
How to Make Immunotherapy an Effective Therapeutic Choice for Uveal Melanoma

 , , , , ,
, , , , ,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Immunobiology of Uveal Melanoma
2.1. Immunosuppressive Mechanisms in the Eye
2.2. Immunosuppressive Mechanisms in the Liver
2.3. Tumor-Infiltrating Lymphocytes
2.4. Alternative Immune Checkpoint
3. Immune Checkpoint Inhibitors: Retrospective, Real-World Studies, and Clinical Trials
4. Immune Signatures
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Diener-West, M.; Reynolds, S.M.; Agugliaro, D.J.; Caldwell, R.; Cumming, K.; Earle, J.D.; Hawkins, B.S.; Hayman, J.A.; Jaiyesimi, I.; Jampol, L.M.; et al. Development of metastatic disease after enrollment in the COMS trials for treatment of choroidal melanoma: Collaborative ocular melanoma study group report no. 26. Arch. Ophthalmol. 2005, 123, 1639–1643. [Google Scholar] [CrossRef] [PubMed]
- Croce, M.; Ferrini, S.; Pfeffer, U.; Gangemi, R. Targeted therapy of uveal melanoma: Recent failures and new perspectives. Cancers 2019, 11, 846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Manson, D.K.; Marr, B.P.; Carvajal, R.D. Treatment of uveal melanoma: Where are we now? Ther. Adv. Med. Oncol. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Rantala, E.S.; Hernberg, M.; Kivelä, T.T. Overall survival after treatment for metastatic uveal melanoma: A systematic review and meta-analysis. Melanoma Res. 2019, 29, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.R.; Ceraudo, E.; Sher, J.J.; Guan, Y.; Shoushtari, A.N.; Chang, M.T.; Zhang, J.Q.; Walczak, E.G.; Kazmi, M.A.; Taylor, B.S.; et al. Recurrent activating mutations of G-protein-coupled receptor CYSLTR2 in uveal melanoma. Nat. Genet. 2016, 48, 675–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, P.; Aoude, L.G.; Wadt, K.; Glasson, W.J.; Warrier, S.K.; Hewitt, A.W.; Kiilgaard, J.F.; Heegaard, S.; Isaacs, T.; Franchina, M.; et al. Deep sequencing of uveal melanoma identifies a recurrent mutation in PLCB4. Oncotarget 2016, 7, 4624–4631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dono, M.; Angelini, G.; Cecconi, M.; Amaro, A.; Esposito, A.I.; Mirisola, V.; Maric, I.; Lanza, F.; Nasciuti, F.; Viaggi, S.; et al. Mutation frequencies of GNAQ, GNA11, BAP1, SF3B1, EIF1AX and TERT in Uveal melanoma: Detection of an activating mutation in the TERT gene promoter in a single case of uveal melanoma. Br. J. Cancer 2014, 110, 1058–1065. [Google Scholar] [CrossRef] [Green Version]
- New, D.C.; Wong, Y.H. Molecular mechanisms mediating the G protein-coupled receptor regulation of cell cycle progression. J. Mol. Signal. 2007, 2, 2. [Google Scholar] [CrossRef] [Green Version]
- Piaggio, F.; Tozzo, V.; Bernardi, C.; Croce, M.; Puzone, R.; Viaggi, S.; Patrone, S.; Barla, A.; Coviello, D.; Jager, M.J.; et al. Secondary somatic mutations in G-protein-related pathways and mutation signatures in uveal melanoma. Cancers 2019, 11, 1688. [Google Scholar] [CrossRef] [Green Version]
- Horsman, D.E.; Sroka, H.; Rootman, J.; White, V.A. Monosomy 3 and isochromosome 8q in a uveal melanoma. Cancer Genet. Cytogenet. 1990, 45, 249–253. [Google Scholar] [CrossRef]
- Prescher, G.; Bornfeld, N.; Hirche, H.; Horsthemke, B.; Jöckel, K.H.; Becher, R. Prognostic implications of monosomy 3 in uveal melanoma. Lancet Lond. Eng. 1996, 347, 1222–1225. [Google Scholar] [CrossRef]
- Onken, M.D.; Worley, L.A.; Person, E.; Char, D.H.; Bowcock, A.M.; Harbour, J.W. Loss of heterozygosity of chromosome 3 detected with single nucleotide polymorphisms is superior to monosomy 3 for predicting metastasis in uveal melanoma. Clin. Cancer Res. Off. J. Am. Assoc. 2007, 13, 2923–2927. [Google Scholar] [CrossRef] [Green Version]
- Harbour, J.W.; Onken, M.D.; Roberson, E.D.O.; Duan, S.; Cao, L.; Worley, L.A.; Council, M.L.; Matatall, K.A.; Helms, C.; Bowcock, A.M. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science 2010, 330, 1410–1413. [Google Scholar] [CrossRef] [Green Version]
- Harbour, J.W.; Roberson, E.D.O.; Anbunathan, H.; Onken, M.D.; Worley, L.A.; Bowcock, A.M. Recurrent mutations at codon 625 of the splicing factor SF3B1 in uveal melanoma. Nat. Genet. 2013, 45, 133–135. [Google Scholar] [CrossRef]
- Martin, M.; Maßhöfer, L.; Temming, P.; Rahmann, S.; Metz, C.; Bornfeld, N.; van de Nes, J.; Klein-Hitpass, L.; Hinnebusch, A.G.; Horsthemke, B.; et al. Exome sequencing identifies recurrent somatic mutations in EIF1AX and SF3B1 in uveal melanoma with disomy 3. Nat. Genet. 2013, 45, 933–936. [Google Scholar] [CrossRef] [Green Version]
- Yavuzyigitoglu, S.; Koopmans, A.E.; Verdijk, R.M.; Vaarwater, J.; Eussen, B.; van Bodegom, A.; Paridaens, D.; Kiliç, E.; de Klein, A. Rotterdam ocular melanoma study group uveal melanomas with SF3B1 mutations: A distinct subclass associated with late-onset metastases. Ophthalmology 2016, 123, 1118–1128. [Google Scholar] [CrossRef]
