
The PARP Enzyme Family and the Hallmarks of Cancer Part 2: Hallmarks Related to Cancer Host Interactions



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Role of PARPs in Hallmarks Related to the Interplay between Cancer and Host
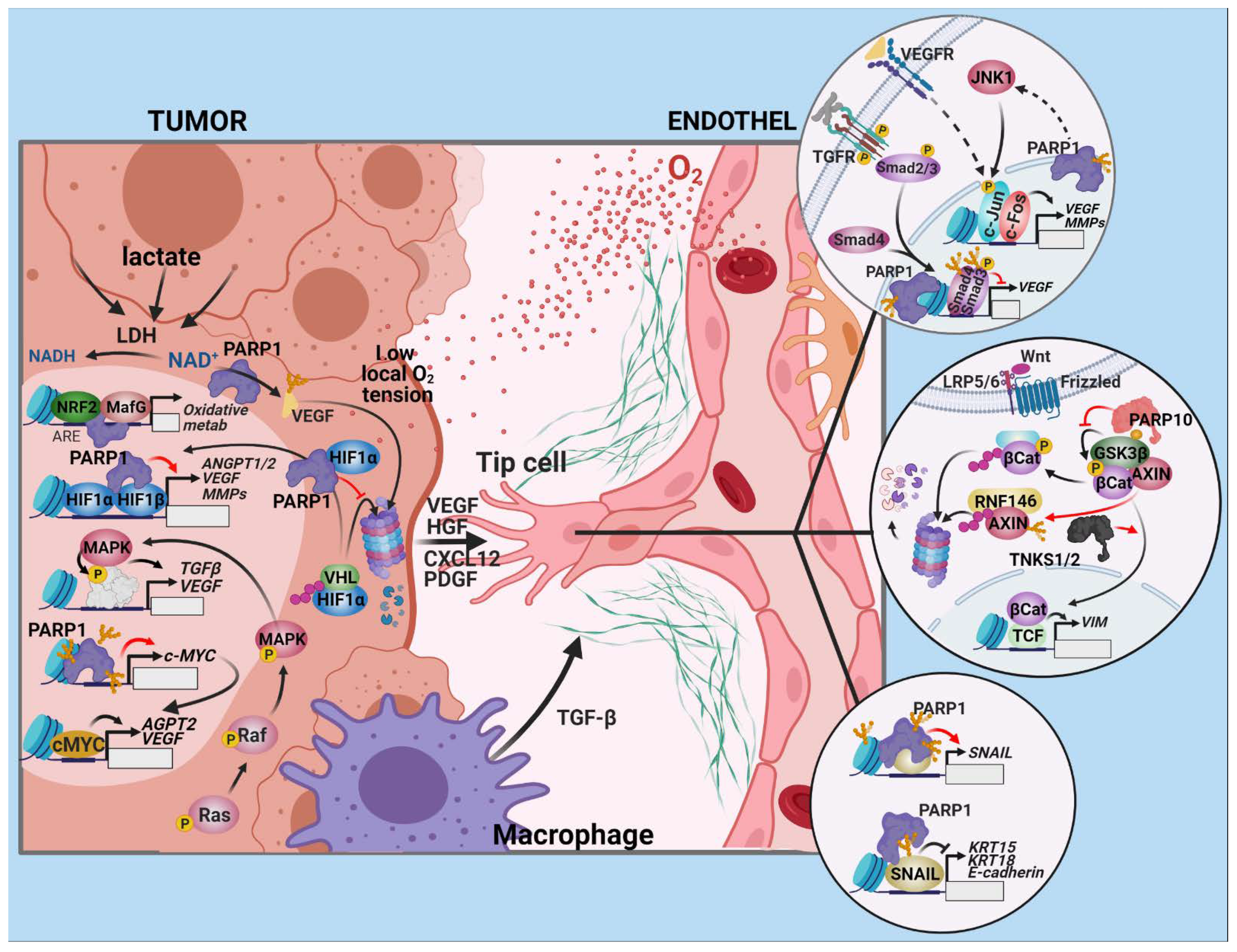
2.1. Angiogenesis
2.1.1. Drivers of Angiogenesis in Tumors
2.1.2. The Endothelial Response
2.1.3. Open Questions and Prospects
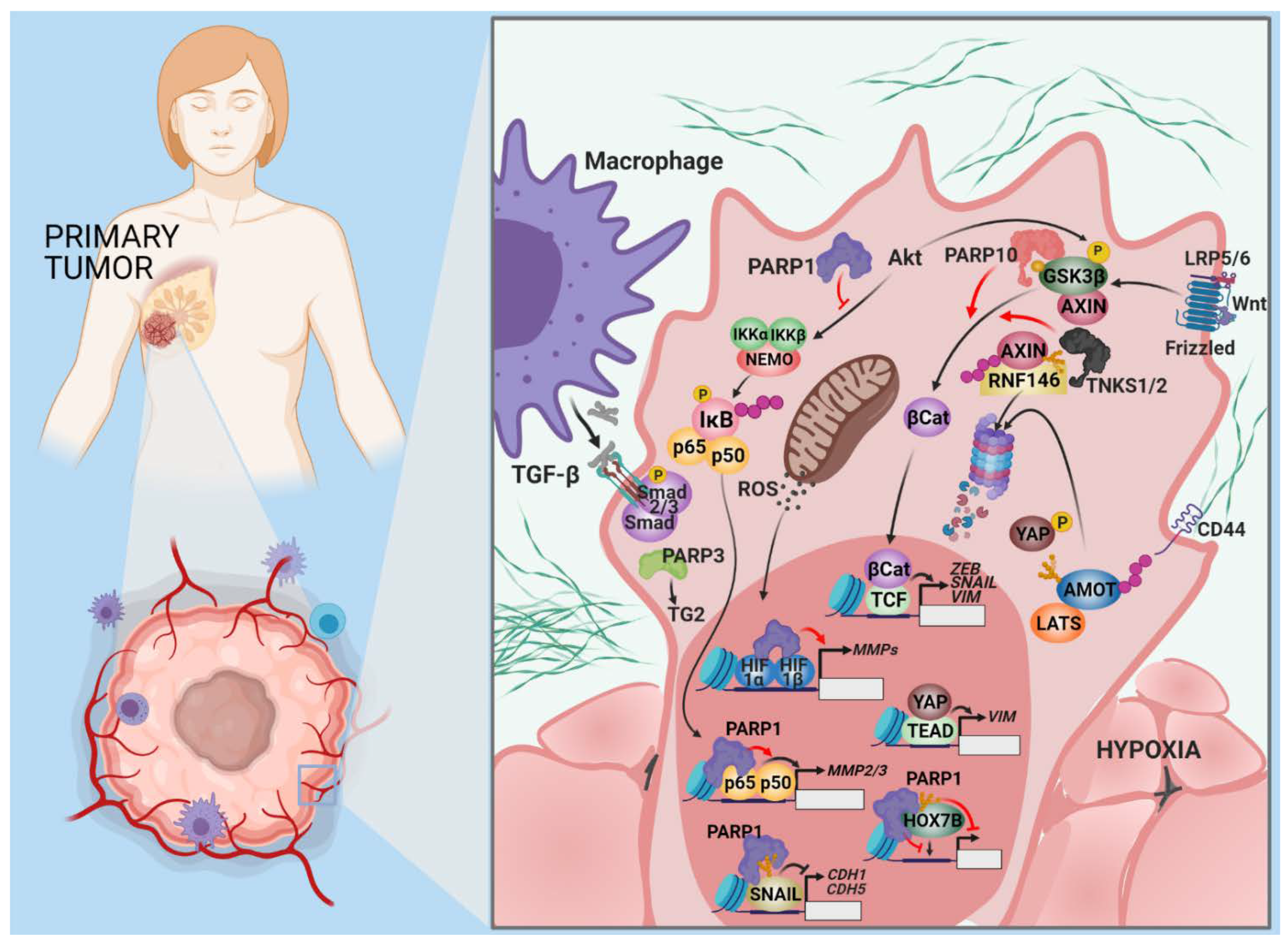
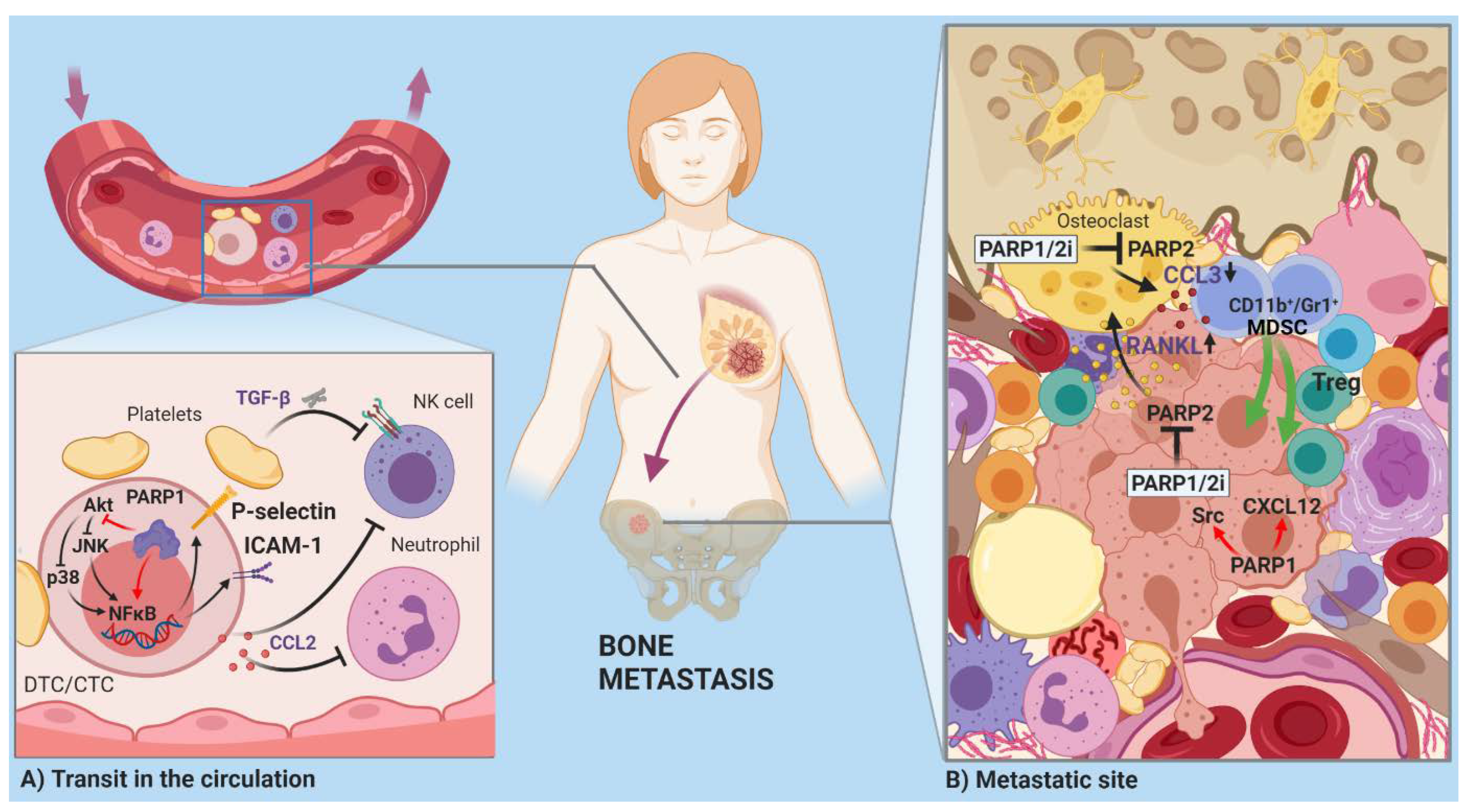
2.2. Invasion and Metastasis
Open Questions and Prospects
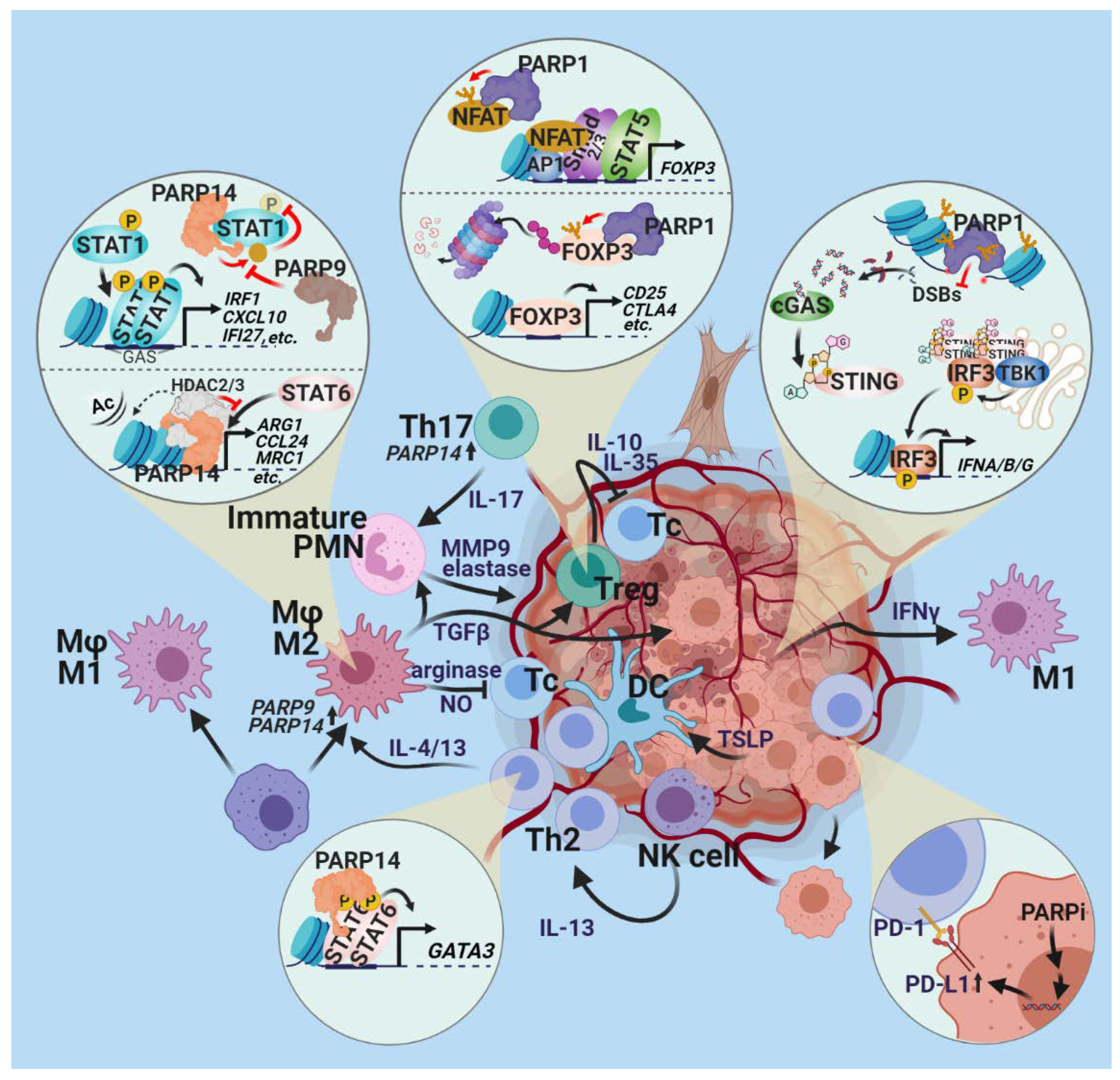
2.3. Tumor-Promoting Inflammation
Open Questions and Prospects
2.4. Evading Immune Destruction
Open Questions and Prospects
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hottiger, M.O.; Hassa, P.O.; Lüscher, B.; Schüler, H.; Koch-Nolte, F. Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem. Sci. 2010, 35, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Hassa, P.O.; Hottiger, M.O. The diverse biological roles of mammalian PARPS, a small but powerful family of poly-ADP-ribose polymerases. Front Biosci. 2008, 13, 3046–3082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burkle, A.; Virag, L. Poly(ADP-ribose): PARadigms and PARadoxes. Mol. Aspects Med. 2013, 34, 1046–1065. [Google Scholar] [CrossRef] [Green Version]
- Hegedus, C.; Virag, L. Inputs and outputs of poly(ADP-ribosyl)ation: Relevance to oxidative stress. Redox Biol. 2014, 2, 978–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, N.; Ganesan, R.; Hegedus, C.; Kovacs, K.; Kufer, T.A.; Virag, L. Programmed necrotic cell death of macrophages: Focus on pyroptosis, necroptosis, and parthanatos. Redox Biol. 2019, 26, 101239. [Google Scholar] [CrossRef] [PubMed]
- Virag, L. The expanding universe of poly(ADP-ribosyl)ation. Cell. Mol. Life Sci. 2005, 62, 719–720. [Google Scholar] [CrossRef] [PubMed]
- Virag, L. 50 Years of poly(ADP-ribosyl)ation. Mol. Aspects Med. 2013, 34, 1043–1045. [Google Scholar] [CrossRef] [PubMed]
- Virag, L.; Robaszkiewicz, A.; Rodriguez-Vargas, J.M.; Oliver, F.J. Poly(ADP-ribose) signaling in cell death. Mol. Aspects Med. 2013, 34, 1153–1167. [Google Scholar] [CrossRef]
- Virag, L.; Szabo, C. The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol. Rev. 2002, 54, 375–429. [Google Scholar] [CrossRef] [PubMed]
- Demény, M.A.; Virág, L. The PARP enzyme family and the hallmarks of cancer. Part 1. Cell intrinsic hallmarks. Cancers (Basel) 2021, 13, 2042. [Google Scholar] [CrossRef]
- Jayson, G.C.; Kerbel, R.; Ellis, L.M.; Harris, A.L. Antiangiogenic therapy in oncology: Current status and future directions. Lancet 2016, 388, 518–529. [Google Scholar] [CrossRef]
- Nishida, N.; Yano, H.; Nishida, T.; Kamura, T.; Kojiro, M. Angiogenesis in cancer. Vasc. Health Risk Manag. 2006, 2, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Liekens, S.; Schols, D.; Hatse, S. CXCL12-CXCR4 axis in angiogenesis, metastasis and stem cell mobilization. Curr. Pharm. Des. 2010, 16, 3903–3920. [Google Scholar] [CrossRef] [PubMed]
- Chu, S.H.; Feng, D.F.; Ma, Y.B.; Zhu, Z.A.; Zhang, H.; Qiu, J.H. Stabilization of hepatocyte growth factor mRNA by hypoxia-inducible factor 1. Mol. Biol. Rep. 2009, 36, 1967–1975. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Lin, Y.; Zhan, T.; Chen, L.; Guo, S. HGF expression induced by HIF-1alpha promote the proliferation and tube formation of endothelial progenitor cells. Cell Biol. Int. 2015, 39, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Rey, S.; Semenza, G.L. Hypoxia-inducible factor-1-dependent mechanisms of vascularization and vascular remodelling. Cardiovasc. Res. 2010, 86, 236–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tentori, L.; Lacal, P.M.; Muzi, A.; Dorio, A.S.; Leonetti, C.; Scarsella, M.; Ruffini, F.; Xu, W.; Min, W.; Stoppacciaro, A.; et al. Poly(ADP-ribose) polymerase (PARP) inhibition or PARP-1 gene deletion reduces angiogenesis. Eur. J. Cancer 2007, 43, 2124–2133. [Google Scholar] [CrossRef] [PubMed]
- Tentori, L.; Muzi, A.; Dorio, A.S.; Bultrini, S.; Mazzon, E.; Lacal, P.M.; Shah, G.M.; Zhang, J.; Navarra, P.; Nocentini, G.; et al. Stable depletion of poly (ADP-ribose) polymerase-1 reduces in vivo melanoma growth and increases chemosensitivity. Eur. J. Cancer 2008, 44, 1302–1314. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Romero, R.; Cañuelo, A.; Martínez-Lara, E.; Javier Oliver, F.; Cárdenas, S.; Siles, E. Poly(ADP-ribose) polymerase-1 modulation of in vivo response of brain hypoxia-inducible factor-1 to hypoxia/reoxygenation is mediated by nitric oxide and factor inhibiting HIF. J. Neurochem. 2009, 111, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Romero, R.; Martínez-Lara, E.; Aguilar-Quesada, R.; Peralta, A.; Oliver, F.J.; Siles, E. PARP-1 modulates deferoxamine-induced HIF-1alpha accumulation through the regulation of nitric oxide and oxidative stress. J. Cell. Biochem. 2008, 104, 2248–2260. [Google Scholar] [CrossRef] [PubMed]
