
Reverse Engineering of Ewing Sarcoma Regulatory Network Uncovers PAX7 and RUNX3 as Master Regulators Associated with Good Prognosis

 ,
,  , , , , ,
, , , , ,  , and
, and 
Abstract
:Simple Summary
Abstract

1. Introduction
2. Materials and Methods
2.1. Data Acquisition and Processing
2.2. Regulatory Network Inference and Master Regulator Analysis
2.3. Differential Methylation Analysis
2.4. Gene Ontology Functional Enrichment
2.5. Network Visualization
2.6. Master Regulators Activity
2.7. Survival Analysis
2.8. RT-qPCR Analysis
3. Results
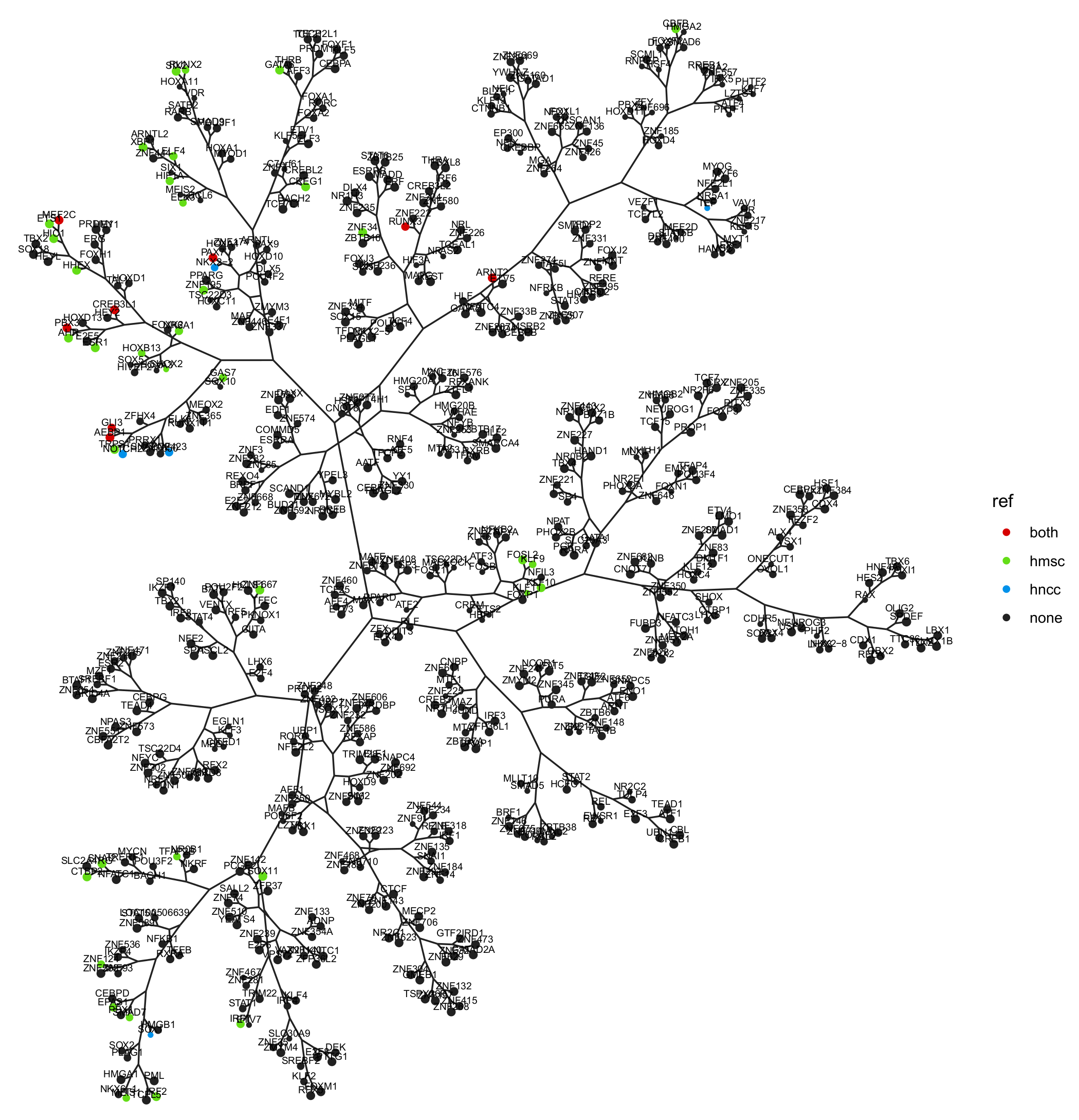
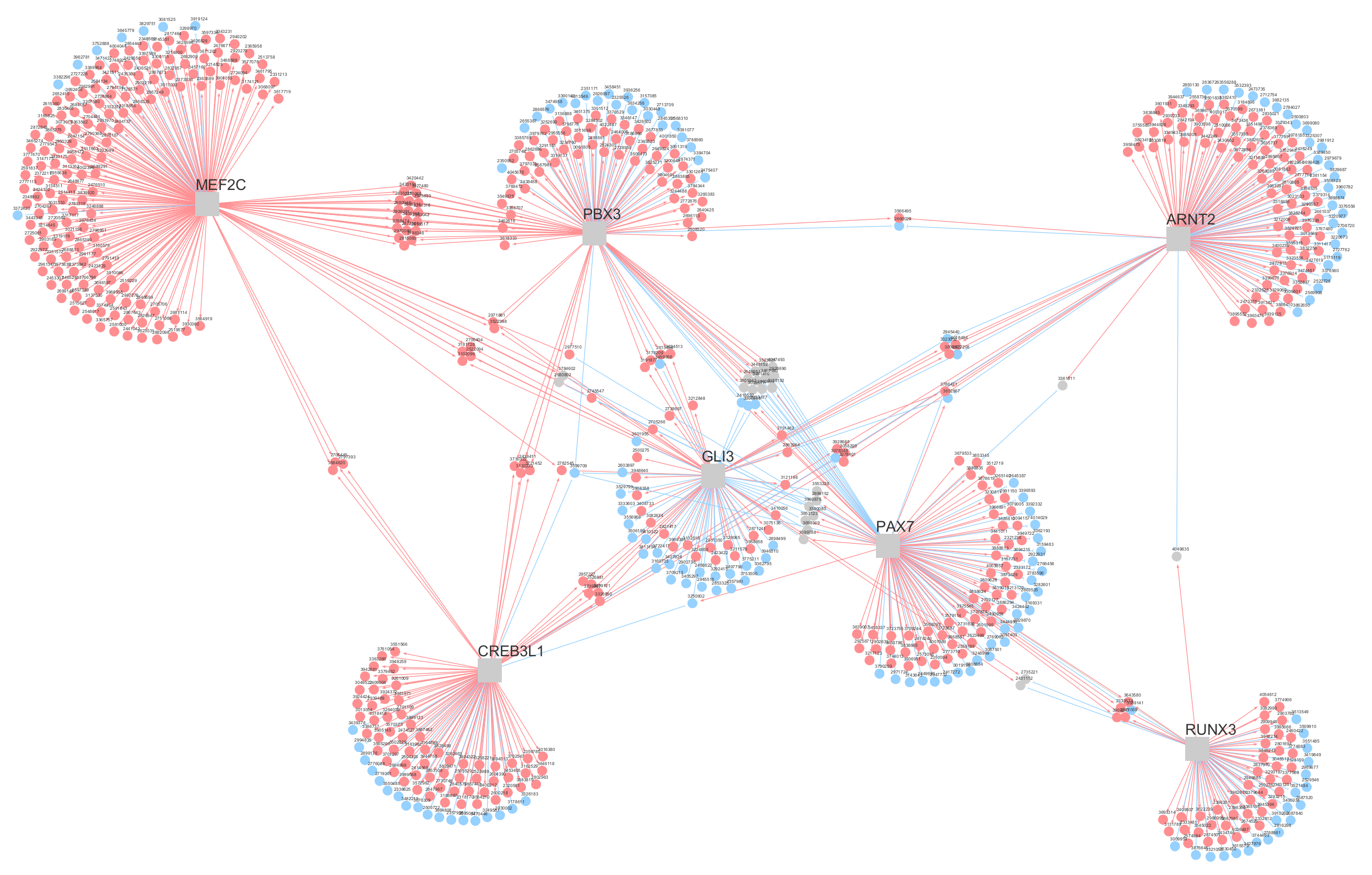
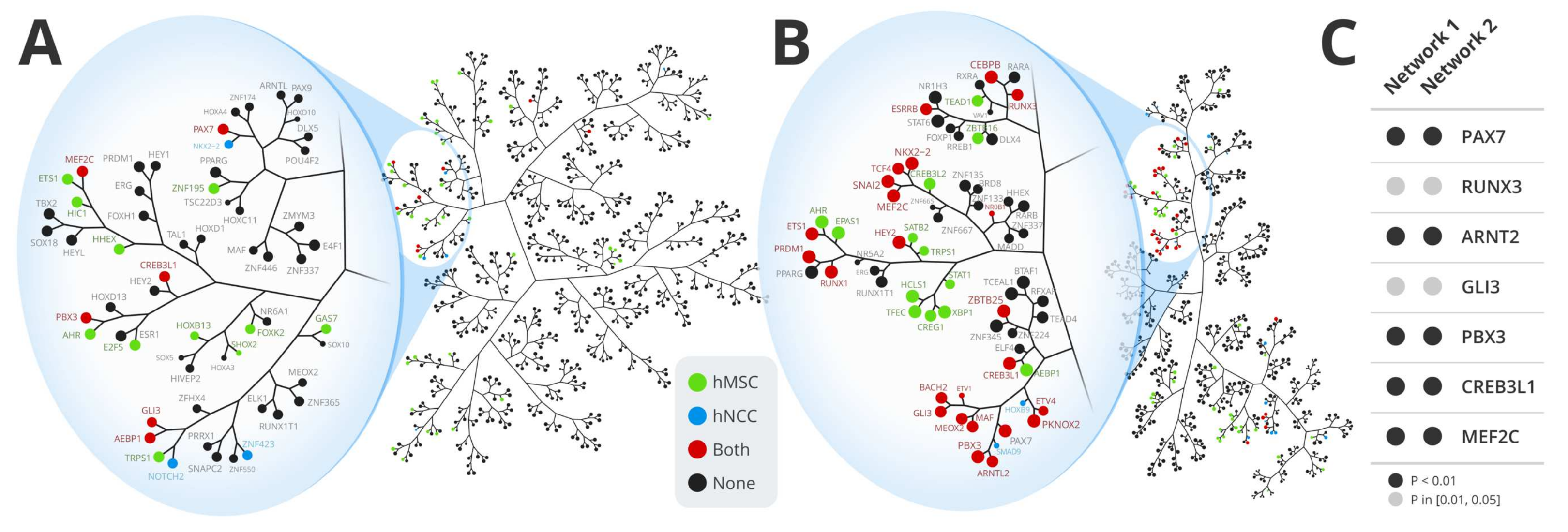
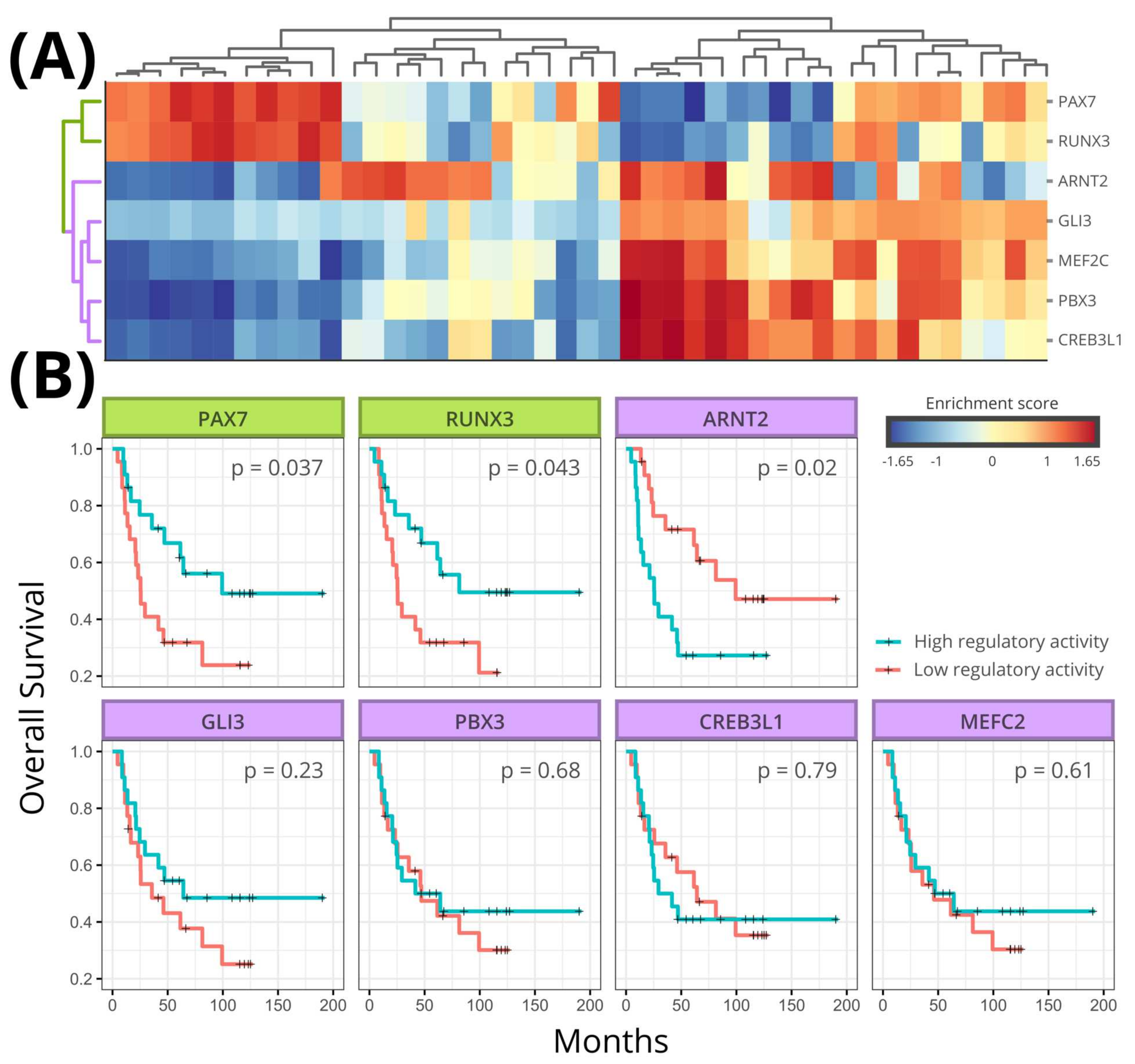
3.1. Regulatory Network Inference and Master Regulator Analysis
3.2. Differential Methylation Analysis
3.3. Biological Function of Master Regulators Associated Genes
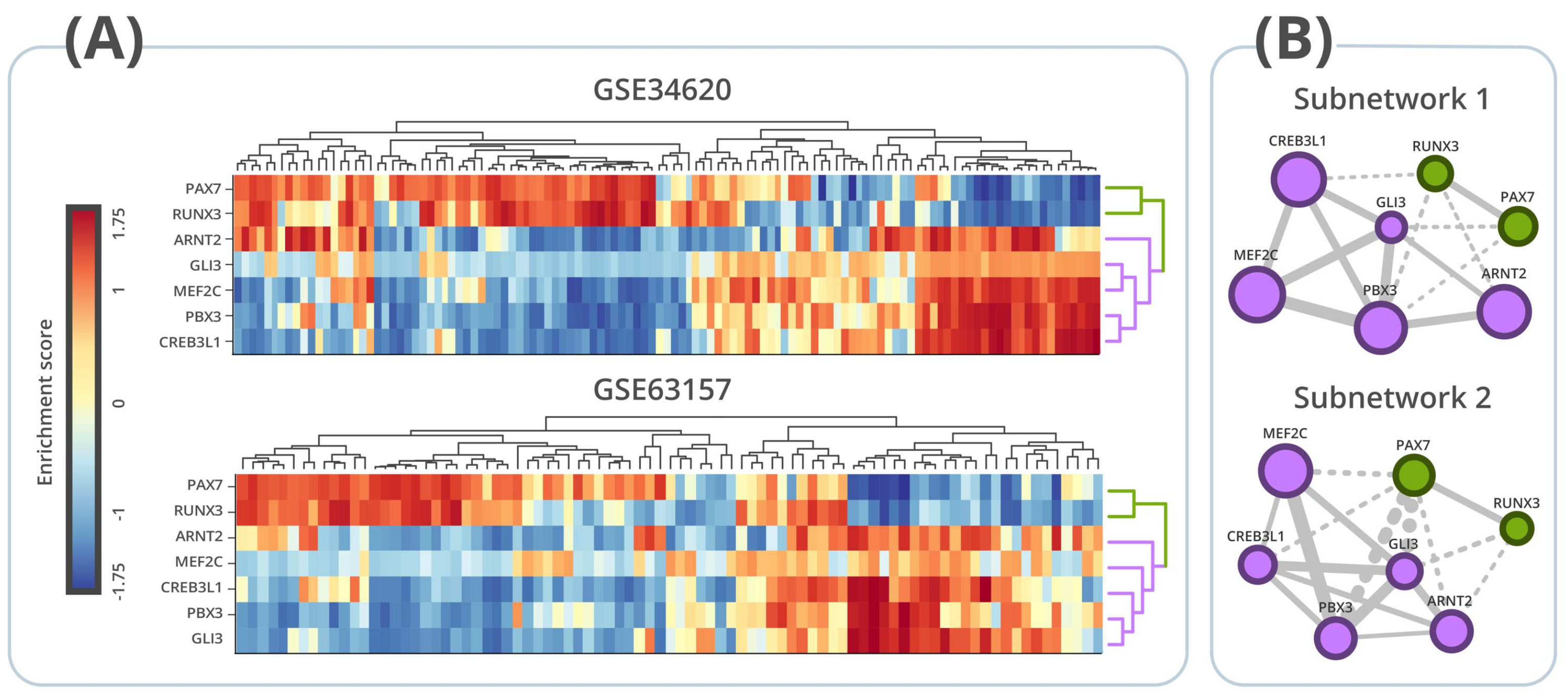
3.4. Regulon Activity
3.5. Survival Analysis
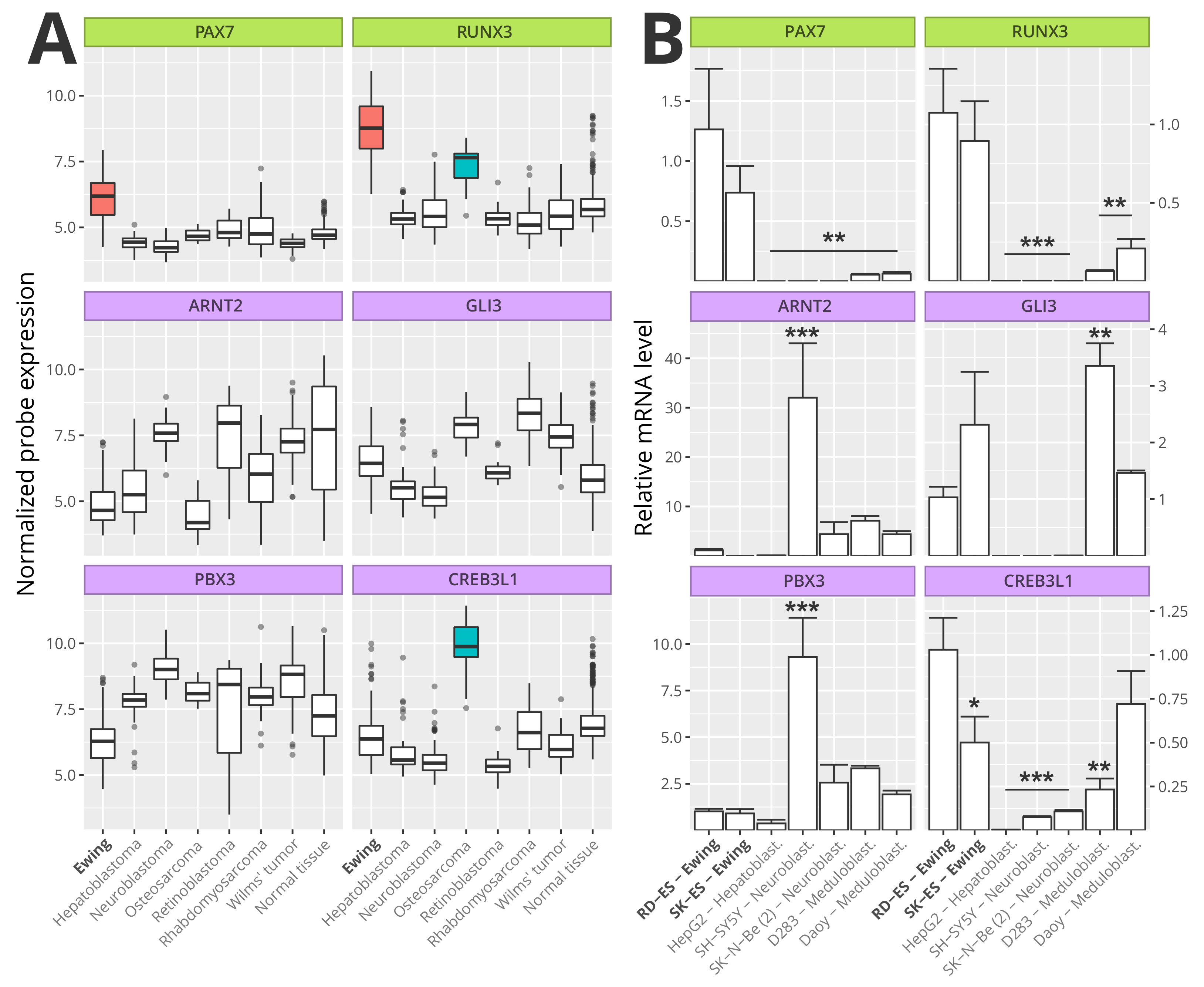
3.6. Master Regulators Expression in Ewing Sarcoma
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 2GSEA | Two-Tailed Gene Set Enrichment Analysis |
| ANOVA | Analysis of Variance |
| ARACNE | Algorithm for the Reconstruction of Accurate Cellular Networks |
| CHTN | Cooperative Human Tissue Network |
| CIT | Cartes d’Identité des Tumeurs research program |
| COG | Children’s Oncology Group |
| EICESS | European Intergroup Cooperative Ewing’s Sarcoma Study |
| ES | Ewing Sarcoma |
| GSEA | Gene Set Enrichment Analysis |
| GSE | GEO Series |
| GSM | GEO Sample |
| hMSC | Human Mesenchymal Stem Cells |
| hNCC | Human Neural Crest Cells |
| iPSC | Induced Pluripotent Stem Cells |
| MR | Master Regulator |
| MRA | Master Regulator Analysis |
| RT-PCR | Real-time polymerase chain reaction |
| TF | Transcription Factor |
Appendix A



Appendix B

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MR | p-Value | Adjusted p-Value |
|---|---|---|
| CREB3L1 | 1.80 × 10 | 1.20 × 10 |
| AEBP1 | 4.80 × 10 | 1.50 × 10 |
| SNAI2 | 8.00 × 10 | 1.70 × 10 |
| PBX3 | 1.10 × 10 | 1.80 × 10 |
| SMAD7 | 6.80 × 10 | 8.70 × 10 |
| NR6A1 | 2.10 × 10 | 2.30 × 10 |
| SOX11 | 5.10 × 10 | 4.30 × 10 |
| HIC1 | 5.40 × 10 | 4.30 × 10 |
| AHR | 8.50 × 10 | 6.10 × 10 |
| CTBP2 | 1.40 × 10 | 8.90 × 10 |
| TFAP2B | 2.60 × 10 | 0.0009 |
| HHEX | 2.70 × 10 | 0.0009 |
| TRPS1 | 3.70 × 10 | 0.00022 |
| HIF1A | 6.00 × 10 | 0.00027 |
| HOXB13 | 5.50 × 10 | 0.00027 |
| SHOX2 | 5.50 × 10 | 0.00027 |
| EPAS1 | 7.80 × 10 | 0.00033 |
| KLF9 | 2.10 × 10 | 0.00083 |
| ELK3 | 2.20 × 10 | 0.00083 |
| KLF10 | 3.20 × 10 | 0.001 |
| IRF2 | 3.30 × 10 | 0.001 |
| FOSL2 | 3.80 × 10 | 0.0011 |
| PAX7 | 6.90 × 10 | 0.0018 |
| XBP1 | 6.90 × 10 | 0.0018 |
| KLF11 | 6.70 × 10 | 0.0018 |
| MEIS1 | 0.00012 | 0.0029 |
| CREG1 | 0.00012 | 0.003 |
| IRF7 | 0.00029 | 0.0067 |
| ELF4 | 0.00038 | 0.0084 |
| SIX2 | 0.00043 | 0.0093 |
| RUNX3 | 0.0008 | 0.016 |
| ESR1 | 0.00076 | 0.016 |
| ZNF667 | 0.00094 | 0.018 |
| MEF2C | 0.0013 | 0.025 |
| GAS7 | 0.0015 | 0.028 |
| ZNF124 | 0.0016 | 0.029 |
| CBFB | 0.0017 | 0.03 |
| GATA6 | 0.0017 | 0.03 |
| GLI3 | 0.0019 | 0.032 |
| RUNX2 | 0.0021 | 0.035 |
| ARNT2 | 0.0026 | 0.042 |
| ZNF34 | 0.0028 | 0.043 |
| ETS1 | 0.003 | 0.045 |
| ZNF195 | 0.0031 | 0.045 |
