Simple Summary
Glucocorticoid therapy resistance in B-cell malignancies is often associated with constitutive activation of tyrosine kinases. Novel anticancer drugs targeting hyperactivated tyrosine kinases, such as Bruton’s tyrosine kinase (BTK), have, therefore, gained much interest over the past few decades and have already been approved for clinical use. In this study, we compared the therapeutic efficacy of the phytochemical kinase inhibitor withaferin A with the clinically approved BTK inhibitor ibrutinib to target hyperactivated tyrosine kinase signaling in glucocorticoid-resistant multiple myeloma cells. Our results demonstrate that withaferin A-induced cell death of glucocorticoid-resistant MM1R cells involves covalent cysteine targeting of multiple Hinge-6 domain type tyrosine kinases of the kinase cysteinome classification, including BTK.
Abstract
Multiple myeloma (MM) is a hematological malignancy characterized by plasma cells’ uncontrolled growth. The major barrier in treating MM is the occurrence of primary and acquired therapy resistance to anticancer drugs. Often, this therapy resistance is associated with constitutive hyperactivation of tyrosine kinase signaling. Novel covalent kinase inhibitors, such as the clinically approved BTK inhibitor ibrutinib (IBR) and the preclinical phytochemical withaferin A (WA), have, therefore, gained pharmaceutical interest. Remarkably, WA is more effective than IBR in killing BTK-overexpressing glucocorticoid (GC)-resistant MM1R cells. To further characterize the kinase inhibitor profiles of WA and IBR in GC-resistant MM cells, we applied phosphopeptidome- and transcriptome-specific tyrosine kinome profiling. In contrast to IBR, WA was found to reverse BTK overexpression in GC-resistant MM1R cells. Furthermore, WA-induced cell death involves covalent cysteine targeting of Hinge-6 domain type tyrosine kinases of the kinase cysteinome classification, including inhibition of the hyperactivated BTK. Covalent interaction between WA and BTK could further be confirmed by biotin-based affinity purification and confocal microscopy. Similarly, molecular modeling suggests WA preferably targets conserved cysteines in the Hinge-6 region of the kinase cysteinome classification, favoring inhibition of multiple B-cell receptors (BCR) family kinases. Altogether, we show that WA’s promiscuous inhibition of multiple BTK family tyrosine kinases represents a highly effective strategy to overcome GC-therapy resistance in MM.
1. Introduction
MM is a hematological malignancy of terminally differentiated plasma cells and is currently the second most common adult blood cancer [1]. MM often results in development of end-organ diseases, such as anemia, hypercalcemia, renal insufficiency, and bone lesions, making it an illness of considerable clinical and social impact [2,3]. In recent years, important therapeutical advancements in the field of MM have been made, increasing the life expectancy of patients by six to ten years [4,5]. These novel therapies have mostly been developed based on an improved understanding of the biology of myeloma cells and their interaction with the bone marrow (BM) environment [6]. They include proteasome inhibitors [7], immunomodulatory drugs [8], glucocorticoids (GCs) [9], monoclonal antibodies [10], and histone deacetylase inhibitors [11]. However, MM still remains an incurable disease as the majority of patients eventually relapse and become refractory to existing therapies [12]. Acquisition of resistance to anticancer drugs, therefore, remains the main barrier in treating MM [4,13].
One of the cellular pathways mediating drug resistance in many B-cell malignancies is the BCR signaling pathway [14]. Under physiological conditions, the BCR is activated upon ligation of antigen and promotes survival, function and development of B-cells [15]. After initial antigen binding, the immune receptor tyrosine activation motif domains CD79A and CD79B are phosphorylated by Src family kinases Lyn and Syk and recruit other adaptor proteins and tyrosine kinases (TK), a key example being Bruton’s tyrosine kinase (BTK) [16]. BTK ultimately orchestrates activation of downstream effectors of BCR signaling, such as nuclear factor-κB (NF-κB) and nuclear factor of activated T cells (NFAT), through PLC-γ2 and phosphoinositide 3-kinase (PI3K) phosphorylation [14]. In MM, BTK is often constitutively activated, thereby modulating survival signals and therapy resistance [17,18,19,20]. As a result, BTK kinase inhibitors have received growing pharmacological interest and have already shown promising therapeutic responses in B-cell malignancies in the clinic [21]. More particularly, the covalent-binding BTK inhibitor IBR has shown to be a potent anticancer drug in chronic lymphocytic leukemia (CLL), mantle cell lymphoma, diffuse large B-cell lymphoma, and MM by interfering with B-cell homing, survival and microenvironment-mediated drug resistance [17,18,22,23,24,25]. Suppression of BTK hyperactivation is also key to the therapeutic efficacy of GCs in B-cell leukemias, where IBR has been shown to improve GC therapy response [26,27,28,29]. In clinical trials of MM, combination therapies investigating IBR efficiency demonstrated encouraging responses and a manageable safety profile [30,31].
Although preliminary clinical data revealed the beneficial effects and acceptable safety profile of IBR in MM, clinical studies in other B-cell malignancies have linked IBR use with adverse effects, including diarrhea, fatigue, nausea, and rashes [32,33]. More important, IBR therapy is associated with a significant increase in the occurrence of ventricular arrhythmias and sudden cardiac death [34,35]. The underlying mechanisms of these severe side effects are not well understood but could be partially explained by the off-targets interaction of IBR with interleukin-2-inducible T-cell kinase (ITK), epidermal growth factor (EGFR), and PI3K [36]. Another pressing issue that has arisen since IBR has been applied in the clinic is the development of therapy resistance (reviewed in [37]). Nearly one-third of patients diagnosed with B-cell malignancies display primary resistance against IBR therapy, while many others acquire resistance over time. These major issues accompanying the therapeutic use of IBR have sparked the development of second and third-generation BTK inhibitors (e.g., acalabrutinib), characterized by higher selectivity and potency profiles [38]. Unfortunately, acquired resistance to these novel BTK inhibitors has already been described in some B-cell malignancies as well [39]. As a result, alternative treatment strategies to suppress BTK hyperactivation in therapy-resistant hematological malignancies are currently being investigated in high-throughput combinatorial screenings of clinically approved and preclinical investigational compound libraries [40]. Given that nearly half of the agents used in cancer therapy today are either natural products or derivatives thereof, novel BTK-targeting lead compounds may be identified from this vast arsenal of chemically active structures [41,42,43]. Interestingly, withaferin A, a withanolide phytochemical isolated from Withania somnifera, is one of the top investigational compounds prioritized for IBR combination therapy to target chronic active BCR signaling [40].
WA reveals broad-spectrum therapeutic activities in several (drug-resistant) cancer cell types [44], including B-cell lymphoma and MM [45,46,47]. Of particular interest, some of WA’s antitumor effects have been attributed to its ability to covalently target kinase activity [48,49,50,51,52]. Accordingly, innovative phosphopeptidome kinome activity profiling, RNA sequencing, in silico docking simulations, and chemo-affinity approaches were combined in this study to characterize BTK hyperactivation and TK inhibitor therapy response of WA and IBR in GC-resistant MM cells.
2. Results
2.1. GC Therapy Resistance in Multiple Myeloma Is Associated with Hyperactivation of Tyrosine Kinases
GC therapy-sensitive MM1S and -resistant MM1R cell lines derived from a single MM patient have previously been described as cell models to study the etiology of GC therapy resistance and to evaluate novel classes of chemotherapeutic drugs [53,54]. To investigate the vulnerability of GC-resistant MM1R cells for specific clinical TK inhibitor drugs, we compared the tyrosine kinome activity profiles of GC-resistant MM1R and GC-sensitive MM1S cell lysates by means of a PTK-specific phosphopeptide array (PamChip), containing 144 conserved peptides corresponding to TK specific substrates [55,56]. Overall, TK activity was consistently higher in MM1R cells compared to MM1S cells (Figure 1a and Figure S1). Identification of the 20 most significant differential hyperphosphorylated peptides (adjusted p-value (FDR) < 0.01) in MM1R compared to MM1S predicted hyperactivation of multiple (non) receptor TK in MM1R, such as SYK, DDR, ABL, ZAP70, FAK2, BRK, BTK, ITK and FGR (Figure 1b). Subsequent MetaCore pathway analysis showed that the hyperactivated kinases in MM1R cells are involved in cell proliferation, cell cycle regulation, cell adhesion, cancer, therapy resistance, immune response, T-cell receptor signaling and BCR signaling (Supplementary Figure S2).
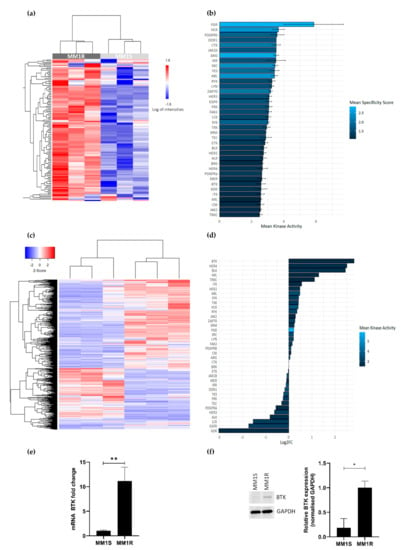
Figure 1.
(a) Heatmap showing phosphorylation intensities of peptides serving as substrates for tyrosine kinases. Figure shows hyperphosphorylated (red) or hypo-phosphorylated (blue) peptides in MM1R (n = 3) and MM1S (n = 3) samples. (b) Ranking of hyperactivated kinases in MM1R versus MM1S cells based on the top 20 significant differentially phosphorylated peptides. Fill color of the bars is based on the kinase specificity score, indicating the specificity of differences in kinase activity with respect to the quantity of peptides used for predicting the corresponding kinase (c) Heatmap representation of differentially expressed genes (logFC >|1|, p < 0.01) in MM1R versus MM1S cells as determined by RNA sequencing. n = 3 biologically independent replicates per cell line. (d) Ranking of the top overexpressed kinases in MM1R versus MM1S cells based on their log2-fold change as determined by RNA sequencing. Fill colors of the bars are a measure for kinase activity as measured via the PTK-specific phosphopeptide array. (e) Relative Bruton’s tyrosine kinase (BTK) mRNA levels in MM1R and MM1S cells. Data are plotted as the mean ± s.d., n = 3 biologically independent replicates (** p = 0.0035, unpaired t-test). (f) Western immunoblot detection and quantification of basal BTK and GAPDH protein levels in MM1R and MM1S cells. Data are plotted as the mean ± s.d., n = 3 biologically independent replicates (* p = 0.0385, unpaired t-test).