- Elser, M.; Borsig, L.; Hassa, P.O.; Erener, S.; Messner, S.; Valovka, T.; Keller, S.; Gassmann, M.; Hottiger, M.O. Poly(ADP-ribose) polymerase 1 promotes tumor cell survival by coactivating hypoxia-inducible factor-1-dependent gene expression. Mol. Cancer Res. 2008, 6, 282–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Flores, A.; Aguilar-Quesada, R.; Siles, E.; Pozo, S.; Rodríguez-Lara, M.I.; López-Jiménez, L.; López-Rodríguez, M.; Peralta-Leal, A.; Villar, D.; Martín-Oliva, D.; et al. Interaction between PARP-1 and HIF-2α in the hypoxic response. Oncogene 2014, 33, 891–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marti, J.M.; Garcia-Diaz, A.; Delgado-Bellido, D.; O’Valle, F.; Gonzalez-Flores, A.; Carlevaris, O.; Rodriguez-Vargas, J.M.; Ame, J.C.; Dantzer, F.; King, G.L.; et al. Selective modulation by PARP-1 of HIF-1alpha-recruitment to chromatin during hypoxia is required for tumor adaptation to hypoxic conditions. Redox Biol. 2021, 41, 101885. [Google Scholar] [CrossRef] [PubMed]
- Martin-Oliva, D.; Aguilar-Quesada, R.; O’Valle, F.; Muñoz-Gámez, J.A.; Martínez-Romero, R.; García Del Moral, R.; Ruiz de Almodóvar, J.M.; Villuendas, R.; Piris, M.A.; Oliver, F.J. Inhibition of poly(ADP-ribose) polymerase modulates tumor-related gene expression, including hypoxia-inducible factor-1 activation, during skin carcinogenesis. Cancer Res. 2006, 66, 5744–5756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quiles-Perez, R.; Munoz-Gamez, J.A.; Ruiz-Extremera, A.; O’Valle, F.; Sanjuan-Nunez, L.; Martin-Alvarez, A.B.; Martin-Oliva, D.; Caballero, T.; Munoz de Rueda, P.; Leon, J.; et al. Inhibition of poly adenosine diphosphate-ribose polymerase decreases hepatocellular carcinoma growth by modulation of tumor-related gene expression. Hepatology 2010, 51, 255–266. [Google Scholar] [CrossRef]
- Martín-Oliva, D.; O’Valle, F.; Muñoz-Gámez, J.A.; Valenzuela, M.T.; Nuñez, M.I.; Aguilar, M.; Ruiz de Almodóvar, J.M.; Garcia del Moral, R.; Oliver, F.J. Crosstalk between PARP-1 and NF-kappaB modulates the promotion of skin neoplasia. Oncogene 2004, 23, 5275–5283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajesh, M.; Mukhopadhyay, P.; Godlewski, G.; Bátkai, S.; Haskó, G.; Liaudet, L.; Pacher, P. Poly(ADP-ribose)polymerase inhibition decreases angiogenesis. Biochem. Biophys. Res. Commun. 2006, 350, 1056–1062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajesh, M.; Mukhopadhyay, P.; Bátkai, S.; Godlewski, G.; Haskó, G.; Liaudet, L.; Pacher, P. Pharmacological inhibition of poly(ADP-ribose) polymerase inhibits angiogenesis. Biochem. Biophys. Res. Commun. 2006, 350, 352–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, J.; Ye, T.; Cui, P.; Hua, Q.; Zeng, H.; Zhao, D. AP-1 transcription factor mediates VEGF-induced endothelial cell migration and proliferation. Microvasc. Res. 2016, 105, 103–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zingarelli, B.; Hake, P.W.; O’Connor, M.; Denenberg, A.; Wong, H.R.; Kong, S.; Aronow, B.J. Differential regulation of activator protein-1 and heat shock factor-1 in myocardial ischemia and reperfusion injury: Role of poly(ADP-ribose) polymerase-1. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H1408–H1415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreone, T.L.; O’Connor, M.; Denenberg, A.; Hake, P.W.; Zingarelli, B. Poly(ADP-ribose) polymerase-1 regulates activation of activator protein-1 in murine fibroblasts. J. Immunol. 2003, 170, 2113–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, S.; Hussain, M.Z.; Beckert, S.; Ghani, Q.P.; Weinreich, J.; Hunt, T.K.; Becker, H.D.; Königsrainer, A. Lactate down-regulates cellular poly(ADP-ribose) formation in cultured human skin fibroblasts. Eur J. Clin. Investig. 2007, 37, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.B.; Viji, R.I.; Kiran, M.S.; Sudhakaran, P.R. Endothelial cell response to lactate: Implication of PAR modification of VEGF. J. Cell. Physiol. 2007, 211, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Tolić, A.; Grdović, N.; Dinić, S.; Rajić, J.; Đorđević, M.; Sinadinović, M.; Arambašić Jovanović, J.; Mihailović, M.; Poznanović, G.; Uskoković, A.; et al. Absence of PARP-1 affects Cxcl12 expression by increasing DNA demethylation. J. Cell. Mol. Med. 2019, 23, 2610–2618. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.H.; Hur, E.G.; Kang, S.J.; Kim, J.A.; Thapa, D.; Lee, Y.M.; Ku, S.K.; Jung, Y.; Kwak, M.K. NRF2 blockade suppresses colon tumor angiogenesis by inhibiting hypoxia-induced activation of HIF-1α. Cancer Res. 2011, 71, 2260–2275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, T.; Wang, X.J.; Tian, W.; Jaramillo, M.C.; Lau, A.; Zhang, D.D. Poly(ADP-ribose) polymerase-1 modulates Nrf2-dependent transcription. Free Radic. Biol. Med. 2014, 67, 69–80. [Google Scholar] [CrossRef] [Green Version]