| MR | p-Value | Adjusted p-Value |
|---|---|---|
| PAX7 | 5.00 × 10 | 6.50 × 10 |
| AEBP1 | 1.50 × 10 | 1.40 × 10 |
| ARNT2 | 8.20 × 10 | 3.80 × 10 |
| PBX3 | 1.70 × 10 | 3.80 × 10 |
| RUNX3 | 5.30 × 10 | 3.40 × 10 |
| NKX2-2 | 2.20 × 10 | 3.80 × 10 |
| MEF2C | 2.40 × 10 | 3.80 × 10 |
| NOTCH2 | 1.90 × 10 | 1.50 × 10 |
| CREB3L1 | 1.30 × 10 | 1.40 × 10 |
| ZNF423 | 6.80 × 10 | 3.70 × 10 |
| TEF | 6.00 × 10 | 0.035 |
| GLI3 | 9.70 × 10 | 0.048 |
| SOX1 | 4.70 × 10 | 0.034 |
| MR | p-Value | Adjusted p-Value |
|---|---|---|
| SNAI2 | 8.30 × 10 | 4.70 × 10 |
| MEF2C | 1.20 × 10 | 3.40 × 10 |
| PAX7 | 5.00 × 10 | 9.50 × 10 |
| RUNX1 | 1.40 × 10 | 2.00 × 10 |
| CREB3L1 | 1.00 × 10 | 1.20 × 10 |
| AEBP1 | 3.30 × 10 | 3.10 × 10 |
| NKX2-2 | 9.00 × 10 | 7.30 × 10 |
| PBX3 | 1.20 × 10 | 8.20 × 10 |
| AHR | 2.50 × 10 | 1.60 × 10 |
| CTNNB1 | 6.50 × 10 | 3.70 × 10 |
| XBP1 | 8.50 × 10 | 4.40 × 10 |
| EPAS1 | 3.50 × 10 | 1.70 × 10 |
| BACH2 | 1.30 × 10 | 5.60 × 10 |
| GLI3 | 1.40 × 10 | 5.60 × 10 |
| SOX11 | 1.80 × 10 | 6.90 × 10 |
| ETV1 | 1.30 × 10 | 4.20 × 10 |
| MAF | 1.20 × 10 | 4.20 × 10 |
| ETS1 | 1.60 × 10 | 4.90 × 10 |
| TCF4 | 2.70 × 10 | 8.20 × 10 |
| CREG1 | 3.60 × 10 | 1.00 × 10 |
| MEOX2 | 5.20 × 10 | 1.40 × 10 |
| RUNX3 | 1.90 × 10 | 4.80 × 10 |
| ETV5 | 2.20 × 10 | 5.40 × 10 |
| TRPS1 | 8.80 × 10 | 2.10 × 10 |
| FOSL2 | 1.50 × 10 | 3.30 × 10 |
| HEY2 | 1.70 × 10 | 3.70 × 10 |
| CEBPB | 7.30 × 10 | 0.00015 |
| PRDM1 | 9.40 × 10 | 0.00019 |
| PKNOX2 | 1.20 × 10 | 0.00023 |
| ZNF74 | 1.30 × 10 | 0.00024 |
| ARNTL2 | 1.80 × 10 | 0.00033 |
| SOX1 | 2.00 × 10 | 0.00035 |
| IRF2 | 2.30 × 10 | 0.00039 |
| ETV4 | 4.50 × 10 | 0.00075 |
| ELF1 | 5.30 × 10 | 0.00086 |
| ZBTB25 | 7.40 × 10 | 0.0011 |
| ARNT2 | 7.60 × 10 | 0.0011 |
| NR0B1 | 7.50 × 10 | 0.0011 |
| RUNX2 | 8.00 × 10 | 0.0012 |
| ESRRB | 0.00016 | 0.0023 |
| ZBTB16 | 0.00017 | 0.0024 |
| KLF9 | 0.0002 | 0.0027 |
| TFEC | 0.00021 | 0.0028 |
| HEYL | 0.00028 | 0.0036 |
| HCLS1 | 0.00031 | 0.0039 |
| HR | 0.0005 | 0.0062 |
| MEIS2 | 0.00064 | 0.0076 |
| ZFP36L2 | 0.0007 | 0.0082 |
| KLF4 | 0.00078 | 0.009 |
| CREB3L2 | 0.00088 | 0.01 |
| HIVEP1 | 0.0012 | 0.013 |
| ZNF516 | 0.0012 | 0.013 |
| ZNF235 | 0.0013 | 0.014 |
| MYT1 | 0.0015 | 0.016 |
| ZNF219 | 0.0016 | 0.017 |
| AFF3 | 0.0021 | 0.021 |
| HIVEP3 | 0.0024 | 0.024 |
| CEBPD | 0.0027 | 0.026 |
| TEAD1 | 0.0028 | 0.027 |
| SMAD5 | 0.0031 | 0.029 |
| NR3C1 | 0.0041 | 0.038 |
| NFE2L1 | 0.0043 | 0.039 |
| NKX6-1 | 0.0049 | 0.043 |
| ZNF550 | 0.0049 | 0.043 |
| STAT1 | 0.0054 | 0.047 |
| SATB2 | 0.0058 | 0.05 |
| MR | p-Value | Adjusted p-Value |
|---|---|---|
| PBX3 | 1.80 × 10 | 1.00 × 10 |
| PKNOX2 | 3.90 × 10 | 1.10 × 10 |
| PAX7 | 3.30 × 10 | 6.20 × 10 |
| CEBPB | 1.60 × 10 | 2.20 × 10 |
| GLI3 | 2.30 × 10 | 2.60 × 10 |
| AFF3 | 9.20 × 10 | 7.40 × 10 |
| ZNF215 | 9.20 × 10 | 7.40 × 10 |
| MYT1 | 1.20 × 10 | 7.60 × 10 |
| RUNX3 | 1.20 × 10 | 7.60 × 10 |
| ARNT2 | 5.00 × 10 | 2.80 × 10 |
| ETV4 | 1.40 × 10 | 7.20 × 10 |
| CREB3L1 | 6.30 × 10 | 3.00 × 10 |
| MYT1L | 1.60 × 10 | 7.00 × 10 |
| ZBTB25 | 1.80 × 10 | 7.40 × 10 |
| MEOX2 | 4.30 × 10 | 0.00016 |
| SNAI2 | 8.70 × 10 | 0.00031 |
| RUNX1 | 9.40 × 10 | 0.00031 |
| NKX2-2 | 1.60 × 10 | 0.00051 |
| MSC | 3.50 × 10 | 0.001 |
| ETV1 | 1.10 × 10 | 0.0031 |
| MAF | 1.10 × 10 | 0.0031 |