- Ventii, K.H.; Devi, N.S.; Friedrich, K.L.; Chernova, T.A.; Tighiouart, M.; Van Meir, E.G.; Wilkinson, K.D. BRCA1-Associated protein-1 is a tumor suppressor that requires deubiquitinating activity and nuclear localization. Cancer Res. 2008, 68, 6953–6962. [Google Scholar] [CrossRef] [Green Version]
- Jensen, D.E.; Proctor, M.; Marquis, S.T.; Gardner, H.P.; Ha, S.I.; Chodosh, L.A.; Ishov, A.M.; Tommerup, N.; Vissing, H.; Sekido, Y.; et al. BAP1: A novel ubiquitin hydrolase which binds to the BRCA1 RING finger and enhances BRCA1-mediated cell growth suppression. Oncogene 1998, 16, 1097–1112. [Google Scholar] [CrossRef] [Green Version]
- Robertson, A.G.; Shih, J.; Yau, C.; Gibb, E.A.; Oba, J.; Mungall, K.L.; Hess, J.M.; Uzunangelov, V.; Walter, V.; Danilova, L.; et al. Integrative analysis identifies four molecular and clinical subsets in uveal melanoma. Cancer Cell 2017, 32, 204–220.e15. [Google Scholar] [CrossRef] [Green Version]
- Gupta, M.P.; Lane, A.M.; DeAngelis, M.M.; Mayne, K.; Crabtree, M.; Gragoudas, E.S.; Kim, I.K. Clinical characteristics of uveal melanoma in patients with germline BAP1 mutations. JAMA Ophthalmol. 2015, 133, 881–887. [Google Scholar] [CrossRef] [Green Version]
- Murali, R.; Wiesner, T.; Scolyer, R.A. Tumours associated with BAP1 mutations. Pathology 2013, 45, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Amaro, A.; Gangemi, R.; Piaggio, F.; Angelini, G.; Barisione, G.; Ferrini, S.; Pfeffer, U. The biology of uveal melanoma. Cancer Metastasis Rev. 2017, 36, 109–140. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vergara, I.A.; Wilmott, J.S.; Long, G.V.; Scolyer, R.A. Genetic drivers of non-cutaneous melanomas: Challenges and opportunities in a heterogeneous landscape. Exp. Dermatol. 2021. [Google Scholar] [CrossRef]
- Saini, N.; Giacobone, C.K.; Klimczak, L.J.; Papas, B.N.; Burkholder, A.B.; Li, J.-L.; Fargo, D.C.; Bai, R.; Gerrish, K.; Innes, C.L.; et al. UV-exposure, endogenous DNA damage, and DNA replication errors shape the spectra of genome changes in human skin. PLoS Genet. 2021, 17, e1009302. [Google Scholar] [CrossRef]
- Karlsson, J.; Nilsson, L.M.; Mitra, S.; Alsén, S.; Shelke, G.V.; Sah, V.R.; Forsberg, E.M.V.; Stierner, U.; All-Eriksson, C.; Einarsdottir, B.; et al. Molecular profiling of driver events in metastatic uveal melanoma. Nat. Commun. 2020, 11, 1894. [Google Scholar] [CrossRef] [Green Version]
- Derrien, A.-C.; Rodrigues, M.; Eeckhoutte, A.; Dayot, S.; Houy, A.; Mobuchon, L.; Gardrat, S.; Lequin, D.; Ballet, S.; Pierron, G.; et al. Germline MBD4 mutations and predisposition to uveal melanoma. J. Natl. Cancer Inst. 2021, 113, 80–87. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, M.; Mobuchon, L.; Houy, A.; Alsafadi, S.; Baulande, S.; Mariani, O.; Marande, B.; Ait Rais, K.; Van der Kooij, M.K.; Kapiteijn, E.; et al. Evolutionary routes in metastatic uveal melanomas depend on MBD4 alterations. Clin. Cancer Res. Off. J. Am. Assoc. 2019, 25, 5513–5524. [Google Scholar] [CrossRef]
- Rodrigues, M.; Mobuchon, L.; Houy, A.; Fiévet, A.; Gardrat, S.; Barnhill, R.L.; Popova, T.; Servois, V.; Rampanou, A.; Mouton, A.; et al. Outlier response to anti-PD1 in uveal melanoma reveals germline MBD4 mutations in hypermutated tumors. Nat. Commun. 2018, 9, 1866. [Google Scholar] [CrossRef] [Green Version]
- Blum, E.S.; Yang, J.; Komatsubara, K.M.; Carvajal, R.D. Clinical management of uveal and conjunctival melanoma. Oncol. Williston Park N 2016, 30, 29–32, 34–43, 48. [Google Scholar]
- Chattopadhyay, C.; Kim, D.W.; Gombos, D.S.; Oba, J.; Qin, Y.; Williams, M.D.; Esmaeli, B.; Grimm, E.A.; Wargo, J.A.; Woodman, S.E.; et al. Uveal melanoma: From diagnosis to treatment and the science in between. Cancer 2016, 122, 2299–2312. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-Year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol. 2018, 19, 1480–1492. [Google Scholar] [CrossRef]
- Jager, M.J.; Shields, C.L.; Cebulla, C.M.; Abdel-Rahman, M.H.; Grossniklaus, H.E.; Stern, M.-H.; Carvajal, R.D.; Belfort, R.N.; Jia, R.; Shields, J.A.; et al. Uveal melanoma. Nat. Rev. Dis. Primer 2020, 6, 24. [Google Scholar] [CrossRef] [PubMed]
- Fallico, M.; Raciti, G.; Longo, A.; Reibaldi, M.; Bonfiglio, V.; Russo, A.; Caltabiano, R.; Gattuso, G.; Falzone, L.; Avitabile, T. Current molecular and clinical insights into uveal melanoma (Review). Int. J. Oncol. 2021, 58, 10. [Google Scholar] [CrossRef]
- Luyten, G.P.; van der Spek, C.W.; Brand, I.; Sintnicolaas, K.; de Waard-Siebinga, I.; Jager, M.J.; de Jong, P.T.; Schrier, P.I.; Luider, T.M. Expression of MAGE, Gp100 and tyrosinase genes in uveal melanoma cell lines. Melanoma Res. 1998, 8, 11–16. [Google Scholar] [CrossRef]
- Kan-Mitchell, J.; Liggett, P.E.; Harel, W.; Steinman, L.; Nitta, T.; Oksenberg, J.R.; Posner, M.R.; Mitchell, M.S. Lymphocytes cytotoxic to uveal and skin melanoma cells from peripheral blood of ocular melanoma patients. Cancer Immunol. Immunother. 1991, 33, 333–340. [Google Scholar] [CrossRef]