- Baudino, T.A.; McKay, C.; Pendeville-Samain, H.; Nilsson, J.A.; Maclean, K.H.; White, E.L.; Davis, A.C.; Ihle, J.N.; Cleveland, J.L. c-Myc is essential for vasculogenesis and angiogenesis during development and tumor progression. Genes Dev. 2002, 16, 2530–2543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mostocotto, C.; Carbone, M.; Battistelli, C.; Ciotti, A.; Amati, P.; Maione, R. Poly(ADP-ribosyl)ation is required to modulate chromatin changes at c-MYC promoter during emergence from quiescence. PLoS ONE 2014, 9, e102575. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, S.; Mickanin, C.; Feng, Y.; Charlat, O.; Michaud, G.A.; Schirle, M.; Shi, X.; Hild, M.; Bauer, A.; et al. RNF146 is a poly(ADP-ribose)-directed E3 ligase that regulates axin degradation and Wnt signalling. Nat. Cell Biol. 2011, 13, 623–629. [Google Scholar] [CrossRef]
- Feijs, K.L.; Kleine, H.; Braczynski, A.; Forst, A.H.; Herzog, N.; Verheugd, P.; Linzen, U.; Kremmer, E.; Lüscher, B. ARTD10 substrate identification on protein microarrays: Regulation of GSK3β by mono-ADP-ribosylation. Cell Commun. Signal 2013, 11, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez, M.I.; Majuelos-Melguizo, J.; Martí Martín-Consuegra, J.M.; Ruiz de Almodóvar, M.; López-Rivas, A.; Javier Oliver, F. Deciphering the insights of poly(ADP-ribosylation) in tumor progression. Med. Res. Rev. 2015, 35, 678–697. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, M.I.; Peralta-Leal, A.; O’Valle, F.; Rodriguez-Vargas, J.M.; Gonzalez-Flores, A.; Majuelos-Melguizo, J.; López, L.; Serrano, S.; de Herreros, A.G.; Rodríguez-Manzaneque, J.C.; et al. PARP-1 regulates metastatic melanoma through modulation of vimentin-induced malignant transformation. PLoS Genet. 2013, 9, e1003531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McPhee, T.R.; McDonald, P.C.; Oloumi, A.; Dedhar, S. Integrin-linked kinase regulates E-cadherin expression through PARP-1. Dev. Dyn. 2008, 237, 2737–2747. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, M.I.; González-Flores, A.; Dantzer, F.; Collard, J.; de Herreros, A.G.; Oliver, F.J. Poly(ADP-ribose)-dependent regulation of Snail1 protein stability. Oncogene 2011, 30, 4365–4372. [Google Scholar] [CrossRef] [Green Version]
- Lönn, P.; van der Heide, L.P.; Dahl, M.; Hellman, U.; Heldin, C.H.; Moustakas, A. PARP-1 attenuates Smad-mediated transcription. Mol. Cell 2010, 40, 521–532. [Google Scholar] [CrossRef]
- Dahl, M.; Maturi, V.; Lönn, P.; Papoutsoglou, P.; Zieba, A.; Vanlandewijck, M.; van der Heide, L.P.; Watanabe, Y.; Söderberg, O.; Hottiger, M.O.; et al. Fine-tuning of Smad protein function by poly(ADP-ribose) polymerases and poly(ADP-ribose) glycohydrolase during transforming growth factor β signaling. PLoS ONE 2014, 9, e103651. [Google Scholar] [CrossRef] [Green Version]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging Biol.ogical Principles of Metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef] [Green Version]
- Polyak, K.; Weinberg, R.A. Transitions between epithelial and mesenchymal states: Acquisition of malignant and stem cell traits. Nat. Rev. Cancer 2009, 9, 265–273. [Google Scholar] [CrossRef]
- Nieto, M.A. Epithelial plasticity: A common theme in embryonic and cancer cells. Science 2013, 342, 1234850. [Google Scholar] [CrossRef] [Green Version]
- Overholtzer, M.; Zhang, J.; Smolen, G.A.; Muir, B.; Li, W.; Sgroi, D.C.; Deng, C.X.; Brugge, J.S.; Haber, D.A. Transforming properties of YAP, a candidate oncogene on the chromosome 11q22 amplicon. Proc. Natl. Acad. Sci. USA 2006, 103, 12405–12410. [Google Scholar] [CrossRef] [Green Version]
- Lamar, J.M.; Stern, P.; Liu, H.; Schindler, J.W.; Jiang, Z.G.; Hynes, R.O. The Hippo pathway target, YAP, promotes metastasis through its TEAD-interaction domain. Proc. Natl. Acad. Sci. USA 2012, 109, E2441–E2450. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Wei, X.; Li, W.; Udan, R.S.; Yang, Q.; Kim, J.; Xie, J.; Ikenoue, T.; Yu, J.; Li, L.; et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007, 21, 2747–2761. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Li, L.; Lu, Q.; Wang, L.H.; Liu, C.Y.; Lei, Q.; Guan, K.L. Angiomotin is a novel Hippo pathway component that inhibits YAP oncoprotein. Genes Dev. 2011, 25, 51–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Li, N.; Li, X.; Tran, M.K.; Han, X.; Chen, J. Tankyrase Inhibitors Target YAP by Stabilizing Angiomotin Family Proteins. Cell Rep. 2015, 13, 524–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Chen, H.; Parker, B.; Rubin, E.; Zhu, T.; Lee, J.S.; Argani, P.; Sukumar, S. HOXB7, a homeodomain protein, is overexpressed in breast cancer and confers epithelial-mesenchymal transition. Cancer Res. 2006, 66, 9527–9534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Ellmann, S.; Rubin, E.; Gil, M.; Jin, K.; Han, L.; Chen, H.; Kwon, E.M.; Guo, J.; Ha, H.C.; et al. ADP ribosylation by PARP-1 suppresses HOXB7 transcriptional activity. PLoS ONE 2012, 7, e40644. [Google Scholar] [CrossRef] [PubMed]