| MYC | 1.40 × 10 | 0.0036 |
| HOXB9 | 1.60 × 10 | 0.0039 |
| MEF2C | 1.80 × 10 | 0.0044 |
| LMO1 | 2.40 × 10 | 0.0055 |
| ETS1 | 0.00029 | 0.0063 |
| MEIS2 | 0.00041 | 0.0086 |
| SOX17 | 0.00068 | 0.014 |
| BACH2 | 0.00069 | 0.014 |
| HEY2 | 0.00076 | 0.014 |
| HIVEP3 | 0.00097 | 0.018 |
| ARNTL2 | 0.001 | 0.018 |
| SMAD9 | 0.001 | 0.018 |
| TCF4 | 0.0011 | 0.019 |
| HOXC11 | 0.0014 | 0.023 |
| NR0B1 | 0.0019 | 0.029 |
| ESRRB | 0.0022 | 0.033 |
| PRDM1 | 0.0024 | 0.035 |
| SMAD6 | 0.0026 | 0.037 |
| NR2F1 | 0.003 | 0.042 |
| SALL1 | 0.0036 | 0.049 |
| TCF7L2 | 0.0037 | 0.05 |
References
- Moch, H. (Ed.) Soft Tissue and Bone Tumours WHO Classification of Tumours, 5th ed.; International Agency for Research on Cancer: Lyon, France, 2020. [Google Scholar]
- Tu, J.; Huo, Z.; Gingold, J.; Zhao, R.; Shen, J.; Lee, D.F. The histogenesis of Ewing Sarcoma. Cancer Rep. Rev. 2017, 1, 1–2. [Google Scholar] [CrossRef]
- Kallen, M.E.; Hornick, J.L. The 2020 WHO classification: What’s new in soft tissue tumor pathology? Am. J. Surg. Pathol. 2021, 45, 1–23. [Google Scholar] [CrossRef]
- Grünewald, T.G.P.; Cidre-Aranaz, F.; Surdez, D.; Tomazou, E.M.; de Álava, E.; Kovar, H.; Sorensen, P.H.; Delattre, O.; Dirksen, U. Ewing sarcoma. Nat. Rev. Dis. Prim. 2018, 4, 5. [Google Scholar] [CrossRef]
- Patel, M.; Simon, J.M.; Iglesia, M.D.; Wu, S.B.; McFadden, A.W.; Lieb, J.D.; Davis, I.J. Tumor-specific retargeting of an oncogenic transcription factor chimera results in dysregulation of chromatin and transcription. Genome Res. 2012, 22, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Riggi, N.; Knoechel, B.; Gillespie, S.M.; Rheinbay, E.; Boulay, G.; Suvà, M.L.; Rossetti, N.E.; Boonseng, W.E.; Oksuz, O.; Cook, E.B.; et al. EWS-FLI1Utilizes Divergent Chromatin Remodeling Mechanisms to Directly Activate or Repress Enhancer Elements in Ewing Sarcoma. Cancer Cell 2014, 26, 668–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaspar, N.; Hawkins, D.S.; Dirksen, U.; Lewis, I.J.; Ferrari, S.; Le Deley, M.C.; Kovar, H.; Grimer, R.; Whelan, J.; Claude, L.; et al. Ewing sarcoma: Current management and future approaches through collaboration. J. Clin. Oncol. 2015, 33, 3036–3046. [Google Scholar] [CrossRef] [PubMed]
- Cotterill, S.J.; Ahrens, S.; Paulussen, M.; Jürgens, H.F.; Voûte, P.A.; Gadner, H.; Craft, A.W. Prognostic factors in Ewing’s tumor of bone: Analysis of 975 patients from the European Intergroup Cooperative Ewing’s Sarcoma Study Group. J. Clin. Oncol. 2000, 18, 3108–3114. [Google Scholar] [CrossRef]
- Cavazzana, A.O.; Miser, J.S.; Jefferson, J.; Triche, T.J. Experimental evidence for a neural origin of Ewing’s sarcoma of bone. Am. J. Pathol. 1987, 127, 507–518. [Google Scholar]
- Von Levetzow, C.; Jiang, X.; Gwye, Y.; von Levetzow, G.; Hung, L.; Cooper, A.; Hsu, J.H.R.; Lawlor, E.R. Modeling initiation of ewing sarcoma in human neural crest cells. PLoS ONE 2011, 6, e19305. [Google Scholar] [CrossRef] [Green Version]
- Carro, M.S.; Lim, W.K.; Alvarez, M.J.; Bollo, R.J.; Zhao, X.; Snyder, E.Y.; Sulman, E.P.; Anne, S.L.; Doetsch, F.; Colman, H.; et al. The transcriptional network for mesenchymal transformation of brain tumours. Nature 2010, 463, 318–325. [Google Scholar] [CrossRef]
