
Ex Vivo High Salt Activated Tumor-Primed CD4+T Lymphocytes Exert a Potent Anti-Cancer Response

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Cell Lines
2.2. Isolation and Ex Vivo Activation of CD4+ T-cell
2.3. FACS Analysis and Intracellular Staining
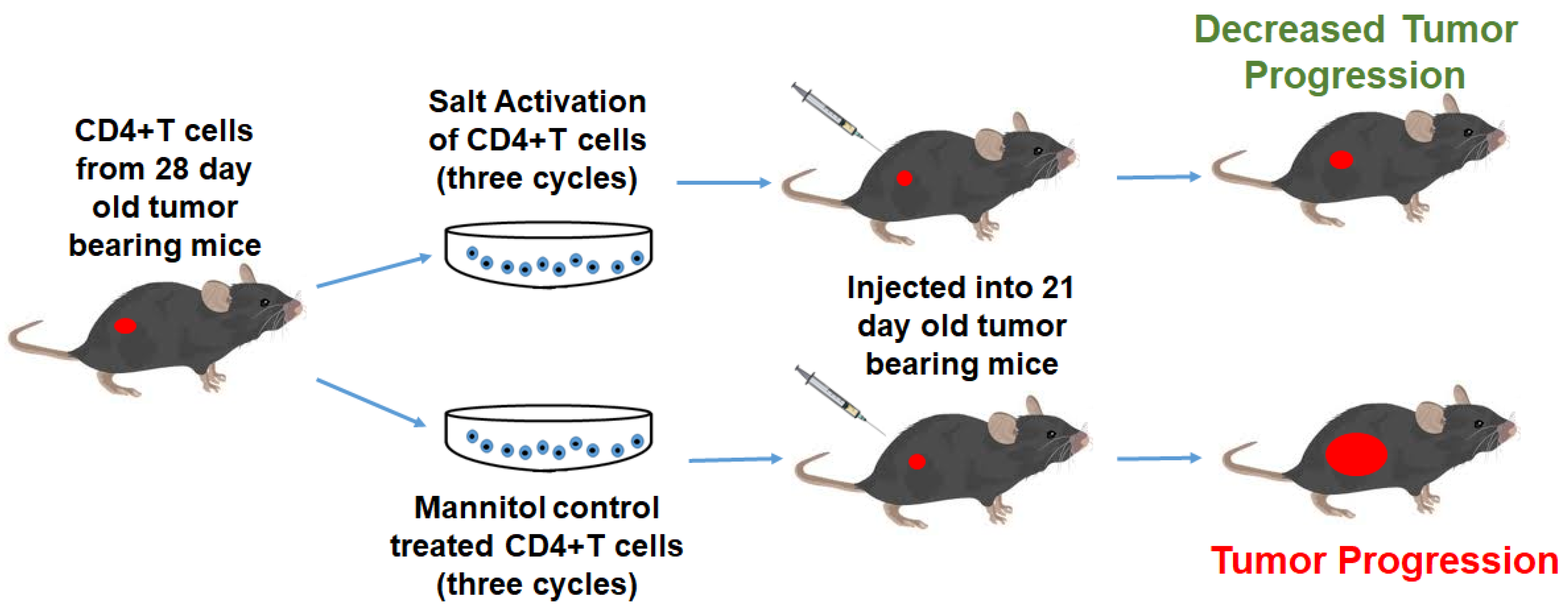
2.4. Adoptive Immunotherapy
2.5. Quantitative Real Time Polymerase Chain Reaction
2.6. Western Blot
2.7. Cytokine Assay
2.8. Cytotoxicity Assay
2.9. Cell Proliferation Assay
2.10. Metabolic Assays
2.11. Statistical Analysis
3. Results
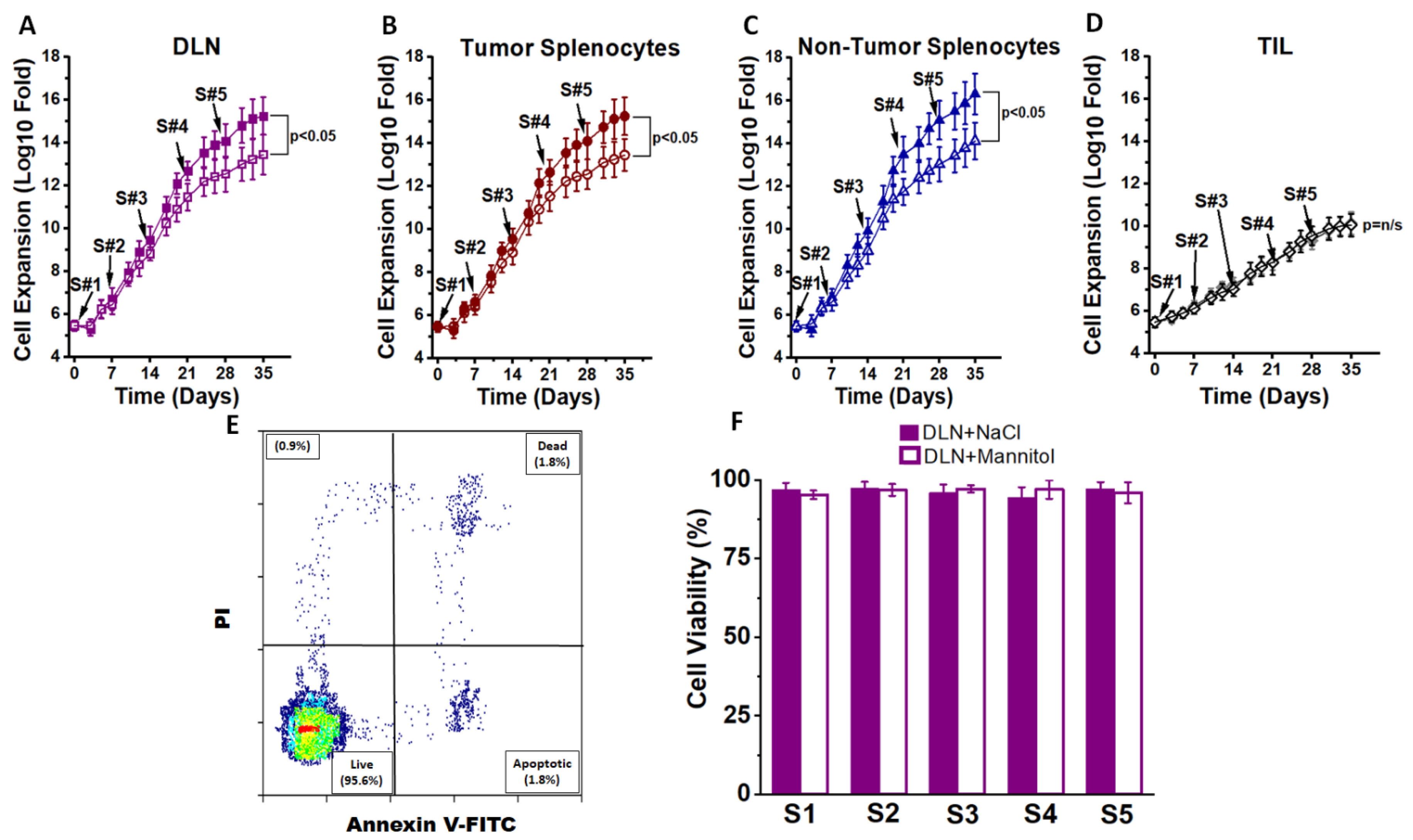
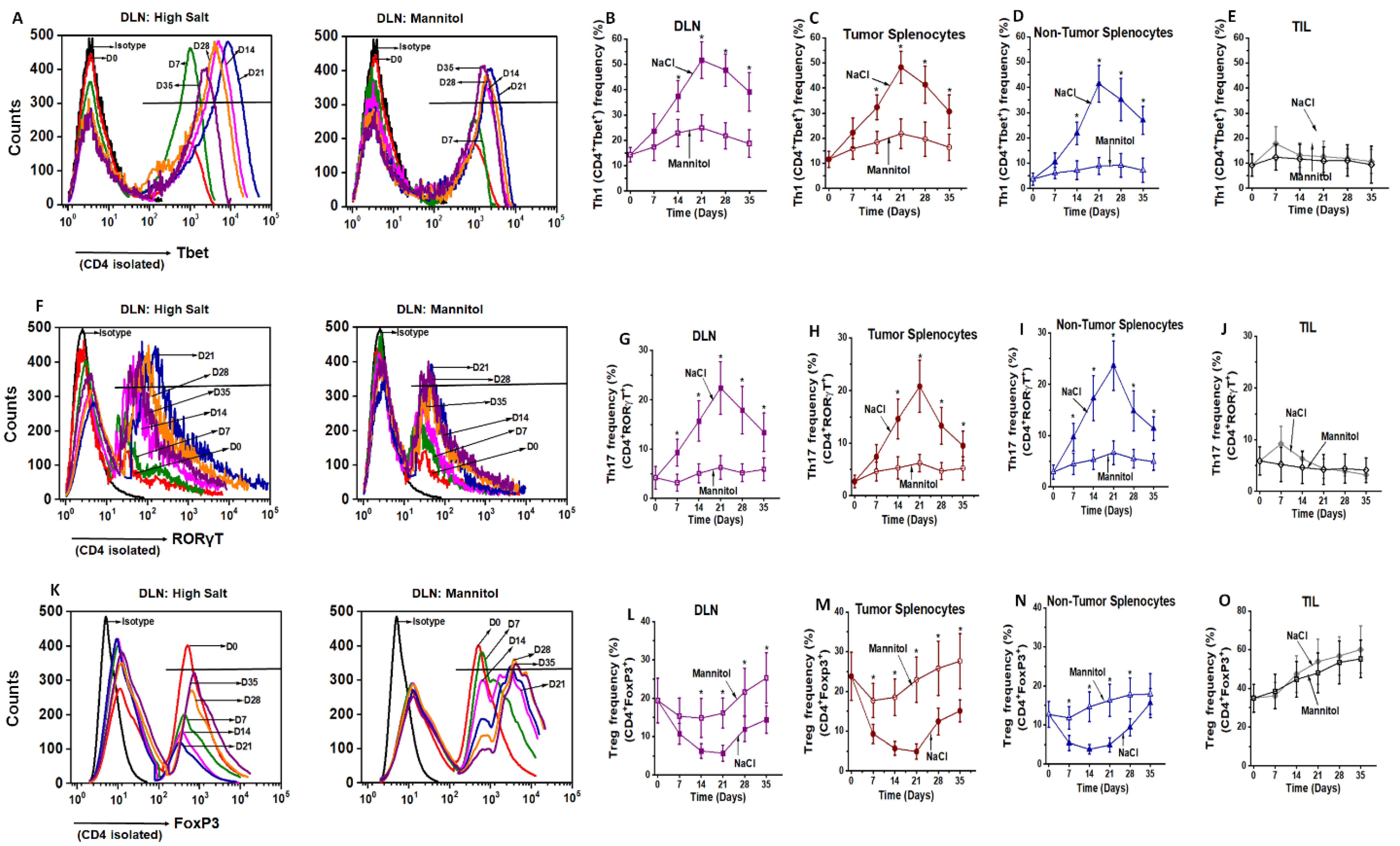
3.1. Ex Vivo High Salt Treatment Induces CD4+T cell Expansion and Effector Differentiation
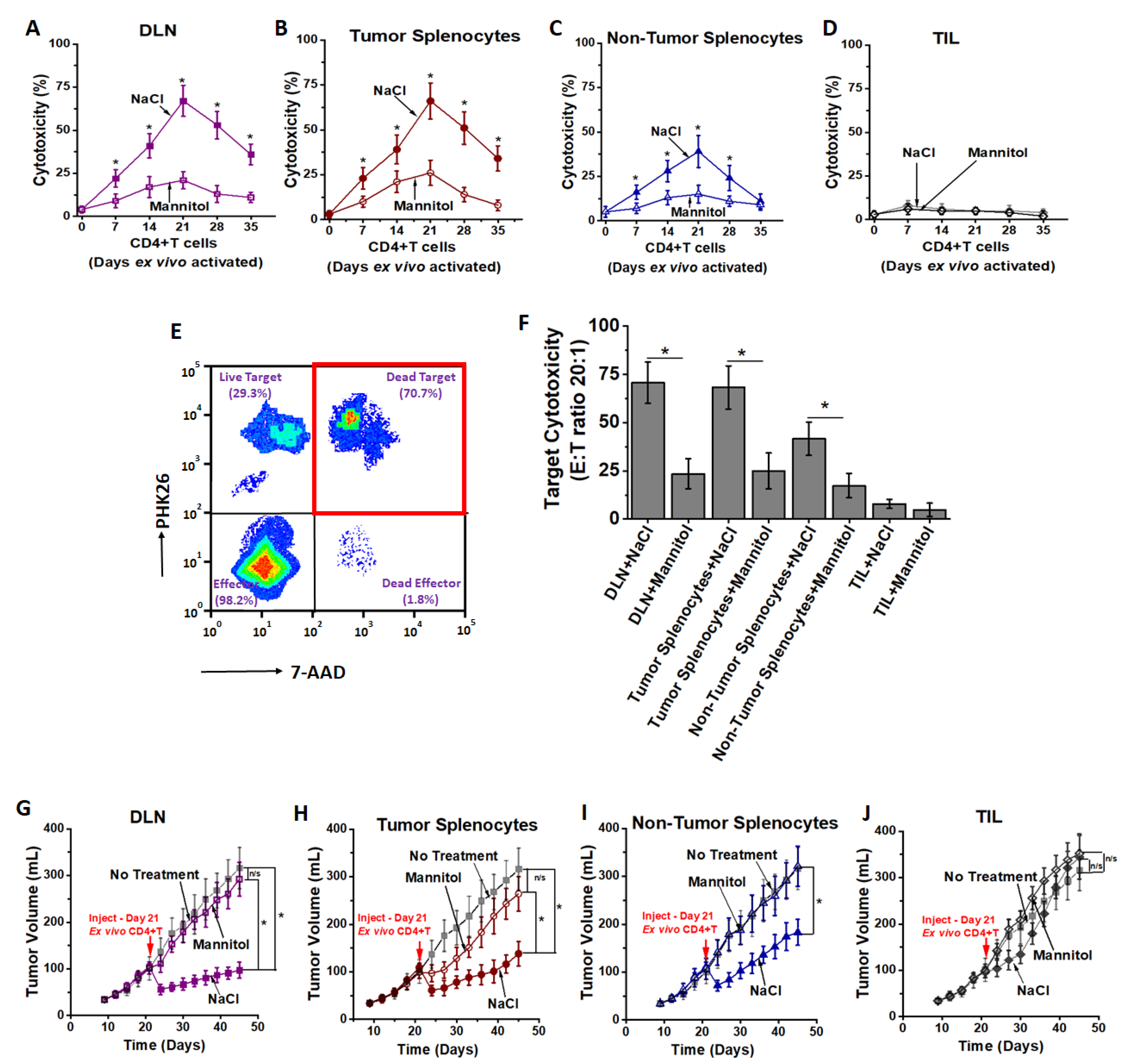
3.2. High Salt Activated CD4+T Cells Induce Tumor Regression
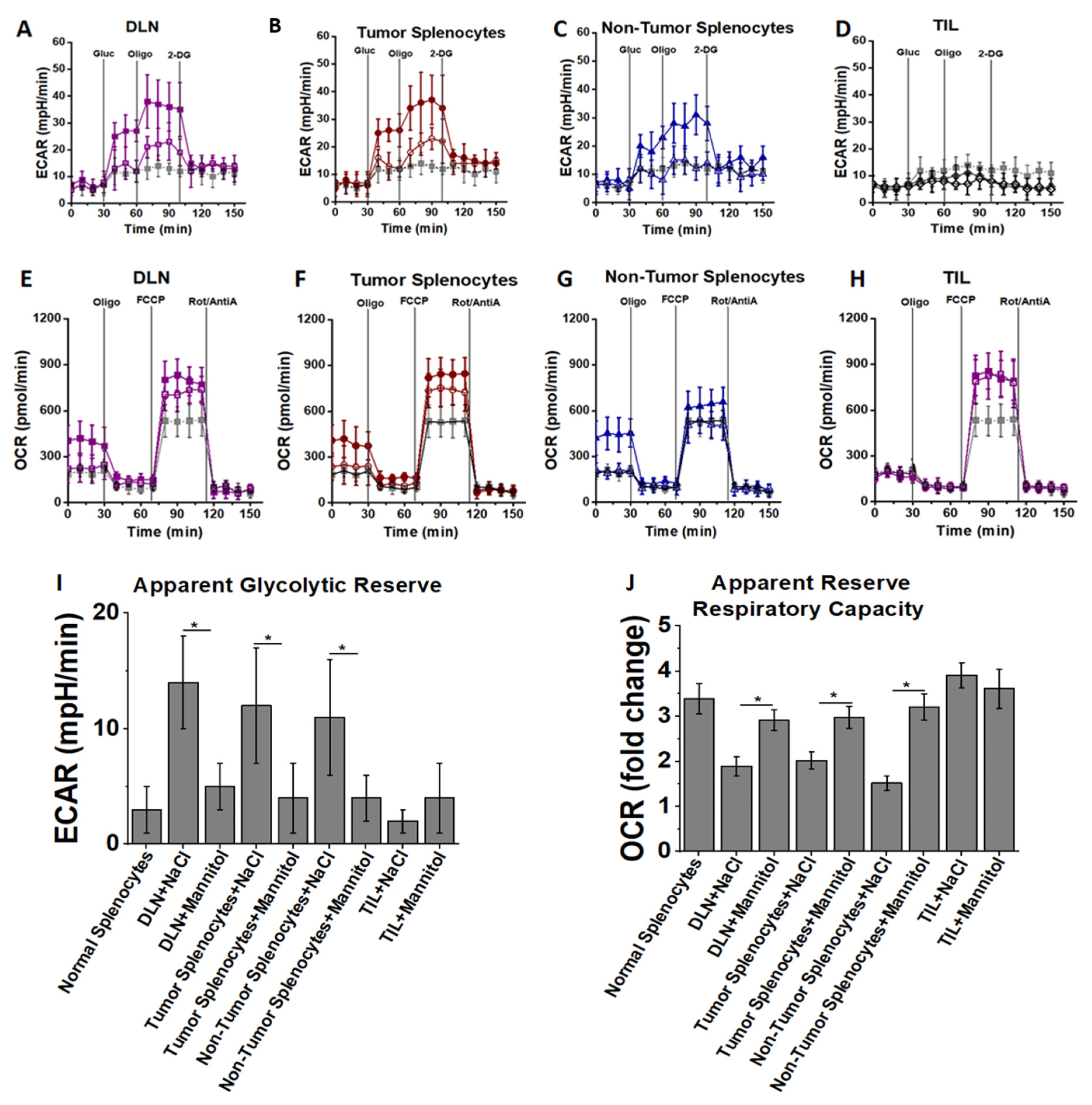
3.3. High Salt Activated CD4+T Cells Utilize Glycolytic Metabolic Pathway
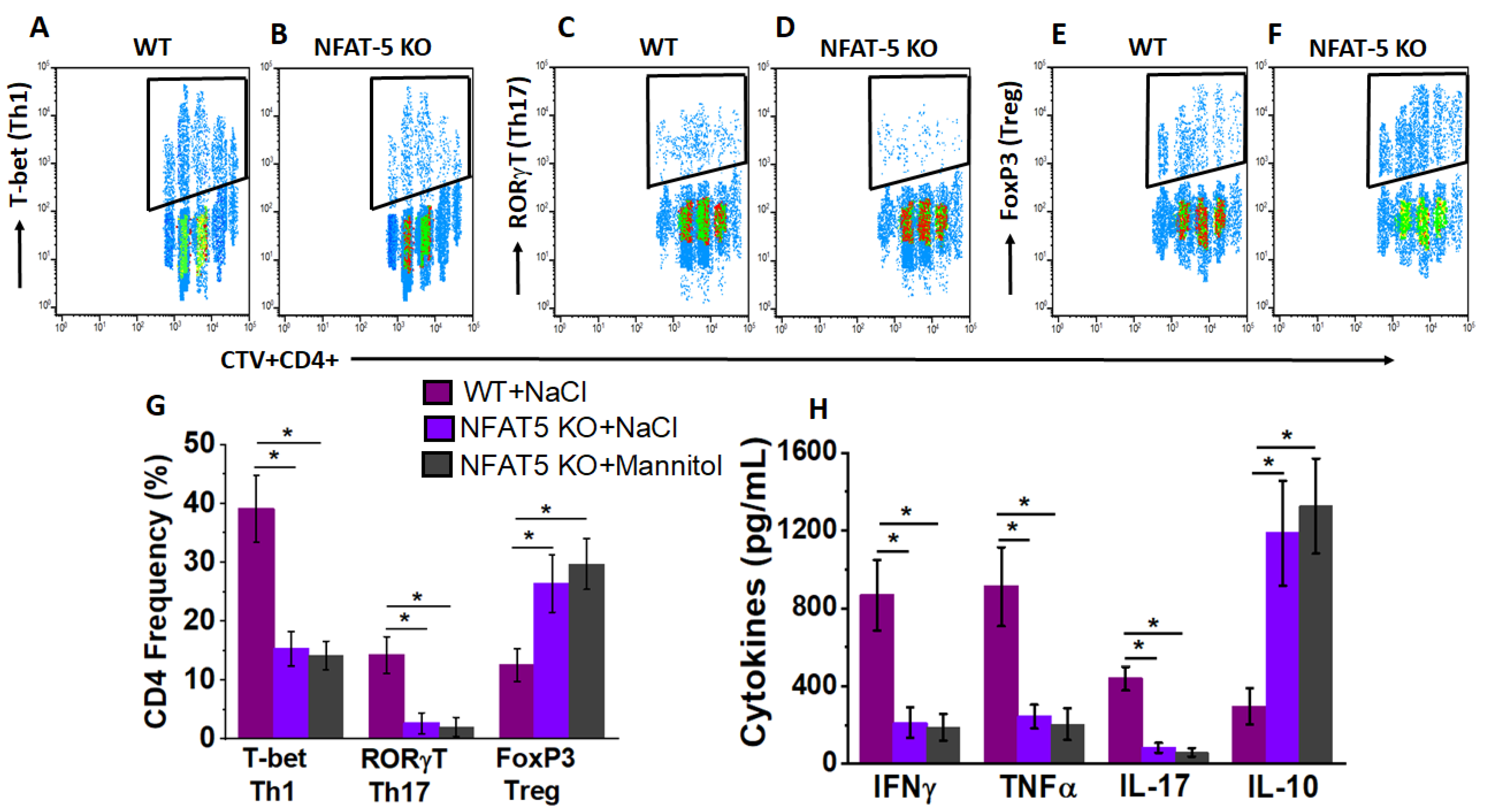
3.4. NFAT5 Mediates High Salt Mediated Effector CD4+T Cell Differentiation
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef] [PubMed]
- Thompson, E.D.; Enriquez, H.L.; Fu, Y.-X.; Engelhard, V.H. Tumor masses support naive T cell infiltration, activation, and differentiation into effectors. J. Exp. Med. 2010, 207, 1791–1804. [Google Scholar] [CrossRef] [PubMed]
- Xia, A.; Zhang, Y.; Xu, J.; Yin, T.; Lu, X.-J. T cell dysfunction in cancer immunity and immunotherapy. Front. Immunol. 2019, 10, 1719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manfredi, F.; Cianciotti, B.C.; Potenza, A.; Tassi, E.; Noviello, M.; Biondi, A.; Ciceri, F.; Bonini, C.; Ruggiero, E. TCR redirected T cells for cancer treatment: Achievements, hurdles, and goals. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, A.P.; Stadtmauer, E.A.; Aqui, N.; Vogl, D.T.; Chew, A.; Fang, H.-B.; Janofsky, S.; Yager, K.; Veloso, E.; Zheng, Z.; et al. Rapid immune recovery and graft-versus-host disease–like engraftment syndrome following adoptive transfer of costimulated autologous T cells. Clin. Cancer Res. 2009, 15, 4499–4507. [Google Scholar] [CrossRef] [Green Version]
