1. Introduction
Lung cancer is the commonest form of cancer and the leading cause of cancer death both in the UK and worldwide [
1]. Non-small cell lung cancer (NSCLC) is the most prevalent type of lung cancer, accounting for approximately 85% of all lung cancers [
2]. In comparison to small cell lung cancer (SCLC), NSCLC tumours contain numerous cancer driver gene mutations which may be targeted for treatment [
3]. Multiple studies have shown that targeted therapies have improved the prognosis and survival of NSCLC patients carrying appropriate driver genes [
4]. In developed countries, all NSCLC patients, regardless of their clinical characteristics, are routinely tested for suspected mutation points, such as epidermal growth factor receptor (EGFR) mutations, anaplastic lymphoma kinase (ALK) fusions, ROS proto-oncogene 1 (ROS1) fusions, and N-methyl-N′-nitroso-guanidine human osteosarcoma transforming gene (MET) mutation and amplification [
5,
6].
High expression of MET (a receptor tyrosine-protein kinase) can be observed in approximately 60% of NSCLC patients and correlates with poor prognosis [
7]. In NSCLC, anomalous expression of MET may work as the driver of tumorigenesis and the cause for drug resistance to other therapies [
8]. MET exon 14-skipping mutations and amplification of MET are the most frequent types of MET mutation found in NSCLC and are already included in the routine tests for driver oncogenes and therapeutic targets [
5,
6]. The
MET gene encodes the protein MET which acts as the receptor for hepatocyte growth factor (HGF) [
9]. MET belongs to the receptor tyrosine kinase (RTK) family with a conservative molecular structure and intracellular downstream signaling pathways [
10]. Usually, HGF activation of MET acts in a paracrine pathway at the cell surface where there is a cycle of degradation and internalization. HGF/MET has been demonstrated to play an essential role in the ability of a tumour to metastasize by regulating cell motility, migration, proliferation and angiogenesis, and in addition, modulating the tight junction (TJs) cell to cell barrier [
11,
12]. TJ, together with anchoring (adherens) junctions and communicating (gap) junctions, constitute the epithelial cell junction, which is the basic structural entity which strengthens the mechanical connection between cells. They also maintain the integrity of tissue architecture and help coordinate the function of cells within human tissues [
11]. TJ are located in the areas between the cell membranes of adjacent cells, and can completely occlude the space between cells and isolate the extracellular space, thereby creating an intercellular barrier which functions to maintain permeability and polarity, prevents migration and motility, mediates cell to cell adhesion, and transduce cell signaling which plays a role in differentiation and growth of epithelial and endothelial cells [
13]. The TJ proteins consist of three major components: integral transmembrane proteins, peripheral or plaque anchoring proteins, and TJ associated or regulatory proteins. It has become increasingly recognized that human cancer is frequently associated with failure of epithelial cells to form TJ and to establish correct apicobasal polarity [
14]. Changes in the expression and/or distribution of TJ proteins may result in complete loss of the TJ structure allowing cancer cell invasion and progression [
11]. Therefore, TJs are an important factor in the progression of tumours. Our previous research has shown that TJ are largely regulated by the HGF/MET signaling pathway [
15]. In human vascular endothelial cells (HUVECs), HGF treatment reduced the transendothelial resistance (TER) and increased paracellular permeability (PCP), which are regarded as key measurements of cell to cell TJ function [
15,
16,
17]. HGF/MET signaling disrupted TJs in the breast cancer cell lines MDA-MB-231 and MCF-7 by downregulating the expression of several important TJ molecules, such as occludin, at both transcript and protein levels [
18]. In prostate cancer, PC-3 cells had reduced TER and increased PCP after treatment with HGF [
19]. Signal induced proliferation associated protein 1 (SIPA1), appears to play a crucial role in the regulation of TJs by HGF/MET.
SIPA1 was first cloned from murine cells in 1995 [
20], and the human SIPA1 cDNA was identified in 1997 [
21]. Initially, SIPA1 was believed to be a specific mitogen induced GTPase activating protein (GAP) for several Ras-related mediating proteins such Rap1 and Rap2 [
20,
21,
22]. Subsequently, SIPA1 was found to be important in the development of various cancers and metastases. SIPA1 promotes the adhesion, migration and invasion of breast cancer MDA-MB-231 cells by binding to the promoter region of ITGB1 (integrin β1) gene and activating it, thus stimulating the downstream integrin mediated focal adhesion kinase/Protein kinase B/matrix metalloproteinase 9 (FAK/Akt-MMP9) pathway [
23]. Higher expression levels of SIPA1 were associated with worse prognosis and increased incidence of metastases for prostate cancer (CaP) patients. SIPA1 has been found to intensify the invasion efficiency, but decrease the attachment ability, of CaP cells by down regulating bromodomain protein 4 (BRD4) and extra cellular matrix (ECM)-related gene expression [
24]. In human oral squamous cell carcinoma (OSCC), SIPA1 interacting with BRD4 increases the level of matrix metalloproteinase 7 (MMP7), leading to increased invasiveness of the OSCC cells and worse prognosis for OSCC patients [
25]. In colorectal cancer (CRC) cell lines HT115 and CaCO-2, knockdown of SIPA1 increased the invasion, adhesion, and migration potential of cancer cells compared to the control group, while the ability to proliferate was decreased [
26]. It may be that SIPA1 can exert similar effects in cells with either high or low/knocked down of SIPA1 in different cancer types due to their tissue specificity, an area that requires further study. Moreover, SIPA1 is considered to be involved in many cancer types, including cervical cancer [
27], gastric cancer [
28], lung cancer [
29,
30], and melanoma [
31], suggesting that SIPA1 may have an active role during progression and metastasis of cancer. While there has been little research focused on the modulating function of SIPA1 on the regulation of HGF/MET in TJs, previous work from the host laboratory indicated that in breast and prostate cancer cells, the presence of SIPA1 was required for the regulation of HGF/MET in TJs. Knockdown of SIPA1 silenced the effect of HGF on TJs in breast and prostate cancer cells [
32,
33]. However, there are limited studies investigating the function of SIPA1 in lung cancer [
29,
30] and the mechanism of SIPA1 in cancer development and metastasis remains largely unknown, particularly the role of SIPA1 in regulating the TJs and reacting to HGF signaling in lung cancer cells. This project, therefore, aimed to evaluate the interaction between SIPA1 and HGF/MET, as well as their influence on lung cancer in terms of molecular activation, cellular behaviour and clinical relevance.
