune system. ALL cells escape from NK cell recognition and cytotoxicity by downregulating the ligands of the NK cell-activating receptors (MICA/B, ULBP1, and NEC-2) [119], resist phagocytosis by upregulating the CD47 “do not eat me signal” [105], and avoid a T cell response by triggering selected inhibitory checkpoints (TIM-3, CD200R) [5]. Moreover, accumulating evidence indicates that ALL-driven alterations in immune cells contribute to escape from immunosurveillance, leukemia progression, and thus affect treatment outcome. It has been recently demonstrated that the efficacy of chemo- and immunotherapies of ALL may be compromised by the accumulation of cells with immunosuppressive function, such as Tregs and non-classical monocytes [6,11,171]. However, more research is needed to assess the functional role of Tregs and to elucidate which properties of non-classical monocytes promote leukemic survival. Importantly, some strategies to target the leukemia-supporting interactions with the cells of the BM microenvironment already have been proposed in preclinical studies (Table 5).
Table 5.
Overcoming immune evasion and targeting leukemia-promoting interactions provided by the cells of the bone marrow niche—preclinical strategies.
The impairment of the numbers and function of effector immune cells (NK cells, T cells, and M1 macrophages) may diminish the efficacy of therapeutic modalities relying on the function of these effector cells. For example, functionally compromised and infrequent NK cells in ALL may negatively affect the outcome of treatment with therapeutic monoclonal antibodies. On the other hand, thanks to carefully optimized protocols of in vitro expansion and activation, healthy donor-derived NK cells have already demonstrated preclinical efficacy [125,126,127] and may pave the way for novel cell-based immunotherapies. However, these NK cell-based adoptive therapies must be further tested for their clinical efficacy.
Although some recent studies showed the occurrence of ALL neoantigen-specific T cells [3], further research is needed to elucidate if their numbers and activity is sufficient to employ strategies aimed at restoring T cell function with immune checkpoint blockade. Nevertheless, some clinical trials testing the efficacy of CTLA-4 and PD-1-mediated blockade have already been launched. Furthermore, the mechanisms behind T cell exhaustion in ALL are not yet fully understood. Preclinical studies indicate that TIM-3 rather than PD-1 or CTLA-4 is the major inhibitory checkpoint in ALL patients’ T cells [5,10]. Further studies are needed to show if TIM-3 expression affects the efficacy of ALL therapies and if TIM-3 can be a novel, useful immunotherapy target in ALL. These studies explaining the mechanisms of ALL T cells’ dysfunction may also contribute to improvements in CAR-based adoptive immunotherapy.
Author Contributions
Conceptualization, M.F. and A.P.; writing—original draft preparation, A.P., K.D., M.P., K.F. and M.F.; writing—review and editing, A.P., K.D., M.P., K.F. and M.F.; visualization, A.P. and K.D.; supervision, M.F.; funding acquisition, M.F. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Polish National Science Centre grant 2019/35/B/NZ5/01428 (M.F.) and by the Ministry of Education and Science within “Regional Initiative of Excellence” program 013/RID/2018/19, project budget 12 000 000 PLN.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Roberts, K.G.; Mullighan, C.G. The Biology of B-Progenitor Acute Lymphoblastic Leukemia. Cold Spring Harb. Perspect. Med. 2020, 10. [Google Scholar] [CrossRef]
- Malard, F.; Mohty, M. Acute lymphoblastic leukaemia. Lancet 2020, 395, 1146–1162. [Google Scholar] [CrossRef]
- Zamora, A.E.; Crawford, J.C.; Allen, E.K.; Guo, X.-z.J.; Bakke, J.; Carter, R.A.; Abdelsamed, H.A.; Moustaki, A.; Li, Y.; Chang, T.-C.; et al. Pediatric patients with acute lymphoblastic leukemia generate abundant and functional neoantigen-specific CD8+ T cell responses. Sci. Transl. Med. 2019, 11, eaat8549. [Google Scholar] [CrossRef] [PubMed]
- Babor, F.; Manser, A.R.; Fischer, J.C.; Scherenschlich, N.; Enczmann, J.; Chazara, O.; Moffett, A.; Borkhardt, A.; Meisel, R.; Uhrberg, M. KIR ligand C2 is associated with increased susceptibility to childhood ALL and confers an elevated risk for late relapse. Blood 2014, 124, 2248–2251. [Google Scholar] [CrossRef] [PubMed]
- Blaeschke, F.; Willier, S.; Stenger, D.; Lepenies, M.; Horstmann, M.A.; Escherich, G.; Zimmermann, M.; Rojas Ringeling, F.; Canzar, S.; Kaeuferle, T.; et al. Leukemia-induced dysfunctional TIM-3+CD4+ bone marrow T cells increase risk of relapse in pediatric B-precursor ALL patients. Leukemia 2020, 34, 2607–2620. [Google Scholar] [CrossRef] [PubMed]
- Duell, J.; Dittrich, M.; Bedke, T.; Mueller, T.; Eisele, F.; Rosenwald, A.; Rasche, L.; Hartmann, E.; Dandekar, T.; Einsele, H.; et al. Frequency of regulatory T cells determines the outcome of the T-cell-engaging antibody blinatumomab in patients with B-precursor ALL. Leukemia 2017, 31, 2181–2190. [Google Scholar] [CrossRef]
- Kang, S.H.; Hwang, H.J.; Yoo, J.W.; Kim, H.; Choi, E.S.; Hwang, S.-H.; Cho, Y.-U.; Jang, S.; Park, C.-J.; Im, H.J.; et al. Expression of Immune Checkpoint Receptors on T-Cells and Their Ligands on Leukemia Blasts in Childhood Acute Leukemia. Anticancer Res. 2019, 39, 5531–5539. [Google Scholar] [CrossRef]
- Brouwer, R.E.; van der Heiden, P.; Schreuder, G.M.T.; Mulder, A.; Datema, G.; Anholts, J.D.H.; Willemze, R.; Claas, F.H.J.; Falkenburg, J.H.F. Loss or downregulation of HLA class I expression at the allelic level in acute leukemia is infrequent but functionally relevant, and can be restored by interferon. Hum. Immunol. 2002, 63, 200–210. [Google Scholar] [CrossRef]
- Anand, P.; Guillaumet-Adkins, A.; Dimitrova, V.; Yun, H.; Drier, Y.; Sotudeh, N.; Rogers, A.J.; Ouseph, M.M.; Nair, M.; Potdar, S.; et al. Single cell RNA-seq reveals developmental plasticity with coexisting oncogenic and immune evasion programs in ETP-ALL. Blood 2020. [Google Scholar] [CrossRef]