- Dudley, M.E.; Wunderlich, J.R.; Robbins, P.F.; Yang, J.C.; Hwu, P.; Schwartzentruber, D.J.; Topalian, S.L.; Sherry, R.; Restifo, N.P.; Hubicki, A.M.; et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 2002, 298, 850–854. [Google Scholar] [CrossRef] [Green Version]
- Laport, G.G.; Levine, B.L.; Stadtmauer, E.A.; Schuster, S.J.; Luger, S.M.; Grupp, S.; Bunin, N.; Strobl, F.J.; Cotte, J.; Zheng, Z.; et al. Adoptive transfer of costimulated T cells induces lymphocytosis in patients with re-lapsed/refractory non-Hodgkin lymphoma following CD34+-selected hematopoietic cell transplantation. Blood 2003, 102, 2004–2013. [Google Scholar] [CrossRef] [Green Version]
- Hay, K.A.; Hanafi, L.-A.; Li, D.; Gust, J.; Liles, W.C.; Wurfel, M.M.; López, J.A.; Chen, J.; Chung, D.; Harju-Baker, S.; et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor–modified T-cell therapy. Blood 2017, 130, 2295–2306. [Google Scholar] [CrossRef] [Green Version]
- Sim, G.C.; Martin-Orozco, N.; Jin, L.; Yang, Y.; Wu, S.; Washington, E.; Sanders, D.; Lacey, C.; Wang, Y.; Vence, L.; et al. IL-2 therapy promotes suppressive ICOS+ Treg expansion in melanoma patients. J. Clin. Investig. 2014, 124, 99–110. [Google Scholar] [CrossRef] [Green Version]
- Pandiyan, P.; Zheng, L.; Ishihara, S.; Reed, J.; Lenardo, M.J. CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation–mediated apoptosis of effector CD4+ T cells. Nat. Immunol. 2007, 8, 1353–1362. [Google Scholar] [CrossRef] [PubMed]
- Chinen, T.; Kannan, A.K.; Levine, A.G.; Fan, X.; Klein, U.; Zheng, Y.; Gasteiger, G.; Feng, Y.; Fontenot, J.D.; Rudensky, A.Y. An essential role for the IL-2 receptor in Treg cell function. Nat. Immunol. 2016, 17, 1322–1333. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T cells and immune tolerance. Cell 2008, 133, 775–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, T.O.; Schluns, K.S. The potential and promise of IL-15 in immuno-oncogenic therapies. Immunol. Lett. 2017, 190, 159–168. [Google Scholar] [CrossRef]