- Ksander, B.R.; Geer, D.C.; Chen, P.W.; Salgaller, M.L.; Rubsamen, P.; Murray, T.G. Uveal melanomas contain antigenically specific and non-specific infiltrating lymphocytes. Curr. Eye Res. 1998, 17, 165–173. [Google Scholar] [CrossRef]
- McMenamin, P.G.; Saban, D.R.; Dando, S.J. Immune cells in the retina and choroid: Two different tissue environments that require different defenses and surveillance. Prog. Retin. Eye Res. 2019, 70, 85–98. [Google Scholar] [CrossRef]
- Forrester, J.V.; Xu, H. Good news-bad news: The Yin and Yang of immune privilege in the eye. Front. Immunol. 2012, 3, 338. [Google Scholar] [CrossRef] [Green Version]
- Vendomèle, J.; Khebizi, Q.; Fisson, S. Cellular and molecular mechanisms of anterior chamber-associated immune deviation (ACAID): What we have learned from knockout mice. Front. Immunol. 2017, 8, 1686. [Google Scholar] [CrossRef] [Green Version]
- Niederkorn, J.Y. Immune escape mechanisms of intraocular tumors. Prog. Retin. Eye Res. 2009, 28, 329–347. [Google Scholar] [CrossRef] [Green Version]
- Taylor, A.W.; Alard, P.; Yee, D.G.; Streilein, J.W. Aqueous humor induces transforming growth factor-beta (TGF-Beta)-producing regulatory T-cells. Curr. Eye Res. 1997, 16, 900–908. [Google Scholar] [CrossRef]
- Ferguson, T.A.; Griffith, T.S. The role of fas ligand and TNF-related apoptosis-inducing ligand (TRAIL) in the ocular immune response. Chem. Immunol. Allergy 2007, 92, 140–154. [Google Scholar] [CrossRef]
- Apte, R.S.; Sinha, D.; Mayhew, E.; Wistow, G.J.; Niederkorn, J.Y. Cutting edge: Role of macrophage migration inhibitory factor in inhibiting NK cell activity and preserving immune privilege. J. Immunol. 1998, 160, 5693–5696. [Google Scholar]
- Yoshida, M.; Takeuchi, M.; Streilein, J.W. Participation of pigment epithelium of iris and ciliary body in ocular immune privilege. 1. Inhibition of T-cell activation in vitro by direct cell-to-cell contact. Invest. Ophthalmol. Vis. Sci. 2000, 41, 811–821. [Google Scholar]
- Wierenga, A.P.A.; Gezgin, G.; van Beelen, E.; Eikmans, M.; Spruyt-Gerritse, M.; Brouwer, N.J.; Versluis, M.; Verdijk, R.M.; van Duinen, S.G.; Marinkovic, M.; et al. Soluble HLA in the aqueous humour of uveal melanoma is associated with unfavourable tumour characteristics. Cancers 2019, 11, 202. [Google Scholar] [CrossRef] [Green Version]
- Javed, A.; Milhem, M. Role of Natural Killer Cells in Uveal Melanoma. Cancers 2020, 12, 694. [Google Scholar] [CrossRef]
- Chen, P.W.; Mellon, J.K.; Mayhew, E.; Wang, S.; He, Y.G.; Hogan, N.; Niederkorn, J.Y. Uveal melanoma expression of indoleamine 2,3-deoxygenase: Establishment of an immune privileged environment by tryptophan depletion. Exp. Eye Res. 2007, 85, 617–625. [Google Scholar] [CrossRef] [Green Version]
- Repp, A.C.; Mayhew, E.S.; Apte, S.; Niederkorn, J.Y. Human uveal melanoma cells produce macrophage migration-inhibitory factor to prevent lysis by NK cells. J. Immunol. 2000, 165, 710–715. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Li, H.; Chen, P.W.; Alizadeh, H.; He, Y.; Hogan, R.N.; Niederkorn, J.Y. PD-L1 Expression on human ocular cells and its possible role in regulating immune-mediated ocular inflammation. Invest. Ophthalmol. Vis. Sci. 2009, 50, 273–280. [Google Scholar] [CrossRef] [Green Version]
- Blanco, P.L.; Lim, L.A.; Miyamoto, C.; Burnier, M.N. Uveal melanoma dormancy: An acceptable clinical endpoint? Melanoma Res. 2012, 22, 334–340. [Google Scholar] [CrossRef]
- Vera-Ramirez, L.; Hunter, K.W. Tumor cell dormancy as an adaptive cell stress response mechanism. F1000Research 2017, 6. [Google Scholar] [CrossRef] [Green Version]
- Grossniklaus, H.E.; Zhang, Q.; You, S.; McCarthy, C.; Heegaard, S.; Coupland, S.E. Metastatic ocular melanoma to the liver exhibits infiltrative and nodular growth patterns. Hum. Pathol. 2016, 57, 165–175. [Google Scholar] [CrossRef] [Green Version]
- Jager, M.J.; Hurks, H.M.H.; Levitskaya, J.; Kiessling, R. HLA Expression in uveal melanoma: There is no rule without some exception. Hum. Immunol. 2002, 63, 444–451. [Google Scholar] [CrossRef]
- Vetter, C.S.; Lieb, W.; Bröcker, E.-B.; Becker, J.C. Loss of nonclassical MHC molecules MIC-A/B expression during progression of uveal melanoma. Br. J. Cancer 2004, 91, 1495–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piquet, L.; Dewit, L.; Schoonjans, N.; Millet, M.; Bérubé, J.; Gerges, P.R.A.; Bordeleau, F.; Landreville, S. Synergic interactions between hepatic stellate cells and uveal melanoma in metastatic growth. Cancers 2019, 11, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whelchel, J.C.; Farah, S.E.; McLean, I.W.; Burnier, M.N. Immunohistochemistry of infiltrating lymphocytes in uveal malignant melanoma. Invest. Ophthalmol. Vis. Sci. 1993, 34, 2603–2606. [Google Scholar] [PubMed]
- Bronkhorst, I.H.G.; Vu, T.H.K.; Jordanova, E.S.; Luyten, G.P.M.; van der Burg, S.H.; Jager, M.J. Different subsets of tumor-infiltrating lymphocytes correlate with macrophage influx and monosomy 3 in uveal melanoma. Invest. Ophthalmol. Vis. Sci. 2012, 53, 5370–5378. [Google Scholar] [CrossRef] [Green Version]