2. Materials and Methods
2.1. Lung Cancer Tissue Samples
A clinical cohort consisting of lung tumours (n = 148) together with adjacent background tissues (n = 148) was obtained from patients at Peking University Cancer Hospital immediately after surgery and stored in a freezer at −80 °C until used. All protocol and procedures were approved by the Peking University Cancer Hospital Research Ethics Committee and informed consent was obtained from the patients.
2.2. Cell Culture
The cell lines used throughout this study to compare different types of lung cancer were the lung cancer cell lines A549 (an epithelial lung carcinoma cell), SK-MES-1 (a squamous cell carcinoma derived from a metastatic site) and COR-L23 (a tumourigenic lung large cell carcinoma). A549 and SK-MES-1 cell lines were purchased from the American Type Culture Collection (ATCC, LGC Standard, Salisbury, UK) and COR-L23 was purchased from the European Collection of Animal Cell Cultures (ECACC, Salisbury, UK). A549 and SK-MES-1 cells were cultured in Dulbecco’s Modified Eagle’s medium (DMEM), and COR-L23 lung cancer cells were cultured in GI1640 containing 2 mM glutamine. Medium was supplemented with heat inactivated fetal calf serum (FCS) (Sigma-Aldrich, Poole, Dorset, UK) and an antibiotic cocktail comprising penicillin, streptomycin and amphotericin B (Sigma-Aldrich).
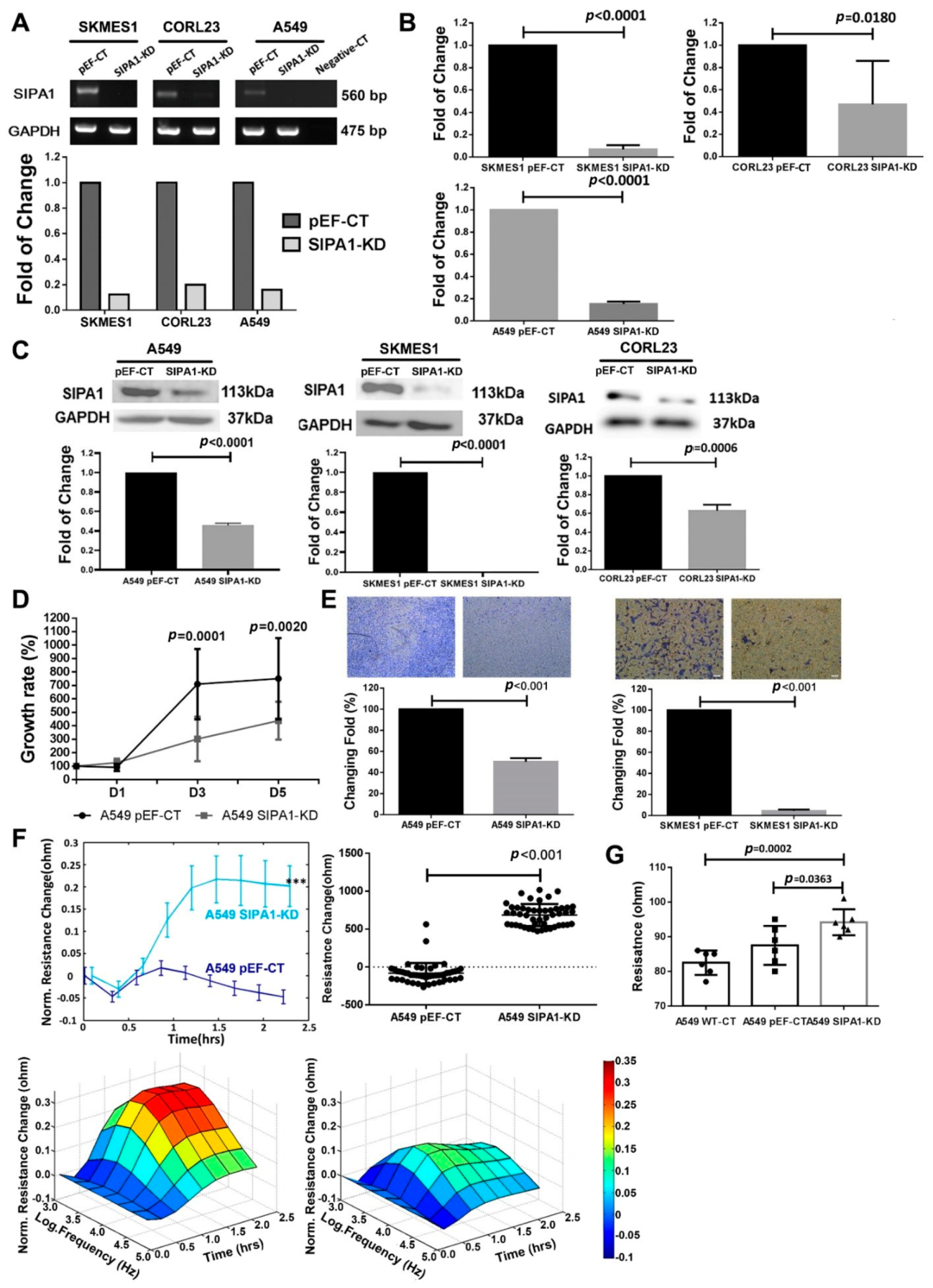
2.3. Establishment of the Stable SIPA1 Knockdown Cell Lines
SIPA1 knockdown in A549, SK-MES-1 and COR-L23 cells was performed using hammerhead ribozyme transgenes. Anti-SIPA1 hammerhead ribozymes targeting the secondary structure of SIPA1 mRNA were designed using the Zuker NA mFold program, according to the protocol provided [
34]. SIPA1 knockdown cells were designated SIPA1-KD, whereas plasmid control cells were designated pEF-CT.
The ribozymes were synthesized using touchdown polymerase chain reaction (PCR) and cloned into the pEF6/V5-His TOPO TA expression plasmid vector (Invitrogen, Loughborough, UK). Ribozyme transgenes and empty plasmids as control were transfected into the lung cancer cell lines using the Easyjet Plus electroporator (EquiBio, Kent, UK). Transfected cells for use in this projected were verified by selection with blasticidin.