- Jeong, G.H.; Lee, K.H.; Lee, I.R.; Oh, J.H.; Kim, D.W.; Shin, J.W.; Kronbichler, A.; Eisenhut, M.; Van Der Vliet, H.J.; Abdel-Rahman, O.; et al. Incidence of capillary leak syndrome as an adverse effect of drugs in cancer patients: A systematic review and meta-analysis. J. Clin. Med. 2019, 8, 143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutcher, J.P.; Schwartzentruber, D.J.; Kaufman, H.L.; Agarwala, S.S.; Tarhini, A.A.; Lowder, J.N.; Atkins, M.B. High dose interleukin-2 (Aldesleukin)—Expert consensus on best management practices-2014. J. Immunother. Cancer 2014, 2, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, R.; Westergaard, M.C.W.; Kjeldsen, J.W.; Muller, A.; Pedersen, N.W.; Hadrup, S.R.; Met, O.; Seliger, B.; Kromann-Andersen, B.; Hasselager, T.; et al. T-cell responses in the microenvironment of primary renal cell carcinoma-implications for adoptive cell therapy. Cancer Immunol. Res. 2018, 6, 222–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohaan, M.W.; Van den Berg, J.H.; Kvistborg, P.; Haanen, J. Adoptive transfer of tumor-infiltrating lymphocytes in melanoma: A viable treatment option. J. Immunother. Cancer 2018, 6, 102. [Google Scholar] [CrossRef] [PubMed]
- Dar, H.Y.; Singh, A.; Shukla, P.; Anupam, R.; Mondal, R.K.; Mishra, P.K.; Srivastava, R.K. High dietary salt intake correlates with modulated Th17-Treg cell balance resulting in enhanced bone loss and im-paired bone-microarchitecture in male mice. Sci. Rep. 2018, 8, 2503. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Yosef, N.; Thalhamer, T.; Zhu, C.; Xiao, S.; Kishi, Y.; Regev, A.; Kuchroo, V.K. Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature 2013, 496, 513–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez, A.L.; Kitz, A.; Wu, C.; Lowther, D.E.; Rodriguez, D.M.; Vudattu, N.; Deng, S.; Herold, K.C.; Kuchroo, V.K.; Kleinewietfeld, M.; et al. Sodium chloride inhibits the suppressive function of FOXP3+ regulatory T cells. J. Clin. Investig. 2015, 125, 4212–4222. [Google Scholar] [CrossRef] [Green Version]