- Pu, H.; Horbinski, C.; Hensley, P.J.; Matuszak, E.A.; Atkinson, T.; Kyprianou, N. PARP-1 regulates epithelial-mesenchymal transition (EMT) in prostate tumorigenesis. Carcinogenesis 2014, 35, 2592–2601. [Google Scholar] [CrossRef] [Green Version]
- Karicheva, O.; Rodriguez-Vargas, J.M.; Wadier, N.; Martin-Hernandez, K.; Vauchelles, R.; Magroun, N.; Tissier, A.; Schreiber, V.; Dantzer, F. PARP3 controls TGFβ and ROS driven epithelial-to-mesenchymal transition and stemness by stimulating a TG2-Snail-E-cadherin axis. Oncotarget 2016, 7, 64109–64123. [Google Scholar] [CrossRef] [Green Version]
- Navas, T.; Kinders, R.J.; Lawrence, S.M.; Ferry-Galow, K.V.; Borgel, S.; Hollingshead, M.G.; Srivastava, A.K.; Alcoser, S.Y.; Makhlouf, H.R.; Chuaqui, R.; et al. Clinical Evolution of Epithelial-Mesenchymal Transition in Human Carcinomas. Cancer Res. 2020, 80, 304–318. [Google Scholar] [CrossRef]
- Ordonez, L.D.; Hay, T.; McEwen, R.; Polanska, U.M.; Hughes, A.; Delpuech, O.; Cadogan, E.; Powell, S.; Dry, J.; Tornillo, G.; et al. Rapid activation of epithelial-mesenchymal transition drives PARP inhibitor resistance in Brca2-mutant mammary tumours. Oncotarget 2019, 10, 2586–2606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shay, G.; Lynch, C.C.; Fingleton, B. Moving targets: Emerging roles for MMPs in cancer progression and metastasis. Matrix Biol. 2015, 44–46, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.; Jiang, Z.; Cheng, L.; Chen, K.; Zhou, C.; Sun, L.; Qian, W.; Li, J.; Cao, J.; Xu, Q.; et al. Paracrine HGF/c-MET enhances the stem cell-like potential and glycolysis of pancreatic cancer cells via activation of YAP/HIF-1α. Exp. Cell Res. 2018, 371, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Matsui, S.; Osada, S.; Tomita, H.; Komori, S.; Mori, R.; Sanada, Y.; Takahashi, T.; Yamaguchi, K.; Yoshida, K. Clinical significance of aggressive hepatectomy for colorectal liver metastasis, evaluated from the HGF/c-Met pathway. Int. J. Oncol. 2010, 37, 289–297. [Google Scholar] [CrossRef] [Green Version]
- Mariani, M.; McHugh, M.; Petrillo, M.; Sieber, S.; He, S.; Andreoli, M.; Wu, Z.; Fiedler, P.; Scambia, G.; Shahabi, S.; et al. HGF/c-Met axis drives cancer aggressiveness in the neo-adjuvant setting of ovarian cancer. Oncotarget 2014, 5, 4855–4867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, W.; Lv, S.; Zhang, C.; Tian, Y. Potential role of HGF-PARP-1 signaling in invasion of ovarian cancer cells. Int. J. Clin. Exp. Pathol. 2018, 11, 3310–3317. [Google Scholar] [PubMed]
- Mishra, M.; Kowluru, R.A. Role of PARP-1 as a novel transcriptional regulator of MMP-9 in diabetic retinopathy. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1761–1769. [Google Scholar] [CrossRef]
- Virág, L. Poly(ADP-ribosyl)ation in asthma and other lung diseases. Pharmacol. Res. 2005, 52, 83–92. [Google Scholar] [CrossRef]
- Ghorai, A.; Sarma, A.; Chowdhury, P.; Ghosh, U. PARP-1 depletion in combination with carbon ion exposure significantly reduces MMPs activity and overall increases TIMPs expression in cultured HeLa cells. Radiat. Oncol. 2016, 11, 126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Hamoly, T.; Hegedűs, C.; Lakatos, P.; Kovács, K.; Bai, P.; El-Ghazaly, M.A.; El-Denshary, E.S.; Szabó, É.; Virág, L. Activation of poly(ADP-ribose) polymerase-1 delays wound healing by regulating keratinocyte migration and production of inflammatory mediators. Mol. Med. 2014, 20, 363–371. [Google Scholar] [CrossRef] [Green Version]
- Nicolescu, A.C.; Holt, A.; Kandasamy, A.D.; Pacher, P.; Schulz, R. Inhibition of matrix metalloproteinase-2 by PARP inhibitors. Biochem. Biophys. Res. Commun. 2009, 387, 646–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Li, M.; Wang, Y.L.; Fauzee, N.J.; Yang, Y.; Pan, J.; Yang, L.; Lazar, A. RNA interference of PARG could inhibit the metastatic potency of colon carcinoma cells via PI3-kinase/Akt pathway. Cell. Physiol. Biochem. 2012, 29, 361–372. [Google Scholar] [CrossRef]
- Kim, J.; Yum, S.; Kang, C.; Kang, S.-J. Gene-gene interactions in gastrointestinal cancer susceptibility. Oncotarget 2016, 7, 67612–67625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Pyun, J.A.; Cho, S.W.; Lee, K.; Kwack, K. Lymph node metastasis of gastric cancer is associated with the interaction between poly (ADP-ribose) polymerase 1 and matrix metallopeptidase 2. DNA Cell Biol. 2011, 30, 1011–1017. [Google Scholar] [CrossRef] [PubMed]
- Schlesinger, M. Role of platelets and platelet receptors in cancer metastasis. J. Hematol. Oncol. 2018, 11, 125. [Google Scholar] [CrossRef]
