
The Tumour Vasculature as a Target to Modulate Leucocyte Trafficking

 ,
, _Qi.png) ,
, 

Abstract
:Simple Summary
Abstract
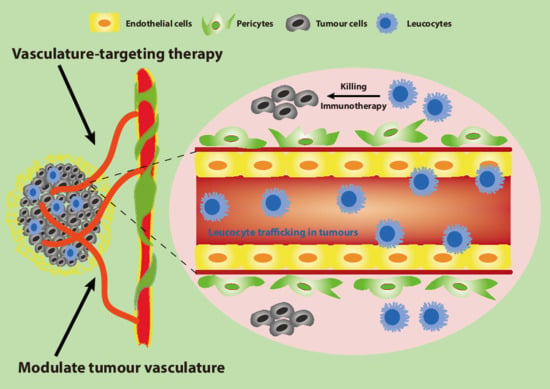
1. Introduction
2. The Tumour Vasculature: An Important Mediator of a Suppressive Tumour Microenvironment
2.1. The Structurally and Functionally Aberrant Tumour Vasculature
2.2. The Immunoresistant Microenvironment
2.3. Vascular Normalisation
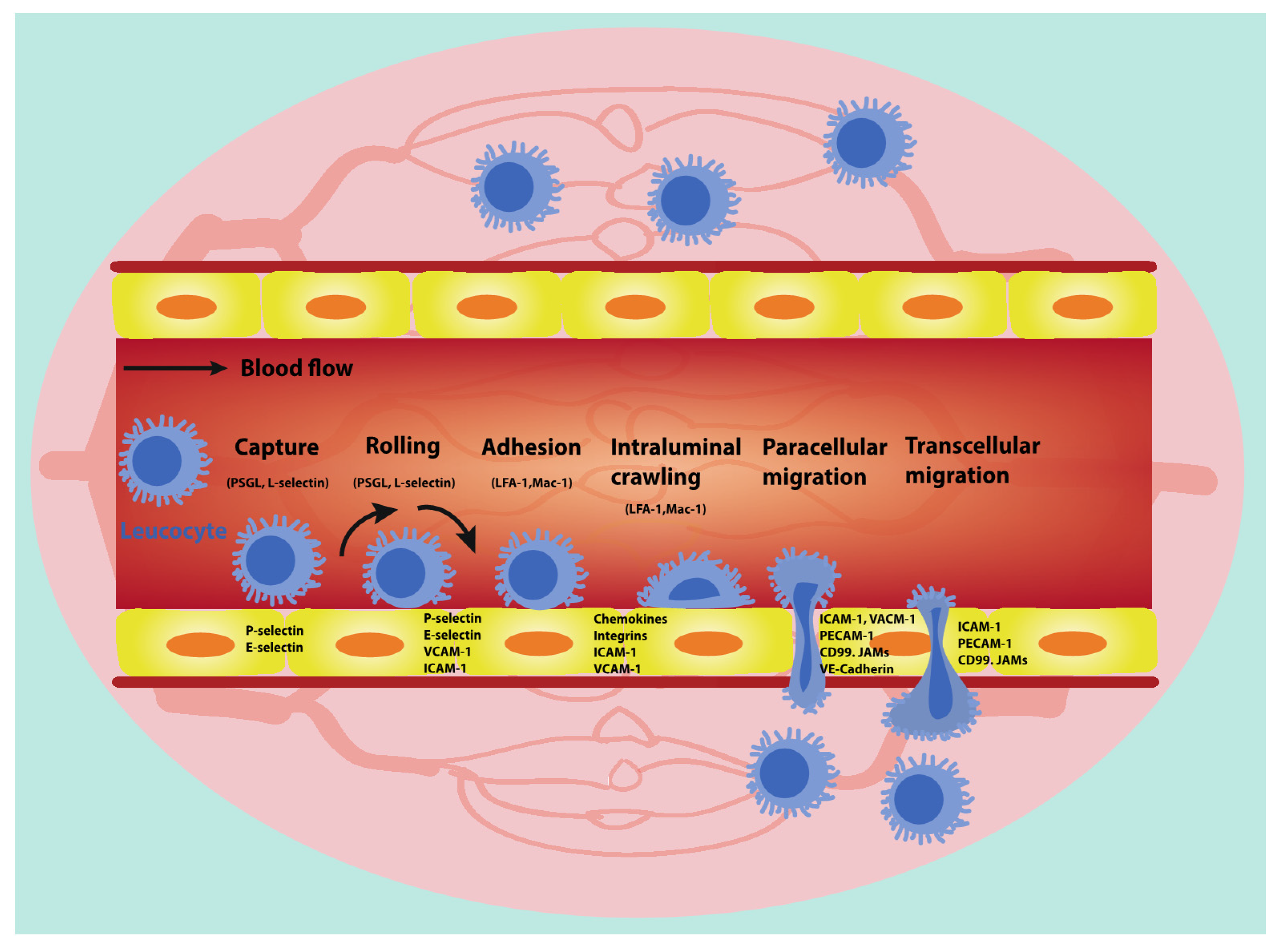
3. Leucocyte Trafficking
3.1. Leucocyte Capture
3.2. Leucocyte Rolling and Adhesion
3.3. Intraluminal Crawling of Leucocytes
3.4. Leucocyte Transendothelial Migration (TEM)
3.4.1. Paracellular Migration
3.4.2. Transcellular Migration
3.4.3. Leucocyte Transmigration in Tumour Blood Vessels
4. Trafficking of Leucocytes Subpopulation in Tumour Blood Vessels

{kind=link}
{kind=link}
{kind=link}
| Leucocyte Subpopulation | Vasculature-Targeting Strategy | Leucocyte Trafficking Phenotype | Reference | ||
|---|---|---|---|---|---|
| T-lymphocyte trafficking | Vascular Normalisation | Deletion of RGS5 | Increased CD8+ T cell infiltration | [126] | |
| Anti-VEGFR2 therapy | Increased CD8+ T cell infiltration | [18] | |||
| Dual inhibition of VEGF-A and Ang-2 | Increased CD8+ T cell infiltration | [127] | |||
| Anti-CTLA4 or anti-PD1 | Increased CD8+ T cell infiltration | [128] | |||
| Increase VE-Cadherin level | Increased CD8+ T cell transcellular migration | [20,34] | |||
| Inhibition of FasL | Protect CD8+ T cells | [2,129] | |||
| HEVs | Combining anti-VEGFR2 and anti-PD-L1 antibodies (activation of LTβR signalling) | Increased CD8+ T cell infiltration | [130] | ||
| LIGHT peptide | Increased CD8+ T cell infiltration | [131] | |||
| Augmented levels of various chemokines (CCL19, CCL21, et al.) | Increased CD8+ T cell adhesion and transmigration | [132,133] | |||
| Inhibit anergy | Angiogenic inhibitors (PF4, anginex, endostatin, angiostatin) | Increased CD8+ T cell adhesion | [58,63] | ||
| Angiostatic factor (16K hPRL et al.) | Increased CD8+ T cell adhesion and transmigration | [134] | |||
| DNMT or HDAC inhibitors | Increased CD8+ T cell adhesion and transmigration | [135] | |||
| Monocyte trafficking | Macrophage | M2-like | Combining anti-PD-L1 and CSF1R | Decreased M2-like macrophage trafficking | [136] |
| Inhibition of hedgehog signalling | Decreased M2-like macrophage trafficking | [137] | |||
| CCL2 inhibitor bindarit | Decreased M2-like macrophage trafficking | [138] | |||
| M1-like | Downregulation of PIGF | Increased M1-like macrophage trafficking | [46] | ||
| Activation of STING | Increased M1-like macrophage trafficking | [139] | |||
| DCs | Hypoxia, oxidized low density lipoprotein or TNF-α | Increased DC adhesion and transmigration | [140] | ||
| Deletion of COX | Increased DC infiltration | [141,142] | |||
| Repression of mTORC1 | Increased DC infiltration | [143] | |||
| Anti-VEGF therapy | Increased DC infiltration | [144] | |||
| Granulocyte trafficking | Neutrophils | Silence of some chemokines (CXCL1, et al.) | Decreased neutrophil infiltration | [145] | |
| Increase VE-Cadherin expression | Decreased neutrophil adhesion and transmigration | [34,146] | |||
| Dual inhibition of VRGF and Ang2 | Decreased neutrophil infiltration | [147] | |||
| RvD1, RvE1 and ATLa | Decreased neutrophil infiltration | [148] | |||
| Suppression of Semaphorin 7A | Decreased neutrophil infiltration | [149,150] | |||
| Eosinophils | Anti-CTLA4 therapy | Increased eosinophil infiltration | [151] | ||
| TNF-α and IFN-γ | Eosinophilic secretion of Th1-type chemokines | [152] | |||
| IL-4 | Eosinophilic production of Th2-type chemokines | [152] | |||
4.1. T-Lymphocyte Trafficking
4.1.1. Normalised Tumour Blood Vessels Affect T Lymphocyte Trafficking
4.1.2. High Endothelial Venules (HEVs) in Tumours Control T Lymphocyte Trafficking
4.1.3. Endothelial Cell Anergy Has Impact on T Lymphocyte Trafficking
4.2. Targeting Tumour Blood Vessels in Governing Monocyte Trafficking
4.2.1. Macrophage Trafficking
4.2.2. DC Trafficking
4.3. Granulocyte Trafficking
4.3.1. Neutrophil Trafficking
4.3.2. Eosinophil Trafficking
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lança, T.; Silva-Santos, B. The split nature of tumor-infiltrating leukocytes: Implications for cancer surveillance and immunotherapy. Oncoimmunology 2012, 1, 717–725. [Google Scholar] [CrossRef] [Green Version]
- Lanitis, E.; Irving, M.; Coukos, G. Targeting the tumor vasculature to enhance t cell activity. Curr. Opin. Immunol. 2015, 33, 53–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, W.A. Getting leukocytes to the site of inflammation. Vet. Pathol. 2013, 50, 7–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wettschureck, N.; Strilic, B.; Offermanns, S. Passing the vascular barrier: Endothelial signaling processes controlling extravasation. Physiol. Rev. 2019, 99, 1467–1525. [Google Scholar] [CrossRef] [PubMed]
- Kerbel, R.S. Tumor angiogenesis. N. Engl. J. Med. 2008, 358, 2039–2049. [Google Scholar] [CrossRef] [Green Version]
- Jain, R.K. Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Science 2005, 307, 58–62. [Google Scholar] [CrossRef]
- Tian, L.; Goldstein, A.; Wang, H.; Ching Lo, H.; Sun Kim, I.; Welte, T.; Sheng, K.; Dobrolecki, L.E.; Zhang, X.; Putluri, N.; et al. Mutual regulation of tumour vessel normalization and immunostimulatory reprogramming. Nature 2017, 544, 250–254. [Google Scholar] [CrossRef]
- Potente, M.; Gerhardt, H.; Carmeliet, P. Basic and therapeutic aspects of angiogenesis. Cell 2011, 146, 873–887. [Google Scholar] [CrossRef] [Green Version]
- Jain, R.K. Normalizing tumor vasculature with anti-angiogenic therapy: A new paradigm for combination therapy. Nat. Med. 2001, 7, 987–989. [Google Scholar] [CrossRef]
- Siemann, D.W. The unique characteristics of tumor vasculature and preclinical evidence for its selective disruption by tumor-vascular disrupting agents. Cancer Treat. Rev. 2011, 37, 63–74. [Google Scholar] [CrossRef] [Green Version]
- Fukumura, D.; Jain, R.K. Tumor microenvironment abnormalities: Causes, consequences, and strategies to normalize. J. Cell Biochem. 2007, 101, 937–949. [Google Scholar] [CrossRef] [PubMed]
- Bergers, G.; Song, S. The role of pericytes in blood-vessel formation and maintenance. NeuroOncology 2005, 7, 452–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baluk, P.; Morikawa, S.; Haskell, A.; Mancuso, M.; McDonald, D.M. Abnormalities of basement membrane on blood vessels and endothelial sprouts in tumors. Am. J. Pathol. 2003, 163, 1801–1815. [Google Scholar] [CrossRef] [Green Version]
- Jain, R.K. Normalizing tumor microenvironment to treat cancer: Bench to bedside to biomarkers. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2013, 31, 2205–2218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stylianopoulos, T.; Jain, R.K. Combining two strategies to improve perfusion and drug delivery in solid tumors. Proc. Natl. Acad. Sci. USA 2013, 110, 18632–18637. [Google Scholar] [CrossRef] [Green Version]
- Goel, S.; Duda, D.G.; Xu, L.; Munn, L.L.; Boucher, Y.; Fukumura, D.; Jain, R.K. Normalization of the vasculature for treatment of cancer and other diseases. Physiol. Rev. 2011, 91, 1071–1121. [Google Scholar] [CrossRef]
- Schaaf, M.B.; Garg, A.D.; Agostinis, P. Defining the role of the tumor vasculature in antitumor immunity and immunotherapy. Cell Death Dis. 2018, 9, 115. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Yuan, J.; Righi, E.; Kamoun, W.; Ancukiewicz, M.; Nezivar, J.; Santosuosso, M.; Martin, J.D.; Martin, M.; Vianello, F.; et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc. Natl. Acad. Sci. USA 2012, 109, 17561–17566. [Google Scholar] [CrossRef] [Green Version]