- Aguiar, S.L.F.; Miranda, M.C.G.; Guimaraes, M.A.F.; Santiago, H.C.; Queiroz, C.P.; Cunha, P.D.S.; Cara, D.C.; Foureaux, G.; Ferreira, A.J.; Cardoso, V.N.; et al. High-salt diet induces IL-17-dependent gut inflammation and exacerbates colitis in mice. Front. Immunol. 2017, 8, 1969. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.; Kim, N.; Kang, J.; Yoon, S.; Lee, H.A.; Jung, H.; Kim, S.H.; Kim, I. Activated pathogenic Th17 lymphocytes induce hypertension following high-fructose intake in Dahl salt-sensitive but not Dahl salt-resistant rats. Dis. Models Mech. 2020, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, N.; Kim, D.; Kim, W.-U. Role of NFAT5 in the immune system and pathogenesis of autoimmune diseases. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Babaer, D.; Amara, S.; Ivy, M.; Zhao, Y.; Lammers, P.E.; Titze, J.M.; Tiriveedhi, V. High salt induces P-glycoprotein mediated treatment resistance in breast cancer cells through store operated calcium influx. Oncotarget 2018, 9, 25193–25205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faustino-Rocha, A.; Oliveira, P.A.; Pinho-Oliveira, J.; Teixeira-Guedes, C.; Soares-Maia, R.; da Costa, R.G.; Colaco, B.; Pires, M.J.; Colaco, J.; Ferreira, R.; et al. Estimation of rat mammary tumor volume using caliper and ultrasonography measurements. Lab. Anim. 2013, 42, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Amara, S.; Majors, C.; Roy, B.; Hill, S.; Rose, K.L.; Myles, E.L.; Tiriveedhi, V. Critical role of SIK3 in me-diating high salt and IL-17 synergy leading to breast cancer cell proliferation. PLoS ONE 2017, 12, e0180097. [Google Scholar] [CrossRef] [Green Version]
- Amara, S.; Ivy, M.T.; Myles, E.L.; Tiriveedhi, V. Sodium channel gammaENaC mediates IL-17 synergized high salt induced inflammatory stress in breast cancer cells. Cell Immunol. 2016, 302, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Babaer, D.; Amara, S.; McAdory, B.S.; Johnson, O.; Myles, E.L.; Zent, R.; Rathmell, J.C.; Tiriveedhi, V. Oli-godeoxynucleotides ODN 2006 and M362 exert potent adjuvant effect through TLR-9/-6 synergy to exaggerate mammaglobin-A peptide specific cytotoxic CD8+T lymphocyte responses against breast cancer cells. Cancers 2019, 11, 672. [Google Scholar] [CrossRef] [Green Version]