- Placke, T.; Kopp, H.G.; Salih, H.R. The wolf in sheep’s clothing: Platelet-derived "pseudo self" impairs cancer cell "missing self" recognition by NK cells. Oncoimmunology 2012, 1, 557–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zingarelli, B.; Salzman, A.L.; Szabó, C. Genetic disruption of poly (ADP-ribose) synthetase inhibits the expression of P-selectin and intercellular adhesion molecule-1 in myocardial ischemia/reperfusion injury. Circ. Res. 1998, 83, 85–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, J.; Li, M.; Threadgill, M.D.; Wang, Y.; Fu, W. Decreasing P-selectin and ICAM-1 via activating Akt: A possible mechanism by which PARG inhibits adhesion of mouse colorectal carcinoma CT26 cells to platelets. Cancer Gene Ther. 2013, 20, 487–492. [Google Scholar] [CrossRef] [Green Version]
- Dutta, P.; Paico, K.; Gomez, G.; Wu, Y.; Vadgama, J.V. Transcriptional Regulation of CCL2 by PARP1 Is a Driver for Invasiveness in Breast Cancer. Cancers (Basel) 2020, 12, 1317. [Google Scholar] [CrossRef]
- Xu, F.; Sun, Y.; Yang, S.Z.; Zhou, T.; Jhala, N.; McDonald, J.; Chen, Y. Cytoplasmic PARP-1 promotes pancreatic cancer tumorigenesis and resistance. Int. J. Cancer 2019, 145, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.H.; Wang, Q.; Gerald, W.; Hudis, C.A.; Norton, L.; Smid, M.; Foekens, J.A.; Massagué, J. Latent bone metastasis in breast cancer tied to Src-dependent survival signals. Cancer Cell 2009, 16, 67–78. [Google Scholar] [CrossRef] [Green Version]
- Marković, J.; Grdović, N.; Dinić, S.; Karan-Djurašević, T.; Uskoković, A.; Arambašić, J.; Mihailović, M.; Pavlović, S.; Poznanović, G.; Vidaković, M. PARP-1 and YY1 are important novel regulators of CXCL12 gene transcription in rat pancreatic beta cells. PLoS ONE 2013, 8, e59679. [Google Scholar] [CrossRef]
- Hsu, E.C.; Rice, M.A.; Bermudez, A.; Marques, F.J.G.; Aslan, M.; Liu, S.; Ghoochani, A.; Zhang, C.A.; Chen, Y.S.; Zlitni, A.; et al. Trop2 is a driver of metastatic prostate cancer with neuroendocrine phenotype via PARP1. Proc. Natl. Acad. Sci. USA 2020, 117, 2032–2042. [Google Scholar] [CrossRef] [Green Version]
- Zuo, H.; Yang, D.; Yang, Q.; Tang, H.; Fu, Y.X.; Wan, Y. Differential regulation of breast cancer bone metastasis by PARP1 and PARP2. Nat. Commun. 2020, 11, 1578. [Google Scholar] [CrossRef] [Green Version]
- D’Alterio, C.; Scala, S.; Sozzi, G.; Roz, L.; Bertolini, G. Paradoxical effects of chemotherapy on tumor relapse and metastasis promotion. Semin. Cancer Biol. 2020, 60, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, M.; Werner, S. Cancer as an overhealing wound: An old hypothesis revisited. Nat. Rev. Mol. Cell Biol. 2008, 9, 628–638. [Google Scholar] [CrossRef]
- Hegedus, C.; Kovacs, K.; Polgar, Z.; Regdon, Z.; Szabo, E.; Robaszkiewicz, A.; Forman, H.J.; Martner, A.; Virag, L. Redox control of cancer cell destruction. Redox Biol. 2018, 16, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Brady, P.N.; Goel, A.; Johnson, M.A. Poly(ADP-Ribose) Polymerases in Host-Pathogen Interactions, Inflammation, and Immunity. Microbiol. Mol. Biol. Rev. 2019, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, P.; Virag, L. Role of poly(ADP-ribose) polymerases in the regulation of inflammatory processes. Febs Lett. 2012, 586, 3771–3777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldfarb, R.D.; Marton, A.; Szabo, E.; Virag, L.; Salzman, A.L.; Glock, D.; Akhter, I.; McCarthy, R.; Parrillo, J.E.; Szabo, C. Protective effect of a novel, potent inhibitor of poly(adenosine 5′-diphosphate-ribose) synthetase in a porcine model of severe bacterial sepsis. Crit. Care Med. 2002, 30, 974–980. [Google Scholar] [CrossRef]
- Liaudet, L.; Soriano, F.G.; Szabo, E.; Virag, L.; Mabley, J.G.; Salzman, A.L.; Szabo, C. Protection against hemorrhagic shock in mice genetically deficient in poly(ADP-ribose)polymerase. Proc. Natl. Acad. Sci. USA 2000, 97, 10203–10208. [Google Scholar] [CrossRef] [Green Version]
- Mabley, J.; Virag, L.; Jagtap, P.; Szabo, E.; Prendergast, R.; Perretti, M.; Getting, S.; Szabo, C. Effect of a novel poly (ADP-ribose) polymerase (PARP) inhibitor in rodent models of local inflammation. Faseb J. 2001, 15, A242. [Google Scholar]
- Pacher, P.; Liaudet, L.; Mabley, J.G.; Soriano, F.G.; Virag, L.; Szabo, C. Activation of poly(ADP-ribose) polymerase-1 is a central mechanism of lipopolysaccharide-induced acute lung inflammation. Faseb J. 2002, 16, A20. [Google Scholar]
- Mabley, J.G.; Jagtap, P.; Perretti, M.; Getting, S.J.; Salzman, A.L.; Virag, L.; Szabo, E.; Soriano, F.G.; Liaudet, L.; Abdelkarim, G.E.; et al. Anti-inflammatory effects of a novel, potent inhibitor of poly (ADP-ribose) polymerase. Inflamm. Res. 2001, 50, 561–569. [Google Scholar] [CrossRef]
- Marton, A.; Szabo, E.; Virag, L.; Salzman, A.; Glock, D.; McCarthy, R.; Parrillo, J.E.; Sabo, C.; Goldfarb, R.D. Protective effect of a novel inhibitor of poly (ADP-ribose) synthetase in a porcine model of peritonitis. Faseb J. 2001, 15, A1123. [Google Scholar]
- Liaudet, L.; Pacher, P.; Mabley, J.G.; Virag, L.; Soriano, F.G.; Hasko, G.; Szabo, C. Activation of poly(ADP-ribose) polymerase-1 is a central mechanism of lipopolysaccharide-induced acute lung inflammation. Am. J. Resp. Crit. Care 2002, 165, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Bakondi, E.; Bai, P.; Bak, I.; Szabo, E.; Gergely, P.; Szabo, C.; Virag, L. Role of poly(ADP-ribose) polymerase in contact hypersensitivity. Faseb J. 2002, 16, A674–A675. [Google Scholar]