2.4. RNA Extraction and PCR
RNA extraction was performed according to the Tri Reagent protocol (Sigma-Aldrich). RT-PCR was performed using the GoScriptTM Reverse Transcription System kit (Promega, Southampton, UK). The reverse transcription conditions were 25 °C for 5 min, 42 °C for 60 min and finally 70 °C for 15 min. GoTaq Green master mix (Promega) was subsequently used to amplify the target genes. The cycling conditions of the procedure were: denaturation at 94 °C for 5 min; 32 cycles of 94 °C for 20 s, 55 °C for 20 s and 72 °C for 15 s; with an extension phase at 72 °C for 10 min followed by storage at 4 °C until use. All the reactions were carried out in a 2720 Thermal Cycler (Applied Biosystems, Paisley, UK). Electrophoresis on agarose gel stained by SYBR Safe Gel Stain (Life Technologies, Paisley, UK) was used to separate the DNA fragments.
Quantitative PCR (qPCR) was performed using the Ampliflour™ Uniprimer™ Universal system (Intergen Company
®, New York, NY, USA). The Ampliflour probe consists of a 3′ region specific to the Z-sequence (ACTGAACCTGACCGTACA) present on the target specific primers and a 5′ hairpin structure labelled with a fluorophore (FAM). The cycling conditions of the procedure were: 94 °C for 5 min; up to 100 cycles of 94 °C for 10 s, 55 °C for 35 s and 72 °C for 20 s. The fluorescent signal is detected to determine a threshold for quantification of genes amplified in the reaction. Copy number of a target transcript is determined using the cycle number of a reaction when its fluorescence signal reaches the threshold. The sequences for the primers used in this project are shown in
supplementary data (Table S1).
2.5. Protein Extraction and Western Blotting
Cells were washed, detached, and lysed using lysis buffer, centrifuged and the concentration of proteins was measured using Bio-Rad DC protein assay kit (Bio-Rad Laboratories, Hemel-Hempstead, UK). Protein detection was performed using the sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and antibody recognition. Information of the antibodies used in this project are shown in
supplementary table (Table S2).
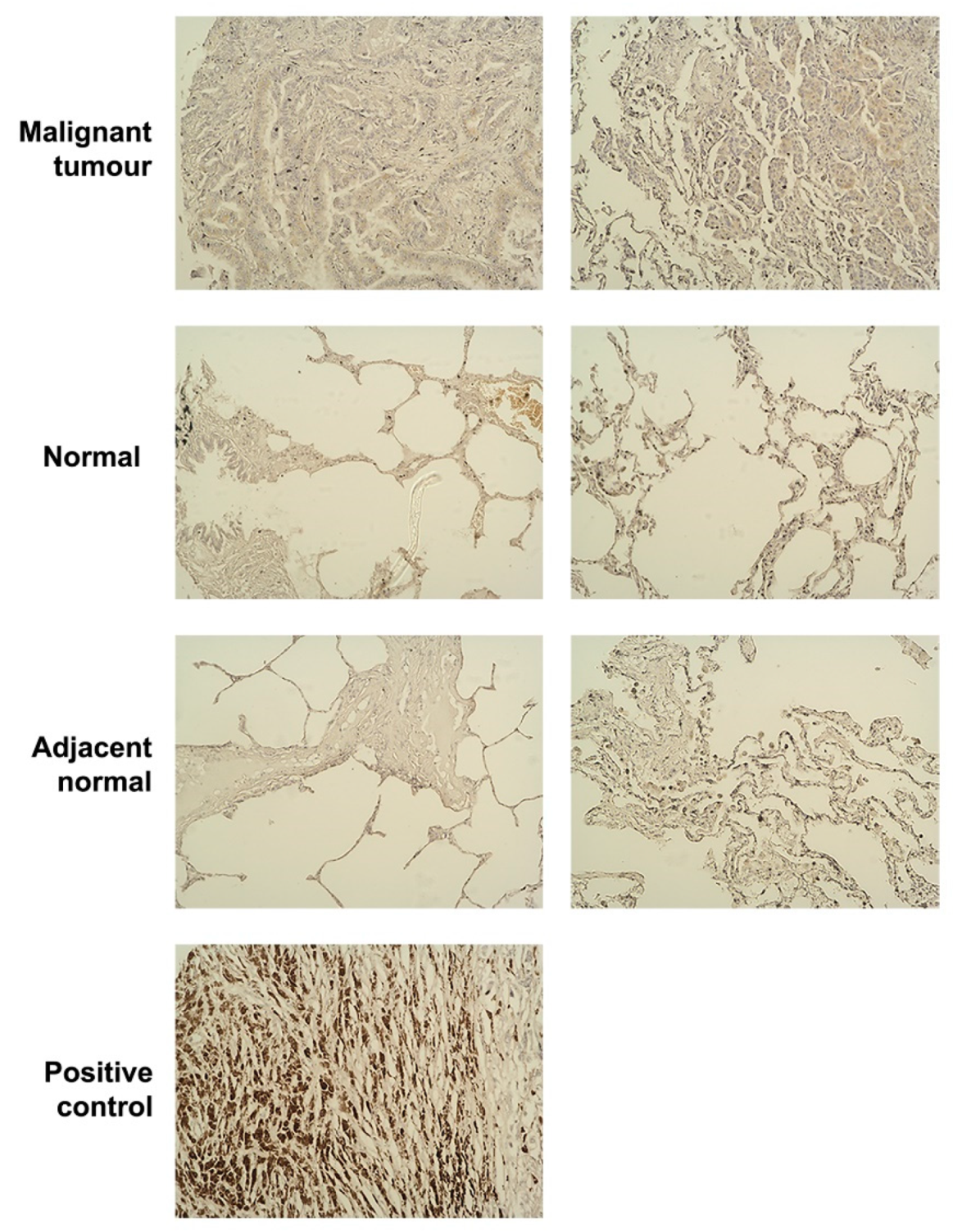
2.6. Tissue Microarray (TMA) and Immunohistochemistry (IHC)
The lung disease spectrum tissue microarray slide LC1201(Biomax, Cheltenham, UK) was used in this project. IHC was performed using the VECTASTAIN® Universal ABC Kit (Vector Laboratories Inc., Upper Heyford, UK). The TMA slide was dewaxed and rehydrated and treated with 30% hydrogen peroxide (H2O2) followed by microwaving for 20 min. After cooling, the slide was blocked with horse serum (1–2 drops) in 5 mL of 1× OptiMax Wash Buffer (BioGenex, Fremont, CA, USA) for 2 h. After washing, the slide was incubated with primary antibody (10 μg/mL) overnight at 4 °C. Following incubation with the corresponding secondary antibody, VECTASTAIN® Universal ABC complex (Vector Laboratories Inc.) was added, then 3,3′-Diaminobenzidine (DAB) chromogen (Vector Laboratories Inc.) and the slide incubated in the dark. The target protein was indicated by brown colouration.