- Babaer, D.; Zheng, M.; Ivy, M.T.; Zent, R.; Tiriveedhi, V. Methylselenol producing selenocompounds enhance the efficiency of mammaglobin-A peptide vaccination against breast cancer cells. Oncol. Lett. 2019, 18, 6891–6898. [Google Scholar] [CrossRef] [Green Version]
- Gerriets, V.A.; Kishton, R.J.; Nichols, A.G.; Macintyre, A.N.; Inoue, M.; Ilkayeva, O.; Winter, P.S.; Liu, X.; Priyadharshini, B.; Slawinska, M.E.; et al. Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J. Clin. Investig. 2015, 125, 194–207. [Google Scholar] [CrossRef]
- Amara, S.; Alotaibi, D.; Tiriveedhi, V. NFAT5/STAT3 interaction mediates synergism of high salt with IL-17 towards induction of VEGF-A expression in breast cancer cells. Oncol. Lett. 2016, 12, 933–943. [Google Scholar] [CrossRef] [Green Version]
- Amara, S.; Zheng, M.; Tiriveedhi, V. Oleanolic acid inhibits high salt-induced exaggeration of war-burg-like metabolism in breast cancer cells. Cell Biochem. Biophys. 2016, 74, 427–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aramburu, J.; López-Rodríguez, C. Regulation of inflammatory functions of macrophages and T lymphocytes by NFAT5. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Cheung, C.Y.K.; Ko, B.C.B. NFAT5 in cellular adaptation to hypertonic stress-regulations and functional significance. J. Mol. Signal. 2013, 8, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, R.; Dumond, J.F.; Khan, S.H.; Thompson, E.B.; He, Y.; Burg, M.B.; Ferraris, J.D. NFAT5, which protects against hypertonicity, is activated by that stress via structuring of its intrinsically disordered domain. Proc. Natl. Acad. Sci. USA 2020, 117, 20292–20297. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, T.; Rezvani, K. Adoptive cell therapy: Living drugs against cancer. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef] [PubMed]