- Hohtari, H.; Brück, O.; Blom, S.; Turkki, R.; Sinisalo, M.; Kovanen, P.E.; Kallioniemi, O.; Pellinen, T.; Porkka, K.; Mustjoki, S. Immune cell constitution in bone marrow microenvironment predicts outcome in adult ALL. Leukemia 2019, 33, 1570–1582. [Google Scholar] [CrossRef]
- Witkowski, M.T.; Dolgalev, I.; Evensen, N.A.; Ma, C.; Chambers, T.; Roberts, K.G.; Sreeram, S.; Dai, Y.; Tikhonova, A.N.; Lasry, A.; et al. Extensive Remodeling of the Immune Microenvironment in B Cell Acute Lymphoblastic Leukemia. Cancer Cell 2020, 37, 867–882.e12. [Google Scholar] [CrossRef]
- Dworzak, M.N.; Buldini, B.; Gaipa, G.; Ratei, R.; Hrusak, O.; Luria, D.; Rosenthal, E.; Bourquin, J.-P.; Sartor, M.; Schumich, A.; et al. AIEOP-BFM Consensus Guidelines 2016 for Flow Cytometric Immunophenotyping of Pediatric Acute Lymphoblastic Leukemia. Cytom. Part B Clin. Cytom. 2018, 94, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Pui, C.-H.; Roberts, K.G.; Yang, J.J.; Mullighan, C.G. Philadelphia Chromosome like Acute Lymphoblastic Leukemia. Clin. Lymphoma Myeloma Leuk. 2017, 17, 464–470. [Google Scholar] [CrossRef]
- Miroslav, D.; Elisabet, B.; Elisabeth, B.; Joanna, M.; Stefan, S.; Anna, P. Overexpression of CD123 correlates with the hyperdiploid genotype in acute lymphoblastic leukemia. Haematologica 2009, 94, 1016–1019. [Google Scholar] [CrossRef]
- Kalina, T.; Vaskova, M.; Mejstrikova, E.; Madzo, J.; Trka, J.; Stary, J.; Hrusak, O. Myeloid antigens in childhood lymphoblastic leukemia: Clinical data point to regulation of CD66c distinct from other myeloid antigens. BMC Cancer 2005, 5, 38. [Google Scholar] [CrossRef] [PubMed]
- De Zen, L.; Orfao, A.; Cazzaniga, G.; Masiero, L.; Cocito, M.G.; Spinelli, M.; Rivolta, A.; Biondi, A.; Zanesco, L.; Basso, G. Quantitative multiparametric immunophenotyping in acute lymphoblastic leukemia: Correlation with specific genotype. I. ETV6/AML1 ALLs identification. Leukemia 2000, 14, 1225–1231. [Google Scholar] [CrossRef][Green Version]
- Tsagarakis, N.J.; Papadhimitriou, S.I.; Pavlidis, D.; Marinakis, T.; Kostopoulos, I.V.; Stiakaki, E.; Polychronopoulou, S.; Paterakis, G. Flow cytometric predictive scoring systems for common fusions ETV6/RUNX1, BCR/ABL1, TCF3/PBX1 and rearrangements of the KMT2A gene, proposed for the initial cytogenetic approach in cases of B-acute lymphoblastic leukemia. Int. J. Lab. Hematol. 2019, 41, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Boer, J.M.; Valsecchi, M.G.; Hormann, F.M.; Antic, Z.; Zaliova, M.; Schwab, C.; Cazzaniga, G.; Arfeuille, C.; Cavé, H.; Attarbaschi, A.; et al. NUTM1-Rearranged Infant and Pediatric B Cell Precursor Acute Lymphoblastic Leukemia: A Good Prognostic Subtype Identified in a Collaborative International Study. Blood 2020, 136, 25–26. [Google Scholar] [CrossRef]
- Shinsuke, H.; Kentaro, O.; Kazuhiko, N.; Hitoshi, I.; Yukihide, M.; Kohji, O.; Akinori, Y.; Kazuki, T.; Yuya, S.; Ai, Y.; et al. ZNF384-related fusion genes define a subgroup of childhood B-cell precursor acute lymphoblastic leukemia with a characteristic immunotype. Haematologica 2017, 102, 118–129. [Google Scholar] [CrossRef]
- Zangrando, A.; Intini, F.; te Kronnie, G.; Basso, G. Validation of NG2 antigen in identifying BP-ALL patients with MLL rearrangements using qualitative and quantitative flow cytometry: A prospective study. Leukemia 2008, 22, 858–861. [Google Scholar] [CrossRef]
- Bugarin, C.; Sarno, J.; Palmi, C.; Savino, A.M.; te Kronnie, G.; Dworzak, M.; Shumich, A.; Buldini, B.; Maglia, O.; Sala, S.; et al. Fine tuning of surface CRLF2 expression and its associated signaling profile in childhood B-cell precursor acute lymphoblastic leukemia. Haematologica 2015, 100, e229–e232. [Google Scholar] [CrossRef]
- Attarbaschi, A.; Mann, G.; Schumich, A.; König, M.; Pickl, W.F.; Haas, O.A.; Gadner, H.; Dworzak, M.N.; on behalf of the Austrian Berlin–Frankfurt–Münster Cooperative Study Group. CD44 deficiency is a consistent finding in childhood Burkitt’s lymphoma and leukemia. Leukemia 2007, 21, 1110–1113. [Google Scholar] [CrossRef][Green Version]
- Gu, Z.; Churchman, M.; Roberts, K.; Li, Y.; Liu, Y.; Harvey, R.C.; McCastlain, K.; Reshmi, S.C.; Payne-Turner, D.; Iacobucci, I.; et al. Genomic analyses identify recurrent MEF2D fusions in acute lymphoblastic leukaemia. Nat. Commun. 2016, 7, 13331. [Google Scholar] [CrossRef] [PubMed]
- Szczepański, T.; van der Velden, V.H.J.; van Dongen, J.J.M. Classification systems for acute and chronic leukaemias. Best Pract. Res. Clin. Haematol. 2003, 16, 561–582. [Google Scholar] [CrossRef]
- Jain, N.; Lamb, A.V.; O’Brien, S.; Ravandi, F.; Konopleva, M.; Jabbour, E.; Zuo, Z.; Jorgensen, J.; Lin, P.; Pierce, S.; et al. Early T-cell precursor acute lymphoblastic leukemia/lymphoma (ETP-ALL/LBL) in adolescents and adults: A high-risk subtype. Blood 2016, 127, 1863–1869. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Wang, X.; Tang, J.; Xue, H.; Chen, J.; Pan, C.; Jiang, H.; Shen, S. Early T-cell precursor leukemia: A subtype of high risk childhood acute lymphoblastic leukemia. Front. Med. 2012, 6, 416–420. [Google Scholar] [CrossRef]
- Raetz, E.A.; Teachey, D.T. T-cell acute lymphoblastic leukemia. Hematology 2016, 2016, 580–588. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Easton, J.; Shao, Y.; Maciaszek, J.; Wang, Z.; Wilkinson, M.R.; McCastlain, K.; Edmonson, M.; Pounds, S.B.; Shi, L.; et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat. Genet. 2017, 49, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- Stock, W.; Luger, S.M.; Advani, A.S.; Yin, J.; Harvey, R.C.; Mullighan, C.G.; Willman, C.L.; Fulton, N.; Laumann, K.M.; Malnassy, G.; et al. A pediatric regimen for older adolescents and young adults with acute lymphoblastic leukemia: Results of CALGB 10403. Blood 2019, 133, 1548–1559. [Google Scholar] [CrossRef] [PubMed]
- Jacoby, E. The role of allogeneic HSCT after CAR T cells for acute lymphoblastic leukemia. Bone Marrow Transplant. 2019, 54, 810–814. [Google Scholar] [CrossRef] [PubMed]