- Krishna, Y.; McCarthy, C.; Kalirai, H.; Coupland, S.E. Inflammatory cell infiltrates in advanced metastatic uveal melanoma. Hum. Pathol. 2017, 66, 159–166. [Google Scholar] [CrossRef]
- Qin, Y.; Bollin, K.; de Macedo, M.P.; Carapeto, F.; Kim, K.B.; Roszik, J.; Wani, K.M.; Reuben, A.; Reddy, S.T.; Williams, M.D.; et al. Immune profiling of uveal melanoma identifies a potential signature associated with response to immunotherapy. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Qin, Y.; Petaccia de Macedo, M.; Reuben, A.; Forget, M.-A.; Haymaker, C.; Bernatchez, C.; Spencer, C.N.; Gopalakrishnan, V.; Reddy, S.; Cooper, Z.A.; et al. Parallel profiling of immune infiltrate subsets in uveal melanoma versus cutaneous melanoma unveils similarities and differences: A pilot study. Oncoimmunology 2017, 6, e1321187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durante, M.A.; Rodriguez, D.A.; Kurtenbach, S.; Kuznetsov, J.N.; Sanchez, M.I.; Decatur, C.L.; Snyder, H.; Feun, L.G.; Livingstone, A.S.; Harbour, J.W. Single-cell analysis reveals new evolutionary complexity in uveal melanoma. Nat. Commun. 2020, 11, 496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishna, Y.; Acha-Sagredo, A.; Sabat-Pośpiech, D.; Kipling, N.; Clarke, K.; Figueiredo, C.R.; Kalirai, H.; Coupland, S.E. Transcriptome profiling reveals new insights into the immune microenvironment and upregulation of novel biomarkers in metastatic uveal melanoma. Cancers 2020, 12, 832. [Google Scholar] [CrossRef] [PubMed]
- Gezgin, G.; Dogrusöz, M.; van Essen, T.H.; Kroes, W.G.M.; Luyten, G.P.M.; van der Velden, P.A.; Walter, V.; Verdijk, R.M.; van Hall, T.; van der Burg, S.H.; et al. Genetic evolution of uveal melanoma guides the development of an inflammatory microenvironment. Cancer Immunol. Immunother. 2017, 66, 903–912. [Google Scholar] [CrossRef] [Green Version]
- Figueiredo, C.R.; Kalirai, H.; Sacco, J.J.; Azevedo, R.A.; Duckworth, A.; Slupsky, J.R.; Coulson, J.M.; Coupland, S.E. Loss of BAP1 expression is associated with an immunosuppressive microenvironment in uveal melanoma, with implications for immunotherapy development. J. Pathol. 2020, 250, 420–439. [Google Scholar] [CrossRef] [Green Version]
- Rothermel, L.D.; Sabesan, A.C.; Stephens, D.J.; Chandran, S.S.; Paria, B.C.; Srivastava, A.K.; Somerville, R.; Wunderlich, J.R.; Lee, C.-C.R.; Xi, L.; et al. Identification of an immunogenic subset of metastatic uveal melanoma. Clin. Cancer Res. Off. J. Am. Assoc. 2016, 22, 2237–2249. [Google Scholar] [CrossRef] [Green Version]
- Tavera, R.J.; Forget, M.-A.; Kim, Y.U.; Sakellariou-Thompson, D.; Creasy, C.A.; Bhatta, A.; Fulbright, O.J.; Ramachandran, R.; Thorsen, S.T.; Flores, E.; et al. Utilizing T-cell activation signals 1, 2, and 3 for tumor-infiltrating lymphocytes (TIL) expansion: The advantage over the sole use of interleukin-2 in cutaneous and uveal melanoma. J. Immunother. 2018, 41, 399–405. [Google Scholar] [CrossRef]
- Chandran, S.S.; Somerville, R.P.T.; Yang, J.C.; Sherry, R.M.; Klebanoff, C.A.; Goff, S.L.; Wunderlich, J.R.; Danforth, D.N.; Zlott, D.; Paria, B.C.; et al. Treatment of metastatic uveal melanoma with adoptive transfer of tumour-infiltrating lymphocytes: A single-centre, two-stage, single-arm, phase 2 study. Lancet Oncol. 2017, 18, 792–802. [Google Scholar] [CrossRef]
- Johansson, J.; Siarov, J.; Kiffin, R.; Mölne, J.; Mattsson, J.; Naredi, P.; Olofsson Bagge, R.; Martner, A.; Lindnér, P. Presence of tumor-infiltrating CD8+ T cells and macrophages correlates to longer overall survival in patients undergoing isolated hepatic perfusion for uveal melanoma liver metastasis. Oncoimmunology 2020, 9, 1854519. [Google Scholar] [CrossRef]
- Khoja, L.; Atenafu, E.G.; Suciu, S.; Leyvraz, S.; Sato, T.; Marshall, E.; Keilholz, U.; Zimmer, L.; Patel, S.P.; Piperno-Neumann, S.; et al. Meta-analysis in metastatic uveal melanoma to determine progression free and overall survival benchmarks: An international rare cancers initiative (IRCI) ocular melanoma study. Off. J. Eur. Soc. Med. Oncol. 2019, 30, 1370–1380. [Google Scholar] [CrossRef]
- Klemen, N.D.; Wang, M.; Rubinstein, J.C.; Olino, K.; Clune, J.; Ariyan, S.; Cha, C.; Weiss, S.A.; Kluger, H.M.; Sznol, M. Survival after checkpoint inhibitors for metastatic acral, mucosal and uveal melanoma. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Bol, K.F.; Ellebaek, E.; Hoejberg, L.; Bagger, M.M.; Larsen, M.S.; Klausen, T.W.; Køhler, U.H.; Schmidt, H.; Bastholt, L.; Kiilgaard, J.F.; et al. Real-world impact of immune checkpoint inhibitors in metastatic uveal melanoma. Cancers 2019, 11, 489. [Google Scholar] [CrossRef] [Green Version]
- Heppt, M.V.; Amaral, T.; Kähler, K.C.; Heinzerling, L.; Hassel, J.C.; Meissner, M.; Kreuzberg, N.; Loquai, C.; Reinhardt, L.; Utikal, J.; et al. Combined immune checkpoint blockade for metastatic uveal melanoma: A retrospective, multi-center study. J. Immunother. Cancer 2019, 7, 299. [Google Scholar] [CrossRef]