To investigate whether the observed TK hyperactivation is also reflected at the transcriptome level, RNA sequencing analysis of basal gene expression in MM1S and MM1R cells was conducted. Results were analyzed with the R-package DESeq2 [57], using a selection criterion of a minimal FDR < 0.01. Upon comparing gene expression patterns in both cell lines, 1383 differentially expressed genes (DEG) (logFC > 1 or logFC < −1) could be identified (Figure 1c). Interestingly, from all hyperactivated TK in MM1R cells listed before, BTK was identified as the strongest upregulated TK in GC-resistant MM1R cells (Figure 1d, is included in the top 20 most significant DEG (Table 1). Upregulation of BTK in MM1R cells was validated by qPCR and Western blot analysis (Figure 1e–f) and is in line with previous observations [17,58]. Taken together, these findings suggest that BCR signaling and BTK in MM1R cells may represent an attractive target to kill GC-resistant MM1R cells.

Table 1.
Overview of top 20 most significantly differentially expressed genes (logFC > 1 or logFC < −1) between glucocorticoid (GC)-resistant MM1R and GC-sensitive MM1S cells.
2.2. The Tyrosine Kinase Inhibitor Profile of Withaferin A and Ibrutinib Show a High Degree of Similarity
Taking into account the hyperactivation of BCR-BTK kinase signaling in MM1R cells (see 2.1.), we next tested their sensitivity for the clinically approved BTK inhibitor IBR as well as WA, a top prioritized investigational phytotherapeutic compound identified in a high-throughput drug screening against chronic BCR signaling [40]. MM1R cells were treated for 24 h with different concentrations of WA or IBR kinase inhibitors, and the relative % cell survival/cell death was evaluated by MTT assay. Both compounds were effective in killing GC-resistant MM1R cells in a dose-dependent manner, although WA is the more potent cell death inducer (IC50 = 1.7 µM) since its IC50 was > 10 times lower than the one of IBR (IC50 = 27.9 µM) (Figure 2a). Remarkably, both WA and IBR also induce cell death in GC-sensitive MM1S cells lacking BTK overexpression, although IC50 values were higher than MM1R cells (IC50WA = 1.9 µM, IC50IBR = 49.3 µM) (Figure 2a).
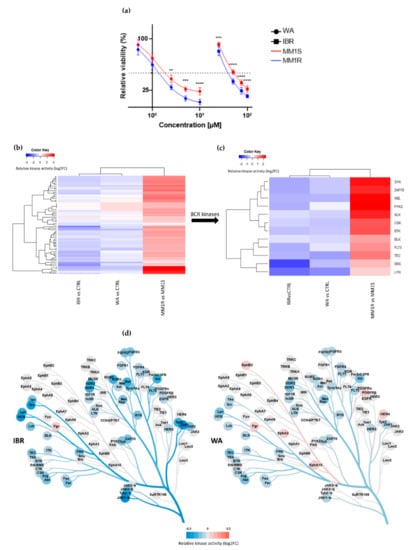
Figure 2.
(a) Relative cell viability of MM1 cells upon 24 h exposure to increasing concentrations of BTK inhibitor ibrutinib (IBR) or withaferin A (WA). Data are plotted as the mean ± s.d., n = 3 biologically independent replicates. (** p < 0.01, *** p < 0.001 **** p < 0.0001, ANOVA). (b) Heatmap representation of hyperactivated or inhibited kinases in MM1R versus MM1S cells, or following 15 min IBR or WA treatment, n = 3 biologically independent replicates per treatment group. (c) Close-up heatmap representation of Figure 2b showing inhibited BCR-related kinases in MM1R versus MM1S or following 15 min IBR or WA treatment, n = 3 biologically independent replicates per treatment group. (d) Kinase trees displaying the tyrosine kinase targets of IBR (left) and WA (right). Kinase trees were generated with the CORAL web tool (http://phanstiel-lab.med.unc.edu/CORAL/, accessed on 30 November 2020).
To explore the mode of action of these compounds, we measured corresponding cellular changes in TK activities by phosphopeptidome based tyrosine kinome profiling of MM1R cells exposed to either WA (1 µM) or IBR (1 µM). Similarly, and as mentioned above, cell lysates of treated cells were analyzed through PTK-specific phospho-peptide arrays, after which the top activated or inhibited kinases were identified based on the significant differences in phospho-intensities of the PTK peptide substrates. We found that WA and IBR both inhibited most of the hyperactivated kinases in MM1R cells, with largely overlapping, though promiscuous, TK inhibitor profiles (Figure 2b, annotation of heatmap rows can be found in Supplementary Figure S3). At the level of BCR signaling, clear inhibition of BTK kinase activity can be observed in the presence of IBR, as expected, and WA (Figure 2c,d). Besides BTK, multiple BCR signaling kinases, such as ZAP70, BLK, FLT3, TEC, and SYK, are also targeted by both WA and IBR (Figure 2c). In line with previous studies, which already revealed that IBR could trigger off-target BCR-BTK independent kinase inhibitor (side) effects [59], we also identified additional IBR-responsive TK in MM1R cells (Figure 2c, Figure S3). Of special note, whereas most binding affinities of IBR have been determined in vitro (Supplementary Figure S4), we provide the first cell-based integrated tyrosine kinome activity map in MM1R cells in the presence of the IBR inhibitor. Although WA inhibits similar BCR family kinases as IBR, variations in kinase inhibitor specificity/potency of WA in comparison to IBR treatment may explain differences in the therapeutic efficacy of both compounds in MM1R cells (Figure 2a,d).
2.3. WA Inhibits BCR-BTK Kinase Activity by Transcriptional Downregulation and Covalent Cysteine-Dependent Targeting of BTK
To further characterize how WA decreases BTK kinase activity in MM1R cells, we next checked whether WA treatment changes BTK mRNA and protein expression levels. Panther pathway enrichment analysis of RNA sequencing data from WA-treated MM1R cells already revealed that DEGs are significantly enriched in B-cell activation (Figure 3a). More particularly, WA significantly decreased BTK mRNA expression (Log2FC = −0.525, FDR = 0.023). This was further confirmed by qPCR and western immunoblot experiments. As can be observed from Figure 3b,c, WA was able to lower BTK expression in a time-dependent manner, both at the mRNA and protein level. Similar WA-specific changes could also be observed in U266 cells, another GC-resistant multiple myeloma cell line sensitive to WA treatment (Figure 3b,c and Figure S5). In line with previous studies [60,61,62], we did not observe any changes in BTK expression after the IBR treatment of MM1R cells indicating that IBR mainly targets BTK (hyper)phosphorylation and not total BTK protein levels (Supplementary Figure S6).
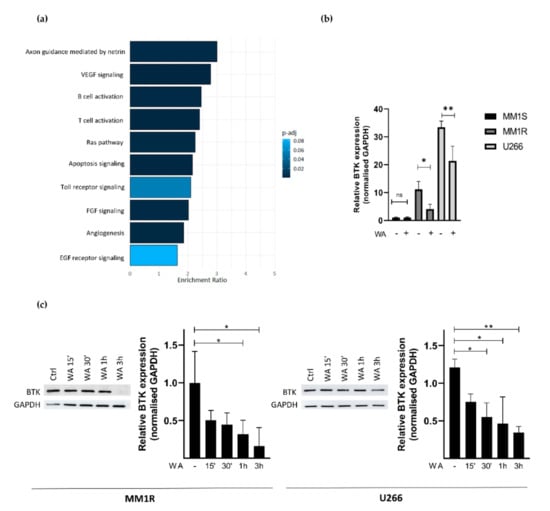
Figure 3.
(a) Panther pathway enrichment analysis of significant (FDR < 0.05) differentially expressed genes of WA-treated MM1R cells as determined by RNA sequencing. (b) Relative BTK mRNA levels of MM1R, MM1S and U266 cells treated with WA for 3 h. Data are plotted as the mean ± s.d., n = 3 biologically independent replicates (* p = 0.0453, ** p = 0.0015, ANOVA) (c) Western blot detection and quantification of BTK and GAPDH expression levels after WA treatment in MM1R and U266 cells. Data are plotted as the mean ± s.d., n = 3 biologically independent replicates. (* p < 0.05, ** p < 0.01, ANOVA).
Since WA contains several reactive nucleophilic groups, which can covalently bind to kinase sulfhydryl groups of cysteines through Michael addition [48,63,64], we also evaluated the potential covalent binding of WA to BTK by pulldown experiments with biotinylated WA (WABI). Pull-down experiments with WABI in MM1R cells indeed confirmed cysteine-dependent binding to BTK, which can be blocked by excess amounts (1 mM) of the reducing agent dithiothreitol (DTT) (Figure 4a). Along the same line, covalent WA-BTK interaction could be confirmed in U266 cells (Figure 4b). The biological relevance of this covalent interaction could further be validated in washout experiments where MM1R cells were exposed to increasing concentrations of WA for 15 min, after which it was washed away with PBS. Although WA cells were only briefly treated with WA, the cell viability of MM1R cells was still affected in a dose-dependent manner 24 h post-treatment (Supplementary Figure S7). The growth inhibition remains the strongest in the unwashed cells, suggesting the contribution of non-covalent interactions of WA as well. H-bond and van der Waals interactions between WA and target molecules, such as Hsp90, have indeed been reported to contribute to the anticancer mechanism of WA [65]. Alternatively, covalent binding by WA may be weaker than a normal covalent binding and become reversed by washout experiments [66,67,68]. Finally, by confocal microscopy, we could also demonstrate colocalization of BTK and WABI in MM1R cells (Figure 4c).
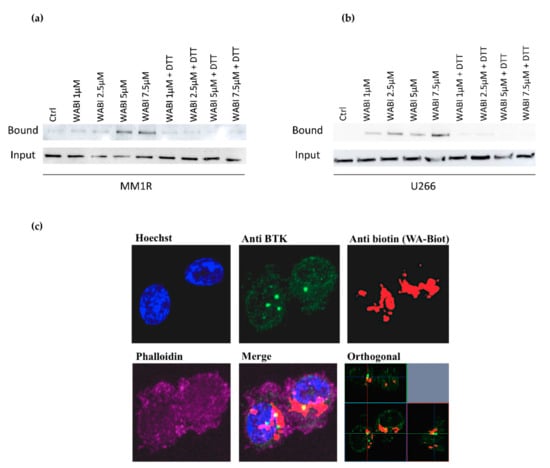
Figure 4.