- Solimando, A.G.; Summa, S.; Vacca, A.; Ribatti, D. Cancer-associated angiogenesis: The endothelial cell as a checkpoint for immunological patrolling. Cancers 2020, 12, 3380. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Li, J.; Ting, K.K.; Chen, J.B.; Coleman, P.R.; Liu, K.; Wan, L.; Moller, T.; Vadas, M.A.; Gamble, J.R. The ve-cadherin/β-catenin signalling axis regulates immune cell infiltration into tumours. Cancer Lett. 2021, 496, 1–15. [Google Scholar] [CrossRef]
- Chouaib, S.; Noman, M.Z.; Kosmatopoulos, K.; Curran, M.A. Hypoxic stress: Obstacles and opportunities for innovative immunotherapy of cancer. Oncogene 2017, 36, 439–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Yan, J.; Liu, B. Targeting vegf/vegfr to modulate antitumor immunity. Front. Immunol. 2018, 9, 978. [Google Scholar] [CrossRef] [Green Version]
- Tan, H.Y.; Wang, N.; Lam, W.; Guo, W.; Feng, Y.; Cheng, Y.C. Targeting tumour microenvironment by tyrosine kinase inhibitor. Mol. Cancer 2018, 17, 43. [Google Scholar] [CrossRef] [Green Version]
- Isomoto, K.; Haratani, K.; Hayashi, H.A.-O.; Shimizu, S.; Tomida, S.; Niwa, T.; Yokoyama, T.; Fukuda, Y.; Chiba, Y.; Kato, R.; et al. Impact of egfr-tki treatment on the tumor immune microenvironment in egfr mutation-positive non-small cell lung cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020, 26, 2037–2046. [Google Scholar] [CrossRef] [PubMed]
- Rodig, N.; Ryan, T.; Allen, J.A.; Pang, H.; Grabie, N.; Chernova, T.; Greenfield, E.A.; Liang, S.C.; Sharpe, A.H.; Lichtman, A.H.; et al. Endothelial expression of pd-l1 and pd-l2 down-regulates cd8+ t cell activation and cytolysis. Eur. J. Immunol. 2003, 33, 3117–3126. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Ding, Y.; Zhou, M.; Rini, B.I.; Petillo, D.; Qian, C.N.; Kahnoski, R.; Futreal, P.A.; Furge, K.A.; Teh, B.T. Interleukin-8 mediates resistance to antiangiogenic agent sunitinib in renal cell carcinoma. Cancer Res. 2010, 70, 1063–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zang, X.; Sullivan, P.S.; Soslow, R.A.; Waitz, R.; Reuter, V.E.; Wilton, A.; Thaler, H.T.; Arul, M.; Slovin, S.F.; Wei, J.; et al. Tumor associated endothelial expression of b7-h3 predicts survival in ovarian carcinomas. Mod. Pathol. 2010, 23, 1104–1112. [Google Scholar] [CrossRef] [PubMed]
- Rossin, A.; Miloro, G.; Hueber, A.O. Trail and fasl functions in cancer and autoimmune diseases: Towards an increasing complexity. Cancers 2019, 11, 639. [Google Scholar] [CrossRef] [Green Version]
- Martin, J.D.; Seano, G.; Jain, R.K. Normalizing function of tumor vessels: Progress, opportunities, and challenges. Annu. Rev. Physiol. 2019, 81, 505–534. [Google Scholar] [CrossRef]
- Willett, C.G.; Boucher, Y.; di Tomaso, E.; Duda, D.G.; Munn, L.L.; Tong, R.T.; Chung, D.C.; Sahani, D.V.; Kalva, S.P.; Kozin, S.V.; et al. Direct evidence that the vegf-specific antibody bevacizumab has antivascular effects in human rectal cancer. Nat. Med. 2004, 10, 145–147. [Google Scholar] [CrossRef]
- Hidalgo, M.; Martinez-Garcia, M.; Le Tourneau, C.; Massard, C.; Garralda, E.; Boni, V.; Taus, A.; Albanell, J.; Sablin, M.; Alt, M.; et al. First-in-human phase i study of single-agent vanucizumab, a first-in-class bispecific anti-angiopoietin-2/anti-vegf-a antibody, in adult patients with advanced solid tumors. Clin. Cancer Res. 2018, 24, 1536–1545. [Google Scholar] [CrossRef] [Green Version]
- Park, J.S.; Kim, I.K.; Han, S.; Park, I.; Kim, C.; Bae, J.; Oh, S.J.; Lee, S.; Kim, J.H.; Woo, D.C.; et al. Normalization of tumor vessels by tie2 activation and ang2 inhibition enhances drug delivery and produces a favorable tumor microenvironment. Cancer Cell 2016, 30, 953–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, A.; Hamzah, J.; Payne, C.J.; Ganss, R. Tumor-targeted tnfα stabilizes tumor vessels and enhances active immunotherapy. Proc. Natl. Acad. Sci. USA 2012, 109, 7841–7846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Ting, K.K.; Li, J.; Cogger, V.C.; Chen, J.; Johansson-Percival, A.; Ngiow, S.F.; Holst, J.; Grau, G.; Goel, S.; et al. Targeting vascular endothelial-cadherin in tumor-associated blood vessels promotes t-cell-mediated immunotherapy. Cancer Res. 2017, 77, 4434–4447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganss, R. Tumour vessel remodelling: New opportunities in cancer treatment. Vasc. Biol. 2020, 2, R35–R43. [Google Scholar] [CrossRef] [Green Version]
- Mpekris, F.; Voutouri, C.; Baish, J.W.; Duda, D.G.; Munn, L.L.; Stylianopoulos, T.; Jain, R.K. Combining microenvironment normalization strategies to improve cancer immunotherapy. Proc. Natl. Acad. Sci. USA 2020, 117, 3728–3737. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Kim, B.Y.S.; Chan, C.K.; Hahn, S.M.; Weissman, I.L.; Jiang, W. Improving immune-vascular crosstalk for cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 195–203. [Google Scholar] [CrossRef]
- Hendry, S.A.; Farnsworth, R.H.; Solomon, B.; Achen, M.G.; Stacker, S.A.; Fox, S.B. The role of the tumor vasculature in the host immune response: Implications for therapeutic strategies targeting the tumor microenvironment. Front. Immunol. 2016, 7, 621. [Google Scholar] [CrossRef]
- Huang, Y.; Stylianopoulos, T.; Duda, D.G.; Fukumura, D.; Jain, R.K. Benefits of vascular normalization are dose and time dependent. Cancer Res. 2013, 73, 7144–7146. [Google Scholar] [CrossRef] [Green Version]
- Pàez-Ribes, M.; Allen, E.; Hudock, J.; Takeda, T.; Okuyama, H.; Viñals, F.; Inoue, M.; Bergers, G.; Hanahan, D.; Casanovas, O. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 2009, 15, 220–231. [Google Scholar] [CrossRef] [Green Version]
- Carmeliet, P.; Jain, R.K. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat. Rev. Drug Discov. 2011, 10, 417–427. [Google Scholar] [CrossRef]
- Petri, B.; Phillipson, M.; Kubes, P. The physiology of leukocyte recruitment: An in vivo perspective. J. Immunol. 2008, 180, 6439–6446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alon, R.; Feigelson, S.W. Chemokine-triggered leukocyte arrest: Force-regulated bi-directional integrin activation in quantal adhesive contacts. Curr. Opin. Cell Biol. 2012, 24, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Vestweber, D. How leukocytes cross the vascular endothelium. Nat. Rev. Immunol. 2015, 15, 692–704. [Google Scholar] [CrossRef] [PubMed]
- Nagarsheth, N.; Wicha, M.S.; Zou, W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol. 2017, 17, 559–572. [Google Scholar] [CrossRef] [Green Version]
- Rolny, C.; Mazzone, M.; Tugues, S.; Laoui, D.; Johansson, I.; Coulon, C.; Squadrito, M.I.; Segura, I.; Li, X.; Knevels, E.; et al. Hrg inhibits tumor growth and metastasis by inducing macrophage polarization and vessel normalization through downregulation of plgf. Cancer Cell 2010, 19, 31–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Peranzoni, E.; Lemoine, J.; Vimeux, L.; Feuillet, V.; Barrin, S.; Kantari-Mimoun, C.; Bercovici, N.; Guérin, M.; Biton, J.; Ouakrim, H.; et al. Macrophages impede cd8 t cells from reaching tumor cells and limit the efficacy of anti–pd-1 treatment. Proc. Natl. Acad. Sci. USA 2018, 115, E4041–E4050. [Google Scholar] [CrossRef] [Green Version]
- Harjunpää, H.; Llort Asens, M.; Guenther, C.; Fagerholm, S.C. Cell adhesion molecules and their roles and regulation in the immune and tumor microenvironment. Front. Immunol. 2019, 10, 1078. [Google Scholar] [CrossRef] [Green Version]
- Eriksson, E.E.; Xie, X.; Werr, J.; Thoren, P.; Lindbom, L. Importance of primary capture and l-selectin-dependent secondary capture in leukocyte accumulation in inflammation and atherosclerosis in vivo. J. Exp. Med. 2001, 194, 205–218. [Google Scholar] [CrossRef] [Green Version]