2.7. Immunofluorescence (IFC)
Cells were seeded into 8 or 16 well glass chamber. Cells were fixed, treated with a permeabilization buffer, blocked by horse serum and incubated with primary antibody overnight at 4 °C. 4′,6-Diamidino-2-phenylindole (DAPI), for nucleus staining, together with secondary antibodies tagged with either fluorescein isothiocyanate (FITC) or tetramethylrhodamine isothiocyanate (TRITC) were added. The procedures were carried out in the dark and slides stored at 4 °C. The slides were examined under a fluorescent microscope and photographs obtained using an Orca digital camera (Hamamatsu, Welwyn Garden City, UK).
2.8. Protein Array
Protein was extracted from A549 Pef-CT and SIPA1-KD cells. After quantification, protein samples were sent to Kinexus™ Bioinformatics (Kinexus Bioinformatics, Vancouver, BC, Canada). Data were analyzed using several parameters including Globally Normalized Signal Intensity, percentage (%) change from control (CFC) and the Z-scores.
2.9. Transendothelial Resistance (TER) Assay
Transwell inserts (Millicell, MerkMillipore, Watford, UK) with 0.4 μm pores were placed into the wells of a 24 well plate. Appropriately 5 × 104 cells in 50 μL medium were seeded into inserts until confluent with 1ml medium in the well outside the insert. Resistance across the membrane was then measured in triplicate using the EVOM2 Epithelial Volt/Ohm Meter (World Precision Instruments, Hitchin, Hertfordshire, UK). Change in resistance was used as the measurement of the change in the TJ function of the cells.
2.10. Electric Cell-Substrate Impedance Sensing (ECIS) Assay
An ECIS instrument (Applied Biophysics Inc., Troy, NJ, USA) was used to asses changes in cell behaviour assay. The 96 well ECIS plate was connected to the Applied Bio Physics-ECIS Software V1.2.135 (Applied Biophysics Inc.), and then stabilized in 200 μL of serum free medium. Cells in appropriate density were seeded on the plate with at least six replicates. After cell seeding, the 96 well ECIS plate was connected to the ECIS station in the incubator. Resistance increases as the cells attach to the electrode and begin spreading and the resistance will continue to increase until the cells reach confluence.
2.11. In-Vitro Cell Proliferation Assay
Medium (200 μL containing appropriate cells (2 × 103) was added to 96 well plates. These plates were incubated at 37 °C, with 5% carbon dioxide (CO2), for periods of 24, 48, 72, and 120 h.
After incubation, the medium was aspirated, and the cells were fixed with 4% formaldehyde for 20 min before being stained with 0.5% crystal violet for 15 min. The plate was then rinsed with tap water and left to air dry. The crystal violet was solubilized using 200 μL of 10% acetic acid, and the number of cells was assessed by measuring the absorbance of the resulting solution at 540 nm using a spectrophotometer (ELx800, BIO-TEK, Swindon, UK). The growth rate was calculated as a percentage, using the absorbance from the corresponding plate collected at 24 h as a baseline.
2.12. In-Vitro Transwell Matrigel Invasion Assays
Transwell inserts containing 8.0 μm pores (Millicell, Merck KGaA, Darmstadt, Germany) were placed into the wells of a 24 well plate. Inserts were coated with 100 μL of serum free medium containing 50 μg Matrigel® Basement Membrane Matrix (Corning Incorporated, Flintshire, UK, stock concentration 0.5 μg/μL) and left to dry for 2 h at 55 °C. After rehydration, 2 × 103 cells in 200 μL culture medium were seeded into each insert. Medium was then added to the lower chamber of each well. The cells were incubated with 5% CO2 at 37 °C for 72 h. After incubation, the Matrigel layer and the non-invasive cells were removed and the invasive cells were then fixed with 4% formalin and stained with 0.5% crystal violet. Stained cells were subsequently counted and photographed under the microscope, and the crystal violet was solubilized using 10% acetic acid to measure the absorbance.
2.13. Statistical Analysis
Statistical analysis was performed using the SPSS 26 (SPSS, Inc., Chicago, IL, USA), GraphPad Prism 8 (GraphPad Software, La Jolla, CA, USA), and Minitab 14 (Minitab Ltd., Coventry, UK). The cumulative survival curves were generated using Kaplan-Meier plots and analyzed using log-rank test. The two-tailed Student’s t-test or Mann–Whitney U test were used for two group comparisons depending on data parameters. For PCR and western blotting, band intensity was quantified using Image J software. Statistical significance was set at p < 0.05.
4. Discussion
Lung cancer is the leading cause of malignant tumour death, independently of country or region [
34]. Extensive evidence has suggested that the HGF/MET signaling pathway is essential in lung cancer tumorigenesis and progression via the alteration of cell apoptosis, growth, migration and morphology [
35]. MET has been proposed as one of the driving oncogenes in lung cancer and the cause of drug resistance to EGFR TKIs [
8]. Meanwhile, an increasing number of studies indicate the importance of TJs in cancer metastasis [
11]. SIPA1 has been shown to work as a driving factor in a variety of tumours [
22]. The potential of SIPA1 in regulating TJs has also been confirmed in breast cancer and prostate cancer in the host laboratory. The effect of the HGF/MET signaling pathway on breast cancer cells requires the presence of SIPA1 [
19,
32,
33]. During this study a series of functional assays were performed in order to find out more about the role of SIPA1 in the function of lung cancer cells.