- Kirchberger, M.C.; Moreira, A.; Erdmann, M.; Schuler, G.; Heinzerling, L. Real world experience in low-dose ipilimumab in combination with PD-1 blockade in advanced melanoma patients. Oncotarget 2018, 9, 28903–28909. [Google Scholar] [CrossRef]
- Piulats, J.M.; Espinosa, E.; de la Cruz Merino, L.; Varela, M.; Alonso Carrión, L.; Martín-Algarra, S.; López Castro, R.; Curiel, T.; Rodríguez-Abreu, D.; Redrado, M.; et al. Nivolumab plus ipilimumab for treatment-naïve metastatic uveal melanoma: An open-label, multicenter, phase II trial by the spanish multidisciplinary melanoma group (GEM-1402). J. Clin. Oncol. Off. J. Am. Soc. 2021, JCO2000550. [Google Scholar] [CrossRef]
- Pelster, M.S.; Gruschkus, S.K.; Bassett, R.; Gombos, D.S.; Shephard, M.; Posada, L.; Glover, M.S.; Simien, R.; Diab, A.; Hwu, P.; et al. Nivolumab and ipilimumab in metastatic uveal melanoma: Results from a single-arm phase II study. J. Clin. Oncol. Off. J. Am. Soc. 2020, JCO2000605. [Google Scholar] [CrossRef]
- Middleton, M.R.; McAlpine, C.; Woodcock, V.K.; Corrie, P.; Infante, J.R.; Steven, N.M.; Evans, T.R.J.; Anthoney, A.; Shoushtari, A.N.; Hamid, O.; et al. Tebentafusp, A TCR/Anti-CD3 bispecific fusion protein targeting Gp100, potently activated antitumor immune responses in patients with metastatic melanoma. Clin. Cancer Res. Off. J. Am. Assoc. 2020, 26, 5869–5878. [Google Scholar] [CrossRef]
- Amaria, R.N.; Reddy, S.M.; Tawbi, H.A.; Davies, M.A.; Ross, M.I.; Glitza, I.C.; Cormier, J.N.; Lewis, C.; Hwu, W.-J.; Hanna, E.; et al. Neoadjuvant immune checkpoint blockade in high-risk resectable melanoma. Nat. Med. 2018, 24, 1649–1654. [Google Scholar] [CrossRef] [PubMed]
- Binkley, E.; Triozzi, P.L.; Rybicki, L.; Achberger, S.; Aldrich, W.; Singh, A. A Prospective trial of adjuvant therapy for high-risk uveal melanoma: Assessing 5-year survival outcomes. Br. J. Ophthalmol. 2020, 104, 524–528. [Google Scholar] [CrossRef] [Green Version]
- Lawson, D.H.; Lee, S.; Zhao, F.; Tarhini, A.A.; Margolin, K.A.; Ernstoff, M.S.; Atkins, M.B.; Cohen, G.I.; Whiteside, T.L.; Butterfield, L.H.; et al. Randomized, placebo-controlled, phase III trial of yeast-derived granulocyte-macrophage colony-stimulating factor (GM-CSF) versus peptide vaccination versus GM-CSF plus peptide vaccination versus placebo in patients with no evidence of disease after complete surgical resection of locally advanced and/or stage IV melanoma: A trial of the Eastern Cooperative Oncology Group-American College of Radiology Imaging Network Cancer Research Group (E4697). J. Clin. Oncol. Off. J. Am. Soc. 2015, 33, 4066–4076. [Google Scholar] [CrossRef]
- Jespersen, H.; Bagge, R.O.; Ullenhag, G.; Carneiro, A.; Helgadottir, H.; Ljuslinder, I.; Levin, M.; All-Eriksson, C.; Andersson, B.; Stierner, U.; et al. Phase II multicenter open label study of pembrolizumab and entinostat in adult patients with metastatic uveal melanoma (PEMDAC Study). Ann. Oncol. 2019, 30, v907. [Google Scholar] [CrossRef]
- Joseph, R.W.; Peddareddigari, V.R.; Liu, P.; Miller, P.W.; Overwijk, W.W.; Bekele, N.B.; Ross, M.I.; Lee, J.E.; Gershenwald, J.E.; Lucci, A.; et al. Impact of clinical and pathologic features on tumor-infiltrating lymphocyte expansion from surgically excised melanoma metastases for adoptive T-cell therapy. Clin. Cancer Res. Off. J. Am. Assoc. 2011, 17, 4882–4891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bol, K.F.; van den Bosch, T.; Schreibelt, G.; Mensink, H.W.; Keunen, J.E.E.; Kiliç, E.; Japing, W.J.; Geul, K.W.; Westdorp, H.; Boudewijns, S.; et al. Adjuvant dendritic cell vaccination in high-risk uveal melanoma. Ophthalmology 2016, 123, 2265–2267. [Google Scholar] [CrossRef] [PubMed]
- Wessely, A.; Steeb, T.; Erdmann, M.; Heinzerling, L.; Vera, J.; Schlaak, M.; Berking, C.; Heppt, M.V. The role of immune checkpoint blockade in uveal melanoma. Int. J. Mol. Sci. 2020, 21, 879. [Google Scholar] [CrossRef] [Green Version]
- Chua, V.; Mattei, J.; Han, A.; Johnston, L.; LiPira, K.; Selig, S.M.; Carvajal, R.D.; Aplin, A.E.; Patel, S.P. The latest on uveal melanoma research and clinical trials: Updates from the cure ocular melanoma (CURE OM) science meeting (2019). Clin. Cancer Res. Off. J. Am. Assoc. 2021, 27, 28–33. [Google Scholar] [CrossRef]
- Tebentafusp Named FDA Breakthrough Therapy for Advanced Eye Cancer. Available online: https://immuno-oncologynews.com/2021/02/24/tebentafusp-wins-fda-breakthrough-therapy-designation-advanced-eye-cancer/ (accessed on 4 March 2021).
- Field, M.G.; Durante, M.A.; Decatur, C.L.; Tarlan, B.; Oelschlager, K.M.; Stone, J.F.; Kuznetsov, J.; Bowcock, A.M.; Kurtenbach, S.; Harbour, J.W. Epigenetic reprogramming and aberrant expression of PRAME are associated with increased metastatic risk in class 1 and class 2 uveal melanomas. Oncotarget 2016, 7, 59209–59219. [Google Scholar] [CrossRef] [Green Version]