- Kunkel, E.J.; Chomas, J.E.; Ley, K. Role of primary and secondary capture for leukocyte accumulation in vivo. Circ. Res. 1998, 82, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Paschall, C.D.; Lawrence, M.B. L-selectin shear thresholding modulates leukocyte secondary capture. Ann. Biomed. Eng. 2008, 36, 622–631. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.Z.; Klitzman, B.; Dodge, R.; Dewhirst, M.W. Diminished leukocyte-endothelium interaction in tumor microvessels. Cancer Res. 1992, 52, 4265–4268. [Google Scholar] [PubMed]
- Dirkx, A.E.; Oude Egbrink, M.G.; Kuijpers, M.J.; van der Niet, S.T.; Heijnen, V.V.; Bouma-ter Steege, J.C.; Wagstaff, J.; Griffioen, A.W. Tumor angiogenesis modulates leukocyte-vessel wall interactions in vivo by reducing endothelial adhesion molecule expression. Cancer Res. 2003, 63, 2322–2329. [Google Scholar] [PubMed]
- Nooijen, P.T.; Westphal, J.R.; Eggermont, A.M.; Schalkwijk, C.; Max, R.; de Waal, R.M.; Ruiter, D.J. Endothelial p-selectin expression is reduced in advanced primary melanoma and melanoma metastasis. Am. J. Pathol. 1998, 152, 679–682. [Google Scholar] [PubMed]
- Georganaki, M.; van Hooren, L.; Dimberg, A. Vascular targeting to increase the efficiency of immune checkpoint blockade in cancer. Front. Immunol. 2018, 9, 3081. [Google Scholar] [CrossRef]
- Ivetic, A.; Hoskins Green, H.L.; Hart, S.J. L-selectin: A major regulator of leukocyte adhesion, migration and signaling. Front. Immunol. 2019, 14, 1068. [Google Scholar] [CrossRef] [Green Version]
- Dirkx, A.E.; Egbrink, M.G.; Castermans, K.; Van, D.W.; Thijssen, V.L.; Dings, R.P.; Kwee, L.; Mayo, K.H.; Bouma-ter Steege, J.C. Anti-angiogenesis therapy can overcome endothelial cell anergy and promote leukocyte-endothelium interactions and infiltration in tumors. FASEB J. 2006, 20, 621–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McEver, R.P. Selectins: Initiators of leucocyte adhesion and signalling at the vascular wall. Cardiovasc. Res. 2015, 107, 331–339. [Google Scholar] [CrossRef] [Green Version]
- da Costa Martins, P.; Garcia-Vallejo, J.J.; van Thienen, J.V.; Fernandez-Borja, M.; van Gils, J.M.; Beckers, C.; Horrevoets, A.J.; Hordijk, P.L.; Zwaginga, J.J. P-selectin glycoprotein ligand-1 is expressed on endothelial cells and mediates monocyte adhesion to activated endothelium. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1023–1029. [Google Scholar] [CrossRef] [Green Version]
- Cinamon, G.; Grabovsky, V.; Winter, E.; Franitza, S.; Feigelson, S.; Shamri, R.; Dwir, O.; Alon, R. Novel chemokine functions in lymphocyte migration through vascular endothelium under shear flow. J. Leukoc. Biol. 2001, 69, 860–866. [Google Scholar] [PubMed]
- Langley, R.R.; Russell, J.; Eppihimer, M.J.; Alexander, S.J.; Gerritsen, M.; Specian, R.D.; Granger, D.N. Quantification of murine endothelial cell adhesion molecules in solid tumors. Am. J. Physiol. 1999, 277, 1156–1166. [Google Scholar] [CrossRef] [PubMed]
- Griffioen, A.W.; Damen, C.A.; Mayo, K.H.; Barendsz-Janson, A.F.; Martinotti, S.; Blijham, G.H.; Groenewegen, G. Angiogenesis inhibitors overcome tumor induced endothelial cell anergy. Int. J. Cancer 1999, 80, 315–319. [Google Scholar] [CrossRef]
- Tromp, S.C.; oude Egbrink, M.G.A.; Dings, R.P.M.; van Velzen, S.; Slaaf, D.W.; Hillen, H.F.P.; Tangelder, G.J.; Reneman, R.S.; Griffioen, A.W. Tumor angiogenesis factors reduce leukocyte adhesion in vivo. Int. Immunol. 2000, 12, 671–676. [Google Scholar] [CrossRef]
- Bessa, X.; Elizalde, J.I.; Mitjans, F.; Pinol, V.; Miquel, R.; Panes, J.; Piulats, J.; Pique, J.M.; Castells, A. Leukocyte recruitment in colon cancer: Role of cell adhesion molecules, nitric oxide, and transforming growth factor beta1. Gastroenterology 2002, 122, 1122–1132. [Google Scholar] [CrossRef]
- Borgström, P.; Hughes, G.K.; Hansell, P.; Wolitsky, B.A.; Sriramarao, P. Leukocyte adhesion in angiogenic blood vessels. Role of e-selectin, p-selectin, and beta2 integrin in lymphotoxin-mediated leukocyte recruitment in tumor microvessels. J. Clin. Investig. 1997, 99, 2246–2253. [Google Scholar] [CrossRef] [Green Version]
- Fukumura, D.; Kashiwagi, S.; Jain, R.K. The role of nitric oxide in tumour progression. Nat. Rev. Cancer 2006, 6, 521–534. [Google Scholar] [CrossRef]
- De Caterina, R.; Libby, P.; Peng, H.B.; Thannickal, V.J.; Rajavashisth, T.B.; Gimbrone, M.A.; Shin, W.S.; Liao, J.K. Nitric oxide decreases cytokine-induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J. Clin. Investig. 1995, 96, 60–68. [Google Scholar] [CrossRef]
- Phillipson, M.; Heit, B.; Colarusso, P.; Liu, L.; Ballantyne, C.M.; Kubes, P. Intraluminal crawling of neutrophils to emigration sites: A molecularly distinct process from adhesion in the recruitment cascade. J. Exp. Med. 2006, 203, 2569–2575. [Google Scholar] [CrossRef]
- Schenkel, A.R.; Mamdouh, Z.; Muller, W.A. Locomotion of monocytes on endothelium is a critical step during extravasation. Nat. Immunol. 2004, 5, 393–400. [Google Scholar] [CrossRef]
- Kameritsch, P.; Renkawitz, J. Principles of leukocyte migration strategies. Trends Cell Biol. 2020, 30, 818–832. [Google Scholar] [CrossRef] [PubMed]
- Ryschich, E.; Kerkadze, V.; Lizdenis, P.; Paskauskas, S.; Knaebel, H.-P.; Gross, W.; Gebhard, M.M.; Büchler, M.W.; Schmidt, J. Active leukocyte crawling in microvessels assessed by digital time-lapse intravital microscopy. J. Surg. Res. 2006, 135, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Tousi, N.; Wang, B.; Pant, K.; Kiani, M.F.; Prabhakarpandian, B. Preferential adhesion of leukocytes near bifurcations is endothelium independent. Microvasc. Res. 2010, 80, 384–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, C.; Slattery, M.J.; Rank, B.M.; You, J. In vitro characterization and micromechanics of tumor cell chemotactic protrusion, locomotion, and extravasation. Ann. Biomed. Eng. 2002, 30, 344–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turk, M.; Naumenko, V.; Mahoney, D.J.; Jenne, C.N. Tracking cell recruitment and behavior within the tumor microenvironment using advanced intravital imaging approaches. Cells 2018, 7, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeichi, T.; Engelmann, G.; Mocevicius, P.; Schmidt, J.; Ryschich, E. 4-dimensional intravital microscopy: A new model for studies of leukocyte recruitment and migration in hepatocellular cancer in mice. J. Gastrointest. Surg. 2010, 14, 867–872. [Google Scholar] [CrossRef]
- Choi, S.H.; Kim, A.R.; Nam, J.K.; Kim, J.M.; Kim, J.Y.; Seo, H.R.; Lee, H.J.; Cho, J.; Lee, Y.J. Tumour-vasculature development via endothelial-to-mesenchymal transition after radiotherapy controls cd44v6(+) cancer cell and macrophage polarization. Nat. Commun. 2018, 9, 5108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clere, N.; Renault, S.; Corre, I. Endothelial-to-mesenchymal transition in cancer. Front. Cell Dev. Biol. 2020, 8, 747. [Google Scholar] [CrossRef]
- Nourshargh, S.; Marelli-Berg, F.M. Transmigration through venular walls: A key regulator of leukocyte phenotype and function. Trends Immunol. 2005, 26, 157–165. [Google Scholar] [CrossRef]
- Pober, J.S.; Sessa, W.C. Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. 2007, 7, 803–815. [Google Scholar] [CrossRef]
- Nagl, L.; Horvath, L.; Pircher, A.; Wolf, D. Tumor endothelial cells (tecs) as potential immune directors of the tumor microenvironment—New findings and future perspectives. Front. Cell Dev. Biol. 2020, 8, 766. [Google Scholar] [CrossRef]
- Huang, H.; Langenkamp, E.; Georganaki, M.; Loskog, A.; Fuchs, P.F.; Dieterich, L.C.; Kreuger, J.; Diberg, A. Vegf suppresses t-lymphocyte infiltration in the tumor microenvironment through inhibition of nf-κb-induced endothelial activation. FASEB J. 2015, 29, 227–238. [Google Scholar] [CrossRef] [Green Version]