- Redeker, A.; Arens, R. Improving adoptive T cell therapy: The particular role of T cell costimulation, cytokines, and post-transfer vaccination. Front. Immunol. 2016, 7, 345. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Li, X.; Zhou, W.-L.; Huang, Y.; Liang, X.; Jiang, L.; Yang, X.; Sun, J.; Li, Z.; Han, W.-D.; et al. Genetically engineered T cells for cancer immunotherapy. Signal Transduct. Target. Ther. 2019, 4, 1–17. [Google Scholar] [CrossRef]
- Jiang, T.; Zhou, C.; Ren, S. Role of IL-2 in cancer immunotherapy. OncoImmunology 2016, 5, e1163462. [Google Scholar] [CrossRef] [Green Version]
- Bechman, N.; Maher, J. Lymphodepletion strategies to potentiate adoptive T-cell immunotherapy—What are we doing; where are we going? Expert Opin. Biol. Ther. 2020, 2020, 1857361. [Google Scholar] [CrossRef]
- Sharpe, M.; Mount, N. Genetically modified T cells in cancer therapy: Opportunities and challenges. Dis. Models Mech. 2015, 8, 337–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Cao, Y.J. Engineered T cell therapy for cancer in the clinic. Front. Immunol. 2019, 10, 2250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furlan, S.N.; Singh, K.; Lopez, C.; Tkachev, V.; Hunt, D.J.; Hibbard, J.; Betz, K.M.; Blazar, B.R.; Trapnell, C.; Kean, L.S. IL-2 enhances ex vivo–expanded regulatory T-cell persistence after adoptive transfer. Blood Adv. 2020, 4, 1594–1605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilck, N.; Matus, M.G.; Kearney, S.M.; Olesen, S.W.; Forslund, K.; Bartolomaeus, H.; Haase, S.; Mähler, A.; Balogh, A.; Markó, L.; et al. Salt-responsive gut commensal modulates TH17 axis and disease. Nature 2017, 551, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Crowley, S.D. Role of T-cell activation in salt-sensitive hypertension. Am. J. Physiol. Circ. Physiol. 2019, 316, H1345–H1353. [Google Scholar] [CrossRef] [PubMed]