(a) Western immunoblot detection of BTK levels before and after pulldown with biotinylated WA (WABI), following 2 h WABI treatment in MM1R in the presence or absence of excess thiol donor DTT (1 mM). (b) Western immunoblot detection of BTK levels before and after pulldown with biotinylated WA (WABI), following 2 h WABI treatment in U266 cells in the presence or absence of excess thiol donor DTT (1 mM). (c) Confocal imaging of colocalization of BTK expression and WABI localization in MM1R cells.
2.4. Covalent C481 Targeting of BTK by WA in Hinge-6 Domain of the Protein Kinase Cysteinome Classification Reduces Survival of Glucocorticoid Resistant Multiple Myeloma MM1 Cells
Human protein kinases are composed of two highly conserved domains, namely catalytic and regulatory domains. It has been shown that the catalytic domain can be further classified based on the positions of the gatekeeper amino acids and cysteines hosted in the catalytic pocket [69,70,71,72]. Based on the positions of cysteines present across the human kinome, Leproult et al. reclassified the human kinome into a cysteinome according to the cysteine positions relative to the ATP binding pocket (Table 2) [69,73]. Interestingly, most kinases inhibited by WA treatment, including BTK, BLK and EGFR, were found to be highly enriched in Hinge-6 domain type kinases (Table 2). Multiple sequence alignment of these Hinge-6 domain orthologs revealed the presence of a conserved glycine-cysteine motif, suggesting that WA covalently binds to this site within its TK targets (Figure 5a). Through molecular docking studies, we found that the conserved cysteine site within BTK (Cys481) is indeed accessible and favorable (−4.93 kcal/mol) for WA binding (Supplementary Table S1). The docking results predict that covalent bond formation occurs via the C4-OH group of WA and the Cys481 residue of BTK and that this interaction is further stabilized via hydrogen bond formation with surrounding Leu482, Tyr485 and Gly480 amino acids (Figure 5b,c). Given that IBR also covalently targets the Cys481 residue of BTK, these results suggest that WA interacts with BTK in a similar manner as IBR [63]. By using the NanoBRET Target Engagement Intracellular Kinase assay, where the affinity of WA for BTK can be analyzed by competitive displacement of a fluorescent NanoBRET Tracer bound to BTK, we could further confirm that WA can displace the WT BTK protein, but not the mutated C481S BTK protein in HEK-293 cells (Figure 5d). This suggests that the Cys481 residue of BTK is the main binding site of WA. Furthermore, we show that silencing of endogenous WT BTK reduces cell MM1R viability and can be rescued upon overexpression of C481S BTK overexpression. (Figure 5e, Figure S8). Of special note, C481S BTK overexpression cannot completely rescue WA-induced cell death in MM1R, confirming that WA kinase effects on cell viability are not limited to BTK alone and may involve additional hinge 6 domain type kinase targets of the cysteinome classification in MM1R cells. Along the same line, GC-sensitive MM1S cells lacking BTK overexpression are also sensitive to WA treatment through promiscuous covalent cysteine targeting of alternative cell survival tyrosine kinases expressed in MM1S cells (Figure S9a–c).
Table 2.
Kinase cysteinome classification. Summarized from [73]. Bold highlights kinases representing the main tyrosine kinase targets of withaferin A.
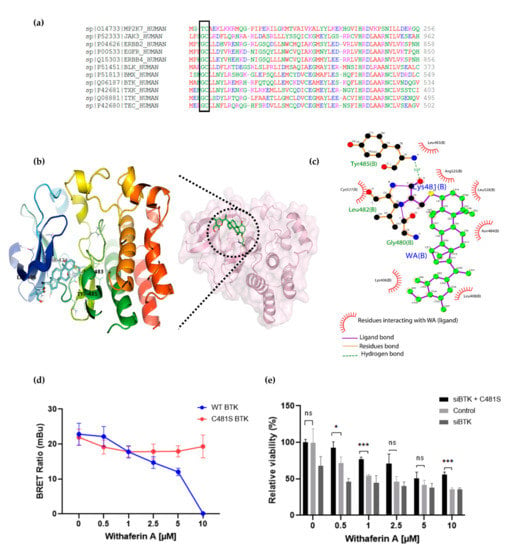
Figure 5.
(a) Multiple sequence alignment of Hinge 6 domain-type kinases. The conserved GC-motif between different orthologs is indicated. Alignment was performed with Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/ accessed on 30 November 2020). (b) Crystal structure of BTK (PBD id: 6TFP) in complex with WA (PubChem CID: 26537) covalently bound to Cys481. (c) Interaction between WA and BTK structure visualized using Ligplot [74], showing the covalent bond formation of the C4-OH group of WA with the SH group of Cys481 from BTK. (d) Inhibition of wild-type (WT) BTK and mutated (C481S) BTK by WA in HEK-293 cells. Data are plotted as mean ± s.d., n = 3 biologically independent replicates. (e) Relative viability of MM1R cells treated for 24 h with increasing concentrations of WA, upon BTK silencing (siBTK) in presence or absence of C481S BTK overexpression. Data are plotted as mean ± s.d., n = 3 biologically independent replicates (ns = p > 0.05, * p = 0.0309, *** p < 0.0001, ANOVA).
3. Discussion
A common feature of many B-cell malignancies is their ability to develop therapy resistance through increased BCR signaling and BTK activity. Along the same line, increased BCR signaling has been shown to reduce the therapeutic efficacy of GCs in B-cell leukemias [26,27,28,29]. Although MM cells are typically lacking active BCR, BTK overexpression and signaling has been associated with increased therapy resistance in the disease [18,75]. Accordingly, we compared the anticancer effect of the clinically approved BTK kinase inhibitor IBR and the preclinical phytotherapeutic kinase inhibitor WA to kill GC therapy-resistant MM cells via suppression of BTK hyperactivation. Phosphopeptidome based tyrosine kinome profiling confirmed hyperactivation of multiple BCR family kinases besides other receptor TK families (FGFR, PDGFR, DDR) in GC therapy-resistant MM1R cells. Furthermore, hyperactivated BCR kinases could completely be suppressed with both kinase inhibitors, WA and IBR. Nonetheless, kinase tree representations of both kinase inhibitor profiles revealed quantitative and qualitative differences in specificity and potency, which may underly the differences in potency of WA and IBR to kill GC-resistant MM cells. Biotin affinity-purification experiments confirmed a cysteine-dependent covalent interaction between WA and BTK, similar to IBR-BTK Cys481 binding [63]. This is supported by molecular modeling studies, which revealed favorable covalent WA binding to the conserved Cys481 residue in BTK. In addition, NanoBRET Target Engagement Intracellular Kinase assays confirm WA-dependent inhibition of WT BTK kinase activity, which is lost upon C481S mutation. Interestingly, most kinases inhibited by WA treatment, including BTK, BLK and EGFR, belong to the Hinge-6 domain-type kinases, according to the kinase cysteinome classification. The latter suggests broad redundant kinase inhibitory effects of WA through covalent cysteine targeting. Accordingly, BTK silencing experiments could only partially mimic the therapeutic cell death effects of WA in MM1R, whereas overexpression of the C481S kinase mutant could only partially protect against WA. Similarly, cancer therapeutic effects of WA could also be observed in GC-sensitive MM1S cells, which lack BTK overexpression, through covalent targeting of alternative cell survival kinases expressed in MM1S. As such, the therapeutic efficacy of WA against different cancer cell types may strongly depend on its promiscuous nucleophilic cysteine reactivity towards the cellular repertoire of hyperactivated tyrosine cell survival kinases. Additionally, the anticancer effects of WA may also partially rely on its non-covalent interactions with target proteins. H-bond interactions with terminal hydroxyl groups of WA were shown to be sufficient to deform protein complexes through naïve hindrance [65]. Alternatively, the covalent interaction of WA may be weaker than that of the normal covalent bond and be reversible in washout experiments [76]. Taunton and coworkers demonstrated that drug efficacy could be optimized by finetuning warhead residence time and exploiting the intrinsic reversibility of the reaction moiety [66,77]. In this respect, partially reversible covalent binding characteristics of WA may promote more promiscuous tyrosine kinase targeting than IBR and explain the higher efficacy of WA than IBR in overcoming MM drug resistance. Finally, WA-mediated inhibition of BTK kinase activity was also accompanied by a time-dependent decrease in BTK mRNA and protein expression, not observed for IBR, suggesting that WA targets BTK hyperactivation at multiple levels [62]. For example, WA may indirectly decrease BTK activity by reducing kinase protein levels by inhibition of Sp1-dependent transcription [78], by post-transcriptional microRNA silencing mechanisms [53,79], or by decreasing kinase stability via heat shock chaperone proteins [45,47].
Although WA clearly suppresses hyperactivated BTK family kinases signaling, some limitations of our study need to be acknowledged. Most notably, mainly GC therapy-resistant MM cell models were studied here. Further preclinical in vitro and in vivo studies are required to fully appreciate the therapeutic efficacy of WA to overcome multidrug resistance by covalent inhibition of hinge 6 cysteine domain BTK family kinases. In addition, small qualitative and/or quantitative variations in specificity/potency observed for the promiscuous BCR TK inhibitor profiles of WA and IBR may not be sufficient to explain the large difference in the therapeutic efficacy of WA as compared to IBR against GC-resistant MM cell lines. Of special note, whereas nM concentrations of IBR are effective against B-cell lymphoma cancer cells, much higher μM concentrations seem to be required to kill BTK overexpressing MM cells [17,80,81,82,83], presumably because of the presence of highly redundant BCR/NFKB survival pathways (PI3K/Akt/mTOR/Syk) in MM, which compensates for pharmacological BTK inhibition [37]. Yet, despite the presence of these compensatory pathways, complete silencing of BTK expression is cytotoxic to MM1R cells, emphasizing BTK’s involvement in myeloma cell survival. This is in line with previous observations made in BTK KO mouse models, where complete loss of BTK expression resulted in a more significant decrease in splenic B-cell numbers compared to mice harboring BTK mutations [84]. Because BTK also possesses crucial shuttling and scaffold activities, complete silencing of BTK is likely more detrimental to B-cells due to additional loss of non-kinase functions [85].