- Giebel, S.; Marks, D.I.; Boissel, N.; Baron, F.; Chiaretti, S.; Ciceri, F.; Cornelissen, J.J.; Doubek, M.; Esteve, J.; Fielding, A.; et al. Hematopoietic stem cell transplantation for adults with Philadelphia chromosome-negative acute lymphoblastic leukemia in first remission: A position statement of the European Working Group for Adult Acute Lymphoblastic Leukemia (EWALL) and the Acute Leukemia Working Party of the European Society for Blood and Marrow Transplantation (EBMT). Bone Marrow Transplant. 2019, 54, 798–809. [Google Scholar] [CrossRef]
- Merli, P.; Algeri, M.; Del Bufalo, F.; Locatelli, F. Hematopoietic Stem Cell Transplantation in Pediatric Acute Lymphoblastic Leukemia. Curr. Hematol. Malig. Rep. 2019, 14, 94–105. [Google Scholar] [CrossRef]
- Wei, G.; Wang, J.; Huang, H.; Zhao, Y. Novel immunotherapies for adult patients with B-lineage acute lymphoblastic leukemia. J. Hematol. Oncol. 2017, 10, 150. [Google Scholar] [CrossRef]
- Jabbour, E.; Kantarjian, H. Immunotherapy in adult acute lymphoblastic leukemia: The role of monoclonal antibodies. Blood Adv. 2016, 1, 260–264. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ronson, A.; Tvito, A.; Rowe, J.M. Treatment of Relapsed/Refractory Acute Lymphoblastic Leukemia in Adults. Curr. Oncol. Rep. 2016, 18, 39. [Google Scholar] [CrossRef] [PubMed]
- Goebeler, M.-E.; Bargou, R.C. T cell-engaging therapies—BiTEs and beyond. Nat. Rev. Clin. Oncol. 2020, 17, 418–434. [Google Scholar] [CrossRef] [PubMed]
- Roselli, E.; Faramand, R.; Davila, M.L. Insight into next-generation CAR therapeutics: Designing CAR T cells to improve clinical outcomes. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef]
- Park, J.H.; Rivière, I.; Gonen, M.; Wang, X.; Sénéchal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef]
- Saadeh, S.S.; Litzow, M.R. Hematopoietic stem cell transplant in adults with acute lymphoblastic leukemia: The present state. Expert Rev. Hematol. 2018, 11, 195–207. [Google Scholar] [CrossRef]
- Kantarjian, H.; Stein, A.; Gökbuget, N.; Fielding, A.K.; Schuh, A.C.; Ribera, J.-M.; Wei, A.; Dombret, H.; Foà, R.; Bassan, R.; et al. Blinatumomab versus Chemotherapy for Advanced Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2017, 376, 836–847. [Google Scholar] [CrossRef] [PubMed]
- Maury, S.; Chevret, S.; Thomas, X.; Heim, D.; Leguay, T.; Huguet, F.; Chevallier, P.; Hunault, M.; Boissel, N.; Escoffre-Barbe, M.; et al. Rituximab in B-Lineage Adult Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2016, 375, 1044–1053. [Google Scholar] [CrossRef] [PubMed]
- Castillo, J.; Milani, C.; Mendez-Allwood, D. Ofatumumab, a second-generation anti-CD20 monoclonal antibody, for the treatment of lymphoproliferative and autoimmune disorders. Expert Opin. Investig. Drugs 2009, 18, 491–500. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; DeAngelo, D.J.; Stelljes, M.; Martinelli, G.; Liedtke, M.; Stock, W.; Gökbuget, N.; O’Brien, S.; Wang, K.; Wang, T.; et al. Inotuzumab Ozogamicin versus Standard Therapy for Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2016, 375, 740–753. [Google Scholar] [CrossRef]
- Mueller, K.T.; Waldron, E.; Grupp, S.A.; Levine, J.E.; Laetsch, T.W.; Pulsipher, M.A.; Boyer, M.W.; August, K.J.; Hamilton, J.; Awasthi, R.; et al. Clinical Pharmacology of Tisagenlecleucel in B-cell Acute Lymphoblastic Leukemia. Clin. Cancer Res. 2018, 24, 6175–6184. [Google Scholar] [CrossRef] [PubMed]
- Advani, A.S.; McDonough, S.; Coutre, S.; Wood, B.; Radich, J.; Mims, M.; O’Donnell, M.; Elkins, S.; Becker, M.; Othus, M.; et al. SWOG S0910: A phase 2 trial of clofarabine/cytarabine/epratuzumab for relapsed/refractory acute lymphocytic leukaemia. Br. J. Haematol. 2014, 165, 504–509. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Turner, M.J.; Shields, J.; Gale, M.S.; Hutto, E.; Roberts, B.L.; Siders, W.M.; Kaplan, J.M. Investigation of the mechanism of action of alemtuzumab in a human CD52 transgenic mouse model. Immunology 2009, 128, 260–270. [Google Scholar] [CrossRef]
- Stock, W.; Sanford, B.; Lozanski, G.; Vij, R.; Byrd, J.C.; Powell, B.L.; Wetzler, M.; Sher, D.; Edwards, C.; Kelly, M.; et al. Alemtuzumab can be Incorporated Into Front-Line Therapy of Adult Acute Lymphoblastic Leukemia (ALL): Final Phase I Results of a Cancer and Leukemia Group B Study (CALGB 10102). Blood 2009, 114, 838. [Google Scholar] [CrossRef]
- Fathi, A.T.; Borate, U.; DeAngelo, D.J.; O’Brien, M.M.; Trippett, T.; Shah, B.D.; Hale, G.A.; Foran, J.M.; Silverman, L.B.; Tibes, R.; et al. A Phase 1 Study of Denintuzumab Mafodotin (SGN-CD19A) in Adults with Relapsed or Refractory B-Lineage Acute Leukemia (B-ALL) and Highly Aggressive Lymphoma. Blood 2015, 126, 1328. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; Lioure, B.; Kim, S.K.; Atallah, E.; Leguay, T.; Kelly, K.; Marolleau, J.-P.; Escoffre-Barbe, M.; Thomas, X.G.; Cortes, J.; et al. A Phase II Study of Coltuximab Ravtansine (SAR3419) Monotherapy in Patients with Relapsed or Refractory Acute Lymphoblastic Leukemia. Clin. Lymphoma Myeloma Leuk. 2016, 16, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.R.; Lee, Y.S.; Yang, S.H.; Ahn, K.H.; Lee, J.-H.; Lee, J.-H.; Kim, D.Y.; Kang, Y.A.; Jeon, M.; Seol, M.; et al. Generation of donor natural killer cells from CD34+ progenitor cells and subsequent infusion after HLA-mismatched allogeneic hematopoietic cell transplantation: A feasibility study. Bone Marrow Transplant. 2010, 45, 1038–1046. [Google Scholar] [CrossRef]
- Ofran, Y.; Ringelstein-Harlev, S.; Slouzkey, I.; Zuckerman, T.; Yehudai-Ofir, D.; Henig, I.; Beyar-Katz, O.; Hayun, M.; Frisch, A. Daratumumab for eradication of minimal residual disease in high-risk advanced relapse of T-cell/CD19/CD22-negative acute lymphoblastic leukemia. Leukemia 2020, 34, 293–295. [Google Scholar] [CrossRef]