- Madsen, C.D.; Sahai, E. Cancer dissemination--lessons from leukocytes. Dev. Cell 2010, 19, 13–26. [Google Scholar] [CrossRef] [Green Version]
- Wittchen, E.S. Endothelial signaling in paracellular and transcellular leukocyte transmigration. Front. Biosci. 2009, 14, 2522–2545. [Google Scholar] [CrossRef] [Green Version]
- Engelhardt, B.; Wolburg, H. Mini-review: Transendothelial migration of leukocytes: Through the front door or around the side of the house? Eur. J. Immunol. 2004, 34, 2955–2963. [Google Scholar] [CrossRef]
- Carman, C.V.; Sage, P.T.; Sciuto, T.E.; Fuente, M.A.D.L.; Springer, T.A. Transcellular diapedesis is initiated by invasive podosomes. Immunity 2007, 26, 784–797. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Voisin, M.; Larbi, K.Y.; Dangerfield, J.; Scheiermann, C.; Tran, M.; Maxwell, P.H.; Sorokin, L.; Nourshargh, S. Venular basement membranes contain specific matrix protein low expression regions that act as exit points for emigrating neutrophils. J. Exp. Med. 2006, 203, 1519–1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, W.A. Mechanisms of leukocyte transendothelial migration. Annu. Rev. Pathol. 2011, 6, 323–344. [Google Scholar] [CrossRef] [Green Version]
- Buul, J.D.; Allingham, M.J.; Samson, T.; Meller, J.; Boulter, E.; Rafael, G.; Keith, B. Rhog regulates endothelial apical cup assembly downstream from icam1 engagement and is involved in leukocyte trans-endothelial migration. J. Cell Biol. 2007, 178, 1279–1293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Kowalski, J.R.; Zhan, X.; Thomas, S.M.; Luscinskas, F.W. Endothelial cell cortactin phosphorylation by src contributes to polymorphonuclear leukocyte transmigration in vitro. Circ. Res. 2006, 98, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Schimmel, L.; Heemskerk, N.; van Buul, J.D. Leukocyte transendothelial migration: A local affair. Small Gtpases 2017, 8, 1–15. [Google Scholar] [CrossRef]
- Filewod, N.C.; Lee, W.L. Inflammation without vascular leakage. Science fiction no longer? Am. J. Rspir. Crit. Care Med. 2019, 200, 1472–1476. [Google Scholar] [CrossRef] [Green Version]
- Dejana, E.; Tournier-Lasserve, E.; Weinstein, B.M. The control of vascular integrity by endothelial cell junctions: Molecular basis and pathological implications. Dev. Cell 2009, 16, 209–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allingham, M.J.; van Buul, J.D.; Burridge, K. Icam-1-mediated, src- and pyk2-dependent vascular endothelial cadherin tyrosine phosphorylation is required for leukocyte transendothelial migration. J. Immunol. 2007, 179, 4053–4064. [Google Scholar] [CrossRef] [Green Version]
- Nottebaum, A.F.; Cagna, G.; Winderlich, M.; Gamp, A.C.; Linnepe, R.; Polaschegg, C.; Filippova, K.; Lyck, R.; Engelhardt, B.; Kamenyeva, O.; et al. Ve-ptp maintains the endothelial barrier via plakoglobin and becomes dissociated from ve-cadherin by leukocytes and by vegf. J. Exp. Med. 2008, 205, 2929–2945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winger, R.C.; Koblinski, J.E.; Kanda, T.; Ransohoff, R.M.; Muller, W.A. Rapid remodeling of tight junctions during paracellular diapedesis in a human model of the blood-brain barrier. J. Immunol. 2014, 193, 2427–2437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, J.N.; Verin, A.D.; Herenyiova, M.; English, D. Adherent neutrophils activate endothelial myosin light chain kinase: Role in transendothelial migration. J. Appl. Physiol. 1998, 84, 1817–1821. [Google Scholar] [CrossRef] [Green Version]
- Muller, W.A. How endothelial cells regulate transmigration of leukocytes in the inflammatory response. Am. J. Pathol. 2014, 184, 886–896. [Google Scholar] [CrossRef] [Green Version]
- Dasgupta, B.; Muller, W.A. Endothelial src kinase regulates membrane recycling from the lateral border recycling compartment during leukocyte transendothelial migration. Eur. J. Immunol. 2010, 38, 3499–3507. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, D.P.; Dalal, P.J.; Jaulin, F.; Sacks, D.B.; Muller, W.A. Endothelial iqgap1 regulates leukocyte transmigration by directing the lbrc to the site of diapedesis. J. Exp. Med. 2019, 216, 2582–2601. [Google Scholar] [CrossRef]
- Sullivan, D.P.; Muller, W.A. Neutrophil and monocyte recruitment by pecam, cd99, and other molecules via the lbrc. Semin. Immunopathol. 2014, 36, 193–209. [Google Scholar] [CrossRef] [Green Version]
- Ostermann, G.; Weber, K.S.C.; Zernecke, A.; Schröder, A.; Weber, C. Jam-1 is a ligand of the β2 integrin lfa-1 involved in transendothelial migration of leukocytes. Nat. Immunol. 2002, 3, 151–158. [Google Scholar] [CrossRef]
- Stan, R.V. Endothelial stomatal and fenestral diaphragms in normal vessels and angiogenesis. J. Cell. Mol. Med. 2010, 11, 621–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, D.; Nagy, J.A.; Hipp, J.; Dvorak, H.F.; Dvorak, A.M. Vesiculo-vacuolar organelles and the regulation of venule permeability to macromolecules by vascular permeability factor, histamine, and serotonin. J. Exp. Med. 1996, 183, 1981–1986. [Google Scholar] [CrossRef] [Green Version]
- Tkachenko, E.; Tse, D.; Sideleva, O.; Deharvengt, S.J.; Luciano, M.R.; Xu, Y.; McGarry, C.L.; Chidlow, J.; Pilch, P.F.; Sessa, W.C.; et al. Caveolae, fenestrae and transendothelial channels retain pv1 on the surface of endothelial cells. PLoS ONE 2012, 7, e32655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Millán, J.; Hewlett, L.; Glyn, M.; Toomre, D.; Clark, P.; Ridley, A.J. Lymphocyte transcellular migration occurs through recruitment of endothelial icam-1 to caveola- and f-actin-rich domains. Nat. Cell Biol. 2006, 8, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Marmon, S.; Hinchey, J.; Oh, P.; Cammer, M.; Almeida, C.J.; Gunther, L.; Raine, C.S.; Lisanti, M.P. Caveolin-1 expression determines the route of neutrophil extravasation through skin microvasculature. Am. J. Pathol. 2009, 174, 684–692. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Froio, R.M.; Sciuto, T.E.; Dvorak, A.M.; Luscinskas, F.W. Icam-1 regulates neutrophil adhesion and transcellular migration of tnf-α-activated vascular endothelium under flow. Blood 2005, 106, 584–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cinamon, G.; Shinder, V.; Shamri, R.; Alon, R. Chemoattractant signals and beta 2 integrin occupancy at apical endothelial contacts combine with shear stress signals to promote transendothelial neutrophil migration. J. Immunol. 2004, 173, 7282–7291. [Google Scholar] [CrossRef] [Green Version]
- Hordijk, P.L. Endothelial signalling events during leukocyte transmigration. FEBS J. 2010, 273, 4408–4415. [Google Scholar] [CrossRef]
- Filippi, M.D. Leukocyte transcellular diapedesis: Rap1b is in control. Tissue Barriers 2015, 3, e1052185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyck, R.; Reiss, Y.; Gerwin, N.; Greenwood, J.; Adamson, P.; Engelhardt, B. T-cell interaction with icam-1/icam-2 double-deficient brain endothelium in vitro: The cytoplasmic tail of endothelial icam-1 is necessary for transendothelial migration of t cells. Blood 2003, 102, 3675–3683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mamdouh, Z.; Mikhailov, A.; Muller, W.A. Transcellular migration of leukocytes is mediated by the endothelial lateral border recycling compartment. J. Exp. Med. 2009, 206, 2795–2808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filippi, M.D. Mechanism of diapedesis: Importance of the transcellular route. Adv. Immunol. 2016, 129, 25–53. [Google Scholar] [PubMed] [Green Version]