- Rooj, A.K.; Bronisz, A.; Godlewski, J. The role of octamer binding transcription factors in glioblastoma multiforme. Biochim. Biophys. Acta 2016, 1859, 805–811. [Google Scholar] [CrossRef] [Green Version]
- Castro, M.A.; De Santiago, I.; Campbell, T.M.; Vaughn, C.; Hickey, T.E.; Ross, E.; Tilley, W.D.; Markowetz, F.; Ponder, B.A.; Meyer, K.B. Regulators of genetic risk of breast cancer identified by integrative network analysis. Nat. Genet. 2015, 48, 12–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albanus, R.D.O.; Dalmolin, R.J.S.; Castro, M.A.A.; De Bittencourt Pasquali, M.A.; De Miranda Ramos, V.; Gelain, D.P.; Moreira, J.C.F. Reverse engineering the neuroblastoma regulatory network uncovers max as one of the master regulators of tumor progression. PLoS ONE 2013, 8, e82457. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, M.N.; Castro, M.A.; Wang, X.; De Santiago, I.; O’Reilly, M.; Chin, S.F.; Rueda, O.M.; Caldas, C.; Ponder, B.A.; Markowetz, F.; et al. Master regulators of FGFR2 signalling and breast cancer risk. Nat. Commun. 2013, 4, 2464. [Google Scholar] [CrossRef] [PubMed]
- Sartor, I.T.S.; Zeidán-Chuliá, F.; Albanus, R.D.; Dalmolin, R.J.S.; Moreira, J.C.F. Computational analyses reveal a prognostic impact of TULP3 as a transcriptional master regulator in pancreatic ductal adenocarcinoma. Mol. BioSystems 2014, 10, 1461–1468. [Google Scholar] [CrossRef] [PubMed]
- Mattick, J.S.; Taft, R.J.; Faulkner, G.J. A global view of genomic information–moving beyond the gene and the master regulator. Trends Genet. 2010, 26, 21–28. [Google Scholar] [CrossRef]
- Chan, S.S.K.; Kyba, M. What is a Master Regulator? J. Stem Cell Res. Ther. 2013, 3. [Google Scholar] [CrossRef] [Green Version]
- Brohl, A.S.; Solomon, D.A.; Chang, W.; Wang, J.; Song, Y.; Sindiri, S.; Patidar, R.; Hurd, L.; Chen, L.; Shern, J.F.; et al. The Genomic Landscape of the Ewing Sarcoma Family of Tumors Reveals Recurrent STAG2 Mutation. PLoS Genet. 2014, 10, e1004475. [Google Scholar] [CrossRef] [Green Version]
- Crompton, B.D.; Stewart, C.; Taylor-Weiner, A.; Alexe, G.; Kurek, K.C.; Calicchio, M.L.; Kiezun, A.; Carter, S.L.; Shukla, S.A.; Mehta, S.S.; et al. The genomic landscape of pediatric Ewing sarcoma. Cancer Discov. 2014, 4, 1326–1341. [Google Scholar] [CrossRef] [Green Version]
- Tirode, F.; Surdez, D.; Ma, X.; Parker, M.; Le Deley, M.C.; Bahrami, A.; Zhang, Z.; Lapouble, E.; Grossetete-Lalami, S.; Rusch, M.; et al. Genomic landscape of ewing sarcoma defines an aggressive subtype with co-association of STAG2 and TP53 mutations. Cancer Discov. 2014, 4, 1342–1353. [Google Scholar] [CrossRef] [Green Version]
- Solomon, D.A.; Kim, T.; Diaz-Martinez, L.A.; Fair, J.; Elkahloun, A.G.; Harris, B.T.; Toretsky, J.A.; Rosenberg, S.A.; Shukla, N.; Ladanyi, M.; et al. Mutational inactivation of STAG2 causes aneuploidy in human cancer. Science 2011, 333, 1039–1043. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Postel-Vinay, S.; Véron, A.S.; Tirode, F.; Pierron, G.; Reynaud, S.; Kovar, H.; Oberlin, O.; Lapouble, E.; Ballet, S.; Lucchesi, C.; et al. Common variants near TARDBP and EGR2 are associated with susceptibility to Ewing sarcoma. Nat. Genet. 