Furthermore, covalent cysteine binding of WA has also been reported to Ser/Thr kinases [86] and phosphatases [87], which were not included in our phosphopeptidome screening approach. Indeed, in addition to BTK, BCR signaling is fine-tuned by Ser/Thr kinases (for example, IKK2, PKCβ) [15,88] and phosphatases (i.e., SHIP1, PTEN) [89,90]. Finally, chemoproteomic strategies have identified additional WA non-kinase target proteins, which may further strengthen the potential cancer therapeutic efficacy of WA [45,91]. Therefore, functional silencing approaches with shRNA libraries will be informative to further distinguish druggable key cancer targets from adverse off-targets to defeat therapy resistance in MM by WA [87,92]. Further research determining the safety and pharmacokinetic profile of WA is needed as well. Preliminary (pre)clinical toxicology studies of WA in different cancer models revealed that WA administration is generally well tolerated with limited to no adverse toxicity reported [93,94,95]. Yet, no toxicology data of WA have been collected in B-cell malignancies, such as MM.
4. Materials and Methods
4.1. Cell Culture and Cell Viability Assays
GC-sensitive MM1S (CRL-2974) and GC-resistant MM1R MM cell lines (CRL-2975) have been described previously and were purchased from ATCC [53]. GC-resistant U266 cells were kindly provided by Dr. Eva Lion, Head of Tumor Immunology Group of the Laboratory of Experimental Hematology (University of Antwerp). Cells were cultivated in RPMI-1640 supplemented with 10% fetal bovine serum (E.U approved; South American origin) and 1% penicillin/streptomycin solution (Invitrogen, CA, USA). The cell lines were additionally supplemented with 1% MEM Non-Essential amino acids and 1% sodium pyruvate (Invitrogen, CA, USA). Each cell line was maintained at 37 °C in 5% CO2 and 95% air atmosphere and 95–98% humidity. Cell viability was assessed by colorimetric assay with 3-(4, 5-dimethylthiozol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) (Sigma-Aldrich, St. Louis, MO, US) as previously described [53].
4.2. Cell Lysis and Peptide Array-Based TK Activity Profiling
WA- or BTK-treated and untreated MM1S and MM1R cells (n = 3 biologically independent samples per cell line per treatment) were lysed in M-PER lysis buffer containing 1:100 Halt’s protease and Pierce™ phosphatase inhibitors (Thermo Scientific, Waltham, MA, USA). The lysates were collected and centrifuged at >13,000× rpm for 15 min at 4 °C. The supernatants were transferred to pre-chilled Eppendorf tubes and flash-frozen immediately on dry ice. Protein concentrations were determined by the Pierce™ BCA method (Thermo Scientific, Waltham, MA, USA) [96]. Cellular kinase activities were measured by TK-specific phosphopeptide arrays (PamChip, PamGene International B.V. the Netherlands) according to the manufacturer’s protocol as previously described [56,97]. The differences in peptide phosphorylation signal intensity between different experimental setups were analyzed in a linear mixed model by using BioNavigator 6.3 software integrated with R (Bioconductor) for statistical analysis (paired two-sided t-test and unpaired t-tests). FDR were calculated using Benjamini–Hochberg method (adjusted p-values < 0.05) [55]. The upstream kinases were correlated with the identified PTK-specific phosphopeptide fingerprints according to the human (phospho)protein reference database [98,99,100,101]. Peptide lists showing differential phosphorylation intensities were further cross-compared with phosphorylation specific datasets of the Kinexus kinase predictor [102]. The kinases’ ranking was based upon scoring penalties of sensitivity and specificity of observed peptide phosphorylation changes in the tested experimental conditions. The significance of the upstream kinase scoring functions was calculated by Fisher’s exact test. Pathway enrichment of differentially phosphorylated protein IDs was analyzed via MetaCore (Clarivate Analytics, Philadelphia, PA, USA) and Ingenuity pathway analysis (IPA) software (Qiagen, Germany) [103].
4.3. cDNA Conversion Quantitative Real-Time PCR
Total RNA (1 µg) extracted from each sample was converted into cDNA with the Go Script reverse transcription system (Promega, Madison, WI, USA) following the manufacturer’s protocol. Next, qPCR analysis was carried out using the GoTaq qPCR master mix (Promega, Madison, WI, USA) according to the manufacturer’s instructions. In brief, a 25 µL reaction volume mix per sample was prepared, containing 12.5 µL GoTaq qPCR master mix, 0.4 µM forward and reverse primer and nuclease-free water. The following PCR program was applied on the Rotor-Gene Q qPCR machine of Qiagen: 95 °C for 2 min, 40 cyclic denaturations (95 °C, 15 s) and annealing/extension (60 °C, 30 s), and dissociation (60–95 °C). Each sample was run in triplicate. The median value of the triplicates was taken to calculate the ΔΔCt-values using GAPDH as the normalization gene. Primer sequences are listed in Supplementary Table S2.
4.4. RNA Extraction and RNA Sequencing
The RNeasy mini kit (Qiagen, Germany) was used to extract total RNA from untreated, or WA exposed MM1S and MM1R cells (n = 3 biologically independent samples per cell line per treatment) according to the manufacturer’s protocol. Isolated, pure total RNA was then quantified and qualified using an EpochTM microplate spectrophotometer (BioTek, Winusky, VT, USA). RNA samples were stored at −80 °C and subsequently shipped to BGI (BGI Group, Beijing, China), where RNA integrity was determined using the 2100 Bioanalyzer system (Agilent Technologies, CA, USA). All 12 samples with acceptable quality levels (RNA content >80 ng/µL, 28 s/18 s ≥ 1.0 and RIN ≥ 7.0) were included for sequencing library preparation. Prepared libraries were 2 × 50 bp paired-end sequenced using the BGISEQ-500 platform (BGI Group, China). RNAseq data were deposited in the NCBI GEO database (GSE162475).
The quality of the RNA sequencing reads was assessed using FastQC (v0.11.5, Babraham Institute, Cambridge, UK) [104]. STAR (v2.7.3a, Cold Spring Harbor Laboratory, CA, USA) [105] was subsequently used to map reads to the human reference genome build 37 (hg19) and to generate a read count table summarizing the counts per gene. Finally, differential gene expression analysis was performed using the DESeq2 package [57], and pathway analysis was performed with Panther (Protein Informatics, Celera Genomics, CA, USA) [106].
4.5. Antibodies and Reagents
WA was purchased from Alta Vista Phytochemicals (Hyderabad, India), and biotinylation of WA was performed by Dr. P. Van der Veken (WA-BT; Universiteit Antwerpen, Belgium). Both formulations were stored as 20 mM stocks in DMSO at −20 °C as previously described [48,107]. IBR (IMBRUVICA®) stock solutions were obtained from Pharmacyclics (Sunnyvale, CA, USA). Antibodies BTK (3533) and GAPDH (2118S) were obtained from Cell Signaling Technology (Danvers, MA, USA).
4.6. Cell Viability after WA Washout
After 15 min treatment of MM1R cells with increasing concentrations of WA (with or without DTT), cells were washed extensively with PBS (3 × 5 min) and left to grow for an additional 24 h in WA-free supplemented RPMI-1640 medium. Once incubation was complete, cell viability was determined by colorimetric MTT assay as described above. MM1R cells continuously treated with WA for 24 h were included to allow for a direct comparison of covalent and non-covalent effects on cell viability.
4.7. Protein Extraction and Western Immunoblot Analysis
For western immunoblot analyses, cell pellets were lysed in 0.5 mL RIPA buffer (150 mM NaCl, 0.1% Triton X-100, 0.1% SDS, 50 mM Tris-HCl pH 8) supplemented with protease inhibitors (Complete Mini®, Roche, CA, USA). Soluble protein extracts were obtained after 15 min incubation on ice, followed by brief sonication and centrifugation at 16 g for 20 min at 4 °C. Samples were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS–PAGE) and transferred onto nitrocellulose membranes (Hybond C, Amersham) following standard protocols. After blocking, membranes were incubated overnight at 4 °C with the primary antibodies, followed by dye-conjugated secondary antibodies (polyclonal goat anti-rabbit HRP, #P0448, Dako). Bound complexes were detected with the Amersham Imager 680 (Cytiva, Austria) and quantified by Image J software (Max Planck Institute, Dresden, Germany) [108]. Original immunoblot images are provided in Supplementary Table S4.
4.8. WA-Biotin-Based Affinity Purification
WA-BT affinity purification and co-precipitation were performed as previously described [48]. In short, MM1R and U266 cells were seeded in 10 cm culture dishes and incubated for 24 h at 37 °C. Cells were treated with biotinylated WA (1 µM, 2.5 µM, 5 µM or 7.5 μM as indicated) or left untreated for 2 h. Cells were then lysed in 1 mL lysis buffer (5 mM Tris pH 7.6, 1% Triton-X100 and 5 mM EDTA, supplemented with Complete™ protease inhibitor cocktail) and incubated with Neutravidin beads overnight. Beads were centrifuged for 5 min at 500 g after which the supernatant was removed. Beads were subsequently washed 5 times with lysis buffer. Total protein lysates, or coprecipitates, were separated by SDS–PAGE and electrotransferred onto a nitrocellulose membrane. Blots were probed using the appropriate antibodies, and the immunoreactive proteins were detected using the Amersham Imager 680 (Cytiva, Austria).
4.9. Immunofluorescence Confocal Microscopy
After treatment with WABI, MM1R cells were fixed in 4% (v/v) formaldehyde. Cells were stained with rhodamine–phalloidin (Molecular Probes, Thermo Fisher Scientific Inc., Waltham, MA, USA) to visualize the cellular morphology, Hoechst 33,258 (Sigma-Aldrich, St. Louis, MO, USA) to visualize the nucleus and streptavidin-Alexa Fluor555 (Molecular Probes, Thermo Scientific, MA, USA) to visualize biotinylated WA. To determine WABI colocalization with specific proteins (BTK), WABI-treated MM1R cells were immunostained with primary (anti-rabbit BTK, Cell Signaling Technology, mAb#8547) and corresponding labeled secondary antibodies (anti-rabbit Alexa Fluor-488, Molecular Probes, Thermo Scientific, MA, USA). Cells were then placed on glass slides and mounted with coverslips using FluoromountTM (Sigma-Aldrich, St. Louis, MO, US)Confocal imaging was carried out by a laser-scanning microscope equipped with a Plan-Apochromat 63X/1.40 Oil DIC objective lens and excitation wavelengths 405, 488, 561 and 640 nm (Zeiss LSM 800, Carl Zeiss, Germany). At least 20 different microscopic fields were analyzed for each sample using ZEN imaging software (Carl Zeiss). ZEN liteTM (Carl Zeiss, Germany) was used to perform image reconstruction and presentation.