- Beck, L.; Harel, M.; Yu, S.; Markovits, E.; Boursi, B.; Markel, G.; Geiger, T. Clinical proteomics of metastatic melanoma reveals profiles of organ specificity and treatment resistance. Clin. Cancer Res. Off. J. Am. Assoc. 2021. [Google Scholar] [CrossRef]
- Zhang, Z.; Ni, Y.; Chen, G.; Wei, Y.; Peng, M.; Zhang, S. Construction of immune-related risk signature for uveal melanoma. Artif. Cells Nanomedicine Biotechnol. 2020, 48, 912–919. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, Y.; Dai, X.; Lin, X.; Shan, Y.; Ye, J. The prognostic landscape of adaptive immune resistance signatures and infiltrating immune cells in the tumor microenvironment of uveal melanoma. Exp. Eye Res. 2020, 196, 108069. [Google Scholar] [CrossRef]
- Li, Y.-Z.; Huang, Y.; Deng, X.-Y.; Tu, C.-S. Identification of an immune-related signature for the prognosis of uveal melanoma. Int. J. Ophthalmol. 2020, 13, 458–465. [Google Scholar] [CrossRef]
- Gong, Q.; Wan, Q.; Li, A.; Yu, Y.; Ding, X.; Lin, L.; Qi, X.; Hu, L. Development and validation of an immune and stromal prognostic signature in uveal melanoma to guide clinical therapy. Aging 2020, 12, 20254–20267. [Google Scholar] [CrossRef]
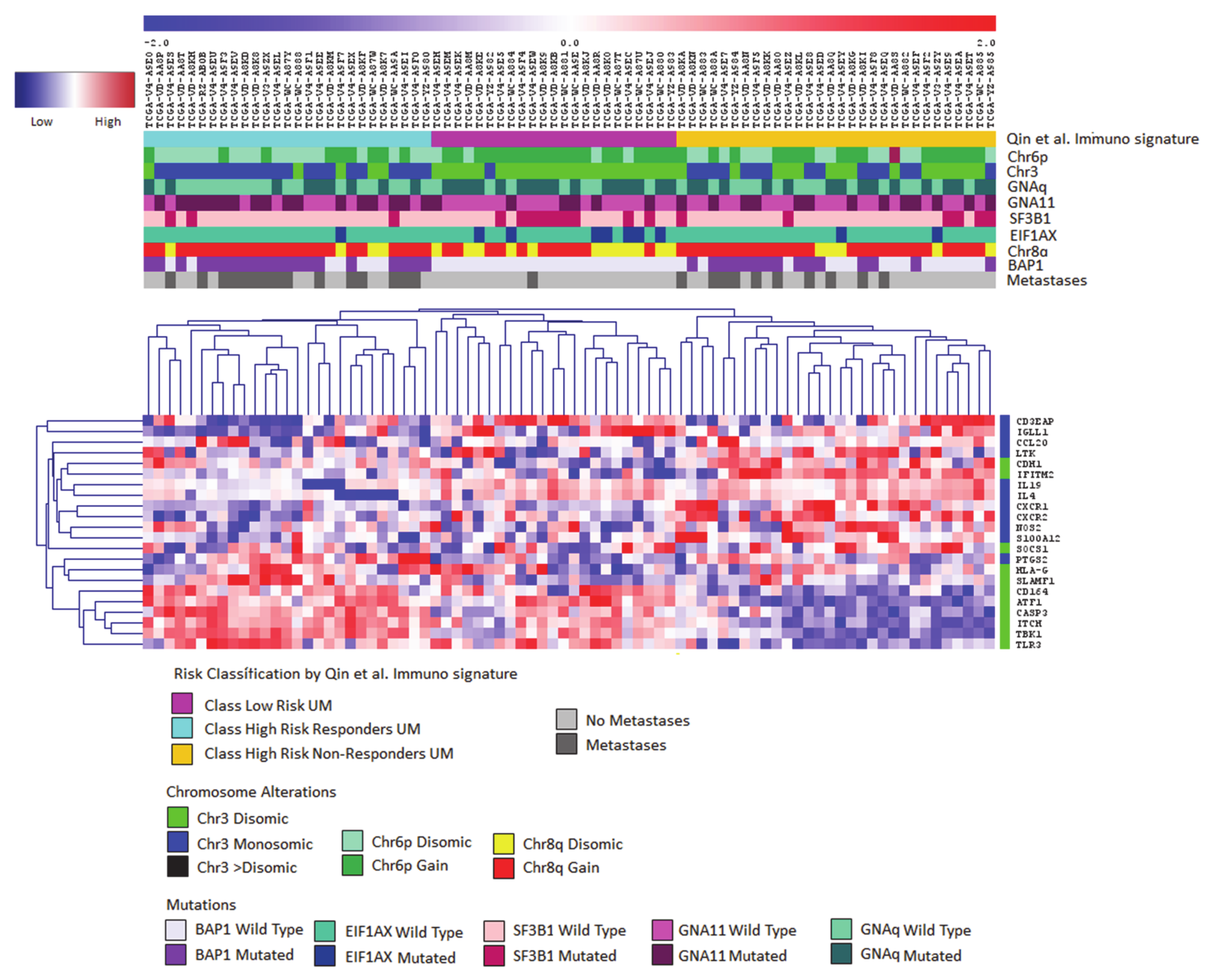

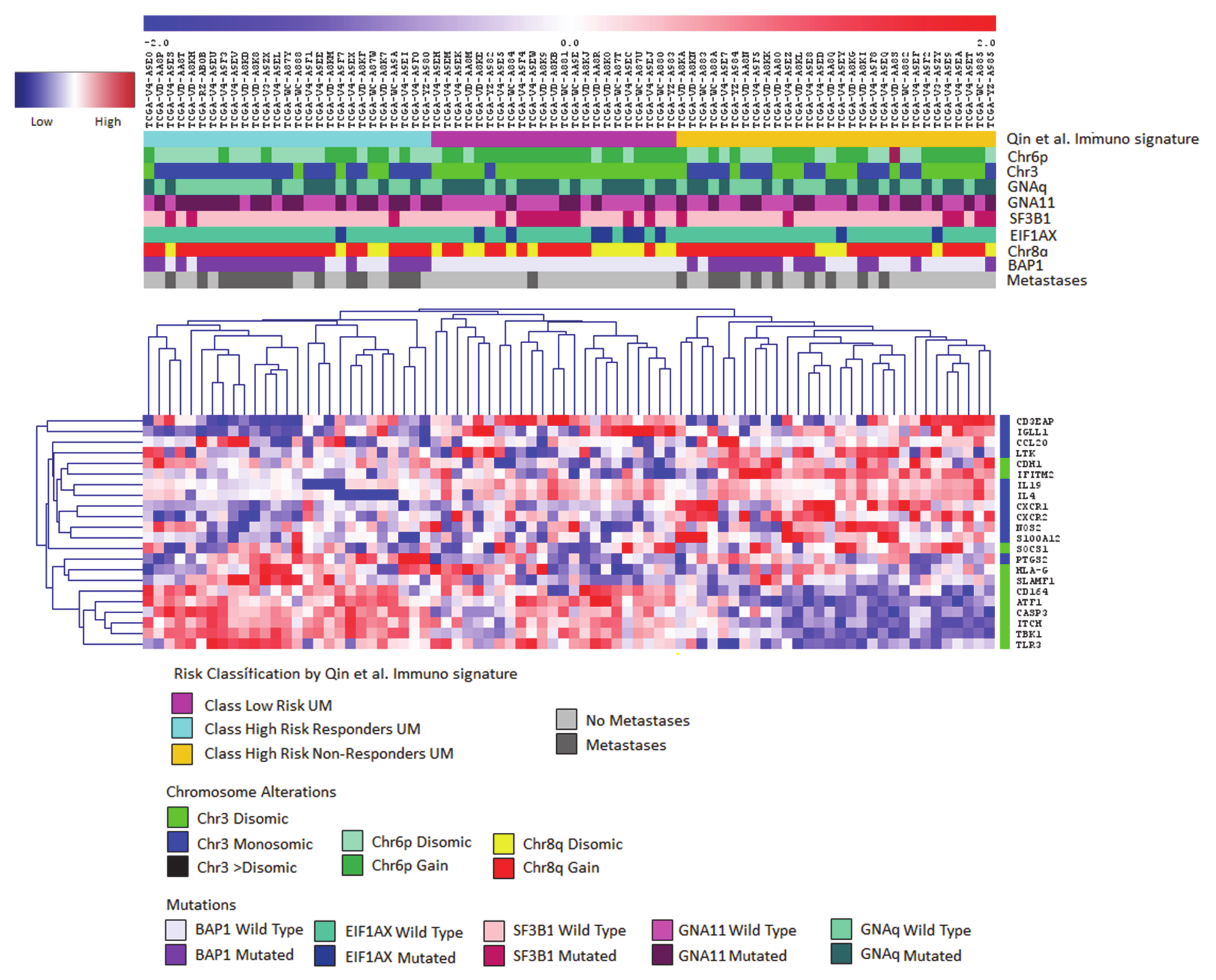{kind=link}
| Study | Type of Study | Targeted Patients | No Patients (UM Patients) | ORR | Median OS | Median PFS | Rate 1-Year Surv | 6 Months PFS | PR | CR | SD |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Khoia [70] | Meta-analysis (2000–2016) | metastatic uveal melanoma | (912) | - | 10.2 | 3.3 | 43% | 27% | - | - | - |
| Chandran [68] | Phase II ClinicalTrials.gov Identifier: NCT01814046 (autologous TILs) | Metastatic Ocular Melanoma Metastatic Uveal Melanoma | (21) | NE | NE | NE | NE | NE | 30% | 5% | NE |
| Klemen [71] | Retrospective(Ipilimumab+Nivolumab) | metastatic melanoma | 428 (30) | - | 12.2 | - | - | - | - | - | - |
| Bol [72] | Retrospective (Ipilimumab+Nivolumab) | metastatic UM | (126) Ipilimumab/Nivolumab n = 19l | - | 18.9 | 3.7 | 57.6% | 3.7 | 21.1% | 0 | 10.5% |
| Heppt [73] | Retrospective (Ipilimumab+Nivolumab) | metastatic or unresectable UM | (64) Ipi+nivo55 | 15.6 | 16.1 | 3 | - | - | - | - | - |
| Kirchberg [74] | Real world (Ipilimumab+Nivolumab) | metastatic melanoma | 33(9) | - | 18.4 | - | - | - | 0 | 0 | 56% |
| Piulats [75] | Phase II ClinicalTrials.gov Identifier: NCT02626962 (Ipilimumab+Nivolumab) | Metastatic uvela melanoma | (52) | - | 12.7 | 3 | 51.9% | - | 9.6% | 1.9% | - |
| Pelster [76] | Phase II ClinicalTrials.gov Identifier: NCT01585194 (Ipilimumab+Nivolumab) | Metastatic uveal melanoma | (35) | 18% | 19.1 | 5.5 | 56% | - | 15% | 3% | 33% |
| Middleton [77] | Phase I/II ClinicalTrials.gov Identifier:NCT01211262 (Tebentafusp) | Advanced melanoma | 84 (19) | - | - | - | 65% | - | 16.6% | 0 | 44.4% |
| ClinicalTrials.gov Identifier: NCT Number | Trial (Adjuvant) | Status | Phase | Targeted Patients | Actual Enrollment (Estimated Enrollment) | Principal Investigator | First Submitted Date | Last Update Posted Date |
|---|---|---|---|---|---|---|---|---|
| NCT02068586 | A Randomized Phase II Study of Adjuvant Sunitinib or Valproic Acid in High-Risk Patients With Uveal Melanoma | Recruiting | Phase II | Ciliary Body and Choroid Melanoma Iris Melanoma Intraocular Melanoma | (150) | Takami Sato | 19 February 2014 | 7 January 2021 |
| NCT02223819 | Phase II Trial of Adjuvant Crizotinib in High-Risk Uveal Melanoma Following Definitive Therapy | Active, not recruiting | Phase II | Uveal Melanoma | 34 (30) | Richard Carvajal | 20 August 2014 | 18 December 2019 |
| NCT01983748 | A non-commercial, multicenter, randomized, two-armed, open-label phase III study to evaluate the adjuvant vaccination with tumor RNA-loaded autologous dendritic cells versus observation of patients with resected monosomy 3 uveal melanoma | Recruiting | Phase III | Uveal Melanoma | (200) | Beatrice Schuler-Thurner | 17 September 2013 | 6 January 2020 |
| NCT01100528 [79] | Adjuvant Therapy for Patients With Primary Uveal Melanoma With Genetic Imbalance (Dacarbazine+IFNa-2B) | Completed | Phase II | Iris, Ciliary Body or Choroidal Melanoma | 38(36) | Yogen Saunthararajah | 7 April 2010 | 26 February 2019 |
| NCT02519322 [78] | Neoadjuvant and Adjuvant Checkpoint Blockade (Ipi+Nivo+Relatlimab) | Recruiting | Phase II | Cutaneous Melanoma Mucosal Melanoma Ocular Melanoma | (53) | Rodabe N Amaria | 4 August 2015 | 30 December 2020 |
| NCT00254397 | Study of the Modulatory Activity of an LHRH-Agonist (Leuprolide) on Melanoma Peptide Vaccines as Adjuvant Therapy in Melanoma Patients | Completed | Phase II | Melanoma | 98 | Patrick Hwu | 14 November 2005 | 16 October 2019 |
| NCT01989572 [80] | A Randomized, Placebo-Controlled Phase III Trial of Yeast Derived GM-CSF Versus Peptide Vaccination Versus GM-CSF Plus Peptide Vaccination Versus Placebo in Patients With “No Evidence of Disease” After Complete Surgical Resection of “Locally Advanced” and/or Stage IV Melanoma | Completed | Phase III | Ocular melanoma Cutaneous Melanoma Mucosal melanoma | 815 | David H Lawson | 18 November 2013 | 7 July 2020 |