- Trinquand, A.; dos Santos, N.R.; Tran Quang, C.; Rocchetti, F.; Zaniboni, B.; Belhocine, M.; Da Costa de Jesus, C.; Lhermitte, L.; Tesio, M.; Dussiot, M.; et al. Triggering the TCR Developmental Checkpoint Activates a Therapeutically Targetable Tumor Suppressive Pathway in T-cell Leukemia. Cancer Discov. 2016, 6, 972–985. [Google Scholar] [CrossRef] [PubMed]
- Gardner, R.; Wu, D.; Cherian, S.; Fang, M.; Hanafi, L.-A.; Finney, O.; Smithers, H.; Jensen, M.C.; Riddell, S.R.; Maloney, D.G.; et al. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood 2016, 127, 2406–2410. [Google Scholar] [CrossRef] [PubMed]
- Landau, D.A.; Carter, S.L.; Getz, G.; Wu, C.J. Clonal evolution in hematological malignancies and therapeutic implications. Leukemia 2014, 28, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Biswas, D.; Swanton, C. Impact of cancer evolution on immune surveillance and checkpoint inhibitor response. Semin. Cancer Biol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Jacoby, E.; Nguyen, S.M.; Fountaine, T.J.; Welp, K.; Gryder, B.; Qin, H.; Yang, Y.; Chien, C.D.; Seif, A.E.; Lei, H.; et al. CD19 CAR immune pressure induces B-precursor acute lymphoblastic leukaemia lineage switch exposing inherent leukaemic plasticity. Nat. Commun. 2016, 7, 12320. [Google Scholar] [CrossRef]
- Mercier, F.E.; Ragu, C.; Scadden, D.T. The bone marrow at the crossroads of blood and immunity. Nat. Rev. Immunol. 2012, 12, 49–60. [Google Scholar] [CrossRef]
- Karantanou, C.; Godavarthy, P.S.; Krause, D.S. Targeting the bone marrow microenvironment in acute leukemia. Leuk. Lymphoma 2018, 59, 2535–2545. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Godavarthy, P.S.; Krause, D.S. The bone marrow microenvironment in health and disease at a glance. J. Cell Sci. 2018, 131, jcs201707. [Google Scholar] [CrossRef]
- Chiarini, F.; Lonetti, A.; Evangelisti, C.; Buontempo, F.; Orsini, E.; Evangelisti, C.; Cappellini, A.; Neri, L.M.; McCubrey, J.A.; Martelli, A.M. Advances in understanding the acute lymphoblastic leukemia bone marrow microenvironment: From biology to therapeutic targeting. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2016, 1863, 449–463. [Google Scholar] [CrossRef]
- Blau, O.; Baldus, C.D.; Hofmann, W.-K.; Thiel, G.; Nolte, F.; Burmeister, T.; Türkmen, S.; Benlasfer, O.; Schümann, E.; Sindram, A.; et al. Mesenchymal stromal cells of myelodysplastic syndrome and acute myeloid leukemia patients have distinct genetic abnormalities compared with leukemic blasts. Blood 2011, 118, 5583–5592. [Google Scholar] [CrossRef] [PubMed]
- Park, M.; Park, C.-J.; Cho, Y.W.; Jang, S.; Lee, J.-H.; Lee, J.-H.; Lee, K.-H.; Lee, Y.H. Alterations in the bone marrow microenvironment may elicit defective hematopoiesis: A comparison of aplastic anemia, chronic myeloid leukemia, and normal bone marrow. Exp. Hematol. 2017, 45, 56–63. [Google Scholar] [CrossRef]
- Bhagat, T.D.; Chen, S.; Bartenstein, M.; Barlowe, A.T.; Von Ahrens, D.; Choudhary, G.S.; Tivnan, P.; Amin, E.; Marcondes, A.M.; Sanders, M.A.; et al. Epigenetically Aberrant Stroma in MDS Propagates Disease via Wnt/β-Catenin Activation. Cancer Res. 2017, 77, 4846–4857. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.H.; Qu, C.-K.; Pauly, M. Germline mutations in the bone marrow microenvironment and dysregulated hematopoiesis. Exp. Hematol. 2018, 66, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Verma, D.; Zanetti, C.; Godavarthy, P.S.; Kumar, R.; Minciacchi, V.R.; Pfeiffer, J.; Metzler, M.; Lefort, S.; Maguer-Satta, V.; Nicolini, F.E.; et al. Bone marrow niche-derived extracellular matrix-degrading enzymes influence the progression of B-cell acute lymphoblastic leukemia. Leukemia 2020, 34, 1540–1552. [Google Scholar] [CrossRef]
- Crazzolara, R.; Kreczy, A.; Mann, G.; Heitger, A.; Eibl, G.; Fink, F.-M.; Möhle, R.; Meister, B. High expression of the chemokine receptor CXCR4 predicts extramedullary organ infiltration in childhood acute lymphoblastic leukaemia. Br. J. Haematol. 2001, 115, 545–553. [Google Scholar] [CrossRef]
- Juarez, J.; Dela Pena, A.; Baraz, R.; Hewson, J.; Khoo, M.; Cisterne, A.; Fricker, S.; Fujii, N.; Bradstock, K.F.; Bendall, L.J. CXCR4 antagonists mobilize childhood acute lymphoblastic leukemia cells into the peripheral blood and inhibit engraftment. Leukemia 2007, 21, 1249–1257. [Google Scholar] [CrossRef]
- Cooper, T.M.; Sison, E.A.R.; Baker, S.D.; Li, L.; Ahmed, A.; Trippett, T.; Gore, L.; Macy, M.E.; Narendran, A.; August, K.; et al. A phase 1 study of the CXCR4 antagonist plerixafor in combination with high-dose cytarabine and etoposide in children with relapsed or refractory acute leukemias or myelodysplastic syndrome: A Pediatric Oncology Experimental Therapeutics Investigators’ Consortium study (POE 10-03). Pediatr. Blood Cancer 2017, 64, e26414. [Google Scholar] [CrossRef]
- Churchman, M.L.; Low, J.; Qu, C.; Paietta, E.M.; Kasper, L.H.; Chang, Y.; Payne-Turner, D.; Althoff, M.J.; Song, G.; Chen, S.-C.; et al. Efficacy of Retinoids in IKZF1-Mutated BCR-ABL1 Acute Lymphoblastic Leukemia. Cancer Cell 2015, 28, 343–356. [Google Scholar] [CrossRef] [PubMed]
- Pal, D.; Blair, H.J.; Elder, A.; Dormon, K.; Rennie, K.J.; Coleman, D.J.L.; Weiland, J.; Rankin, K.S.; Filby, A.; Heidenreich, O.; et al. Long-term in vitro maintenance of clonal abundance and leukaemia-initiating potential in acute lymphoblastic leukaemia. Leukemia 2016, 30, 1691–1700. [Google Scholar] [CrossRef]
- Fidyt, K.; Pastorczak, A.; Goral, A.; Szczygiel, K.; Fendler, W.; Muchowicz, A.; Bartlomiejczyk, M.A.; Madzio, J.; Cyran, J.; Graczyk-Jarzynka, A.; et al. Targeting the thioredoxin system as a novel strategy against B-cell acute lymphoblastic leukemia. Mol. Oncol. 2019, 13, 1180–1195. [Google Scholar] [CrossRef]
- Julius, J.; Rana, B.; Shivashni, G.; Kenneth, B.; Linda, B. Interaction of interleukin-7 and interleukin-3 with the CXCL12-induced proliferation of B-cell progenitor acute lymphoblastic leukemia. Haematologica 2007, 92, 450–459. [Google Scholar] [CrossRef]
- Maria, T.S.; Omar, P.; Mauro, K.; Fabrizio, V.; Federica, C.; Giovanni, P. Interleukin 7 requirement for survival of T-cell acute lymphoblastic leukemia and human thymocytes on bone marrow stroma. Haematologica 2007, 92, 264–266. [Google Scholar] [CrossRef]