- Willebrand, R.; Hamad, I.; Van Zeebroeck, L.; Kiss, M.; Bruderek, K.; Geuzens, A.; Swinnen, D.; Côrte-Real, B.F.; Markó, L.; Lebegge, E.; et al. High salt inhibits tumor growth by enhancing anti-tumor immunity. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- He, W.; Xu, J.; Mu, R.; Li, Q.; Lv, D.-L.; Huang, Z.; Zhang, J.; Wang, C.; Dong, L. High-salt diet inhibits tumour growth in mice via regulating myeloid-derived suppressor cell differentiation. Nat. Commun. 2020, 11, 1–17. [Google Scholar] [CrossRef]
- Gerriets, V.A.; Kishton, R.J.; Johnson, M.O.; Cohen, S.; Siska, P.J.; Nichols, A.G.; Warmoes, M.O.; de Cubas, A.A.; MacIver, N.J.; Locasale, J.W.; et al. Foxp3 and Toll-like receptor signaling balance Treg cell anabolic metabolism for suppression. Nat. Immunol. 2016, 17, 1459–1466. [Google Scholar] [CrossRef]
- Salmond, R.J. mTOR regulation of glycolytic metabolism in T cells. Front. Cell Dev. Biol. 2018, 6, 122. [Google Scholar] [CrossRef]
- Chi, H. Regulation and function of mTOR signalling in T cell fate decisions. Nat. Rev. Immunol. 2012, 12, 325–338. [Google Scholar] [CrossRef]
- Saravia, J.; Raynor, J.L.; Chapman, N.M.; Lim, S.A.; Chi, H. Signaling networks in immunometabolism. Cell Res. 2020, 30, 328–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alberdi, M.; Iglesias, M.; Tejedor, S.; Merino, R.; Lopez-Rodriguez, C.; Aramburu, J. Context-dependent regulation of Th17-associated genes and IFNgamma expression by the transcription factor NFAT5. Immunol. Cell Biol. 2017, 95, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Kleinewietfeld, M.; Manzel, A.; Titze, J.; Kvakan, H.; Yosef, N.; Linker, R.A.; Muller, D.N.; Hafler, D.A. Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature 2013, 496, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Ritz, E.; Koleganova, N.; Piecha, G. Role of sodium intake in the progression of chronic kidney disease. J. Ren. Nutr. 2009, 19, 61–62. [Google Scholar] [CrossRef] [PubMed]
- Tellechea, M.; Buxade, M.; Tejedor, S.; Aramburu, J.; Lopez-Rodriguez, C. NFAT5-regulated macrophage polarization supports the proinflammatory function of macrophages and T lymphocytes. J. Immunol. 2018, 200, 305–315. [Google Scholar] [CrossRef] [Green Version]