| ClinicalTrials.gov Identifier: NCT number | Trial (other not adjuvant immunological therapies) | Status | Phase | Targeted Patients | Actual enrollment (Estimated enrollment) | Principal Investigator | First Submitted Date | Last Update Posted Date |
| NCT03070392 | A Phase II Randomized, Open-label, Multi-center Study of the Safety and Efficacy of IMCgp100 Compared With Investigator Choice in HLA-A*0201 Positive Patients With Previously Untreated Advanced Uveal Melanoma | Active, not recruiting | Phase II | Uveal Melanoma | 378 (327) | Mohammed Dar | 14 February 2017 | 6 January 2021 |
| NCT02570308 | A Study of the Intra-Patient Escalation Dosing Regimen With IMCgp100 in Patients With Advanced Uveal Melanoma | Active, not recruiting | Phase IPhase II | Uveal Melanoma | (150) | Not Provided | 6 October 2015 | 6 January 2021 |
| NCT03467516 | A Phase II Study to Evaluate the Efficacy and Safety of Adoptive Transfer of Autologous Tumor-Infiltrating Lymphocytes in Patients With Metastatic Uveal Melanoma | Recruiting | Phase II | Uveal Neoplasms Melanoma, Uveal | (59) | Udai S Kammula | 9 March 2018 | 18February 2020 |
| NCT00986661 | A Phase I Study to Assess the Safety, Tolerability, and Pharmacokinetics of PV-10 Chemoablation of Cancer Metastatic to the Liver or Hepatocellular Carcinoma Not Amenable to Resection or Transplant | Recruiting | Phase I | Cancer Metastatic to the Liver Hepatocellular Carcinoma Metastatic Melanoma Metastatic Ocular Melanoma Metastatic Uveal Melanoma Metastatic Lung Cancer Metastatic Colon Cancer Metastatic Colorectal Cancer Metastatic Breast Cancer Metastatic Pancreatic Cancer | (78) | Eric Wachter | 24 September 2009 | 5 March 2020 |
| NCT01211262 [77] | A Phase I, Open-Label, Dose-Finding Study to Assess the Safety and Tolerability of IMCgp100, a Monoclonal T Cell Receptor Anti-CD3 scFv Fusion Protein in Patients With Advanced Malignant Melanoma | Completed | Phase I | Malignant Melanoma | 84(50) | Namir Hassan | 28 September 2010 | 8 July 2020 |
| NCT04262466 | Phase I/II Study of IMC-F106C in Advance PRAME-Positive Cancers | Recruiting | Phase IPhase II | Select Advanced Solid Tumors | (170) | Shaad Abdullah, FACP | 30 January 2020 | 16 February 2021 |
| NCT02743611 | A Phase I/II Dose-Finding Study to Evaluate the Safety, Feasibility, and Activity of BPX-701, a Controllable PRAME T-Cell Receptor Therapy, in HLA-A2+ Subjects With AML, Previously Treated MDS, or Metastatic Uveal Melanoma | Active, not recruiting | Phase IPhase II | Acute Myeloid Leukemia Myelodysplastic Syndrome Uveal Melanoma | 28 (36) | Bellicum Pharmaceuticals Senior Director | 11 April 2016 | 27 April 2020 |
| NCT02697630 [81] | A Multicenter Phase II Open-Label Study to Evaluate Efficacy of Concomitant Use of Pembrolizumab and Entinostat in Adult Patients With Metastatic Uveal Melanoma | Active, not recruiting | Phase II | Metastatic Uveal Melanoma | (29) | Not Provided | 22 February 2016 | 16 October 2019 |
| NCT00338377 [82] | Lymphodepletion Plus Adoptive Cell Transfer With or Without Dendritic Cell Immunization in Patients With Metastatic Melanoma | Recruiting | Phase II | Melanoma | (189) 5 MU(primary site choroid) | Rodabe N. Amaria | 10 February 2006 | 9 December 2020 |
| NCT03635632 | Phase I Study of Autologous T Lymphocytes Expressing GD2-specific Chimeric Antigen and Constitutively Active IL-7 Receptors for the Treatment of Patients With Relapsed or Refractory Neuroblastoma and Other GD2 Positive Solid Cancers(GAIL-N) | Recruiting | Phase I | Relapsed Neuroblastoma Refractory Neuroblastoma Relapsed Osteosarcoma Relapsed Ewing Sarcoma Relapsed Rhabdomyosarcoma Uveal Melanoma Phyllodes Breast Tumor | (94) | Bilal Omer | 13 August 2018 | 9 December 2020 |
| NCT03865212 | Phase I Trial to Evaluate the Safety and Efficacy of Intratumoral and Intravenous Injection of Vesicular Stomatitis Virus Expressing Human Interferon Beta and Tyrosinase Related Protein 1 (VSV-IFNb-TYRP1) in Patients With Metastatic Ocular Melanoma and Previously Treated Patients With Unresectable Stage III/IV Cutaneous Melanoma | Recruiting | Phase I | Clinical Stage III Cutaneous Melanoma AJCC v8 Clinical Stage IV Cutaneous Melanoma AJCC v8 Metastatic Choroid Melanoma Metastatic Melanoma Metastatic Mucosal Melanoma Metastatic Uveal Melanoma Pathologic Stage III Cutaneous Melanoma AJCC v8 Pathologic Stage IIIA Cutaneous Melanoma AJCC v8 Pathologic Stage IIIB Cutaneous Melanoma AJCC v8 Pathologic Stage IIIC Cutaneous Melanoma AJCC v8 Pathologic Stage IIID Cutaneous Melanoma AJCC v8 Pathologic Stage IV Cutaneous Melanoma AJCC v8 Unresectable Melanoma | (72) | Roxana S Dronca | 6 March 2019 | 18 November 2020 |