- Almeida, R.d.S.; Ramos, A.M.d.L.; Luna, C.F.; Pedrosa, F.; Donadi, E.A.; Lucena-Silva, N. Cytokines and soluble HLA-G levels in bone marrow stroma and their association with the survival rate of patients exhibiting childhood T-cell acute lymphoblastic leukemia. Cytokine 2018, 102, 94–101. [Google Scholar] [CrossRef]
- Polak, R.; de Rooij, B.; Pieters, R.; den Boer, M.L. B-cell precursor acute lymphoblastic leukemia cells use tunneling nanotubes to orchestrate their microenvironment. Blood 2015, 126, 2404–2414. [Google Scholar] [CrossRef]
- Yu, K.; Wang, J.; Lu, T.; Ma, D.; Wei, D.; Guo, Y.; Cheng, B.; Wang, W.; Fang, Q. Overexpression of heme oxygenase-1 in microenvironment mediates vincristine resistance of B-cell acute lymphoblastic leukemia by promoting vascular endothelial growth factor secretion. J. Cell. Biochem. 2019, 120, 17791–17810. [Google Scholar] [CrossRef]
- Fei, F.; Joo, E.J.; Tarighat, S.S.; Schiffer, I.; Paz, H.; Fabbri, M.; Abdel-Azim, H.; Groffen, J.; Heisterkamp, N. B-cell precursor acute lymphoblastic leukemia and stromal cells communicate through Galectin-3. Oncotarget 2015, 6, 11378. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Zhao, X.; Deng, M.; Huang, Z.; Wang, J.; Wu, Y.; Cui, D.; Liu, Y.; Liu, R.; Ouyang, G. Bone Marrow Mesenchymal Stromal Cell-Derived Periostin Promotes B-ALL Progression by Modulating CCL2 in Leukemia Cells. Cell Rep. 2019, 26, 1533–1543.e34. [Google Scholar] [CrossRef]
- Boyerinas, B.; Zafrir, M.; Yesilkanal, A.E.; Price, T.T.; Hyjek, E.M.; Sipkins, D.A. Adhesion to osteopontin in the bone marrow niche regulates lymphoblastic leukemia cell dormancy. Blood 2013, 121, 4821–4831. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, S.; Mihara, K.; Downing, J.R.; Pui, C.-H.; Campana, D. Mesenchymal cells regulate the response of acute lymphoblastic leukemia cells to asparaginase. J. Clin. Investig. 2007, 117, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Lambert, C.; Preijers, F.W.M.B.; Yanikkaya Demirel, G.; Sack, U. Monocytes and macrophages in flow: An ESCCA initiative on advanced analyses of monocyte lineage using flow cytometry. Cytom. Part B Clin. Cytom. 2017, 92, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Veglia, F.; Sanseviero, E.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev. Immunol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Collin, M.; Bigley, V. Human dendritic cell subsets: An update. Immunology 2018, 154, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, J.; Fišer, K.; Liebetrau, C.; Staubach, N.; Kost, D.; Voss, S.; Heiden, A.z.; Dörr, O.; Lipps, C.; Nef, H.M.; et al. High-Content Immunophenotyping and Hierarchical Clustering Reveal Sources of Heterogeneity and New Surface Markers of Human Blood Monocyte Subsets. Thromb. Haemost. 2020, 120, 141–155. [Google Scholar] [CrossRef]
- Yáñez, A.; Coetzee, S.G.; Olsson, A.; Muench, D.E.; Berman, B.P.; Hazelett, D.J.; Salomonis, N.; Grimes, H.L.; Goodridge, H.S. Granulocyte-Monocyte Progenitors and Monocyte-Dendritic Cell Progenitors Independently Produce Functionally Distinct Monocytes. Immunity 2017, 47, 890–902.e4. [Google Scholar] [CrossRef]
- De Veirman, K.; Van Valckenborgh, E.; Lahmar, Q.; Geeraerts, X.; De Bruyne, E.; Menu, E.; Van Riet, I.; Vanderkerken, K.; Van Ginderachter, J.A. Myeloid-Derived Suppressor Cells as Therapeutic Target in Hematological Malignancies. Front. Oncol. 2014, 4. [Google Scholar] [CrossRef]
- Law, A.M.K.; Valdes-Mora, F.; Gallego-Ortega, D. Myeloid-Derived Suppressor Cells as a Therapeutic Target for Cancer. Cells 2020, 9, 561. [Google Scholar] [CrossRef] [PubMed]
- Ribechini, E.; Greifenberg, V.; Sandwick, S.; Lutz, M.B. Subsets, expansion and activation of myeloid-derived suppressor cells. Med. Microbiol. Immunol. 2010, 199, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Bronte, V.; Brandau, S.; Chen, S.-H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7, 12150. [Google Scholar] [CrossRef]
- Ziegler-Heitbrock, L.; Ancuta, P.; Crowe, S.; Dalod, M.; Grau, V.; Hart, D.N.; Leenen, P.J.M.; Liu, Y.-J.; MacPherson, G.; Randolph, G.J.; et al. Nomenclature of monocytes and dendritic cells in blood. Blood 2010, 116, e74–e80. [Google Scholar] [CrossRef] [PubMed]
- Olingy, C.E.; San Emeterio, C.L.; Ogle, M.E.; Krieger, J.R.; Bruce, A.C.; Pfau, D.D.; Jordan, B.T.; Peirce, S.M.; Botchwey, E.A. Non-classical monocytes are biased progenitors of wound healing macrophages during soft tissue injury. Sci. Rep. 2017, 7, 447. [Google Scholar] [CrossRef] [PubMed]
- Davies, L.C.; Taylor, P.R. Tissue-resident macrophages: Then and now. Immunology 2015, 144, 541–548. [Google Scholar] [CrossRef]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef]
- DeNardo, D.G.; Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 2019, 19, 369–382. [Google Scholar] [CrossRef]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-Y.; Yang, X.; Feng, W.-L.; Liao, J.-F.; Wang, L.-N.; Feng, L.; Lin, Y.-M.; Ren, Q.; Zheng, G.-G. Organ-Specific Microenvironment Modifies Diverse Functional and Phenotypic Characteristics of Leukemia-Associated Macrophages in Mouse T Cell Acute Lymphoblastic Leukemia. J. Immunol. 2015, 194, 2919–2929. [Google Scholar] [CrossRef] [PubMed]
- Patente, T.A.; Pinho, M.P.; Oliveira, A.A.; Evangelista, G.C.M.; Bergami-Santos, P.C.; Barbuto, J.A.M. Human Dendritic Cells: Their Heterogeneity and Clinical Application Potential in Cancer Immunotherapy. Front. Immunol. 2019, 9. [Google Scholar] [CrossRef]
- Salem, M.L.; El-Shanshory, M.R.; Abdou, S.H.; Attia, M.S.; Sobhy, S.M.; Zidan, M.F.; Zidan, A.-A.A. Chemotherapy alters the increased numbers of myeloid-derived suppressor and regulatory T cells in children with acute lymphoblastic leukemia. Immunopharmacol. Immunotoxicol. 2018, 40, 158–167. [Google Scholar] [CrossRef]