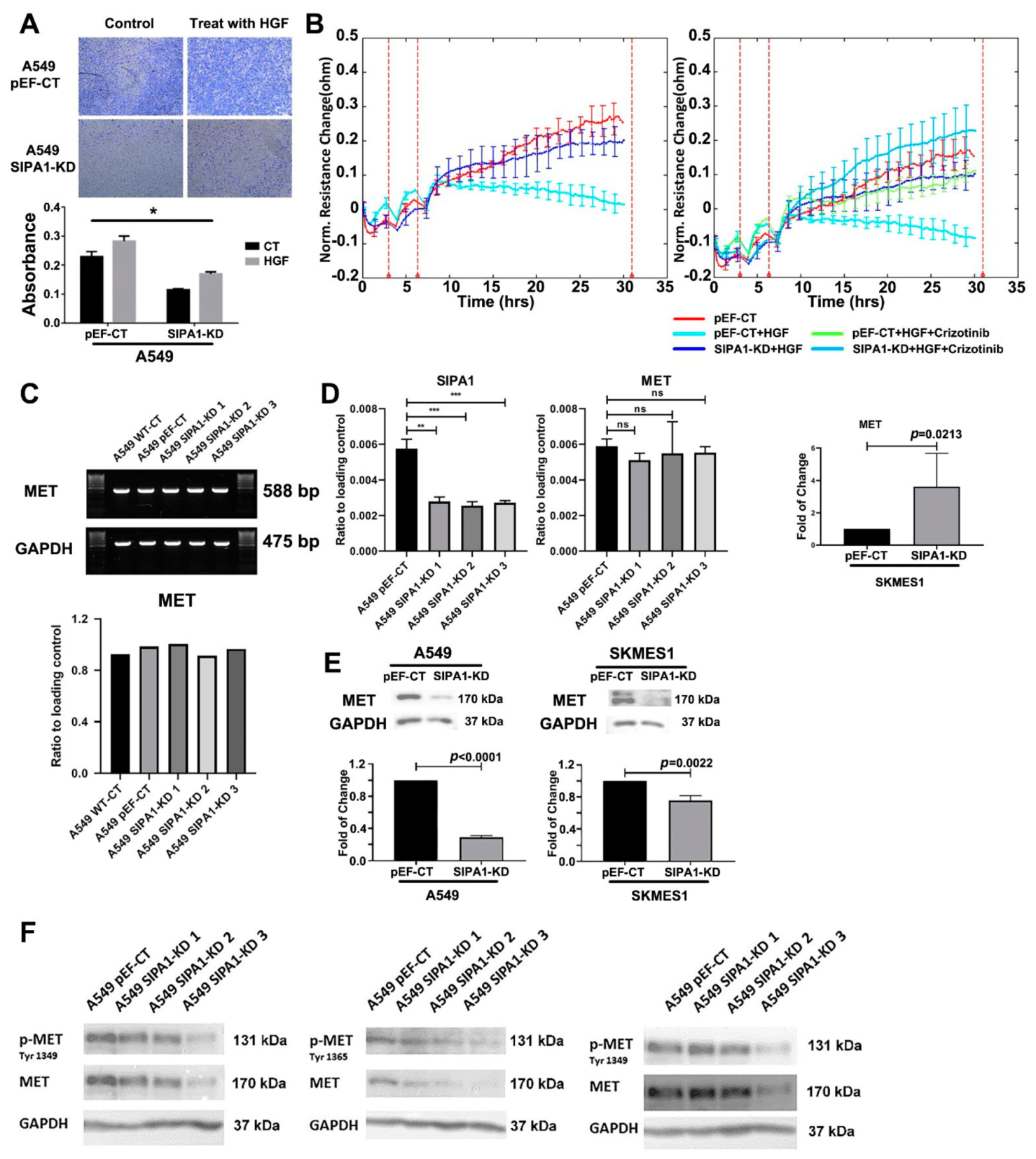
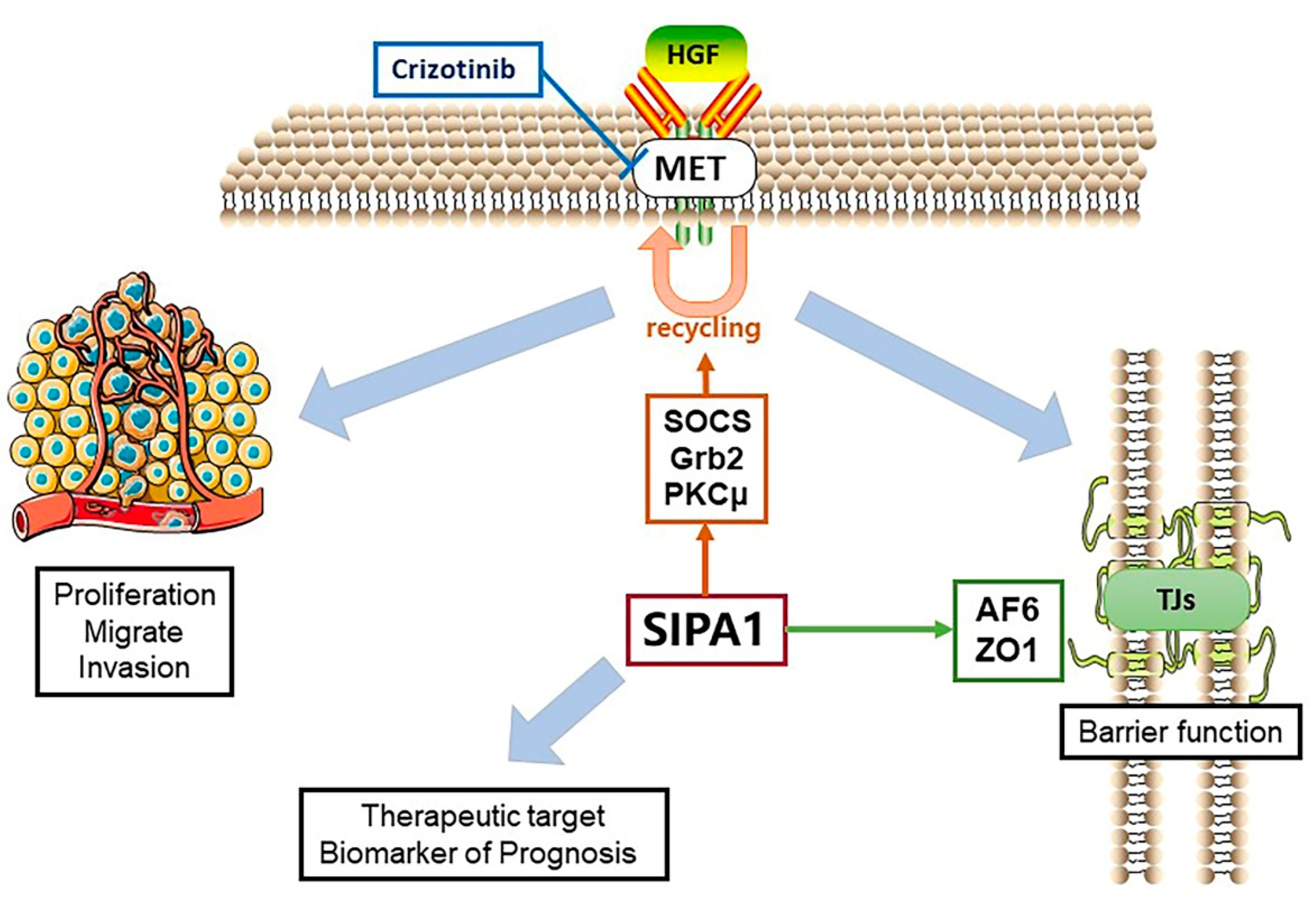
This study showed that knockdown of SIPA1 reduced the response of lung cancer cells to HGF in terms of invasion and barrier function. HGF enhanced the invasion of pEF control cells, while the enhancement could be blocked by SIPA1 knockdown. HGF decreased the barrier function of lung cancer cells, which was counteracted by SIPA1 knockdown with similarity to the small molecule MET targeted inhibitor, Crizotinib. Since knockdown of SIPA1 has a similar effect to pharmacological MET inhibition on blocking the regulation of HGF signaling on tight junctions, it is reasonable to assume that SIPA1 could have some potential interaction with MET.