- Spinella, F.; Rosano, L.; Di Castro, V.; Natali, P.G.; Bagnato, A. Endothelin-1 induces vascular endothelial growth factor by increasing hypoxia-inducible factor-1α in ovarian carcinoma cells. J. Biol. Chem. 2002, 277, 27850–27855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wülfing, P.; Kersting, C.; Tio, J.; Fischer, R.J.; Wülfing, C.; Poremba, C.; Diallo, R.; Böcker, W.; Kiesel, L. Endothelin-1-, endothelin-a-, and endothelin-b-receptor expression is correlated with vascular endothelial growth factor expression and angiogenesis in breast cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2004, 10, 2393–2400. [Google Scholar] [CrossRef] [Green Version]
- Buckanovich, R.J.; Facciabene, A.; Kim, S.; Benencia, F.; Sasaroli, D.; Balint, K.; Katsaros, D.; O’Brien-Jenkins, A.; Gimotty, P.A.; Coukos, G. Endothelin b receptor mediates the endothelial barrier to t cell homing to tumors and disables immune therapy. Nat. Med. 2008, 14, 28–36. [Google Scholar] [CrossRef]
- Stroka, K.M.; Aranda-Espinoza, H. Endothelial cell substrate stiffness influences neutrophil transmigration via myosin light chain kinase-dependent cell contraction. Blood 2011, 118, 1632–1640. [Google Scholar] [CrossRef] [Green Version]
- Roberts, W.G.; Palade, G.E. Neovasculature induced by vascular endothelial growth factor is fenestrated. Cancer Res. 1997, 57, 765–772. [Google Scholar]
- Burns, A.R.; Walker, D.C.; Brown, E.S.; Thurmon, L.T.; Bowden, R.A.; Keese, C.R.; Simon, S.I.; Entman, M.I.; Smith, C.W. Neutrophil transendothelial migration is independent of tight junctions and occurs preferentially at tricellular corners. J. Immunol. 1997, 159, 2893–2903. [Google Scholar]
- Tazzyman, S.; Lewis, C.E.; Murdoch, C. Neutrophils: Key mediators of tumour angiogenesis. Int. J. Exp. Pathol. 2009, 90, 222–231. [Google Scholar] [CrossRef]
- Levine, B.L.; Miskin, J.; Wonnacott, K.; Keir, C. Global manufacturing of car t cell therapy. Mol. Ther. Methods Clin. Dev. 2017, 4, 92–101. [Google Scholar] [CrossRef] [Green Version]
- Urbansky, A.; Olm, F.; Scheding, S.; Laurell, T.; Lenshof, A. Label-free separation of leukocyte subpopulations using high throughput multiplex acoustophoresis. Lab. Chip 2019, 19, 1406–1416. [Google Scholar] [CrossRef] [Green Version]
- Pachynski, R.; Nazha, J.; Kohrt, H. Leukocyte trafficking: Can we bring the fight to the tumor? Discov. Med. 2016, 21, 205–212. [Google Scholar] [PubMed]
- Prete, A.D.; Schioppa, T.; Tiberio, L.; Stabile, H.; Sozzani, S. Leukocyte trafficking in tumor microenvironment. Curr. Opin. Pharmacol. 2017, 35, 40–47. [Google Scholar] [CrossRef]
- Hamzah, J.; Jugold, M.; Kiessling, F.; Rigby, P.; Manzur, M.; Marti, H.H.; Rabie, T.; Kaden, S.; Gröne, H.J.; Hämmerling, G.J.; et al. Vascular normalization in rgs5-deficient tumours promotes immune destruction. Nature 2008, 453, 410–414. [Google Scholar] [CrossRef]
- Schmittnaegel, M.; Rigamonti, N.; Kadioglu, E.; Cassará, A.; Wyser Rmili, C.; Kiialainen, A.; Kienast, Y.; Mueller, H.J.; Ooi, C.H.; Laoui, D. Dual angiopoietin-2 and vegfa inhibition elicits antitumor immunity that is enhanced by pd-1 checkpoint blockade. Sci. Transl. Med. 2017, 9, eaak9670. [Google Scholar] [CrossRef]
- Zheng, X.; Fang, Z.; Liu, X.; Deng, S.; Zhou, P.; Wang, X.; Zhang, C.; Yin, R.; Hu, H.; Chen, X.; et al. Increased vessel perfusion predicts the efficacy of immune checkpoint blockade. J. Clin. Investig. 2018, 128, 2104–2115. [Google Scholar] [CrossRef] [Green Version]
- Motz, G.T.; Santoro, S.P.; Wang, L.P.; Garrabrant, T.; Lastra, R.R.; Hagemann, I.S.; Lal, P.; Feldman, M.D.; Benencia, F.; Coukos, G. Tumor endothelium fasl establishes a selective immune barrier promoting tolerance in tumors. Nat. Med. 2014, 20, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Allen, E.; Jabouille, A.; Rivera, L.B.; Lodewijckx, I.; Missiaen, R.; Steri, V.; Feyen, K.; Tawney, J.; Hanahan, D.; Michael, I.P.; et al. Combined antiangiogenic and anti–pd-l1 therapy stimulates tumor immunity through hev formation. Sci. Transl. Med. 2017, 9, eaak9679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson-Percival, A.; He, B.; Li, Z.J.; Kjellen, A.; Russell, K.; Li, J.; Larma, I.; Ganss, R. De novo induction of intratumoral lymphoid structures and vessel normalization enhances immunotherapy in resistant tumors. Nat. Immunol. 2017, 18, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Vassileva, G.; Soto, H.A.; Nakano, H.; Kakiuchi, T.; Hedrick, J.A.; Lira, S.A. The reduced expression of 6ckine in the plt mouse results from the deletion of one of two 6ckine genes. J. Exp. Med. 1999, 190, 1183–1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakano, H.; Tamura, T.; Yoshimoto, T.; Yagita, H.; Miyasaka, M.; Butcher, E.C.; Nariuchi, H.; Kakiuchi, T.; Matsuzawa, A. Genetic defect in t lymphocyte-specific homing into peripheral lymph nodes. Eur. J. Immunol. 1997, 27, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Tabruyn, S.P.; Sabatel, C.; Nguyen, N.Q.N.; Verhaeghe, C.; Castermans, K.; Malvaux, L.; Griffioen, A.W.; Martial, J.A.; Struman, I. The angiostatic 16k human prolactin overcomes endothelial cell anergy and promotes leukocyte infiltration via nuclear factor-kappab activation. Mol. Endocrinol. 2007, 21, 1422–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellebrekers, D.; Castermans, K.; Viré, E.; Dings, R.P.M.; Hoebers, N.T.H.; Mayo, K.H.; oude Egbrink, M.G.A.; Molema, G.; Fuks, F.; van Engeland, M.; et al. Epigenetic regulation of tumor endothelial cell anergy: Silencing of intercellular adhesion molecule-1 by histone modifications. Cancer Res. 2006, 66, 10770–10777. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Yang, J.; Xu, D.; Gao, X.; Zhang, Z.; Hsu, J.L.; Li, C.; Lim, S.; Sheng, Y.; Zhang, Y.; et al. Disruption of tumour-associated macrophage trafficking by the osteopontin-induced colony-stimulating factor-1 signalling sensitises hepatocellular carcinoma to anti-pd-l1 blockade. Gut 2019, 68, 1653–1666. [Google Scholar] [CrossRef]
- Petty, A.J.; Li, A.; Wang, X.; Dai, R.; Heyman, B.; Hsu, D.; Huang, X.; Yang, Y. Hedgehog signaling promotes tumor-associated macrophage polarization to suppress intratumoral cd8+ t cell recruitment. J. Clin. Investig. 2019, 129, 5151–5162. [Google Scholar] [CrossRef] [Green Version]
- Gazzaniga, S.; Bravo, A.I.; Guglielmotti, A.; van Rooijen, N.; Maschi, F.; Vecchi, A.; Mantovani, A.; Mordoh, J.; Wainstok, R. Targeting tumor-associated macrophages and inhibition of mcp-1 reduce angiogenesis and tumor growth in a human melanoma xenograft. J. Investig. Dermatol. 2007, 127, 2031–2041. [Google Scholar] [CrossRef] [Green Version]
- Downey, C.M.; Aghaei, M.; Schwendener, R.A.; Jirik, F.R. Dmxaa causes tumor site-specific vascular disruption in murine non-small cell lung cancer, and like the endogenous non-canonical cyclic dinucleotide sting agonist, 2′3′-cgamp, induces m2 macrophage repolarization. PLoS ONE 2014, 9, e99988. [Google Scholar] [CrossRef] [Green Version]
- Weis, M.; Schlichting, C.L.; Engleman, E.G.; Cooke, J.P. Endothelial determinants of dendritic cell adhesion and migration. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1817–1823. [Google Scholar] [CrossRef] [Green Version]
- Zelenay, S.; van der Veen, A.G.; Böttcher, J.P.; Snelgrove, K.J.; Rogers, N.; Acton, S.E.; Chakravarty, P.; Girotti, M.R.; Marais, R.; Quezada, S.A.; et al. Cyclooxygenase-dependent tumor growth through evasion of immunity. Cell 2015, 162, 1257–1270. [Google Scholar] [CrossRef] [Green Version]
- Böttcher, J.P.; Bonavita, E.; Chakravarty, P.; Blees, H.; Cabeza-Cabrerizo, M.; Sammicheli, S.; Rogers, N.C.; Sahai, E.; Zelenay, S.; Reis, E.S.C. Nk cells stimulate recruitment of cdc1 into the tumor microenvironment promoting cancer immune control. Cell 2018, 172, 1022–1037. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Raybuck, A.; Shiuan, E.; Cho, S.H.; Wang, Q.; Brantley-Sieders, D.M.; Edwards, D.; Allaman, M.M.; Nathan, J.; Wilson, K.T.; et al. Selective inhibition of mtorc1 in tumor vessels increases antitumor immunity. JCI Insight 2020, 5, e139237. [Google Scholar] [CrossRef]