4.10. Covalent Docking of WA with BTK
In silico molecular docking studies of the BTK protein (PDB id: 6TFP) with WA (PubChem CID: 26537) were performed as previously described [109]. Briefly, BTK and WA structures were energy minimized with Swiss-PdbViewer (v4.1) [110] and UCSF Chimera (Geneva Biomedical Institute, Geneva, Switzerland) [111], respectively. Molecular docking and calculation of the docking scores were performed with Autodock4 (v4.2.6, University of California, Berkeley, CA, USA). The obtained docking solutions were evaluated based on their scoring, and the generated poses were clustered based on the ligand’s cluster ranks. Finally, the clusters were differentiated based on the covalent bond lengths and prime energies. Final docking results were visualized with LIGPLOT (v.4.5.3, University College London, UK) [74] and PyMOL (v.2.4, Schrödinger, NY, USA) [112] software.
4.11. Cell Transfections and BRET Measurements
BTK- and BTK(C481S)-NanoLuc® fusion vectors were purchased from Promega and used in BRET target engagement experiments. HEK-293 cells were transfected with the NanoLuc® fusion vectors using FuGENE HD (Promega, Madison, WI, USA) according to the manufacturer’s protocol. Briefly, fusion vectors were diluted into transfection carrier DNA (Promega, Madison, Wisconsin, USA) at a mass ratio of 1:10, after which FuGENE HD was added at a ratio of 1:3. The FuGENE HD complexes were then added to HEK-293 cells (1:20 ratio) at a density of 2 × 105 per mL. Subsequently, cells were plated onto white, 96-well plates (Corning, NY, USA) at a density of 2 × 104 cells/well and left to incubate for 24 h at 37 °C and 5% CO2. After incubation, cells were equilibrated for 2 h with the NanoBRET tracer reagent (K11, Promega, Madison, WI, USA) and increasing WA concentrations (0.5–10 µM). To measure BRET, NanoBRET NanoGlo substrate and extracellular NanoLuc inhibitor (Promega, Madison, WI, USA) was added according to the manufacturer’s protocol, and filtered luminescence was measured on a GloMax Discover luminometer equipped with 450 nm BP filter and 600 nm LP filter. MilliBRET units (mBU) were calculated by multiplying raw BRET values by 1000 and plotted with GraphPad Prism.
4.12. Nucleofection of siBTK and C481S BTK
The BTK(C481S)-NanoLuc® Fusion Vector was purchased from Promega, and siBTK was purchased from GE-Healthcare Bio-sciences (Accell Human BTK siRNA, EQ-003107–00–0005, 29121299, sequence supplied in Table S3). MM1R cells were transfected with siBTK alone or siBTK combined with BTK(C481S) using the Nucleofector IIb device (Lonza, Switzerland) according to the manufacturer’s instructions. In short, 2.106 MM1R cells were resuspended in supplemented nucleofector solution, after which 300 nM siBTK was added with or without 2 µg BTK(C481S). To each nucleofection reaction, an additional 2 µg of pmaxGFP™ Vector was added as an internal positive control (transfection efficiency = 51.3 ± 2.4%). Resuspended cells were subsequently transferred to a cuvette and transfected using the O-020 nucleofector program. After nucleofection, a pre-equilibrated medium was added, and cells were transferred to a 96-well plate at a density of 80.000 cells/well. Eight hours post-transfection, transfection efficiency and GFP expression were assessed with fluorescence microscopy. If a GFP signal was present, cells were treated with increasing concentrations of WA and cell viability was measured after 24 h using the MTT colorimetric method. BTK mRNA expression of nucleofected cells was further confirmed by quantitative real-time PCR.
5. Conclusions
Although cysteine code classification has been developed to explain the selectivity of different electrophiles classes, many cysteine-reactive natural products have shown promiscuous therapeutic anticancer activities with poorly characterized targets [87,113,114]. In a high-throughput compound library screening, the cysteine reactive phytochemical WA was identified as a top prioritized investigational compound to suppress hyperactivated BCR cancer signaling, together with IBR [63]. Given that WA possesses broad-spectrum therapeutic activities in several (drug-resistant) cancer cell types, including B-cell lymphoma and MM [46,51], we further characterized its molecular mechanisms of action in GC-resistant MM cells by combining innovative transcriptomic and phosphor-peptidomic kinase (activity) profiling approaches. Our findings demonstrate that the TK inhibitory profile of WA in GC-resistant MM cells is similar to that of IBR and that hyperphosphorylation of many BCR-related kinases, including BTK, is inhibited. Because WA also covalently targets BTK in a cysteine-dependent manner, we hypothesize that WA inhibits BTK autophosphorylation and activation in an IBR-like fashion. We additionally found that WA silences BTK expression in GC-resistant MM cells at the mRNA and protein level, suggesting that WA suppresses BTK cancer signaling via a dual mechanism. Since WA displays high a similarity with IBR mode of action, but with a more potent effect on cell death of GC-resistant MM cells, further structure-function analysis of WA-analogs may allow (pre)clinical optimization of highly effective withanolide BCR inhibitors to expand the arsenal of cancer drugs against therapy-resistant B-cell malignancies.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/cancers13071618/s1, Figure S1: Heatmap showing phosphorylation intensities of peptides serving as substrates for tyrosine kinases. Figure S2: MetaCore pathway analysis of differentially phosphorylated protein peptides (p < 0.05) in MM1R versus MM1S cells. Figure S3: Row-annotated heatmap representation of hyperactivated or inhibited kinases in MM1R versus MM1S cells. Figure S4: Target-binding affinities of Ibrutinib. Figure S5: Relative viability of U266 cells after 24 h treatment with WA or DEX. Figure S6: Relative BTK mRNA and protein levels of MM1R cells treated with IBR. Figure S7: Relative viability of MM1R cells treated with increasing concentrations of WA for 15 min. Figure S8: Relative BTK mRNA and protein expression of siBTK-transfected MM1R cells with or without the C481S BTK mutant. Figure S9: The biological targets of WA treatment strongly depends on cellular context. Table S1: Summary of the binding energy of the covalent bond of Cys481 with WA as predicted by covalent docking. Table S2: Overview of qPCR primers used in this study. Table S3: Overview of siRNA sequences used in this study. Table S3: Overview of siRNA sequences used in this study. Table S4: Original images western immunoblots.
Author Contributions
Conceptualization, E.L., C.S.C., C.P.-N. and W.V.B.; methodology, E.L., C.S.C., A.P., S.R., B.C., N.D.N., F.M.H.U.T., P.S. and N.K.V.; software, C.P.-N., K.D., B.C. and N.D.N.; validation, E.L. and C.S.C.; formal analysis, E.L., C.S.C., K.D. and C.P.-N.; investigation, E.L., C.S.C. and W.V.B.; resources, W.V.B. and A.B.; data curation, E.L., K.D., B.C. and N.D.N.; writing—original draft preparation, E.L.; writing—review and editing, E.L., P.V.V., X.V.O., C.P.-N., S.R., N.K.V., P.S., F.M.H.U.T., A.B. and W.V.B.; visualization, E.L. and K.D.; supervision, K.L. and W.V.B.; project administration, W.V.B., F.O., P.V.V., X.V.O.; funding acquisition, E.L. and W.V.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Foundation Against Cancer (Belgium), grant number 7872, Hercules Foundation, grant number AUHA/13/012 and Research Foundation Flanders, grant number FWO G1179120N, G059713N. N.K.V. acknowledges funding support from the Singapore Ministry of Education (MOE) under its MOE Academic Research Fund (AcRF) Tier 2 Grant (grant number MOE2017-T2-2-004) and the National Research Foundation Singapore under its Open Fund Large Collaborative Grant (grant number OFLCG18May-0028) and administered by the Singapore Ministry of Health’s National Medical Research Council (NMRC).
Data Availability Statement
The data presented in this study are openly available in the NCBI GEO database, GEO accession number GSE162475.