2012, 44, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Volchenboum, S.L.; Andrade, J.; Huang, L.; Barkauskas, D.A.; Krailo, M.; Womer, R.B.; Ranft, A.; Potratz, J.; Dirksen, U.; Triche, T.J.; et al. Gene expression profiling of Ewing sarcoma tumours reveals the prognostic importance of tumour-stromal interactions: A report from the Children’s Oncology Group. J. Pathol. Clin. Res. 2015, 1, 83–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeltner, N.; Fattahi, F.; Dubois, N.C.; Saurat, N.; Lafaille, F.; Shang, L.; Zimmer, B.; Tchieu, J.; Soliman, M.A.; Lee, G.; et al. Capturing the biology of disease severity in a PSC-based model of familial dysautonomia. Nat. Med. 2016, 22, 1421–1427. [Google Scholar] [CrossRef] [Green Version]
- Town, J.; Pais, H.; Harrison, S.; Stead, L.F.; Bataille, C.; Bunjobpol, W.; Zhang, J.; Rabbitts, T.H. Exploring the surfaceome of Ewing sarcoma identifies a new and unique therapeutic target. Proc. Natl. Acad. Sci. USA 2016, 113, 3603–3608. [Google Scholar] [CrossRef] [Green Version]
- Gautier, L.; Cope, L.; Bolstad, B.M.; Irizarry, R.A. Affy—Analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 2004, 20, 307–315. [Google Scholar] [CrossRef]
- Carvalho, B.S.; Irizarry, R.A. A framework for oligonucleotide microarray preprocessing. Bioinformatics 2010, 26, 2363–2367. [Google Scholar] [CrossRef]
- Savola, S.; Klami, A.; Myllykangas, S.; Manara, C.; Scotlandi, K.; Picci, P.; Knuutila, S.; Vakkila, J. High Expression of Complement Component 5 (C5) at Tumor Site Associates with Superior Survival in Ewing’s Sarcoma Family of Tumour Patients. Int. Sch. Res. Not. Oncol. 2011, 2011, 168712. [Google Scholar] [CrossRef] [Green Version]
- Puerto-Camacho, P.; Teresa Amaral, A.; Lamhamedi-Cherradi, S.E.; Menegaz, B.A.; Castillo-Ecija, H.; Luis Ord, J.; Domínguez, S.; Jordan-Perez, C.; Diaz-Martin, J.; Romero-Perez, L.; et al. Preclinical Efficacy of Endoglin-Targeting Antibody-Drug Conjugates for the Treatment of Ewing Sarcoma. Clin. Cancer Res. 2019, 25. [Google Scholar] [CrossRef]
- Maksimovic, J.; Phipson, B.; Oshlack, A. A cross-package Bioconductor workflow for analysing methylation array data. F1000Research 2017, 5. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. ClusterProfiler: An R package for comparing biological themes among gene clusters. OMICS A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Castro, M.A.A.; Wang, X.; Fletcher, M.N.C.; Meyer, K.B.; Markowetz, F. RedeR: R/Bioconductor package for representing modular structures, nested networks and multiple levels of hierarchical associations. Genome Biol. 2012, 13, R29. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Allen-Rhoades, W.; Whittle, S.B.; Rainusso, N. Pediatric Solid Tumors of Infancy: An Overview. Pediatr. Rev. 2018, 39, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Riggi, N.; Suvà, M.L.; Suvà, D.; Cironi, L.; Provero, P.; Tercier, S.; Joseph, J.M.; Stehle, J.C.; Baumer, K.; Kindler, V.; et al. EWS-FLI-1 Expression Triggers a Ewing’s Sarcoma Initiation Program in Primary Human Mesenchymal Stem Cells. Cancer Res. 2008, 68, 2176–2185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinsey, M.; Smith, R.; Lessnick, S.L. NR0B1 Is Required for the Oncogenic Phenotype Mediated by EWS/FLI in Ewing’s Sarcoma. Mol. Cancer Res. 2006, 4, 851–859. [Google Scholar] [CrossRef] [Green Version]