- Virag, L.; Szabo, E. Complex Role of Poly(Adp-Ribosyl)Ation in Shock and Other Oxidative Stress-Related Pathologies. Shock 2011, 36, 34. [Google Scholar]
- Virag, L.; Bai, P.; Bak, I.; Pacher, P.; Mabley, J.; Liaudet, L.; Bakondl, E.; Gergely, P.; Kollai, M.; Szabo, C. Effects of poly(ADP-ribose) polymerase inhibition on inflammatory cell migration in a murine model of asthma. Med. Sci. Monitor 2004, 10, Br77–Br83. [Google Scholar]
- Soriano, F.G.; Liaudet, L.; Szabo, E.; Virag, L.; Mabley, J.G.; Pacher, P.; Szabo, C. Resistance to acute septic peritonitis in poly(ADP-ribose) polymerase-1 deficient mice. Faseb J. 2002, 16, A1120. [Google Scholar] [CrossRef]
- Scott, G.S.; Hake, P.; Kean, R.B.; Virag, L.; Szabo, C.; Hooper, D.C. Role of poly(ADP-ribose) synthetase activation in the development of experimental allergic encephalomyelitis. J. Neuroimmunol. 2001, 117, 78–86. [Google Scholar] [CrossRef]
- Szabo, E.; Bodnar, E.; Erdelyi, K.; Hegedus, C.; Bakondi, E.; Remenyik, E.; Virag, L. Possible role of the nitric oxide-peroxynitrite-poly(ADP-ribose) polymerase pathway in chronic wounds. J. Investig. Dermatol. 2009, 129, S96. [Google Scholar]
- El-Hamoly, T.; Hajnady, Z.; Nagy-Penzes, M.; Bakondi, E.; Regdon, Z.; Demeny, M.A.; Kovacs, K.; Hegedus, C.; Abd El-Rahman, S.S.; Szabo, E.; et al. Poly(ADP-Ribose) Polymerase 1 Promotes Inflammation and Fibrosis in a Mouse Model of Chronic Pancreatitis. Int. J. Mol. Sci. 2021, 22, 3593. [Google Scholar] [CrossRef]
- Hassa, P.O.; Hottiger, M.O. The functional role of poly(ADP-ribose)polymerase 1 as novel coactivator of NF-kappaB in inflammatory disorders. Cell. Mol. Life Sci. 2002, 59, 1534–1553. [Google Scholar] [CrossRef]
- Oliver, F.J.; Ménissier-de Murcia, J.; Nacci, C.; Decker, P.; Andriantsitohaina, R.; Muller, S.; de la Rubia, G.; Stoclet, J.C.; de Murcia, G. Resistance to endotoxic shock as a consequence of defective NF-kappaB activation in poly (ADP-ribose) polymerase-1 deficient mice. Embo J. 1999, 18, 4446–4454. [Google Scholar] [CrossRef]
- Oei, S.L.; Shi, Y. Poly(ADP-ribosyl)ation of transcription factor Yin Yang 1 under conditions of DNA damage. Biochem. Biophys. Res. Commun. 2001, 285, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Laugesen, A.; Hojfeldt, J.W.; Helin, K. Role of the Polycomb Repressive Complex 2 (PRC2) in Transcriptional Regulation and Cancer. Cold Spring Harb. Perspect. Med. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.Y.; Choi, E.B.; So Park, M.; Kim, S.K.; Park, M.S.; Kim, M.Y. PARP1 and PRC2 double deficiency promotes BRCA-proficient breast cancer growth by modification of the tumor microenvironment. Febs J. 2020. [Google Scholar] [CrossRef] [PubMed]
- Iwata, H.; Goettsch, C.; Sharma, A.; Ricchiuto, P.; Goh, W.W.; Halu, A.; Yamada, I.; Yoshida, H.; Hara, T.; Wei, M.; et al. PARP9 and PARP14 cross-regulate macrophage activation via STAT1 ADP-ribosylation. Nat. Commun. 2016, 7, 12849. [Google Scholar] [CrossRef]
- Locati, M.; Curtale, G.; Mantovani, A. Diversity, Mechanisms, and Significance of Macrophage Plasticity. Annu. Rev. Pathol. 2020, 15, 123–147. [Google Scholar] [CrossRef] [Green Version]
- Levaot, N.; Voytyuk, O.; Dimitriou, I.; Sircoulomb, F.; Chandrakumar, A.; Deckert, M.; Krzyzanowski, P.M.; Scotter, A.; Gu, S.; Janmohamed, S.; et al. Loss of Tankyrase-mediated destruction of 3BP2 is the underlying pathogenic mechanism of cherubism. Cell 2011, 147, 1324–1339. [Google Scholar] [CrossRef] [Green Version]
- Verheugd, P.; Forst, A.H.; Milke, L.; Herzog, N.; Feijs, K.L.; Kremmer, E.; Kleine, H.; Luscher, B. Regulation of NF-kappaB signalling by the mono-ADP-ribosyltransferase ARTD10. Nat. Commun. 2013, 4, 1683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, S.O.; Khan, F.A.; Galindo-Campos, M.A.; Yelamos, J. Understanding specific functions of PARP-2: New lessons for cancer therapy. Am. J. Cancer Res. 2016, 6, 1842–1863. [Google Scholar] [PubMed]
- Popoff, I.; Jijon, H.; Monia, B.; Tavernini, M.; Ma, M.; McKay, R.; Madsen, K. Antisense oligonucleotides to poly(ADP-ribose) polymerase-2 ameliorate colitis in interleukin-10-deficient mice. J. Pharmacol. Exp. Ther. 2002, 303, 1145–1154. [Google Scholar] [CrossRef] [PubMed]
- Kamboj, A.; Lu, P.; Cossoy, M.B.; Stobart, J.L.; Dolhun, B.A.; Kauppinen, T.M.; de Murcia, G.; Anderson, C.M. Poly(ADP-ribose) polymerase 2 contributes to neuroinflammation and neurological dysfunction in mouse experimental autoimmune encephalomyelitis. J. Neuroinflamm. 2013, 10, 49. [Google Scholar] [CrossRef] [Green Version]
- Kickhoefer, V.A.; Siva, A.C.; Kedersha, N.L.; Inman, E.M.; Ruland, C.; Streuli, M.; Rome, L.H. The 193-kD vault protein, VPARP, is a novel poly(ADP-ribose) polymerase. J. Cell Biol. 1999, 146, 917–928. [Google Scholar] [CrossRef] [Green Version]