Acknowledgments
The authors thank Eva Lion, Head of Tumor Immunology Group of the Laboratory of Experimental Hematology (University of Antwerp), for kindly providing GC-resistant U266 cells.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef]
- Hussein, M.A. Multiple myeloma: Most common end-organ damage and management. J. Natl. Compr. Cancer Netw. 2007, 5, 170–178. [Google Scholar] [CrossRef]
- Robak, P.; Drozdz, I.; Szemraj, J.; Robak, T. Drug resistance in multiple myeloma. Cancer Treat. Rev. 2018, 70, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V. Myeloma today: Disease definitions and treatment advances. Am. J. Hematol. 2016, 91, 90–100. [Google Scholar] [CrossRef]
- Yang, W.C.; Lin, S.F. Mechanisms of Drug Resistance in Relapse and Refractory Multiple Myeloma. Biomed. Res. Int. 2015, 2015, 341430. [Google Scholar] [CrossRef] [PubMed]
- Ito, S. Proteasome Inhibitors for the Treatment of Multiple Myeloma. Cancers 2020, 12, 265. [Google Scholar] [CrossRef]
- Abe, Y.; Ishida, T. Immunomodulatory drugs in the treatment of multiple myeloma. Jpn. J. Clin. Oncol. 2019, 49, 695–702. [Google Scholar] [CrossRef]
- Burwick, N.; Sharma, S. Glucocorticoids in multiple myeloma: Past, present, and future. Ann. Hematol. 2019, 98, 19–28. [Google Scholar] [CrossRef]
- Thanendrarajan, S.; Davies, F.E.; Morgan, G.J.; Schinke, C.; Mathur, P.; Heuck, C.J.; Zangari, M.; Epstein, J.; Yaccoby, S.; Weinhold, N.; et al. Monoclonal antibody therapy in multiple myeloma: Where do we stand and where are we going? Immunotherapy 2016, 8, 367–384. [Google Scholar] [CrossRef]
- Imai, Y.; Hirano, M.; Kobayashi, M.; Futami, M.; Tojo, A. HDAC Inhibitors Exert Anti-Myeloma Effects through Multiple Modes of Action. Cancers 2019, 11, 475. [Google Scholar] [CrossRef]
- Kumar, S.K.; Therneau, T.M.; Gertz, M.A.; Lacy, M.Q.; Dispenzieri, A.; Rajkumar, S.V.; Fonseca, R.; Witzig, T.E.; Lust, J.A.; Larson, D.R.; et al. Clinical course of patients with relapsed multiple myeloma. Mayo Clin. Proc. 2004, 79, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Issa, M.E.; Takhsha, F.S.; Chirumamilla, C.S.; Perez-Novo, C.; Vanden Berghe, W.; Cuendet, M. Epigenetic strategies to reverse drug resistance in heterogeneous multiple myeloma. Clin. Epigenet. 2017, 9, 17. [Google Scholar] [CrossRef] [PubMed]
- Shain, K.H.; Tao, J. The B-cell receptor orchestrates environment-mediated lymphoma survival and drug resistance in B-cell malignancies. Oncogene 2014, 33, 4107–4113. [Google Scholar] [CrossRef]
- Woyach, J.A.; Johnson, A.J.; Byrd, J.C. The B-cell receptor signaling pathway as a therapeutic target in CLL. Blood 2012, 120, 1175–1184. [Google Scholar] [CrossRef]
- Takata, M.; Sabe, H.; Hata, A.; Inazu, T.; Homma, Y.; Nukada, T.; Yamamura, H.; Kurosaki, T. Tyrosine kinases Lyn and Syk regulate B cell receptor-coupled Ca2+ mobilization through distinct pathways. EMBO J. 1994, 13, 1341–1349. [Google Scholar] [CrossRef]
- Tai, Y.T.; Chang, B.Y.; Kong, S.Y.; Fulciniti, M.; Yang, G.; Calle, Y.; Hu, Y.; Lin, J.; Zhao, J.J.; Cagnetta, A.; et al. Bruton tyrosine kinase inhibition is a novel therapeutic strategy targeting tumor in the bone marrow microenvironment in multiple myeloma. Blood 2012, 120, 1877–1887. [Google Scholar] [CrossRef]
- Yang, Y.; Shi, J.; Gu, Z.; Salama, M.E.; Das, S.; Wendlandt, E.; Xu, H.; Huang, J.; Tao, Y.; Hao, M.; et al. Bruton tyrosine kinase is a therapeutic target in stem-like cells from multiple myeloma. Cancer Res. 2015, 75, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wei, R.; Liu, S.; Qiao, L.; Hou, J.; Gu, C.; Yang, Y. BTK induces CAM-DR through regulation of CXCR4 degradation in multiple myeloma. Am. J. Transl. Res. 2019, 11, 4139–4150. [Google Scholar] [PubMed]
- Murray, M.Y.; Zaitseva, L.; Auger, M.J.; Craig, J.I.; MacEwan, D.J.; Rushworth, S.A.; Bowles, K.M. Ibrutinib inhibits BTK-driven NF-kappaB p65 activity to overcome bortezomib-resistance in multiple myeloma. Cell Cycle 2015, 14, 2367–2375. [Google Scholar] [CrossRef] [PubMed]
- Young, R.M.; Staudt, L.M. Targeting pathological B cell receptor signalling in lymphoid malignancies. Nat. Rev. Drug Discov. 2013, 12, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Dasmahapatra, G.; Patel, H.; Dent, P.; Fisher, R.I.; Friedberg, J.; Grant, S. The Bruton tyrosine kinase (BTK) inhibitor PCI-32765 synergistically increases proteasome inhibitor activity in diffuse large-B cell lymphoma (DLBCL) and mantle cell lymphoma (MCL) cells sensitive or resistant to bortezomib. Br. J. Haematol. 2013, 161, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Herman, S.E.; Gordon, A.L.; Hertlein, E.; Ramanunni, A.; Zhang, X.; Jaglowski, S.; Flynn, J.; Jones, J.; Blum, K.A.; Buggy, J.J.; et al. Bruton tyrosine kinase represents a promising therapeutic target for treatment of chronic lymphocytic leukemia and is effectively targeted by PCI-32765. Blood 2011, 117, 6287–6296. [Google Scholar] [CrossRef]
- Herman, S.E.; Sun, X.; McAuley, E.M.; Hsieh, M.M.; Pittaluga, S.; Raffeld, M.; Liu, D.; Keyvanfar, K.; Chapman, C.M.; Chen, J.; et al. Modeling tumor-host interactions of chronic lymphocytic leukemia in xenografted mice to study tumor biology and evaluate targeted therapy. Leukemia 2013, 27, 2311–2321. [Google Scholar] [CrossRef]
- Yang, Y.; Shaffer, A.L., 3rd; Emre, N.C.; Ceribelli, M.; Zhang, M.; Wright, G.; Xiao, W.; Powell, J.; Platig, J.; Kohlhammer, H.; et al. Exploiting synthetic lethality for the therapy of ABC diffuse large B cell lymphoma. Cancer Cell 2012, 21, 723–737. [Google Scholar] [CrossRef]
- Franco, L.M.; Gadkari, M.; Howe, K.N.; Sun, J.; Kardava, L.; Kumar, P.; Kumari, S.; Hu, Z.; Fraser, I.D.C.; Moir, S.; et al. Immune regulation by glucocorticoids can be linked to cell type-dependent transcriptional responses. J. Exp. Med. 2019, 216, 384–406. [Google Scholar] [CrossRef]
- Kruth, K.A.; Fang, M.; Shelton, D.N.; Abu-Halawa, O.; Mahling, R.; Yang, H.; Weissman, J.S.; Loh, M.L.; Muschen, M.; Tasian, S.K.; et al. Suppression of B-cell development genes is key to glucocorticoid efficacy in treatment of acute lymphoblastic leukemia. Blood 2017, 129, 3000–3008. [Google Scholar] [CrossRef]
- Shi, Y.; Wang, G.; Muhowski, E.M.; McCaw, L.; Wang, C.; Bjarnason, G.; Woyach, J.A.; Spaner, D.E. Ibrutinib reprograms the glucocorticoid receptor in chronic lymphocytic leukemia cells. Leukemia 2019, 33, 1650–1662. [Google Scholar] [CrossRef] [PubMed]
- Manzoni, D.; Catallo, R.; Chebel, A.; Baseggio, L.; Michallet, A.S.; Roualdes, O.; Magaud, J.P.; Salles, G.; Ffrench, M. The ibrutinib B-cell proliferation inhibition is potentiated in vitro by dexamethasone: Application to chronic lymphocytic leukemia. Leuk Res. 2016, 47, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Chari, A.; Larson, S.; Holkova, B.; Cornell, R.F.; Gasparetto, C.; Karanes, C.; Matous, J.V.; Niesvizky, R.; Valent, J.; Lunning, M.; et al. Phase 1 trial of ibrutinib and carfilzomib combination therapy for relapsed or relapsed and refractory multiple myeloma. Leuk Lymphoma 2018, 59, 2588–2594. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Bensinger, W.I.; Huff, C.A.; Costello, C.L.; Lendvai, N.; Berdeja, J.G.; Anderson, L.D., Jr.; Siegel, D.S.; Lebovic, D.; Jagannath, S.; et al. Ibrutinib alone or with dexamethasone for relapsed or relapsed and refractory multiple myeloma: Phase 2 trial results. Br. J. Haematol. 2018, 180, 821–830. [Google Scholar] [CrossRef]
- Chi, J.; Park, J.; Saif, M.W. Ibrutinib-Induced Vasculitis in a Patient with Metastatic Colon Cancer Treated in Combination with Cetuximab. Case Rep. Oncol. Med. 2020, 2020, 6154213. [Google Scholar] [CrossRef]
- Burger, J.A.; Tedeschi, A.; Barr, P.M.; Robak, T.; Owen, C.; Ghia, P.; Bairey, O.; Hillmen, P.; Bartlett, N.L.; Li, J.; et al. Ibrutinib as Initial Therapy for Patients with Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2015, 373, 2425–2437. [Google Scholar] [CrossRef]
- Guha, A.; Derbala, M.H.; Zhao, Q.; Wiczer, T.E.; Woyach, J.A.; Byrd, J.C.; Awan, F.T.; Addison, D. Ventricular Arrhythmias Following Ibrutinib Initiation for Lymphoid Malignancies. J. Am. Coll. Cardiol. 2018, 72, 697–698. [Google Scholar] [CrossRef] [PubMed]
- Salem, J.E.; Manouchehri, A.; Bretagne, M.; Lebrun-Vignes, B.; Groarke, J.D.; Johnson, D.B.; Yang, T.; Reddy, N.M.; Funck-Brentano, C.; Brown, J.R.; et al. Cardiovascular Toxicities Associated with Ibrutinib. J. Am. Coll. Cardiol. 2019, 74, 1667–1678. [Google Scholar] [CrossRef]
- Barf, T.; Covey, T.; Izumi, R.; van de Kar, B.; Gulrajani, M.; van Lith, B.; van Hoek, M.; de Zwart, E.; Mittag, D.; Demont, D.; et al. Acalabrutinib (ACP-196): A Covalent Bruton Tyrosine Kinase Inhibitor with a Differentiated Selectivity and In Vivo Potency Profile. J. Pharmacol. Exp. Ther. 2017, 363, 240–252. [Google Scholar] [CrossRef]
- George, B.; Chowdhury, S.M.; Hart, A.; Sircar, A.; Singh, S.K.; Nath, U.K.; Mamgain, M.; Singhal, N.K.; Sehgal, L.; Jain, N. Ibrutinib Resistance Mechanisms and Treatment Strategies for B-Cell lymphomas. Cancers 2020, 12, 1328. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Liu, C.; Tsui, S.T.; Liu, D. Second-generation inhibitors of Bruton tyrosine kinase. J. Hematol. Oncol. 2016, 9, 80. [Google Scholar] [CrossRef] [PubMed]