- Shi, X.; Zheng, Y.; Jiang, L.; Zhou, B.; Yang, W.; Li, L.; Ding, L.; Huang, M.; Gery, S.; Lin, D.C.; et al. EWS-FLI1 regulates and cooperates with core regulatory circuitry in Ewing sarcoma. Nucleic Acids Res. 2020, 48, 11434–11451. [Google Scholar] [CrossRef] [PubMed]
- Vidya Rani, P.S.; Shyamala, K.; Girish, H.C.; Murgod, S. Pathogenesis of Ewing sarcoma: A review. J. Adv. Clin. Res. Insights 2015, 2, 164–168. [Google Scholar] [CrossRef]
- Riggi, N.; Suvá, M.L.; Stamenkovic, I. Ewing’s Sarcoma. N. Engl. J. Med. 2021, 384, 154–164. [Google Scholar] [CrossRef]
- Von Heyking, K.; Roth, L.; Ertl, M.; Schmidt, O.; Calzada-Wack, J.; Neff, F.; Lawlor, E.R.; Burdach, S.; Richter, G.H. The posterior HOXD locus: Its contribution to phenotype and malignancy of Ewing sarcoma. Oncotarget 2016, 7, 41767–41780. [Google Scholar] [CrossRef] [Green Version]
- Selvanathan, S.P.; Graham, G.T.; Erkizan, H.V.; Dirksen, U.; Natarajan, T.G.; Dakic, A.; Yu, S.; Liu, X.; Paulsen, M.T.; Ljungman, M.E.; et al. Oncogenic fusion protein EWS-FLI1 is a network hub that regulates alternative splicing. Proc. Natl. Acad. Sci. USA 2015, 112, E1307–E1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svoboda, L.K.; Harris, A.; Bailey, N.J.; Schwentner, R.; Tomazou, E.; von Levetzow, C.; Magnuson, B.; Ljungman, M.; Kovar, H.; Lawlor, E.R. Overexpression of HOX genes is prevalent in Ewing sarcoma and is associated with altered epigenetic regulation of developmental transcription programs. Epigenetics 2014, 9, 1613–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheffield, N.C.; Pierron, G.; Klughammer, J.; Datlinger, P.; Schoenegger, A.; Schuster, M.; Hadler, J.; Surdez, D.; Guillemot, D.; Lapouble, E.; et al. DNA methylation heterogeneity defines a disease spectrum in Ewing sarcoma. Nat. Med. 2017, 23, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Mellor, P.; Deibert, L.; Calvert, B.; Bonham, K.; Carlsen, S.A.; Anderson, D.H. CREB3L1 Is a Metastasis Suppressor That Represses Expression of Genes Regulating Metastasis, Invasion, and Angiogenesis. Mol. Cell. Biol. 2013, 33, 4985–4995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.X.; Jin, D.X.; Sokol, E.S.; Reinhardt, F.; Miller, D.H.; Gupta, P.B. Cancer-specific PERK signaling drives invasion and metastasis through CREB3L1. Nat. Commun. 2017, 8, 1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogeas, A.; Morvan-Dubois, G.; El-Habr, E.A.; Lejeune, F.X.; Defrance, M.; Narayanan, A.; Kuranda, K.; Burel-Vandenbos, F.; Sayd, S.; Delaunay, V.; et al. Changes in chromatin state reveal ARNT2 at a node of a tumorigenic transcription factor signature driving glioblastoma cell aggressiveness. Acta Neuropathol. 2018, 135, 267–283. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Sun, G.; Liu, C.; Wang, J.; Jing, R.; Wang, J.; Zhao, X.; Xu, X.; Yang, Y. PBX3 is associated with proliferation and poor prognosis in patients with cervical cancer. OncoTargets Ther. 2017, 10, 5685–5694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Wu, J.; Qiu, Z.; Ge, X.; Liu, X.; Zhang, C.; Xu, W.; Wang, F.; Hua, D.; Qi, X.; et al. ACOT1 expression is associated with poor prognosis in gastric adenocarcinoma. Hum. Pathol. 2018, 77, 35–44. [Google Scholar] [CrossRef]
- Li, J.; Qiu, M.; An, Y.; Huang, J.; Gong, C. miR-7-5p acts as a tumor suppressor in bladder cancer by regulating the hedgehog pathway factor Gli3. Biochem. Biophys. Res. Commun. 2018, 503, 2101–2107. [Google Scholar] [CrossRef]