The mechanism of interaction between SIPA1 and MET is an important issue to be determined in this study. SIPA1 gene expression was studied in the Peking lung cancer cohort and the TCGA database. The results showed that SIPA1 expression is highly correlated with some key markers in the HGF/MET signaling pathways and with TJ components. At the same time, protein phosphorylation array revealed that SIPA1 is involved in the regulation of complex signal transduction and RTK activation. This confirms what has previously been described-that SIPA1 and MET interact to effect changes in cell barrier function. It is known that the regulation of RTKs is an extremely complicated process involving multiple signaling pathways. Although protein array analysis has shown that there exists a co-relationship between SIPA1 and the RTKs family, to which MET belongs, the specific mechanism of interaction has yet to be demonstrated. However, the protein expression level of MET was suppressed by SIPA1 knockdown, as detected by western blotting; we can infer from this that the cell surface therefor lacked sufficient MET receptors to transmit extracellular HGF signals. Moreover, reduced MET expression as effected by SIPA1 knockdown led to the downregulation in MET phosphorylation (Tyr 1349 and 1365). The phosphorylation status of key sites of the MET receptor’s domain used to anchor downstream molecules was inhibited, limiting the function of MET to down-stream transmission signals.
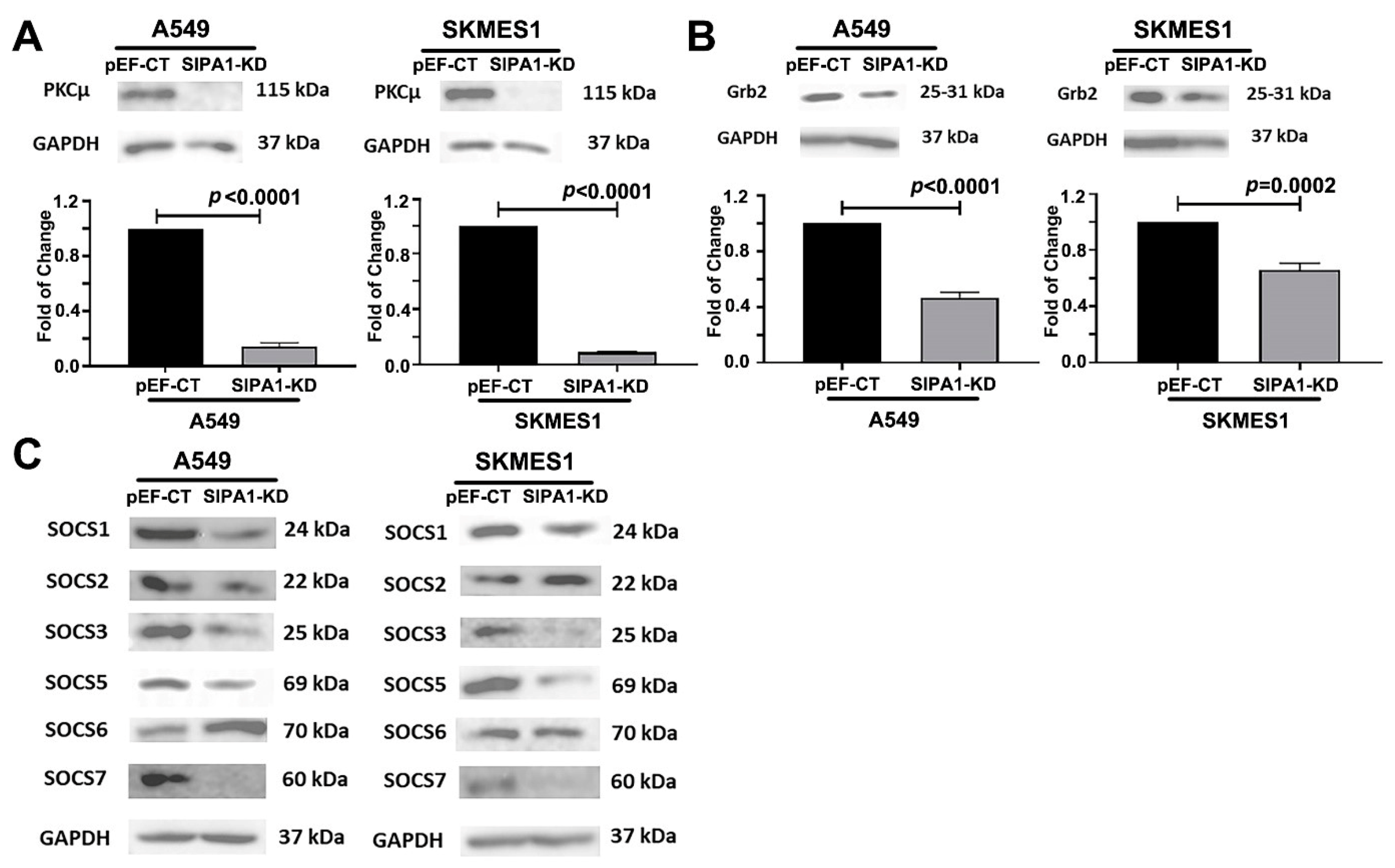
MET regulation involves multiple different processes from transcription to degradation, which include MET oncogene mutations, MET gene methylation, transcription factors regulation, alternative splicing, microRNA regulation, MET translational regulation, proteolysis of MET, glycosylation and phosphorylation on MET, internalization, degradation and recycling of MET, nuclear localization of MET, and autoregulation of MET [
36,
37]. This project has shown that SIPA1 can influence the expression of MET, but the specific process (or processes) by which SIPA1 acts is still an area for future study. Some key regulatory molecules during the MET recycling process, such as Grb2, SOCS, and PKCμ, were selected for further analysis. Western blot results for these molecules showed that the protein expression of these molecules decreased after SIPA1 knockdown. In previous publications, Grb2, SOCS, and PKCμ all promoted internalization of MET, leading the process towards recycling to the membrane rather than degradation [
37]. Knockdown of SIPA1 decreased the expression of these molecules which could be a potential approach to regulate MET receptors. Therefore, we can conclude that in lung cancer cells, Grb2, SCOS and PKCμ are the potential target molecules for SIPA1, thus promoting the recycling of MET, in order to maintain the MET receptor at a high level, thereby enabling the transmission of the HGF signal within the cells.