- Woyach, J.; Huang, Y.; Rogers, K.; Bhat, S.A.; Grever, M.R.; Lozanski, A.; Doong, T.-J.; Blachly, J.S.; Lozanski, G.; Jones, D.; et al. Resistance to Acalabrutinib in CLL Is Mediated Primarily By BTK Mutations. Blood 2019, 134, 504. [Google Scholar] [CrossRef]
- Mathews Griner, L.A.; Guha, R.; Shinn, P.; Young, R.M.; Keller, J.M.; Liu, D.; Goldlust, I.S.; Yasgar, A.; McKnight, C.; Boxer, M.B.; et al. High-throughput combinatorial screening identifies drugs that cooperate with ibrutinib to kill activated B-cell-like diffuse large B-cell lymphoma cells. Proc. Natl. Acad. Sci. USA 2014, 111, 2349–2354. [Google Scholar] [CrossRef]
- Lucas, D.M.; Still, P.C.; Perez, L.B.; Grever, M.R.; Kinghorn, A.D. Potential of plant-derived natural products in the treatment of leukemia and lymphoma. Curr. Drug Targets 2010, 11, 812–822. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; Supuran, C.T.; International Natural Product Sciences Taskforce. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021, 20, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Hassannia, B.; Logie, E.; Vandenabeele, P.; Vanden Berghe, T.; Vanden Berghe, W. Withaferin A: From ayurvedic folk medicine to preclinical anti-cancer drug. Biochem. Pharmacol. 2020, 173, 113602. [Google Scholar] [CrossRef] [PubMed]
- Dom, M.; Offner, F.; Vanden Berghe, W.; Van Ostade, X. Proteomic characterization of Withaferin A-targeted protein networks for the treatment of monoclonal myeloma gammopathies. J. Proteom. 2018, 179, 17–29. [Google Scholar] [CrossRef]
- Issa, M.E.; Cuendet, M. Withaferin A induces cell death and differentiation in multiple myeloma cancer stem cells. Medchemcomm 2017, 8, 112–121. [Google Scholar] [CrossRef] [PubMed]
- McKenna, M.K.; Gachuki, B.W.; Alhakeem, S.S.; Oben, K.N.; Rangnekar, V.M.; Gupta, R.C.; Bondada, S. Anti-cancer activity of withaferin A in B-cell lymphoma. Cancer Biol. Ther. 2015, 16, 1088–1098. [Google Scholar] [CrossRef] [PubMed]
- Heyninck, K.; Lahtela-Kakkonen, M.; Van der Veken, P.; Haegeman, G.; Vanden Berghe, W. Withaferin A inhibits NF-kappaB activation by targeting cysteine 179 in IKKbeta. Biochem. Pharmacol. 2014, 91, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Hahm, E.R.; Lee, J.; Singh, S.V. Role of mitogen-activated protein kinases and Mcl-1 in apoptosis induction by withaferin A in human breast cancer cells. Mol. Carcinog. 2014, 53, 907–916. [Google Scholar] [CrossRef] [PubMed]
- Grogan, P.T.; Sleder, K.D.; Samadi, A.K.; Zhang, H.; Timmermann, B.N.; Cohen, M.S. Cytotoxicity of withaferin A in glioblastomas involves induction of an oxidative stress-mediated heat shock response while altering Akt/mTOR and MAPK signaling pathways. Investig. New Drugs 2013, 31, 545–557. [Google Scholar] [CrossRef]
- Mandal, C.; Dutta, A.; Mallick, A.; Chandra, S.; Misra, L.; Sangwan, R.S.; Mandal, C. Withaferin A induces apoptosis by activating p38 mitogen-activated protein kinase signaling cascade in leukemic cells of lymphoid and myeloid origin through mitochondrial death cascade. Apoptosis 2008, 13, 1450–1464. [Google Scholar] [CrossRef]
- Oh, J.H.; Lee, T.J.; Kim, S.H.; Choi, Y.H.; Lee, S.H.; Lee, J.M.; Kim, Y.H.; Park, J.W.; Kwon, T.K. Induction of apoptosis by withaferin A in human leukemia U937 cells through down-regulation of Akt phosphorylation. Apoptosis 2008, 13, 1494–1504. [Google Scholar] [CrossRef] [PubMed]
- Palagani, A.; Op de Beeck, K.; Naulaerts, S.; Diddens, J.; Sekhar Chirumamilla, C.; Van Camp, G.; Laukens, K.; Heyninck, K.; Gerlo, S.; Mestdagh, P.; et al. Ectopic microRNA-150-5p transcription sensitizes glucocorticoid therapy response in MM1S multiple myeloma cells but fails to overcome hormone therapy resistance in MM1R cells. PLoS ONE 2014, 9, e113842. [Google Scholar] [CrossRef]
- Greenstein, S.; Krett, N.L.; Kurosawa, Y.; Ma, C.; Chauhan, D.; Hideshima, T.; Anderson, K.C.; Rosen, S.T. Characterization of the MM.1 human multiple myeloma (MM) cell lines: A model system to elucidate the characteristics, behavior, and signaling of steroid-sensitive and -resistant MM cells. Exp. Hematol. 2003, 31, 271–282. [Google Scholar] [CrossRef]
- Hilhorst, R.; Houkes, L.; van den Berg, A.; Ruijtenbeek, R. Peptide microarrays for detailed, high-throughput substrate identification, kinetic characterization, and inhibition studies on protein kinase A. Anal. Biochem. 2009, 387, 150–161. [Google Scholar] [CrossRef] [PubMed]
- Chirumamilla, C.S.; Fazil, M.; Perez-Novo, C.; Rangarajan, S.; de Wijn, R.; Ramireddy, P.; Verma, N.K.; Vanden Berghe, W. Profiling Activity of Cellular Kinases in Migrating T-Cells. Methods Mol. Biol. 2019, 1930, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Chauhan, D.; Auclair, D.; Robinson, E.K.; Hideshima, T.; Li, G.; Podar, K.; Gupta, D.; Richardson, P.; Schlossman, R.L.; Krett, N.; et al. Identification of genes regulated by dexamethasone in multiple myeloma cells using oligonucleotide arrays. Oncogene 2002, 21, 1346–1358. [Google Scholar] [CrossRef]
- Berglof, A.; Hamasy, A.; Meinke, S.; Palma, M.; Krstic, A.; Mansson, R.; Kimby, E.; Osterborg, A.; Smith, C.I. Targets for Ibrutinib Beyond B Cell Malignancies. Scand. J. Immunol. 2015, 82, 208–217. [Google Scholar] [CrossRef]
- Cheng, S.; Ma, J.; Guo, A.; Lu, P.; Leonard, J.P.; Coleman, M.; Liu, M.; Buggy, J.J.; Furman, R.R.; Wang, Y.L. BTK inhibition targets in vivo CLL proliferation through its effects on B-cell receptor signaling activity. Leukemia 2014, 28, 649–657. [Google Scholar] [CrossRef]
- Patel, V.; Balakrishnan, K.; Bibikova, E.; Ayres, M.; Keating, M.J.; Wierda, W.G.; Gandhi, V. Comparison of Acalabrutinib, A Selective Bruton Tyrosine Kinase Inhibitor, with Ibrutinib in Chronic Lymphocytic Leukemia Cells. Clin. Cancer Res. 2017, 23, 3734–3743. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Lee, S.; Shah, T.; Yin, C.; Barth, M.; Miles, R.R.; Ayello, J.; Morris, E.; Harrison, L.; Van de Ven, C.; et al. Ibrutinib significantly inhibited Bruton’s tyrosine kinase (BTK) phosphorylation,in-vitro proliferation and enhanced overall survival in a preclinical Burkitt lymphoma (BL) model. Oncoimmunology 2019, 8, e1512455. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.; Scheerens, H.; Li, S.-J.; Schultz, B.E.; Sprengeler, P.A.; Burrill, L.C.; Mendonca, R.V.; Sweeney, M.D.; Scott, K.C.K.; Grothaus, P.G.; et al. Discovery of Selective Irreversible Inhibitors for Bruton’s Tyrosine Kinase. ChemMedChem 2007, 2, 58–61. [Google Scholar] [CrossRef]
- Vanden Berghe, W.; Sabbe, L.; Kaileh, M.; Haegeman, G.; Heyninck, K. Molecular insight in the multifunctional activities of Withaferin A. Biochem. Pharmacol. 2012, 84, 1282–1291. [Google Scholar] [CrossRef]
- Grover, A.; Shandilya, A.; Agrawal, V.; Pratik, P.; Bhasme, D.; Bisaria, V.S.; Sundar, D. Hsp90/Cdc37 chaperone/co-chaperone complex, a novel junction anticancer target elucidated by the mode of action of herbal drug Withaferin A. BMC Bioinform. 2011, 12 (Suppl. S1), S30. [Google Scholar] [CrossRef]
- Bradshaw, J.M.; McFarland, J.M.; Paavilainen, V.O.; Bisconte, A.; Tam, D.; Phan, V.T.; Romanov, S.; Finkle, D.; Shu, J.; Patel, V.; et al. Prolonged and tunable residence time using reversible covalent kinase inhibitors. Nat. Chem. Biol. 2015, 11, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.U.; Grossmann, T.N. Reversible covalent inhibition of a protein target. Angew. Chem. Int. Ed. Engl. 2012, 51, 8699–8700. [Google Scholar] [CrossRef] [PubMed]
- Serafimova, I.M.; Pufall, M.A.; Krishnan, S.; Duda, K.; Cohen, M.S.; Maglathlin, R.L.; McFarland, J.M.; Miller, R.M.; Frodin, M.; Taunton, J. Reversible targeting of noncatalytic cysteines with chemically tuned electrophiles. Nat. Chem. Biol. 2012, 8, 471–476. [Google Scholar] [CrossRef]
- Leproult, E.; Barluenga, S.; Moras, D.; Wurtz, J.M.; Winssinger, N. Cysteine mapping in conformationally distinct kinase nucleotide binding sites: Application to the design of selective covalent inhibitors. J. Med. Chem. 2011, 54, 1347–1355. [Google Scholar] [CrossRef]
- Chaikuad, A.; Koch, P.; Laufer, S.A.; Knapp, S. The Cysteinome of Protein Kinases as a Target in Drug Development. Angew. Chem. Int. Ed. 2018, 57, 4372–4385. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Gray, N.S. Rational design of inhibitors that bind to inactive kinase conformations. Nat. Chem. Biol. 2006, 2, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, P.L.; Gray, N.S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Sabnis, Y.; Zhao, Z.; Zhang, T.; Buhrlage, S.J.; Jones, L.H.; Gray, N.S. Developing irreversible inhibitors of the protein kinase cysteinome. Chem. Biol. 2013, 20, 146–159. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Gu, C.; Peng, H.; Lu, Y.; Yang, H.; Tian, Z.; Yin, G.; Zhang, W.; Lu, S.; Zhang, Y.; Yang, Y. BTK suppresses myeloma cellular senescence through activating AKT/P27/Rb signaling. Oncotarget 2017, 8, 56858–56867. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Bourne, P.E. Progress with covalent small-molecule kinase inhibitors. Drug Discov. Today 2018, 23, 727–735. [Google Scholar] [CrossRef]