- Wang, S.; Li, C.; Wang, W.; Xing, C. PBX3 promotes gastric cancer invasion and metastasis by inducing epithelial-mesenchymal transition. Oncol. Lett. 2016, 12, 3485–3491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sannino, G.; Marchetto, A.; Kirchner, T.; Grünewald, T.G. Epithelial-to-mesenchymal and mesenchymal-to-epithelial transition in mesenchymal tumors: A paradox in sarcomas? Cancer Res. 2017, 77, 4556–4561. [Google Scholar] [CrossRef] [Green Version]
- Maltepe, E.; Keith, B.; Arsham, A.M.; Brorson, J.R.; Simon, M.C. The role of ARNT2 in tumor angiogenesis and the neural response to hypoxia. Biochem. Biophys. Res. Commun. 2000, 273, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Rankin, E.B.; Giaccia, A.J. The role of hypoxia-inducible factors in tumorigenesis. Cell Death Differ. 2008, 15, 678–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, T.D.; Ashby, W.J.; Lewis, J.D.; Zijlstra, A. Targeting tumor cell motility to prevent metastasis. Adv. Drug Deliv. Rev. 2011, 63, 568–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velez, D.O.; Tsui, B.; Goshia, T.; Chute, C.L.; Han, A.; Carter, H.; Fraley, S.I. 3D collagen architecture induces a conserved migratory and transcriptional response linked to vasculogenic mimicry. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Winkler, J.; Abisoye-Ogunniyan, A.; Metcalf, K.J.; Werb, Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat. Commun. 2020, 11, 1–19. [Google Scholar] [CrossRef]
- Saaristo, A.; Karpanen, T.; Alitalo, K. Mechanisms of angiogenesis and their use in the inhibition of tumor growth and metastasis. Oncogene 2000, 19, 6122–6129. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Zheng, H.; Zhan, Y.; Fan, S. An emerging tumor invasion mechanism about the collective cell migration. Am. J. Transl. Res. 2019, 11, 5301–5312. [Google Scholar]
- Franzetti, G.A.; Laud-Duval, K.; Van Der Ent, W.; Brisac, A.; Irondelle, M.; Aubert, S.; Dirksen, U.; Bouvier, C.; De Pinieux, G.; Snaar-Jagalska, E.; et al. Cell-to-cell heterogeneity of EWSR1-FLI1 activity determines proliferation/migration choices in Ewing sarcoma cells. Oncogene 2017, 36, 3505–3514. [Google Scholar] [CrossRef] [Green Version]
- Charville, G.W.; Wang, W.L.; Ingram, D.R.; Roy, A.; Thomas, D.; Patel, R.M.; Hornick, J.L.; Van De Rijn, M.; Lazar, A.J. EWSR1 fusion proteins mediate PAX7 expression in Ewing sarcoma. Mod. Pathol. 2017, 30, 1312–1320. [Google Scholar] [CrossRef] [PubMed]
- Bledsoe, K.L.; Mcgee-Lawrence, M.E.; Camilleri, E.T.; Wang, X.; Riester, S.M.; van Wijnen, A.J.; Oliveira, A.M.; Westendorf, J.J. RUNX3 facilitates growth of Ewing sarcoma cells. J. Cell. Physiol. 2014, 229, 2049–2056. [Google Scholar] [CrossRef] [Green Version]
- Basch, M.L.; Bronner-Fraser, M.; García-Castro, M.I. Specification of the neural crest occurs during gastrulation and requires Pax7. Nature 2006, 441, 218–222. [Google Scholar] [CrossRef]
- Charville, G.W.; Varma, S.; Forgó, E.; Dumont, S.N.; Zambrano, E.; Trent, J.C.; Lazar, A.J.; Van De Rijn, M. PAX7 expression in rhabdomyosarcoma, related soft tissue tumors, and small round blue cell neoplasms. Am. J. Surg. Pathol. 2016, 40, 1305–1315. [Google Scholar] [CrossRef] [Green Version]