| ClinicalTrials.gov Identifier: NCT Number | Trial | Status | Phase | Targeted Patients | Actual Enrollment (Estimated Enrollment) | Principal Investigator | First Submitted Date | Last Update Posted Date |
|---|---|---|---|---|---|---|---|---|
| NCT01585194 [76] | Phase II Study of Nivolumab in Combination With Ipilimumab for Uveal Melanoma | Active, not recruiting | Phase II | Metastatic Uveal Melanoma Stage IV Uveal Melanoma AJCC v7 | 67 (141) | Sapna Patel | 23 April 2012 | 10 December 2020 |
| NCT02626962 [75] | Phase II Multicenter, Non-Randomized, Open-Label Trial of Nivolumab in Combination With Ipilimumab in Subjects With Previously Untreated Metastatic Uveal Melanoma | Active, not recruiting | Phase II | Uveal Melanoma | 48 (48) | Josep Maria Piulats | 1 December 2015 | 19 October 2020 |
| NCT03922880 | Pilot Study Combining Arginine Depletion and Checkpoint Inhibition in Uveal Melanomas | Active, not recruiting | Phase I | Uveal Melanoma | 9 (9) | Alexander Shoushtari | 18 April 2019 | 11 January 2021 |
| NCT02913417 | A Feasibility Study of Sequential Hepatic Internal Radiation and Systemic Ipilimumab and Nivolumab in Patients With Uveal Melanoma Metastatic to Liver | Recruiting | Phase IPhase II | Uveal Melanoma Hepatic Metastases | (26) | David R. Minor | 21 September, 2016 | 25 August 2020 |
| NCT04463368 | SCANDIUM II Trial—A Phase I Randomized Controlled Multicentre Trial of Isolated Hepatic Perfusion in Combination With Ipilimumab and Nivolumab in Patients With Uveal Melanoma Metastases | Not yet recruiting | Phase I | Uveal Melanoma Liver Metastases | (18) | Roger Olofsson Bagge | 5 July 2020 | 1 September 2020 |
| NCT04283890 | Phase Ib/2 Study Combining Hepatic Percutaneous Perfusion With Ipilimumab Plus Nivolumab in Advanced Uveal Melanoma | Recruiting | Phase IPhase II | Uveal Melanoma, Metastatic | (88) | Ellen W. Kapiteijn | 21 February 2020 | 25 February 2020 |
| NCT03472586 | Ipilimumab and Nivolumab in Combination With Immunoembolization for the Treatment of Metastatic Uveal Melanoma | Recruiting | Phase II | Metastatic Uveal Melanoma | (35) | Marlana Orloff | 14 March 2018 | 28 May 2020 |
| Ref. | Signature | Aim of the Study |
|---|---|---|
| Li [91] | Immune-related gene signature based on two immune-related genes for predicting survival in UM. | Development of an immune-related prognostic and predictive signature to identify those patients who could benefit from immunotherapy. The signature is built on the TCGA-UM dataset and is significantly associated with tumor T stage and tumor basal diameter. |
| Wang [90] | Adaptive Immune Resistance Signature based on fifteen markers, to predict prognosis in UM. | Analysis of the immune and stromal infiltrate on gene expression data of the TCGA-UM and TCGA-CM datasets using different digital cytometry algorithms for significant prognostic marker selection. This signature could identify UM subgroups with a characteristic tumor microenvironment. |
| Zhang [89] | Immune cell-based prognosis signature to predict overall survival in UM. The signature is based on the contribution of CD8+, CD4+ T cells, monocytes, and Mast cells. | Tumor microenvironment landscape analysis by the CYBERSORT algorithm to classify the immune cell type profiles in the TCGA-UM patients. This signature highlights the impact of immune infiltrate components in the development of metastases. |
| Gong [92] | Immune and stromal prognostic signature based on published datasets. The signature is developed on a four-cell model (cytotoxic, Th1, Th2 cells, and myocytes). | Tumor microenvironment analysis by ESTIMATE algorithm for the identification of a four-cell model as a biomarker of overall survival in UM. This prognostic signature can stratify subgroups of patients with different classes of risk. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marseglia, M.; Amaro, A.; Solari, N.; Gangemi, R.; Croce, E.; Tanda, E.T.; Spagnolo, F.; Filaci, G.; Pfeffer, U.; Croce, M. How to Make Immunotherapy an Effective Therapeutic Choice for Uveal Melanoma. Cancers 2021, 13, 2043. https://doi.org/10.3390/cancers13092043
Marseglia M, Amaro A, Solari N, Gangemi R, Croce E, Tanda ET, Spagnolo F, Filaci G, Pfeffer U, Croce M. How to Make Immunotherapy an Effective Therapeutic Choice for Uveal Melanoma. Cancers. 2021; 13(9):2043. https://doi.org/10.3390/cancers13092043
Chicago/Turabian StyleMarseglia, Mariarosaria, Adriana Amaro, Nicola Solari, Rosaria Gangemi, Elena Croce, Enrica Teresa Tanda, Francesco Spagnolo, Gilberto Filaci, Ulrich Pfeffer, and Michela Croce. 2021. "How to Make Immunotherapy an Effective Therapeutic Choice for Uveal Melanoma" Cancers 13, no. 9: 2043. https://doi.org/10.3390/cancers13092043
APA StyleMarseglia, M., Amaro, A., Solari, N., Gangemi, R., Croce, E., Tanda, E. T., Spagnolo, F., Filaci, G., Pfeffer, U., & Croce, M. (2021). How to Make Immunotherapy an Effective Therapeutic Choice for Uveal Melanoma. Cancers, 13(9), 2043. https://doi.org/10.3390/cancers13092043