- Rao, S.; Gurbani, D.; Du, G.; Everley, R.A.; Browne, C.M.; Chaikuad, A.; Tan, L.; Schroder, M.; Gondi, S.; Ficarro, S.B.; et al. Leveraging Compound Promiscuity to Identify Targetable Cysteines within the Kinome. Cell Chem. Biol. 2019, 26, 818–829.e9. [Google Scholar] [CrossRef] [PubMed]
- Prasanna, K.S.P.; Bharathi, P.S. Withaferin A suppresses the expression of vascular endothelial growth factor in Ehrlich ascites tumor cells via Sp1 transcription factor. Curr. Trends Biotechnol. Pharm. 2010, 1, 138–148. [Google Scholar]
- Bottoni, A.; Rizzotto, L.; Lai, T.H.; Liu, C.; Smith, L.L.; Mantel, R.; Reiff, S.; El-Gamal, D.; Larkin, K.; Johnson, A.J.; et al. Targeting BTK through microRNA in chronic lymphocytic leukemia. Blood 2016, 128, 3101–3112. [Google Scholar] [CrossRef] [PubMed]
- Terzi, H.E.M. In vitro comparison of cytotoxic effects of bortezomib resistant U266 myeloma cell line (U266/VELR) on combination of ibrutinib with carfilzomib and lenalidomid drugs. Cumhur. Med. J. 2019, 41, 698–702. [Google Scholar] [CrossRef]
- Kraus, J.; Kraus, M.; Liu, N.; Besse, L.; Bader, J.; Geurink, P.P.; de Bruin, G.; Kisselev, A.F.; Overkleeft, H.; Driessen, C. The novel beta2-selective proteasome inhibitor LU-102 decreases phosphorylation of I kappa B and induces highly synergistic cytotoxicity in combination with ibrutinib in multiple myeloma cells. Cancer Chemother. Pharmacol. 2015, 76, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Gong, W.; Liu, S.; Li, Q.; Guo, M.; Wang, J.; Wang, S.; Chen, N.; Wang, Y.; Liu, Q.; et al. Ibrutinib targets microRNA-21 in multiple myeloma cells by inhibiting NF-kappaB and STAT3. Tumour. Biol. 2018, 40, 1010428317731369. [Google Scholar] [CrossRef]
- Rushworth, S.A.; Bowles, K.M.; Barrera, L.N.; Murray, M.Y.; Zaitseva, L.; MacEwan, D.J. BTK inhibitor ibrutinib is cytotoxic to myeloma and potently enhances bortezomib and lenalidomide activities through NF-kappaB. Cell Signal 2013, 25, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Lindvall, J.M.; Blomberg, K.E.; Berglof, A.; Yang, Q.; Smith, C.I.; Islam, T.C. Gene expression profile of B cells from Xid mice and Btk knockout mice. Eur. J. Immunol. 2004, 34, 1981–1991. [Google Scholar] [CrossRef]
- Roman-Garcia, S.; Merino-Cortes, S.V.; Gardeta, S.R.; de Bruijn, M.J.W.; Hendriks, R.W.; Carrasco, Y.R. Distinct Roles for Bruton’s Tyrosine Kinase in B Cell Immune Synapse Formation. Front. Immunol. 2018, 9, 2027. [Google Scholar] [CrossRef] [PubMed]
- Chirumamilla, C.S.; Perez-Novo, C.; Van Ostade, X.; Vanden Berghe, W. Molecular insights into cancer therapeutic effects of the dietary medicinal phytochemical withaferin A. Proc. Nutr. Soc. 2017, 76, 96–105. [Google Scholar] [CrossRef]
- Grossman, E.A.; Ward, C.C.; Spradlin, J.N.; Bateman, L.A.; Huffman, T.R.; Miyamoto, D.K.; Kleinman, J.I.; Nomura, D.K. Covalent Ligand Discovery against Druggable Hotspots Targeted by Anti-cancer Natural Products. Cell Chem. Biol. 2017, 24, 1368–1376.e4. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, H.; Yasuda, T.; Aiba, Y.; Sanjo, H.; Hamadate, M.; Watarai, H.; Sakurai, H.; Kurosaki, T. PKC beta regulates BCR-mediated IKK activation by facilitating the interaction between TAK1 and CARMA1. J. Exp. Med. 2005, 202, 1423–1431. [Google Scholar] [CrossRef]
- Mohammad, D.K.; Nore, B.F.; Smith, C.I.E. Terminating B cell receptor signaling. Oncotarget 2017, 8, 109857–109858. [Google Scholar] [CrossRef] [PubMed]
- Franks, S.E.; Cambier, J.C. Putting on the Brakes: Regulatory Kinases and Phosphatases Maintaining B Cell Anergy. Front. Immunol. 2018, 9, 665. [Google Scholar] [CrossRef] [PubMed]
- Dom, M.; Vanden Berghe, W.; Van Ostade, X. Broad-spectrum antitumor properties of Withaferin A: A proteomic perspective. RSC Med. Chem. 2020, 11, 30–50. [Google Scholar] [CrossRef] [PubMed]
- Widodo, N.; Priyandoko, D.; Shah, N.; Wadhwa, R.; Kaul, S.C. Selective killing of cancer cells by Ashwagandha leaf extract and its component Withanone involves ROS signaling. PLoS ONE 2010, 5, e13536. [Google Scholar] [CrossRef] [PubMed]
- Pires, N.; Gota, V.; Gulia, A.; Hingorani, L.; Agarwal, M.; Puri, A. Safety and pharmacokinetics of Withaferin-A in advanced stage high grade osteosarcoma: A phase I trial. J. Ayurveda Integr. Med. 2020, 11, 68–72. [Google Scholar] [CrossRef]
- Patel, S.B.; Rao, N.J.; Hingorani, L.L. Safety assessment of Withania somnifera extract standardized for Withaferin A: Acute and sub-acute toxicity study. J. Ayurveda Integr. Med. 2016, 7, 30–37. [Google Scholar] [CrossRef]
- Thaiparambil, J.T.; Bender, L.; Ganesh, T.; Kline, E.; Patel, P.; Liu, Y.; Tighiouart, M.; Vertino, P.M.; Harvey, R.D.; Garcia, A.; et al. Withaferin A inhibits breast cancer invasion and metastasis at sub-cytotoxic doses by inducing vimentin disassembly and serine 56 phosphorylation. Int. J. Cancer 2011, 129, 2744–2755. [Google Scholar] [CrossRef]
- Smith, P.K.; Krohn, R.I.; Hermanson, G.T.; Mallia, A.K.; Gartner, F.H.; Provenzano, M.D.; Fujimoto, E.K.; Goeke, N.M.; Olson, B.J.; Klenk, D.C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985, 150, 76–85. [Google Scholar] [CrossRef]
- Hilhorst, R.; Houkes, L.; Mommersteeg, M.; Musch, J.; van den Berg, A.; Ruijtenbeek, R. Peptide microarrays for profiling of serine/threonine kinase activity of recombinant kinases and lysates of cells and tissue samples. Methods Mol. Biol. 2013, 977, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Folkvord, S.; Flatmark, K.; Dueland, S.; de Wijn, R.; Groholt, K.K.; Hole, K.H.; Nesland, J.M.; Ruijtenbeek, R.; Boender, P.J.; Johansen, M.; et al. Prediction of response to preoperative chemoradiotherapy in rectal cancer by multiplex kinase activity profiling. Int. J. Radiat. Oncol. Biol. Phys. 2010, 78, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Maat, W.; el Filali, M.; Dirks-Mulder, A.; Luyten, G.P.; Gruis, N.A.; Desjardins, L.; Boender, P.; Jager, M.J.; van der Velden, P.A. Episodic Src activation in uveal melanoma revealed by kinase activity profiling. Br. J. Cancer 2009, 101, 312–319. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Safaei, J.; Manuch, J.; Gupta, A.; Stacho, L.; Pelech, S. Prediction of 492 human protein kinase substrate specificities. Proteome Sci. 2011, 9 (Suppl. S1), S6. [Google Scholar] [CrossRef]
- Versele, M.; Talloen, W.; Rockx, C.; Geerts, T.; Janssen, B.; Lavrijssen, T.; King, P.; Gohlmann, H.W.; Page, M.; Perera, T. Response prediction to a multitargeted kinase inhibitor in cancer cell lines and xenograft tumors using high-content tyrosine peptide arrays with a kinetic readout. Mol. Cancer Ther. 2009, 8, 1846–1855. [Google Scholar] [CrossRef] [PubMed]
- Kinexus|PhosphoNET. Available online: http://www.phosphonet.ca/ (accessed on 21 May 2019).
- IPA, Q. Ingenuity Pathway Analysis. Available online: http://www.ingenuity.com/products/ipa (accessed on 15 September 2020).
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data; Babraham Bioinformatics, Babraham Institute: Cambridge, UK, 2010. [Google Scholar]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Thomas, P.D.; Campbell, M.J.; Kejariwal, A.; Mi, H.; Karlak, B.; Daverman, R.; Diemer, K.; Muruganujan, A.; Narechania, A. PANTHER: A library of protein families and subfamilies indexed by function. Genome Res. 2003, 13, 2129–2141. [Google Scholar] [CrossRef]
- Szarc vel Szic, K.; Op de Beeck, K.; Ratman, D.; Wouters, A.; Beck, I.M.; Declerck, K.; Heyninck, K.; Fransen, E.; Bracke, M.; De Bosscher, K.; et al. Pharmacological levels of Withaferin A (Withania somnifera) trigger clinically relevant anticancer effects specific to triple negative breast cancer cells. PLoS ONE 2014, 9, e87850. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Heyninck, K.; Sabbe, L.; Chirumamilla, C.S.; Szarc Vel Szic, K.; Vander Veken, P.; Lemmens, K.J.A.; Lahtela-Kakkonen, M.; Naulaerts, S.; Op de Beeck, K.; Laukens, K.; et al. Withaferin A induces heme oxygenase (HO-1) expression in endothelial cells via activation of the Keap1/Nrf2 pathway. Biochem. Pharmacol. 2016, 109, 48–61. [Google Scholar] [CrossRef]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- PyMOL. The PyMOL Molecular Graphics System, Version 2.0; PyMOL: Schrödinger, NY, USA, 2017. [Google Scholar]
- Dayalan Naidu, S.; Dinkova-Kostova, A.T. KEAP1, a cysteine-based sensor and a drug target for the prevention and treatment of chronic disease. Open Biol. 2020, 10, 200105. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Li, L.; Iwamoto, N.; Nakajima-Takagi, Y.; Kaneko, H.; Nakayama, Y.; Eguchi, M.; Wada, Y.; Kumagai, Y.; Yamamoto, M. The antioxidant defense system Keap1-Nrf2 comprises a multiple sensing mechanism for responding to a wide range of chemical compounds. Mol. Cell Biol. 2009, 29, 493–502. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).