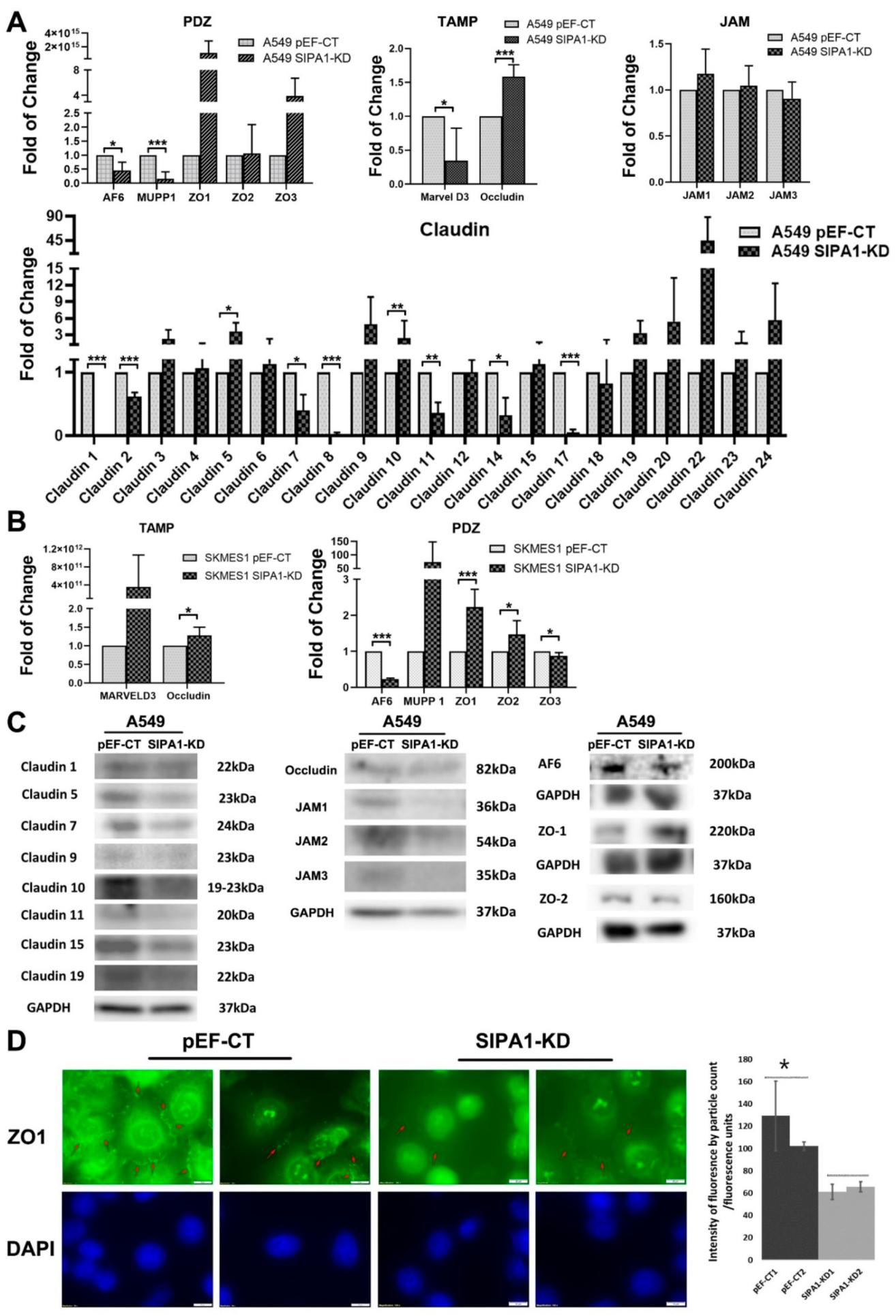
The findings of this study suggest that the expression of SIPA1 in lung cancer cells is closely related to the expression of TJ molecules and the cell’s barrier function. Gene expression correlation analysis showed the potential regulatory effect of SIPA1 on TJs. Data from the Peking cohort and the TGCA database (available from the website
https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga, accessed on 19 January 2019) were analyzed. The components of TJ proteins that are highly correlated with expression of SIPA1 include JAM1, JAM2, ZO family, occludin, Marvel D2, Marvel D3, nectin2, and claudin4, claudin7, claudin12, and claudin15, which are proven in at least one database from the Peking cohort and/or the TGCA database. qPCR and/or western blotting demonstrated that claudins 1, 2, 7, 8, 11, 14 and 17 from the claudin family, and MUPP1 which contains a PDZ domain were downregulated after knockdown of SIPA1. Claudin5, and the PDZ domain containing protein AF6 was downregulated and ZO family proteins were upregulated in SIPA1 knockdown cells compared to pEF control cells. Previous studies have shown that SIPA1 contains a PDZ domain [
38], as do other TJ markers such as the ZO family, AF6, MAGI1, MAGI2, MAGI3, PAR3, PAR6 and MUPP1. The common feature that SIPA1, AF6, and the ZO family all contain the PDZ domain, suggests that this could be a potential interaction site. It is possible that SIPA1 could interact with AF6 and the ZO family in the regulation of TJ function.