- Monteleone, I.; Marafini, I.; Dinallo, V.; Di Fusco, D.; Troncone, E.; Zorzi, F.; Laudisi, F.; Monteleone, G. Sodium chloride-enriched diet enhanced inflammatory cytokine production and exacerbated experimental colitis in mice. J. Crohns Colitis 2017, 11, 237–245. [Google Scholar] [CrossRef] [Green Version]
- Moon, E.K.; Wang, L.-C.S.; Bekdache, K.; Lynn, R.C.; Lo, A.; Thorne, S.H.; Albelda, S.M. Intra-tumoral delivery of CXCL11 via a vaccinia virus, but not by modified T cells, enhances the efficacy of adoptive T cell therapy and vaccines. OncoImmunology 2018, 7, e1395997. [Google Scholar] [CrossRef]
- Moon, E.K.; Carpenito, C.; Sun, J.; Wang, L.C.; Kapoor, V.; Predina, J.; Powell, D.J.; Jr Riley, J.L.; June, C.H.; Albelda, S.M. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin. Cancer Res. 2011, 17, 4719–4730. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.C. Toxicities associated with adoptive T-cell transfer for cancer. Cancer J. 2015, 21, 506–509. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tiriveedhi, V.; Ivy, M.T.; Myles, E.L.; Zent, R.; Rathmell, J.C.; Titze, J. Ex Vivo High Salt Activated Tumor-Primed CD4+T Lymphocytes Exert a Potent Anti-Cancer Response. Cancers 2021, 13, 1690. https://doi.org/10.3390/cancers13071690
Tiriveedhi V, Ivy MT, Myles EL, Zent R, Rathmell JC, Titze J. Ex Vivo High Salt Activated Tumor-Primed CD4+T Lymphocytes Exert a Potent Anti-Cancer Response. Cancers. 2021; 13(7):1690. https://doi.org/10.3390/cancers13071690
Chicago/Turabian StyleTiriveedhi, Venkataswarup, Michael T. Ivy, Elbert L. Myles, Roy Zent, Jeffrey C. Rathmell, and Jens Titze. 2021. "Ex Vivo High Salt Activated Tumor-Primed CD4+T Lymphocytes Exert a Potent Anti-Cancer Response" Cancers 13, no. 7: 1690. https://doi.org/10.3390/cancers13071690
APA StyleTiriveedhi, V., Ivy, M. T., Myles, E. L., Zent, R., Rathmell, J. C., & Titze, J. (2021). Ex Vivo High Salt Activated Tumor-Primed CD4+T Lymphocytes Exert a Potent Anti-Cancer Response. Cancers, 13(7), 1690. https://doi.org/10.3390/cancers13071690