- Ding, Z.-C.; Munn, D.H.; Zhou, G. Chemotherapy-induced myeloid suppressor cells and antitumor immunity: The Janus face of chemotherapy in immunomodulation. OncoImmunology 2014, 3, e954471. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-F.; Chen, Y.-Y.; He, Y.-Y.; Wang, J.-Y.; Yang, J.-P.; Zhong, S.-L.; Jiang, N.; Zhou, P.; Jiang, H.; Zhou, J. Expansion and activation of granulocytic, myeloid-derived suppressor cells in childhood precursor B cell acute lymphoblastic leukemia. J. Leukoc. Biol. 2017, 102, 449–458. [Google Scholar] [CrossRef]
- Song, J.-X.; Wen, Y.; Li, R.-W.; Dong, T.; Tang, Y.-F.; Zhang, J.-J.; Sa, Y.-L. Phenotypic characterization of macrophages in the BMB sample of human acute leukemia. Ann. Hematol. 2020, 99, 539–547. [Google Scholar] [CrossRef]
- Valencia, J.; M. Fernández-Sevilla, L.; Fraile-Ramos, A.; Sacedón, R.; Jiménez, E.; Vicente, A.; Varas, A. Acute Lymphoblastic Leukaemia Cells Impair Dendritic Cell and Macrophage Differentiation: Role of BMP4. Cells 2019, 8, 722. [Google Scholar] [CrossRef]
- Lyu, A.; Triplett, T.A.; Nam, S.H.; Hu, Z.; Arasappan, D.; Godfrey, W.H.; Ames, R.Y.; Sarang, A.; Selden, H.J.; Lee, C.-H.; et al. Tumor-associated myeloid cells provide critical support for T-ALL. Blood 2020, 136, 1837–1850. [Google Scholar] [CrossRef] [PubMed]
- Chao, M.P.; Alizadeh, A.A.; Tang, C.; Jan, M.; Weissman-Tsukamoto, R.; Zhao, F.; Park, C.Y.; Weissman, I.L.; Majeti, R. Therapeutic Antibody Targeting of CD47 Eliminates Human Acute Lymphoblastic Leukemia. Cancer Res. 2011, 71, 1374–1384. [Google Scholar] [CrossRef] [PubMed]
- Escobar, G.; Barbarossa, L.; Barbiera, G.; Norelli, M.; Genua, M.; Ranghetti, A.; Plati, T.; Camisa, B.; Brombin, C.; Cittaro, D.; et al. Interferon gene therapy reprograms the leukemia microenvironment inducing protective immunity to multiple tumor antigens. Nat. Commun. 2018, 9, 2896. [Google Scholar] [CrossRef]
- Moretta, L.; Montaldo, E.; Vacca, P.; Del Zotto, G.; Moretta, F.; Merli, P.; Locatelli, F.; Mingari, M.C. Human Natural Killer Cells: Origin, Receptors, Function, and Clinical Applications. Int. Arch. Allergy Immunol. 2014, 164, 253–264. [Google Scholar] [CrossRef]
- Moretta, A.; Bottino, C.; Mingari, M.C.; Biassoni, R.; Moretta, L. What is a natural killer cell? Nat. Immunol. 2002, 3, 6–8. [Google Scholar] [CrossRef] [PubMed]
- Arthur, R.P.; Mason, D. T cells that help B cell responses to soluble antigen are distinguishable from those producing interleukin 2 on mitogenic or allogeneic stimulation. J. Exp. Med. 1986, 163, 774–786. [Google Scholar] [CrossRef] [PubMed]
- López-Botet, M.; Bellón, T. Natural killer cell activation and inhibition by receptors for MHC class I. Curr. Opin. Immunol. 1999, 11, 301–307. [Google Scholar] [CrossRef]
- Kumar, S. Natural killer cell cytotoxicity and its regulation by inhibitory receptors. Immunology 2018, 154, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Barrow, A.D.; Martin, C.J.; Colonna, M. The Natural Cytotoxicity Receptors in Health and Disease. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Parham, P.; Norman, P.J.; Abi-Rached, L.; Guethlein, L.A. Human-specific evolution of killer cell immunoglobulin-like receptor recognition of major histocompatibility complex class I molecules. Philos. Trans. R. Soc. B Biol. Sci. 2012, 367, 800–811. [Google Scholar] [CrossRef]
- Lima, D.S.; Lemes, R.P.G.; Matos, D.M. Immunosuppressive monocytes (CD14+/HLA-DRlow/−) increase in childhood precursor B-cell acute lymphoblastic leukemia after induction chemotherapy. Med. Oncol. 2018, 35, 36. [Google Scholar] [CrossRef] [PubMed]
- Romanski, A.; Bug, G.; Becker, S.; Kampfmann, M.; Seifried, E.; Hoelzer, D.; Ottmann, O.G.; Tonn, T. Mechanisms of resistance to natural killer cell-mediated cytotoxicity in acute lymphoblastic leukemia. Exp. Hematol. 2005, 33, 344–352. [Google Scholar] [CrossRef]
- Reusing, S.B.; Manser, A.R.; Enczmann, J.; Mulder, A.; Claas, F.H.; Carrington, M.; Fischer, J.C.; Borkhardt, A.; Babor, F.; Uhrberg, M. Selective downregulation of HLA-C and HLA-E in childhood acute lymphoblastic leukaemia. Br. J. Haematol. 2016, 174, 477–480. [Google Scholar] [CrossRef] [PubMed]
- Makanga, D.R.; Da Rin de Lorenzo, F.; David, G.; Willem, C.; Dubreuil, L.; Legrand, N.; Guillaume, T.; Peterlin, P.; Lebourgeois, A.; Béné, M.C.; et al. Genetic and Molecular Basis of Heterogeneous NK Cell Responses against Acute Leukemia. Cancers 2020, 12, 1927. [Google Scholar] [CrossRef] [PubMed]
- Rouce, R.H.; Shaim, H.; Sekine, T.; Weber, G.; Ballard, B.; Ku, S.; Barese, C.; Murali, V.; Wu, M.F.; Liu, H.; et al. The TGF-β/SMAD pathway is an important mechanism for NK cell immune evasion in childhood B-acute lymphoblastic leukemia. Leukemia 2016, 30, 800–811. [Google Scholar] [CrossRef]
- Giovanni, F.T.; Nadia, P.; Sara, R.; Daria, P.; Maria, S.D.P.; Antonella, V.; Alice, B.; Walter, B.; Lorenzo, M.; Giuseppe, B.; et al. Recognition of adult and pediatric acute lymphoblastic leukemia blasts by natural killer cells. Haematologica 2014, 99, 1248–1254. [Google Scholar] [CrossRef][Green Version]
- Parrado, A.; Casares, S.; Rodríguez-Fernández, J.-M. Natural killer cytotoxicity and lymphocyte subpopulations in patients with acute leukemia. Leuk. Res. 1994, 18, 191–197. [Google Scholar] [CrossRef]
- Valenzuela-Vazquez, L.; Núñez-Enríquez, J.C.; Sánchez-Herrera, J.; Jiménez-Hernández, E.; Martín-Trejo, J.A.; Espinoza-Hernández, L.E.; Medina-Sanson, A.; Flores-Villegas, L.V.; Peñaloza-González, J.G.; Refugio Torres-Nava, J.; et al. Functional characterization of NK cells in Mexican pediatric patients with acute lymphoblastic leukemia: Report from the Mexican Interinstitutional Group for the Identification of the Causes of Childhood Leukemia. PLoS ONE 2020, 15, e0227314. [Google Scholar] [CrossRef]
- Gabrilovac, J.; Rajić, L.; Martin-Kleiner, I.; Osmak, M.; Batinić, D.; Tiefenbach, A.; Boranić, M. Defect of NK activity in children with untreated acute lymphocytic leukemia (ALL). I. Dependence on the blast count and phenotype, and response to exogenous and endogenous alpha-interferon. J. Clin. Lab. Immunol. 1989, 29, 9–15. [Google Scholar]