- Lee, W.S.; Yang, H.; Chon, H.J.; Kim, C. Combination of anti-angiogenic therapy and immune checkpoint blockade normalizes vascular-immune crosstalk to potentiate cancer immunity. Exp. Mol. Med. 2020, 52, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Zhu, H.; Xu, J.; Zheng, Y.; Cao, X.; Liu, Q. Tumor-derived cxcl1 promotes lung cancer growth via recruitment of tumor-associated neutrophils. J. Immunol. Res. 2016, 2016, 6530410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Zhao, Y.; Choi, J.; Ting, K.K.; Coleman, P.R.; Chen, J.; Cogger, V.C.; Wan, L.; Shi, Z.; Moller, T.; et al. Targeting mir-27a/ve-cadherin interactions rescues cerebral cavernous malformations in mice. PLoS Biol. 2020, 18, e3000734. [Google Scholar] [CrossRef] [PubMed]
- Schiffmann, L.M.; Fritsch, M.; Gebauer, F.; Günther, S.D.; Stair, N.R.; Seeger, J.M.; Thangarajah, F.; Dieplinger, G.; Bludau, M.; Alakus, H.; et al. Tumour-infiltrating neutrophils counteract anti-vegf therapy in metastatic colorectal cancer. Br. J. Cancer 2019, 120, 69–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Y.; Arita, M.; Zhang, Q.; Saban, D.R.; Chauhan, S.K.; Chiang, N.; Serhan, C.N.; Dana, R. Anti-angiogenesis effect of the novel anti-inflammatory and pro-resolving lipid mediators. Investig. Ophthalmol. Vis. Sci. 2009, 50, 4743–4752. [Google Scholar] [CrossRef] [PubMed]
- Morote-Garcia, J.C.; Napiwotzky, D.; Köhler, D.; Rosenberger, P. Endothelial semaphorin 7a promotes neutrophil migration during hypoxia. Proc. Natl. Acad. Sci. USA 2012, 109, 14146–14151. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Areas, R.; Libreros, S.; Simoes, M.; Castro-Silva, C.; Gazaniga, N.; Amat, S.; Jaczewska, J.; Keating, P.; Schilling, K.; Brito, M.; et al. Suppression of tumor-derived semaphorin 7a and genetic ablation of host-derived semaphorin 7a impairs tumor progression in a murine model of advanced breast carcinoma. Int. J. Oncol. 2017, 51, 1395–1404. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Zhang, N.; Qian, L.; Wang, X.; Fan, P.; Kuai, J.; Lin, S.; Liu, C.; Jiang, W.; Qin, S.; et al. Ctla4 blockade promotes vessel normalization in breast tumors via the accumulation of eosinophils. Int. J. Cancer 2020, 146, 1730–1740. [Google Scholar] [CrossRef]
- Yang, J.; Lagana, S.M.; Saenger, Y.M.; Carvajal, R.D. Dual checkpoint inhibitor-associated eosinophilic enteritis. J. Immunother. Cancer 2019, 7, 310. [Google Scholar] [CrossRef]
- Hedayat, K.M.; Lapraz, J.C. A new approach to biological modeling: Introduction to the biology of functions. Theory Endobiogeny 2019, 15, 215–254. [Google Scholar]
- McKee, A.S.; MacLeod, K.L.; Kappler, J.W.; Marrack, P. Immune mechanisms of protection: Can adjuvants rise to the challenge? BMC Biol. 2010, 8, 37. [Google Scholar] [CrossRef] [Green Version]
- Asadzadeh, Z.; Safarzadeh, E.; Safaei, S.; Baradaran, A.; Baradaran, B. Current approaches for combination therapy of cancer: The role of immunogenic cell death. Cancers 2020, 12, 1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonaventura, P.; Shekarian, T.; Alcazer, V.; Valladeau-Guilemond, J.; Valsesia-Wittmann, S.; Amigorena, S.; Caux, C.; Depil, S. Cold tumors: A therapeutic challenge for immunotherapy. Front. Immunol. 2019, 10, 168. [Google Scholar] [CrossRef] [Green Version]
- Zemek, R.M.; Chin, W.L.; Nowak, A.K.; Millward, M.J.; Lake, R.A.; Lesterhuis, W.J. Sensitizing the tumor microenvironment to immune checkpoint therapy. Front. Immunol. 2020, 11, 223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellone, M.; Calcinotto, A. Ways to enhance lymphocyte trafficking into tumors and fitness of tumor infiltrating lymphocytes. Front. Oncol. 2013, 3, 231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Lee, W.S.; Kong, S.J.; Kim, C.G.; Kim, J.H.; Chang, S.K.; Kim, S.; Kim, G.; Chon, H.J.; Kim, C. Sting activation reprograms tumor vasculatures and synergizes with vegfr2 blockade. J. Clin. Investig. 2019, 129, 4350–4364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jing, X.; Yang, F.; Shao, C.; Wei, K.; Xie, M.; Shen, H.; Shu, Y. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol. Cancer 2019, 18, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maione, F.; Capano, S.; Regano, D.; Zentilin, L.; Giacca, M.; Casanovas, O.; Bussolino, F.; Serini, G.; Giraudo, E. Semaphorin 3a overcomes cancer hypoxia and metastatic dissemination induced by antiangiogenic treatment in mice. J. Clin. Investig. 2012, 122, 1832–1848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayaprakash, P.; Ai, M.; Liu, A.; Budhani, P.; Bartkowiak, T.; Sheng, J.; Ager, C.; Nicholas, C.; Jaiswal, A.R.; Sun, Y.; et al. Targeted hypoxia reduction restores t cell infiltration and sensitizes prostate cancer to immunotherapy. J. Clin. Investig. 2018, 128, 5137–5149. [Google Scholar] [CrossRef] [PubMed]
- Viallard, C.; Audiger, C.; Popovic, N.; Akla, N.; Lanthier, K.; Legault-Navarrete, I.; Melichar, H.; Costantino, S.; Lesage, S.; Larrivée, B. Bmp9 signaling promotes the normalization of tumor blood vessels. Oncogene 2020, 39, 2996–3014. [Google Scholar] [CrossRef] [PubMed]
- Skuli, N.; Liu, L.; Runge, A.; Wang, T.; Yuan, L.; Patel, S.; Iruela-Arispe, L.; Simon, M.C.; Keith, B. Endothelial deletion of hypoxia-inducible factor-2alpha (hif-2alpha) alters vascular function and tumor angiogenesis. Blood 2009, 114, 469–477. [Google Scholar] [CrossRef] [Green Version]
- Mazzone, M.; Dettori, D.; de Oliveira, R.L.; Loges, S.; Schmidt, T.; Jonckx, B.; Tian, Y.M.; Lanahan, A.A.; Pollard, P.; de Almodovar, C.R.; et al. Heterozygous deficiency of phd2 restores tumor oxygenation and inhibits metastasis via endothelial normalization. Cell 2009, 136, 839–851. [Google Scholar] [CrossRef] [Green Version]
- Carmona-Rodríguez, L.; Martínez-Rey, D.; Fernández-Aceñero, M.J.; González-Martín, A.; Paz-Cabezas, M.; Rodríguez-Rodríguez, N.; Pérez-Villamil, B.; Sáez, M.E.; Díaz-Rubio, E.; Mira, E.; et al. Sod3 induces a hif-2α-dependent program in endothelial cells that provides a selective signal for tumor infiltration by t cells. J. Immunother. Cancer 2020, 8, e000432. [Google Scholar] [CrossRef]
- Ann, A. High endothelial venules and other blood vessels: Critical regulators of lymphoid organ development and function. Front. Immunol. 2017, 8, 45. [Google Scholar]
- Bento, D.C.; Jones, E.; Junaid, S.; Tull, J.; Williams, G.T.; Godkin, A.; Ager, A.; Gallimore, A. High endothelial venules are rare in colorectal cancers but accumulate in extra-tumoral areas with disease progression. OncoImmunology 2015, 4, e974374. [Google Scholar] [CrossRef]
- Hindley, J.P.; Jones, E.; Smart, K.; Bridgeman, H.; Lauder, S.N.; Ondondo, B.; Cutting, S.; Ladell, K.; Wynn, K.K.; Withers, D. T-cell trafficking facilitated by high endothelial venules is required for tumor control after regulatory t-cell depletion. Cancer Res. 2012, 72, 5473–5482. [Google Scholar] [CrossRef] [Green Version]
- Martinet, L.; Garrido, I.; Filleron, T.; Le Guellec, S.; Bellard, E.; Fournie, J.J.; Rochaix, P.; Girard, J.P. Human solid tumors contain high endothelial venules: Association with t- and b-lymphocyte infiltration and favorable prognosis in breast cancer. Cancer Res. 2011, 71, 5678–5687. [Google Scholar] [CrossRef] [Green Version]
- Skeate, J.G.; Otsmaa, M.E.; Prins, R.; Fernandez, D.J.; Da Silva, D.M.; Kast, W.M. Tnfsf14: Lighting the way for effective cancer immunotherapy. Front. Immunol. 2020, 11, 922. [Google Scholar] [CrossRef]