- Toki, S.; Wakai, S.; Sekimizu, M.; Mori, T.; Ichikawa, H.; Kawai, A.; Yoshida, A. PAX7 immunohistochemical evaluation of Ewing sarcoma and other small round cell tumours. Histopathology 2018, 73, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Baldauf, M.C.; Gerke, J.S.; Orth, M.F.; Dallmayer, M.; Baumhoer, D.; De Alava, E.; Hartmann, W.; Kirchner, T.; Grunewald, T.G. Are EWSR1-NFATc2-positive sarcomas really Ewing sarcomas? Mod. Pathol. 2018, 31, 997–999. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Bae, S.C.; Chuang, L.S.H. The RUNX family: Developmental regulators in cancer. Nat. Rev. Cancer 2015, 15, 81–95. [Google Scholar] [CrossRef]
- Lau, Q.C.; Raja, E.; Salto-Tellez, M.; Liu, Q.; Ito, K.; Inoue, M.; Putti, T.C.; Loh, M.; Ko, T.K.; Huang, C.; et al. RUNX3 is frequently inactivated by dual mechanisms of protein mislocalization and promoter hypermethylation in breast cancer. Cancer Res. 2006, 66, 6512–6520. [Google Scholar] [CrossRef] [Green Version]
- Mei, P.J.; Bai, J.; Liu, H.; Li, C.; Wu, Y.P.; Yu, Z.Q.; Zheng, J.N. RUNX3 expression is lost in glioma and its restoration causes drastic suppression of tumor invasion and migration. J. Cancer Res. Clin. Oncol. 2011, 137, 1823–1830. [Google Scholar] [CrossRef]
- Zhang, Z.; Chen, G.; Cheng, Y.; Martinka, M.; Li, G. Prognostic significance of RUNX3 expression in human melanoma. Cancer 2011, 117, 2719–2727. [Google Scholar] [CrossRef]
- Ahlquist, T.; Lind, G.E.; Costa, V.L.; Meling, G.I.; Vatn, M.; Hoff, G.S.; Rognum, T.O.; Skotheim, R.I.; Thiis-Evensen, E.; Lothe, R.A. Gene methylation profiles of normal mucosa, and benign and malignant colorectal tumors identify early onset markers. Mol. Cancer 2008, 7, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ribeiro-Dantas, M.d.C.; Oliveira Imparato, D.; Dalmolin, M.G.S.; de Farias, C.B.; Brunetto, A.T.; da Cunha Jaeger, M.; Roesler, R.; Sinigaglia, M.; Siqueira Dalmolin, R.J. Reverse Engineering of Ewing Sarcoma Regulatory Network Uncovers PAX7 and RUNX3 as Master Regulators Associated with Good Prognosis. Cancers 2021, 13, 1860. https://doi.org/10.3390/cancers13081860
Ribeiro-Dantas MdC, Oliveira Imparato D, Dalmolin MGS, de Farias CB, Brunetto AT, da Cunha Jaeger M, Roesler R, Sinigaglia M, Siqueira Dalmolin RJ. Reverse Engineering of Ewing Sarcoma Regulatory Network Uncovers PAX7 and RUNX3 as Master Regulators Associated with Good Prognosis. Cancers. 2021; 13(8):1860. https://doi.org/10.3390/cancers13081860
Chicago/Turabian StyleRibeiro-Dantas, Marcel da Câmara, Danilo Oliveira Imparato, Matheus Gibeke Siqueira Dalmolin, Caroline Brunetto de Farias, André Tesainer Brunetto, Mariane da Cunha Jaeger, Rafael Roesler, Marialva Sinigaglia, and Rodrigo Juliani Siqueira Dalmolin. 2021. "Reverse Engineering of Ewing Sarcoma Regulatory Network Uncovers PAX7 and RUNX3 as Master Regulators Associated with Good Prognosis" Cancers 13, no. 8: 1860. https://doi.org/10.3390/cancers13081860
APA StyleRibeiro-Dantas, M. d. C., Oliveira Imparato, D., Dalmolin, M. G. S., de Farias, C. B., Brunetto, A. T., da Cunha Jaeger, M., Roesler, R., Sinigaglia, M., & Siqueira Dalmolin, R. J. (2021). Reverse Engineering of Ewing Sarcoma Regulatory Network Uncovers PAX7 and RUNX3 as Master Regulators Associated with Good Prognosis. Cancers, 13(8), 1860. https://doi.org/10.3390/cancers13081860