- Sullivan, E.M.; Jeha, S.; Kang, G.; Cheng, C.; Rooney, B.; Holladay, M.; Bari, R.; Schell, S.; Tuggle, M.; Pui, C.-H.; et al. NK Cell Genotype and Phenotype at Diagnosis of Acute Lymphoblastic Leukemia Correlate with Postinduction Residual Disease. Clin. Cancer Res. 2014, 20, 5986–5994. [Google Scholar] [CrossRef] [PubMed]
- Gleason, M.K.; Verneris, M.R.; Todhunter, D.A.; Zhang, B.; McCullar, V.; Zhou, S.X.; Panoskaltsis-Mortari, A.; Weiner, L.M.; Vallera, D.A.; Miller, J.S. Bispecific and Trispecific Killer Cell Engagers Directly Activate Human NK Cells through CD16 Signaling and Induce Cytotoxicity and Cytokine Production. Mol. Cancer Ther. 2012, 11, 2674–2684. [Google Scholar] [CrossRef] [PubMed]
- Peragine, N.; Torelli, G.F.; Mariglia, P.; Pauselli, S.; Vitale, A.; Guarini, A.; Foà, R. Immunophenotypic and functional characterization of ex vivo expanded natural killer cells for clinical use in acute lymphoblastic leukemia patients. Cancer Immunol. Immunother. 2015, 64, 201–211. [Google Scholar] [CrossRef]
- Boieri, M.; Ulvmoen, A.; Sudworth, A.; Lendrem, C.; Collin, M.; Dickinson, A.M.; Kveberg, L.; Inngjerdingen, M. IL-12, IL-15, and IL-18 pre-activated NK cells target resistant T cell acute lymphoblastic leukemia and delay leukemia development in vivo. OncoImmunology 2017, 6, e1274478. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.L.; Béziat, V.; Oei, V.Y.S.; Pfefferle, A.; Schaffer, M.; Lehmann, S.; Hellström-Lindberg, E.; Söderhäll, S.; Heyman, M.; Grandér, D.; et al. Ex Vivo Expanded Adaptive NK Cells Effectively Kill Primary Acute Lymphoblastic Leukemia Cells. Cancer Immunol. Res. 2017, 5, 654–665. [Google Scholar] [CrossRef]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef]
- Müller, S.; Bexte, T.; Gebel, V.; Kalensee, F.; Stolzenberg, E.; Hartmann, J.; Koehl, U.; Schambach, A.; Wels, W.S.; Modlich, U.; et al. High Cytotoxic Efficiency of Lentivirally and Alpharetrovirally Engineered CD19-Specific Chimeric Antigen Receptor Natural Killer Cells Against Acute Lymphoblastic Leukemia. Front. Immunol. 2020, 10. [Google Scholar] [CrossRef]
- Pende, D.; Marcenaro, S.; Falco, M.; Martini, S.; Bernardo, M.E.; Montagna, D.; Romeo, E.; Cognet, C.; Martinetti, M.; Maccario, R.; et al. Anti-leukemia activity of alloreactive NK cells in KIR ligand-mismatched haploidentical HSCT for pediatric patients: Evaluation of the functional role of activating KIR and redefinition of inhibitory KIR specificity. Blood 2009, 113, 3119–3129. [Google Scholar] [CrossRef] [PubMed]
- Matthias, M.P.; Tobias, F.; Heiko-Manuel, T.; Michael, S.; Ingo, M.; Rupert, H.; Peter, L. Reconstitution of natural killer cell receptors influences natural killer activity and relapse rate after haploidentical transplantation of T- and B-cell depleted grafts in children. Haematologica 2010, 95, 1381–1388. [Google Scholar] [CrossRef]
- Locatelli, F.; Pende, D.; Falco, M.; Della Chiesa, M.; Moretta, A.; Moretta, L. NK Cells Mediate a Crucial Graft-versus-Leukemia Effect in Haploidentical-HSCT to Cure High-Risk Acute Leukemia. Trends Immunol. 2018, 39, 577–590. [Google Scholar] [CrossRef] [PubMed]
- Plitas, G.; Rudensky, A.Y. Regulatory T Cells: Differentiation and Function. Cancer Immunol. Res. 2016, 4, 721–725. [Google Scholar] [CrossRef] [PubMed]
- Rezvani, K.; Yong, A.S.M.; Tawab, A.; Jafarpour, B.; Eniafe, R.; Mielke, S.; Savani, B.N.; Keyvanfar, K.; Li, Y.; Kurlander, R.; et al. Ex vivo characterization of polyclonal memory CD8+ T-cell responses to PRAME-specific peptides in patients with acute lymphoblastic leukemia and acute and chronic myeloid leukemia. Blood 2009, 113, 2245–2255. [Google Scholar] [CrossRef] [PubMed]
- Boublikova, L.; Kalinova, M.; Ryan, J.; Quinn, F.; O’Marcaigh, A.; Smith, O.; Browne, P.; Stary, J.; McCann, S.R.; Trka, J.; et al. Wilms’ tumor gene 1 (WT1) expression in childhood acute lymphoblastic leukemia: A wide range of WT1 expression levels, its impact on prognosis and minimal residual disease monitoring. Leukemia 2006, 20, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Weber, G.; Caruana, I.; Rouce, R.H.; Barrett, A.J.; Gerdemann, U.; Leen, A.M.; Rabin, K.R.; Bollard, C.M. Generation of Tumor Antigen-Specific T Cell Lines from Pediatric Patients with Acute Lymphoblastic Leukemia—Implications for Immunotherapy. Clin. Cancer Res. 2013, 19, 5079–5091. [Google Scholar] [CrossRef]
- Chang, T.-C.; Carter, R.A.; Li, Y.; Li, Y.; Wang, H.; Edmonson, M.N.; Chen, X.; Arnold, P.; Geiger, T.L.; Wu, G.; et al. The neoepitope landscape in pediatric cancers. Genome Med. 2017, 9, 78. [Google Scholar] [CrossRef]
- Bayón-Calderón, F.; Toribio, M.L.; González-García, S. Facts and Challenges in Immunotherapy for T-Cell Acute Lymphoblastic Leukemia. Int. J. Mol. Sci. 2020, 21, 7685. [Google Scholar] [CrossRef]
- He, X.; Xu, C. Immune checkpoint signaling and cancer immunotherapy. Cell Res. 2020, 30, 660–669. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, S.; Zhang, B.; Qiao, L.; Zhang, Y. T Cell Dysfunction and Exhaustion in Cancer. Front. Cell Dev. Biol. 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Andersen, M.H. The targeting of immunosuppressive mechanisms in hematological malignancies. Leukemia 2014, 28, 1784–1792. [Google Scholar] [CrossRef]
- Curran, E.K.; Godfrey, J.; Kline, J. Mechanisms of Immune Tolerance in Leukemia and Lymphoma. Trends Immunol. 2017, 38, 513–525. [Google Scholar] [CrossRef]
- Krummel, M.F.; Allison, J.P. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J. Exp. Med. 1995, 182, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of Antitumor Immunity by CTLA-4 Blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef]
- Rowshanravan, B.; Halliday, N.; Sansom, D.M. CTLA-4: A moving target in immunotherapy. Blood 2018, 131, 58–67. [Google Scholar] [CrossRef]