- Peng, N.; Liu, S.; Xia, Z.; Ren, S.; Feng, J.; Jing, M.; Gao, X.; Wiemer, E.A.; Zhu, Y. Inducible Major Vault Protein Plays a Pivotal Role in Double-Stranded RNA- or Virus-Induced Proinflammatory Response. J. Immunol. 2016, 196, 2753–2766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben, J.; Zhang, Y.; Zhou, R.; Zhang, H.; Zhu, X.; Li, X.; Zhang, H.; Li, N.; Zhou, X.; Bai, H.; et al. Major vault protein regulates class A scavenger receptor-mediated tumor necrosis factor-α synthesis and apoptosis in macrophages. J. Biol. Chem. 2013, 288, 20076–20084. [Google Scholar] [CrossRef] [Green Version]
- Ben, J.; Jiang, B.; Wang, D.; Liu, Q.; Zhang, Y.; Qi, Y.; Tong, X.; Chen, L.; Liu, X.; Zhang, Y.; et al. Major vault protein suppresses obesity and atherosclerosis through inhibiting IKK-NF-κB signaling mediated inflammation. Nat. Commun. 2019, 10, 1801. [Google Scholar] [CrossRef] [PubMed]
- Bindesbøll, C.; Tan, S.; Bott, D.; Cho, T.; Tamblyn, L.; MacPherson, L.; Grønning-Wang, L.; Nebb, H.I.; Matthews, J. TCDD-inducible poly-ADP-ribose polymerase (TIPARP/PARP7) mono-ADP-ribosylates and co-activates liver X receptors. Biochem. J. 2016, 473, 899–910. [Google Scholar] [CrossRef] [PubMed]
- Fowler, A.J.; Sheu, M.Y.; Schmuth, M.; Kao, J.; Fluhr, J.W.; Rhein, L.; Collins, J.L.; Willson, T.M.; Mangelsdorf, D.J.; Elias, P.M.; et al. Liver X receptor activators display anti-inflammatory activity in irritant and allergic contact dermatitis models: Liver-X-receptor-specific inhibition of inflammation and primary cytokine production. J. Investig. Dermatol. 2003, 120, 246–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welsby, I.; Hutin, D.; Gueydan, C.; Kruys, V.; Rongvaux, A.; Leo, O. PARP12, an interferon-stimulated gene involved in the control of protein translation and inflammation. J. Biol. Chem. 2014, 289, 26642–26657. [Google Scholar] [CrossRef] [Green Version]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. Emerging role of NF-kappaB signaling in the induction of senescence-associated secretory phenotype (SASP). Cell Signal 2012, 24, 835–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohanna, M.; Giuliano, S.; Bonet, C.; Imbert, V.; Hofman, V.; Zangari, J.; Bille, K.; Robert, C.; Bressac-de Paillerets, B.; Hofman, P.; et al. Senescent cells develop a PARP-1 and nuclear factor-{kappa}B-associated secretome (PNAS). Genes Dev. 2011, 25, 1245–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [Green Version]
- Svane, I.M.; Engel, A.M.; Nielsen, M.B.; Ljunggren, H.G.; Rygaard, J.; Werdelin, O. Chemically induced sarcomas from nude mice are more immunogenic than similar sarcomas from congenic normal mice. Eur. J. Immunol. 1996, 26, 1844–1850. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Pang, Y.; Moses, H.L. TGF-beta and immune cells: An important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010, 31, 220–227. [Google Scholar] [CrossRef] [Green Version]
- Shields, J.D.; Kourtis, I.C.; Tomei, A.A.; Roberts, J.M.; Swartz, M.A. Induction of lymphoidlike stroma and immune escape by tumors that express the chemokine CCL21. Science 2010, 328, 749–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arrieta, V.A.; Cacho-Díaz, B.; Zhao, J.; Rabadan, R.; Chen, L.; Sonabend, A.M. The possibility of cancer immune editing in gliomas. A critical review. Oncoimmunology 2018, 7, e1445458. [Google Scholar] [CrossRef] [PubMed]
- Waldhauer, I.; Steinle, A. NK cells and cancer immunosurveillance. Oncogene 2008, 27, 5932–5943. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Allison, J.P. Immune checkpoint targeting in cancer therapy: Toward combination strategies with curative potential. Cell 2015, 161, 205–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chamoto, K.; Hatae, R.; Honjo, T. Current issues and perspectives in PD-1 blockade cancer immunotherapy. Int. J. Clin. Oncol. 2020, 25, 790–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nasta, F.; Laudisi, F.; Sambucci, M.; Rosado, M.M.; Pioli, C. Increased Foxp3+ regulatory T cells in poly(ADP-Ribose) polymerase-1 deficiency. J. Immunol. 2010, 184, 3470–3477. [Google Scholar] [CrossRef] [PubMed]
- Galindo-Campos, M.A.; Bedora-Faure, M.; Farres, J.; Lescale, C.; Moreno-Lama, L.; Martinez, C.; Martin-Caballero, J.; Ampurdanes, C.; Aparicio, P.; Dantzer, F.; et al. Coordinated signals from the DNA repair enzymes PARP-1 and PARP-2 promotes B-cell development and function. Cell Death Differ. 2019, 26, 2667–2681. [Google Scholar] [CrossRef]
- Navarro, J.; Gozalbo-Lopez, B.; Mendez, A.C.; Dantzer, F.; Schreiber, V.; Martinez, C.; Arana, D.M.; Farres, J.; Revilla-Nuin, B.; Bueno, M.F.; et al. PARP-1/PARP-2 double deficiency in mouse T cells results in faulty immune responses and T lymphomas. Sci. Rep. 2017, 7, 41962. [Google Scholar] [CrossRef] [Green Version]
- Szabó, É.; Kovács, I.; Grune, T.; Haczku, A.; Virág, L. PARP-1: A new player in the asthma field? Allergy 2011, 66, 811–814. [Google Scholar] [CrossRef]