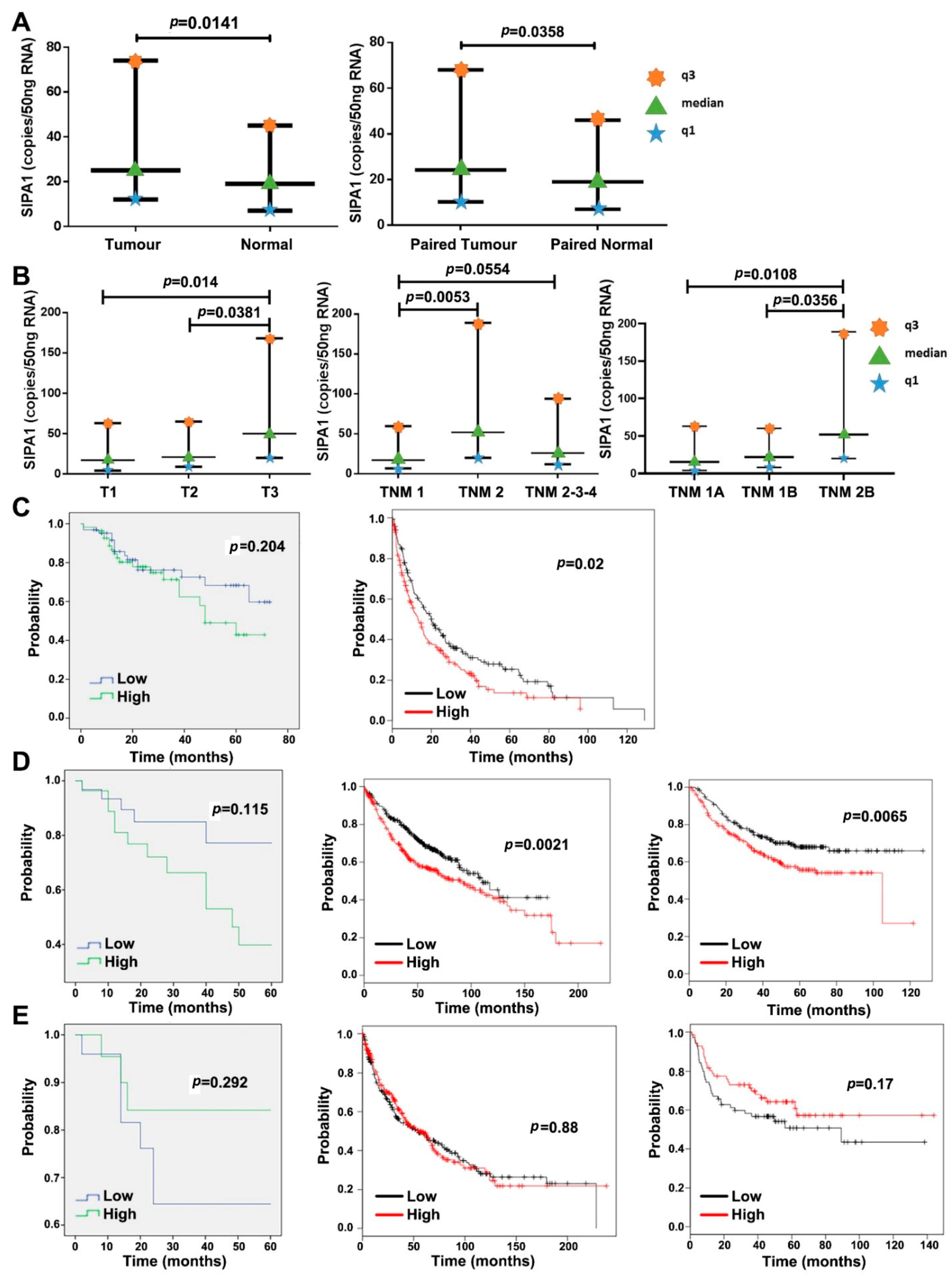
The transcriptional expression level of SIPA1 was found to be higher in cancer than adjacent normal tissue or normal tissue in the Peking clinical cohort. The IHC staining score of the lung tumour TMA lacks statistical significance. The protein level of SIPA1 in tumour tissue samples was higher than that in normal lung tissue and adjacent normal lung tissue, but the difference was not statistically significant (data not shown). There may be inconsistencies between SIPA1 gene transcripts and SIPA1 protein analysis. As previously discussed, there is a possibility that difference exists between the transcription level of SIPA1 gene and the protein level of translation, but the Peking clinical cohort had a larger sample number than the TMA cohort, and importantly, it has a higher number of normal tissues. The clinical cohort samples are fresh frozen from surgery and it was easy to quantify the expression of SIPA1, whilst the TMA samples are could only be used for qualitative or semi-quantitative analysis. In terms of tumorigenesis, the Peking clinical cohort data showed that the transcription level of SIPA1 was significantly higher in the advanced T stage and TNM stage tumours. From the prognostic analysis of lung cancer database, the median survival time of patients with high SIPA1 expression was significantly shorter than that of patients with low SIPA1 expression, and the difference is particularly significant in patients with adenocarcinoma. Transcription of SIPA1 was high in lung cancer tumour and more advanced TNM stages of lung cancer, and the high level of SIPA1 correlated with the worst prognosis of lung cancer patients, which all indicate that SIPA1 can be used as a potential independent clinical biomarker to predict the prognosis and recurrence of lung cancer. MET has been reported as a driving factor and therapeutic target for NSCLC. Crizotinib (Xalkori
®, Pfizer, Surrey, UK) is approved by the FDA as a MET targeted inhibitor used in NSCLC patients with MET amplification and/or exon 14 skipping mutation [
8]. Since SIPA1 can interact with MET, and SIPA1 can regulate the MET receptor protein, and the inhibition of SIPA1 has similar effects on the TJs of lung cancer cells as the MET inhibitor Crizotinib, it is possible that SIPA1 may be a potential therapeutic target in NSCLC and a potential substitute target for MET targeted therapy resistance in NSCLC.














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}