- Martinet, L.; Le Guellec, S.; Filleron, T.; Lamant, L.; Meyer, N.; Rochaix, P.; Garrido, I.; Girard, J.P. High endothelial venules (hevs) in human melanoma lesions. OncoImmunology 2012, 1, 829–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moussion, C.; Girard, J.P. Dendritic cells control lymphocyte entry to lymph nodes through high endothelial venules. Nature 2011, 479, 542–546. [Google Scholar] [CrossRef] [PubMed]
- Cedervall, J.; Dimberg, A.; Olsson, A.K. Tumor-induced local and systemic impact on blood vessel function. Mediat. Inflamm. 2015, 2015, 418290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffioen, A.W.; Damen, C.A.; Blijham, G.H.; Groenewegen, G. Tumor angiogenesis is accompanied by a decreased inflammatory response of tumor-associated endothelium. Blood 1996, 88, 667–673. [Google Scholar] [CrossRef] [PubMed]
- Yona, S.; Jung, S. Monocytes: Subsets, origins, fates and functions. Curr. Opin. Hematol. 2010, 17, 53–59. [Google Scholar] [CrossRef]
- Ginhoux, F.; Jung, S. Monocytes and macrophages: Developmental pathways and tissue homeostasis. Nat. Rev. Immunol. 2014, 14, 392–404. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Figueroa Velez, D.X.; Azevedo, R.; Hoover, E.M.; Tran, C.J.; Lo, C.; Vadpey, O.; Gandhi, S.P.; Lodoen, M.B. Imaging the dynamic recruitment of monocytes to the blood-brain barrier and specific brain regions during toxoplasma gondii infection. Proc. Natl. Acad. Sci. USA 2019, 116, 24796–24807. [Google Scholar] [CrossRef] [PubMed]
- Imhof, B.A.; Aurrand-Lions, M. Adhesion mechanisms regulating the migration of monocytes. Nat. Rev. Immunol. 2004, 4, 432–444. [Google Scholar] [CrossRef]
- Jin, H.; Su, J.; Garmy-Susini, B.; Kleeman, J.; Varner, J. Integrin alpha4beta1 promotes monocyte trafficking and angiogenesis in tumors. Cancer Res. 2006, 66, 2146–2152. [Google Scholar] [CrossRef] [Green Version]
- Shaw, S.K.; Bamba, P.S.; Perkins, B.N.; Luscinskas, F.W. Real-time imaging of vascular endothelial-cadherin during leukocyte transmigration across endothelium. J. Immunol. 2001, 167, 2323–2330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bazzoni, G. The jam family of junctional adhesion molecules. Curr. Opin. Cell Biol. 2003, 15, 525–530. [Google Scholar] [CrossRef]
- Laviron, M.; Boissonnas, A. Ontogeny of tumor-associated macrophages. Front. Immunol. 2019, 10, 1799. [Google Scholar] [CrossRef] [Green Version]
- Sidibe, A.; Ropraz, P.; Jemelin, S.; Emre, Y.; Poittevin, M.; Pocard, M.; Bradfield, P.F.; Imhof, B.A. Angiogenic factor-driven inflammation promotes extravasation of human proangiogenic monocytes to tumours. Nat. Commun. 2018, 9, 355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erja, K.; Ala-Aho, R.; Jeskanen, L.; Rechardt, O.; Saarialho-Kere, U. Expression of human macrophage metalloelastase (mmp-12) by tumor cells in skin cancer. J. Investig. Dermatol. 2000, 114, 1113–1119. [Google Scholar]
- Ojalvo, L.S.; Whittaker, C.A.; Condeelis, J.S.; Pollard, J.W. Gene expression analysis of macrophages that facilitate tumor invasion supports a role for wnt-signaling in mediating their activity in primary mammary tumors. J. Immunol. 2010, 184, 702–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beatty, G.L.; Chiorean, E.G.; Fishman, M.P.; Saboury, B.; Teitelbaum, U.R.; Sun, W.F.; Huhn, R.D.; Song, W.; Li, D.; Sharp, L.I.; et al. Cd40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 2011, 331, 1612–1616. [Google Scholar] [CrossRef] [Green Version]
- Willingham, S.B.; Volkmer, J.; Gentles, A.J.; Sahoo, D.; Dalerba, P.; Mitra, S.S.; Wang, J.; Contreras-Trujillo, H.; Martin, R.; Cohen, J.D.; et al. The cd47-signal regulatory protein alpha (sirpa) interaction is a therapeutic target for human solid tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 6662–6667. [Google Scholar] [CrossRef] [Green Version]
- Jarosz-Biej, M.; Kamińska, N.; Matuszczak, S.; Cichoń, T.; Pamuła-Piłat, J.; Czapla, J.; Smolarczyk, R.; Skwarzyńska, D.; Kulik, K.; Szala, S. M1-like macrophages change tumor blood vessels and microenvironment in murine melanoma. PLoS ONE 2018, 13, e0191012. [Google Scholar] [CrossRef]
- Ono, M.; Torisu, H.; Fukushi, J.; Nishie, A.; Kuwano, M. Biological implications of macrophage infiltration in human tumor angiogenesis. Cancer Chemother. Pharm. 1999, 43, 69–71. [Google Scholar] [CrossRef] [PubMed]
- Harney, A.S.; Arwert, E.N.; Entenberg, D.; Wang, Y.; Guo, P.; Qian, B.; Oktay, M.H.; Pollard, J.W.; Jones, J.G.; Condeelis, J.S. Real-time imaging reveals local, transient vascular permeability, and tumor cell intravasation stimulated by TIE2hi macrophage—Derived VEGFA. Cancer Discov. 2015, 5, 932–943. [Google Scholar] [PubMed] [Green Version]
- Loberg, R.D.; Ying, C.; Craig, M.; Day, L.I.; Sargent, E.; Neeley, C.; Wojno, K.; Snyder, L.A.; Yan, L.; Pienta, K.J. Targeting ccl2 with systemic delivery of neutralizing antibodies induces prostate cancer tumor regression in vivo. Cancer Res. 2007, 67, 9417–9424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coscia, M.; Quaglino, E.; Iezzi, M.; Curcio, C.; Pantaleoni, F.; Riganti, C.; Holen, I.; Mönkkönen, H.; Boccadoro, M.; Forni, G.; et al. Zoledronic acid repolarizes tumour-associated macrophages and inhibits mammary carcinogenesis by targeting the mevalonate pathway. J. Cell Mol. Med. 2010, 14, 2803–2815. [Google Scholar] [CrossRef] [PubMed]
- Tang-Huau, T.; Gueguen, P.; Goudot, C.; Durand, M.; Bohec, M.; Baulande, S.; Pasquier, B.; Amigorena, S.; Segura, E. Human in vivo-generated monocyte-derived dendritic cells and macrophages cross-present antigens through a vacuolar pathway. Nat. Commun. 2018, 9, 2570. [Google Scholar] [CrossRef] [PubMed]
- Laoui, D.; Keirsse, J.; Morias, Y.; Van Overmeire, E.; Geeraerts, X.; Elkrim, Y.; Kiss, M.; Bolli, E.; Lahmar, Q.; Sichien, D.; et al. The tumour microenvironment harbours ontogenically distinct dendritic cell populations with opposing effects on tumour immunity. Nat. Commun. 2016, 7, 13720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segura, E.; Amigorena, S. Inflammatory dendritic cells in mice and humans. Trends Immunol. 2013, 34, 440–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardner, A.; Ruffell, B. Dendritic cells and cancer immunity. Trends Immunol. 2016, 37, 855–865. [Google Scholar] [CrossRef] [Green Version]
- Verdijk, P.; Aarntzen, E.H.; Punt, C.J.; de Vries, I.J.; Figdor, C.G. Maximizing dendritic cell migration in cancer immunotherapy. Expert Opin. Biol. 2008, 8, 865–874. [Google Scholar] [CrossRef]
- Dastmalchi, F.; Karachi, A.; Yang, C.; Azari, H.; Sayour, E.J.; Dechkovskaia, A.; Vlasak, A.L.; Saia, M.E.; Lovaton, R.E.; Mitchell, D.A.; et al. Sarcosine promotes trafficking of dendritic cells and improves efficacy of anti-tumor dendritic cell vaccines via cxc chemokine family signaling. J. Immunother. Cancer 2019, 7, 321. [Google Scholar] [CrossRef]
- Williford, J.; Ishihara, J.; Ishihara, A.; Mansurov, A.; Hosseinchi, P.; Marchell, T.M.; Potin, L.; Swartz, M.A.; Hubbell, J.A. Recruitment of cd103+ dendritic cells via tumor-targeted chemokine delivery enhances efficacy of checkpoint inhibitor immunotherapy. Sci. Adv. 2019, 5, eaay1357. [Google Scholar] [CrossRef] [Green Version]
- Kashyap, A.S.; Schmittnaegel, M.; Rigamonti, N.; Pais-Ferreira, D.; Mueller, P.; Buchi, M.; Ooi, C.H.; Kreuzaler, M.; Hirschmann, P.; Guichard, A.; et al. Optimized antiangiogenic reprogramming of the tumor microenvironment potentiates cd40 immunotherapy. Proc. Natl. Acad. Sci. USA 2020, 117, 541–551. [Google Scholar] [CrossRef] [Green Version]
- Gabrilovich, D.I.; Chen, H.L.; Girgis, K.R.; Cunningham, H.T.; Meny, G.M.; Nadaf, S.; Kavanaugh, D.; Carbone, D.P. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat. Med. 1996, 2, 1096–1103. [Google Scholar] [CrossRef] [PubMed]
- Geering, B.; Stoeckle, C.; Conus, S.; Simon, H.U. Living and dying for inflammation: Neutrophils, eosinophils, basophils. Trends Immunol. 2013, 34, 398–409. [Google Scholar] [CrossRef]