- Ghonim, M.A.; Pyakurel, K.; Ibba, S.V.; Al-Khami, A.A.; Wang, J.; Rodriguez, P.; Rady, H.F.; El-Bahrawy, A.H.; Lammi, M.R.; Mansy, M.S.; et al. PARP inhibition by olaparib or gene knockout blocks asthma-like manifestation in mice by modulating CD4(+) T cell function. J. Transl. Med. 2015, 13, 225. [Google Scholar] [CrossRef] [Green Version]
- Scott, G.S.; Kean, R.B.; Mikheeva, T.; Fabis, M.J.; Mabley, J.G.; Szabó, C.; Hooper, D.C. The therapeutic effects of PJ34 [N-(6-oxo-5,6-dihydrophenanthridin-2-yl)-N,N-dimethylacetamide.HCl], a selective inhibitor of poly(ADP-ribose) polymerase, in experimental allergic encephalomyelitis are associated with immunomodulation. J. Pharmacol. Exp. Ther. 2004, 310, 1053–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiarugi, A. Inhibitors of poly(ADP-ribose) polymerase-1 suppress transcriptional activation in lymphocytes and ameliorate autoimmune encephalomyelitis in rats. Br. J. Pharmacol. 2002, 137, 761–770. [Google Scholar] [CrossRef] [Green Version]
- Valdor, R.; Schreiber, V.; Saenz, L.; Martínez, T.; Muñoz-Suano, A.; Dominguez-Villar, M.; Ramírez, P.; Parrilla, P.; Aguado, E.; García-Cózar, F.; et al. Regulation of NFAT by poly(ADP-ribose) polymerase activity in T cells. Mol. Immunol. 2008, 45, 1863–1871. [Google Scholar] [CrossRef]
- Zhang, P.; Maruyama, T.; Konkel, J.E.; Abbatiello, B.; Zamarron, B.; Wang, Z.Q.; Chen, W. PARP-1 controls immunosuppressive function of regulatory T cells by destabilizing Foxp3. PLoS ONE 2013, 8, e71590. [Google Scholar] [CrossRef] [Green Version]
- Luo, X.; Nie, J.; Wang, S.; Chen, Z.; Chen, W.; Li, D.; Hu, H.; Li, B. Poly(ADP-ribosyl)ation of FOXP3 protein mediated by PARP-1 regulates the function of regulatory T cells. J. Biol. Chem. 2016, 291, 1201. [Google Scholar] [CrossRef] [Green Version]
- Kunze, F.A.; Bauer, M.; Komuczki, J.; Lanzinger, M.; Gunasekera, K.; Hopp, A.K.; Lehmann, M.; Becher, B.; Müller, A.; Hottiger, M.O. ARTD1 in Myeloid Cells Controls the IL-12/18-IFN-γ Axis in a Model of Sterile Sepsis, Chronic Bacterial Infection, and Cancer. J. Immunol. 2019, 202, 1406–1416. [Google Scholar] [CrossRef]
- Shou, Q.; Fu, H.; Huang, X.; Yang, Y. PARP-1 controls NK cell recruitment to the site of viral infection. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Echeverri Tirado, L.C.; Ghonim, M.A.; Wang, J.; Al-Khami, A.A.; Wyczechowska, D.; Luu, H.H.; Kim, H.; Sanchez-Pino, M.D.; Yélamos, J.; Yassin, L.M.; et al. PARP-1 Is Critical for Recruitment of Dendritic Cells to the Lung in a Mouse Model of Asthma but Dispensable for Their Differentiation and Function. Mediators Inflamm. 2019, 2019, 1656484. [Google Scholar] [CrossRef] [Green Version]
- Cavone, L.; Aldinucci, A.; Ballerini, C.; Biagioli, T.; Moroni, F.; Chiarugi, A. PARP-1 inhibition prevents CNS migration of dendritic cells during EAE, suppressing the encephalitogenic response and relapse severity. Mult. Scler. 2011, 17, 794–807. [Google Scholar] [CrossRef]
- Moreno-Lama, L.; Galindo-Campos, M.A.; Martínez, C.; Comerma, L.; Vazquez, I.; Vernet-Tomas, M.; Ampurdanés, C.; Lutfi, N.; Martin-Caballero, J.; Dantzer, F.; et al. Coordinated signals from PARP-1 and PARP-2 are required to establish a proper T cell immune response to breast tumors in mice. Oncogene 2020, 39, 2835–2843. [Google Scholar] [CrossRef] [PubMed]
- Chacon-Cabrera, A.; Fermoselle, C.; Salmela, I.; Yelamos, J.; Barreiro, E. MicroRNA expression and protein acetylation pattern in respiratory and limb muscles of Parp-1(-/-) and Parp-2(-/-) mice with lung cancer cachexia. Biochim. Biophys. Acta 2015, 1850, 2530–2543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, S.; Xia, W.; Yamaguchi, H.; Wei, Y.; Chen, M.K.; Hsu, J.M.; Hsu, J.L.; Yu, W.H.; Du, Y.; Lee, H.H.; et al. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin. Cancer Res. 2017, 23, 3711–3720. [Google Scholar] [CrossRef] [Green Version]
- Navas, L.E.; Carnero, A. NAD(+) metabolism, stemness, the immune response, and cancer. Signal Transduct. Target Ther. 2021, 6, 2. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Demény, M.A.; Virág, L. The PARP Enzyme Family and the Hallmarks of Cancer Part 2: Hallmarks Related to Cancer Host Interactions. Cancers 2021, 13, 2057. https://doi.org/10.3390/cancers13092057
Demény MA, Virág L. The PARP Enzyme Family and the Hallmarks of Cancer Part 2: Hallmarks Related to Cancer Host Interactions. Cancers. 2021; 13(9):2057. https://doi.org/10.3390/cancers13092057
Chicago/Turabian StyleDemény, Máté A., and László Virág. 2021. "The PARP Enzyme Family and the Hallmarks of Cancer Part 2: Hallmarks Related to Cancer Host Interactions" Cancers 13, no. 9: 2057. https://doi.org/10.3390/cancers13092057
APA StyleDemény, M. A., & Virág, L. (2021). The PARP Enzyme Family and the Hallmarks of Cancer Part 2: Hallmarks Related to Cancer Host Interactions. Cancers, 13(9), 2057. https://doi.org/10.3390/cancers13092057