- Agata, Y.; Kawasaki, A.; Nishimura, H.; Ishida, Y.; Tsubat, T.; Yagita, H.; Honjo, T. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int. Immunol. 1996, 8, 765–772. [Google Scholar] [CrossRef]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, T.; Chikuma, S.; Iwai, Y.; Fagarasan, S.; Honjo, T. A rheostat for immune responses: The unique properties of PD-1 and their advantages for clinical application. Nat. Immunol. 2013, 14, 1212–1218. [Google Scholar] [CrossRef] [PubMed]
- Patsoukis, N.; Wang, Q.; Strauss, L.; Boussiotis, V.A. Revisiting the PD-1 pathway. Sci. Adv. 2020, 6, eabd2712. [Google Scholar] [CrossRef] [PubMed]
- Monney, L.; Sabatos, C.A.; Gaglia, J.L.; Ryu, A.; Waldner, H.; Chernova, T.; Manning, S.; Greenfield, E.A.; Coyle, A.J.; Sobel, R.A.; et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature 2002, 415, 536–541. [Google Scholar] [CrossRef]
- Wolf, Y.; Anderson, A.C.; Kuchroo, V.K. TIM3 comes of age as an inhibitory receptor. Nat. Rev. Immunol. 2020, 20, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Ohashi, P.S. Clinical blockade of PD1 and LAG3—Potential mechanisms of action. Nat. Rev. Immunol. 2015, 15, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Solinas, C.; Migliori, E.; De Silva, P.; Willard-Gallo, K. LAG3: The Biological Processes That Motivate Targeting This Immune Checkpoint Molecule in Human Cancer. Cancers 2019, 11, 1213. [Google Scholar] [CrossRef]
- Solomon, B.L.; Garrido-Laguna, I. TIGIT: A novel immunotherapy target moving from bench to bedside. Cancer Immunol. Immunother. 2018, 67, 1659–1667. [Google Scholar] [CrossRef]
- Hoek, R.M.; Ruuls, S.R.; Murphy, C.A.; Wright, G.J.; Goddard, R.; Zurawski, S.M.; Blom, B.; Homola, M.E.; Streit, W.J.; Brown, M.H.; et al. Down-Regulation of the Macrophage Lineage Through Interaction with OX2 (CD200). Science 2000, 290, 1768–1771. [Google Scholar] [CrossRef]
- Rygiel, T.P.; Meyaard, L. CD200R signaling in tumor tolerance and inflammation: A tricky balance. Curr. Opin. Immunol. 2012, 24, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Pilch, Z.; Tonecka, K.; Braniewska, A.; Sas, Z.; Skorzynski, M.; Boon, L.; Golab, J.; Meyaard, L.; Rygiel, T.P. Antitumor Activity of TLR7 Is Potentiated by CD200R Antibody Leading to Changes in the Tumor Microenvironment. Cancer Immunol. Res. 2018, 6, 930–940. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, H.; Chen, L.; Feng, Z.; Gao, L.; Li, Q. TIGIT expression is upregulated in T cells and causes T cell dysfunction independent of PD-1 and Tim-3 in adult B lineage acute lymphoblastic leukemia. Cell. Immunol. 2019, 344, 103958. [Google Scholar] [CrossRef]
- Qin, H.; Ishii, K.; Nguyen, S.; Su, P.P.; Burk, C.R.; Kim, B.-H.; Duncan, B.B.; Tarun, S.; Shah, N.N.; Kohler, M.E.; et al. Murine pre–B-cell ALL induces T-cell dysfunction not fully reversed by introduction of a chimeric antigen receptor. Blood 2018, 132, 1899–1910. [Google Scholar] [CrossRef] [PubMed]
- Coles, S.J.; Hills, R.K.; Wang, E.C.Y.; Burnett, A.K.; Man, S.; Darley, R.L.; Tonks, A. Increased CD200 expression in acute myeloid leukemia is linked with an increased frequency of FoxP3+ regulatory T cells. Leukemia 2012, 26, 2146–2148. [Google Scholar] [CrossRef]
- Wong, K.K.; Khatri, I.; Shaha, S.; Spaner, D.E.; Gorczynski, R.M. The role of CD200 in immunity to B cell lymphoma. J. Leukoc. Biol. 2010, 88, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Feucht, J.; Kayser, S.; Gorodezki, D.; Hamieh, M.; Döring, M.; Blaeschke, F.; Schlegel, P.; Bösmüller, H.; Quintanilla-Fend, L.; Ebinger, M.; et al. T-cell responses against CD19 + pediatric acute lymphoblastic leukemia mediated by bispecific T-cell engager (BiTE) are regulated contrarily by PD-L1 and CD80/CD86 on leukemic blasts. Oncotarget 2016, 7. [Google Scholar] [CrossRef]
- Alapat, D.; Coviello-Malle, J.; Owens, R.; Qu, P.; Barlogie, B.; Shaughnessy, J.D.; Lorsbach, R.B. Diagnostic Usefulness and Prognostic Impact of CD200 Expression in Lymphoid Malignancies and Plasma Cell Myeloma. Am. J. Clin. Pathol. 2012, 137, 93–100. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T Cells and Immune Tolerance. Cell 2008, 133, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Togashi, Y.; Shitara, K.; Nishikawa, H. Regulatory T cells in cancer immunosuppression—Implications for anticancer therapy. Nat. Rev. Clin. Oncol. 2019, 16, 356–371. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Munger, M.E.; Highfill, S.L.; Tolar, J.; Weigel, B.J.; Riddle, M.; Sharpe, A.H.; Vallera, D.A.; Azuma, M.; Levine, B.L.; et al. Program death-1 signaling and regulatory T cells collaborate to resist the function of adoptively transferred cytotoxic T lymphocytes in advanced acute myeloid leukemia. Blood 2010, 116, 2484–2493. [Google Scholar] [CrossRef]
- DeLeeuw, R.J.; Kost, S.E.; Kakal, J.A.; Nelson, B.H. The Prognostic Value of FoxP3+ Tumor-Infiltrating Lymphocytes in Cancer: A Critical Review of the Literature. Clin. Cancer Res. 2012, 18, 3022–3029. [Google Scholar] [CrossRef] [PubMed]
- Manlove, L.S.; Berquam-Vrieze, K.E.; Pauken, K.E.; Williams, R.T.; Jenkins, M.K.; Farrar, M.A. Adaptive Immunity to Leukemia Is Inhibited by Cross-Reactive Induced Regulatory T Cells. J. Immunol. 2015, 195, 4028–4037. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Feng, W.; Wang, H.; Wang, L.; Yang, X.; Yang, F.; Zhang, Y.; Liu, X.; Zhang, D.; Ren, Q.; et al. Blocking migration of regulatory T cells to leukemic hematopoietic microenvironment delays disease progression in mouse leukemia model. Cancer Lett. 2020, 469, 151–161. [Google Scholar] [CrossRef]
- Niedźwiecki, M.; Budziło, O.; Adamkiewicz-Drożyńska, E.; Pawlik-Gwozdecka, D.; Zieliński, M.; Maciejka-Kembłowska, L.; Szczepański, T.; Trzonkowski, P. CD4+CD25highCD127low/-FoxP3+Regulatory T-Cell Population in Acute Leukemias: A Review of the Literature. J. Immunol. Res. 2019, 2019, 2816498. [Google Scholar] [CrossRef]
- An, F.; Wang, H.; Liu, Z.; Wu, F.; Zhang, J.; Tao, Q.; Li, Y.; Shen, Y.; Ruan, Y.; Zhang, Q.; et al. Influence of patient characteristics on chimeric antigen receptor T cell therapy in B-cell acute lymphoblastic leukemia. Nat. Commun. 2020, 11, 5928. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).