- Jaakkola, K.; Jalkanen, S.; Kaunismäki, K.; Vänttinen, E.; Saukko, P.; Alanen, K.; Kallajoki, M.; Voipio-Pulkki, L.; Salmi, M. Vascular adhesion protein-1, intercellular adhesion molecule-1 and p-selectin mediate leukocyte binding to ischemic heart in humans. J. Am. Coll. Cardiol. 2000, 36, 122–129. [Google Scholar] [CrossRef] [Green Version]
- Koskinen, K.; Vainio, P.J.; Smith, D.J.; Pihlavisto, M.; Ylä-Herttuala, S.; Jalkanen, S.; Salmi, M. Granulocyte transmigration through the endothelium is regulated by the oxidase activity of vascular adhesion protein-1 (vap-1). Blood 2004, 103, 3388–3395. [Google Scholar] [CrossRef] [Green Version]
- Jovic, S.; Linge, H.M.; Shikhagaie, M.M.; Olin, A.I.; Lannefors, L.; Erjefält, J.S.; Mörgelin, M.; Egesten, A. The neutrophil-recruiting chemokine gcp-2/cxcl6 is expressed in cystic fibrosis airways and retains its functional properties after binding to extracellular DNA. Mucosal Immunol. 2016, 9, 112–123. [Google Scholar] [CrossRef]
- Bian, Z.; Shi, L.; Venkataramani, M.; Abdelaal, A.M.; Culpepper, C.; Kidder, K.; Liang, H.; Zen, K.; Liu, Y. Tumor conditions induce bone marrow expansion of granulocytic, but not monocytic, immunosuppressive leukocytes with increased cxcr2 expression in mice. Eur. J. Immunol. 2018, 48, 532–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gentles, A.J.; Newman, A.M.; Liu, C.L.; Bratman, S.V.; Feng, W.; Kim, D.; Nair, V.S.; Xu, Y.; Khuong, A.; Hoang, C.D.; et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 2015, 21, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Xu, W. Neutrophils diminish t-cell immunity to foster gastric cancer progression: The role of gm-csf/pd-l1/pd-1 signalling pathway. Gut 2017, 66, 1878–1880. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.; Kloecker, G.; Fleming, C.; Bousamra, M.; Hansen, R.; Hu, X.; Ding, C.; Cai, Y.; Xiang, D.; Donninger, H.; et al. Human polymorphonuclear neutrophils specifically recognize and kill cancerous cells. OncoImmunology 2014, 3, e950163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossaint, J.; Zarbock, A. Tissue-specific neutrophil recruitment into the lung, liver, and kidney. J. Innate Immun. 2013, 5, 348–357. [Google Scholar] [CrossRef]
- Hyun, Y.M.; Hong, C.W. Deep insight into neutrophil trafficking in various organs. J. Leukoc. Biol. 2017, 102, 617–629. [Google Scholar] [CrossRef] [Green Version]
- Sundd, P.; Gutierrez, E.; Koltsova, E.K.; Kuwano, Y.; Fukuda, S.; Pospieszalska, M.K.; Groisman, A.; Ley, K. ‘Slings’ enable neutrophil rolling at high shear. Nature 2012, 488, 399–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Mao, D.; Lü, S.; Tong, C.; Zhang, Y.; Long, M. Distinct binding affinities of mac-1 and lfa-1 in neutrophil activation. J. Immunol. 2013, 190, 4371–4381. [Google Scholar] [CrossRef] [Green Version]
- Woodfin, A.; Voisin, M.B.; Beyrau, M.; Colom, B.; Caille, D.; Diapouli, F.; Nash, G.B.; Chavakis, T.; Albelda, S.M.; Rainger, G.E.; et al. The junctional adhesion molecule jam-c regulates polarized transendothelial migration of neutrophils in vivo. Nat. Immunol. 2011, 12, 761–769. [Google Scholar] [CrossRef] [Green Version]
- de Oliveira, S.; Reyes-Aldasoro, C.C.; Candel, S.; Renshaw, S.A.; Mulero, V.; Calado, A. Cxcl8 (il-8) mediates neutrophil recruitment and behavior in the zebrafish inflammatory response. J. Immunol. 2013, 190, 4349–4359. [Google Scholar] [CrossRef] [PubMed]
- Metzemaekers, M.; Gouwy, M.; Proost, P. Neutrophil chemoattractant receptors in health and disease: Double-edged swords. Cell. Mol. Immunol. 2020, 17, 433–450. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Kawada, K.; Itatani, Y.; Inamoto, S.; Okamura, R.; Iwamoto, M.; Miyamoto, E.; Chen-Yoshikawa, T.F.; Hirai, H.; Hasegawa, S.; et al. Loss of smad4 promotes lung metastasis of colorectal cancer by accumulation of ccr1+ tumor-associated neutrophils through ccl15-ccr1 axis. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 833–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granot, Z. Neutrophils as a therapeutic target in cancer. Front. Immunol. 2019, 10, 1710. [Google Scholar] [CrossRef] [Green Version]
- Mukaida, N.; Sasaki, S.; Baba, T. Two-faced roles of tumor-associated neutrophils in cancer development and progression. Int. J. Mol. Sci. 2020, 21, 3457. [Google Scholar] [CrossRef]
- Jaillon, S.; Ponzetta, A.; Di Mitri, D.; Santoni, A.; Bonecchi, R.; Mantovani, A. Neutrophil diversity and plasticity in tumour progression and therapy. Nat. Rev. Cancer 2020, 20, 485–503. [Google Scholar] [CrossRef]
- Rothenberg, M.E. Eotaxin: An essential mediator of eosinophil trafficking into mucosal tissues. Am. J. Respir. Cell Mol. Biol. 1999, 21, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Grisaru-Tal, S.; Itan, M.; Klion, A.D.; Munitz, A. A new dawn for eosinophils in the tumour microenvironment. Nat. Rev. Cancer 2020, 20, 594–607. [Google Scholar] [CrossRef]
- Hollande, C.; Boussier, J.; Ziai, J.; Nozawa, T.; Bondet, V.; Phung, W.; Lu, B.; Duffy, D.; Paradis, V.; Mallet, V.; et al. Inhibition of the dipeptidyl peptidase dpp4 (cd26) reveals il-33-dependent eosinophil-mediated control of tumor growth. Nat. Immunol. 2019, 20, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Carretero, R.; Sektioglu, I.M.; Garbi, N.; Salgado, O.C.; Beckhove, P.; Hämmerling, G.J. Eosinophils orchestrate cancer rejection by normalizing tumor vessels and enhancing infiltration of cd8+ t cells. Nat. Immunol. 2015, 16, 609–617. [Google Scholar] [CrossRef] [PubMed]

| Mechanisms Involved in Leucocyte Capture | |||
|---|---|---|---|
| Primary Capture | Secondary Capture | ||
| Molecules on ECs | Molecules on Leucocytes | Molecules on ECs | Molecules on Leucocytes |
| P-selectin | PSGL-1 | N/A | L-selectin, PSGL-1 |
| E-selectin | PSGL-1 | ||
| Mechanisms Involved in Leucocyte Rolling and Adhesion | |
|---|---|
| Molecules on ECs | Molecules on Leucocytes |
| P-selectin | PSGL-1 |
| E-selectin | PSGL-1 |
| ICAM-1 | LFA-1, Mac-1 |
| VCAM-1 | VLA-4, α4β1-integrin |
| MAdCAM-1 | α4β7-integrin |
| Mechanisms Involved in Intraluminal Crawling of Leucocytes | |
|---|---|
| Molecules on ECs | Molecules on Leucocytes |
| Chemokines | Chemokine receptors |
| ICAM-1 | LFA-1, Mac-1 |
| VCAM-1 | VLA-4, α4β1-integrin |
| Mechanisms Involved in Leucocyte TEM | |||
|---|---|---|---|
| Paracellular Migration | Transcellular Migration | ||
| Molecules on ECs | Molecules on Leucocytes | Molecules on ECs | Molecules on Leucocytes |
| VE-cadherin | N/A | PECAM-1 | PECAM-1 |
| PECAM-1 | PECAM-1 | JAM-A | LFA-1 |
| JAM-A | LFA-1 | CD99 | CD99 |
| CD99 | CD99 | ICAM-1 | LFA-1, Mac-1 |
| ICAM-1 | LFA-1, Mac-1 | Caveolin-1 | N/A |
| Chemokines | Chemokine receptors | Chemokines | Chemokine receptors |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Y.; Ting, K.K.; Coleman, P.; Qi, Y.; Chen, J.; Vadas, M.; Gamble, J. The Tumour Vasculature as a Target to Modulate Leucocyte Trafficking. Cancers 2021, 13, 1724. https://doi.org/10.3390/cancers13071724
Zhao Y, Ting KK, Coleman P, Qi Y, Chen J, Vadas M, Gamble J. The Tumour Vasculature as a Target to Modulate Leucocyte Trafficking. Cancers. 2021; 13(7):1724. https://doi.org/10.3390/cancers13071724
Chicago/Turabian StyleZhao, Yang, Ka Ka Ting, Paul Coleman, Yanfei Qi, Jinbiao Chen, Mathew Vadas, and Jennifer Gamble. 2021. "The Tumour Vasculature as a Target to Modulate Leucocyte Trafficking" Cancers 13, no. 7: 1724. https://doi.org/10.3390/cancers13071724
APA StyleZhao, Y., Ting, K. K., Coleman, P., Qi, Y., Chen, J., Vadas, M., & Gamble, J. (2021). The Tumour Vasculature as a Target to Modulate Leucocyte Trafficking. Cancers, 13(7), 1724. https://doi.org/10.3390/cancers13071724