
The Ubiquitin Proteasome System in Genome Stability and Cancer


Abstract
:Simple Summary
Abstract
1. Introduction
2. The Ubiquitin Proteasome System
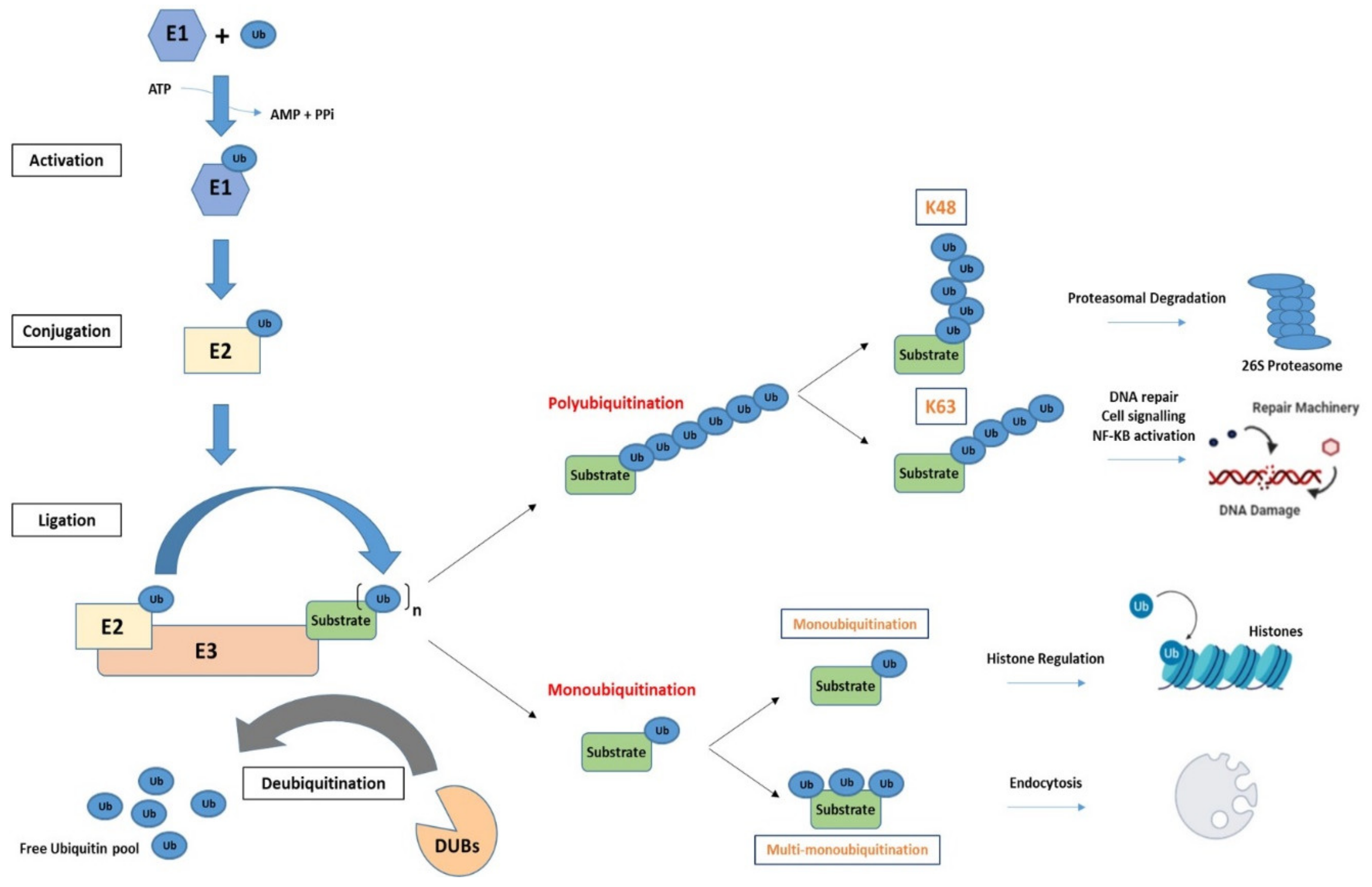
2.1. The Process of Ubiquitination
2.2. Ubiquitination Is a Diverse Modification
3. DNA Replication and Replicative Stress: UPS Surveillance of the Genome
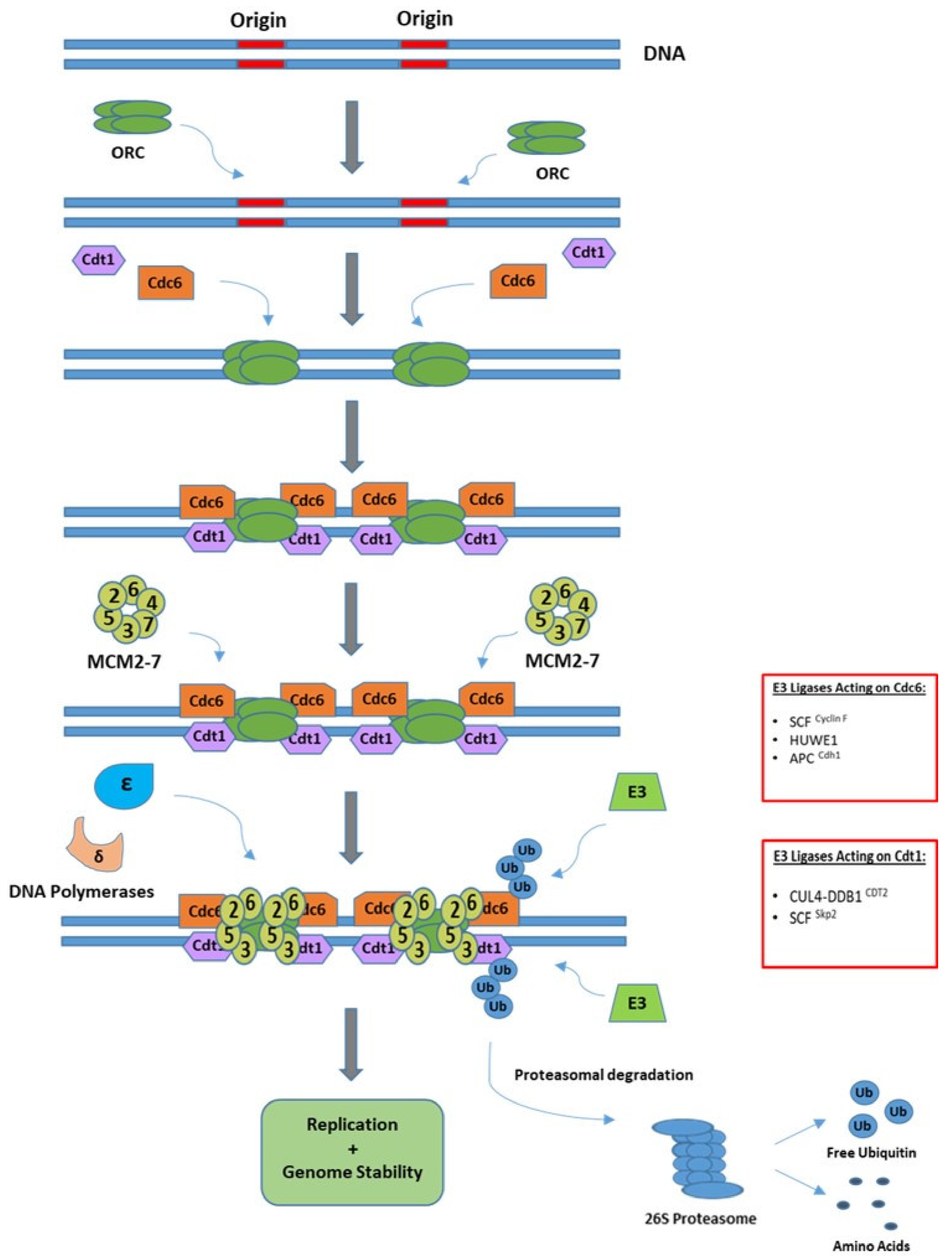
3.1. Initiation of Replication
3.2. Elongation and Termination of Replication
3.3. Ubiquitination at Stalled Replication Forks
3.3.1. Regulation of RPA
3.3.2. PCNA Ubiquitination
3.3.3. TRAIP-Mediated Regulation of Replisome Stability
3.3.4. R-Loop-Induced Stress
4. The UPS Facilitates the DNA Damage Response
4.1. UPS Regulation of ATR-Mediated Repair
4.2. UPS Regulation of ATM-Mediated Repair
5. Therapeutic Interventions
5.1. The Proteasome
5.2. MDM2
5.3. The Anaphase Promoting Complex
5.4. Cullin-RING Ligases
5.5. Hijacking an E3 Ligase
6. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nat. Cell Biol. 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [Green Version]
- Torgovnick, A.; Schumacher, B. DNA repair mechanisms in cancer development and therapy. Front. Genet. 2015, 6, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myung, J.; Kim, K.B.; Crews, C.M. The ubiquitin-proteasome pathway and proteasome inhibitors. Med. Res. Rev. 2001, 21, 245–273. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Meng, T.; Chen, L.; Wei, W.; Wang, P. The role of ubiquitination in tumorigenesis and targeted drug discovery. Signal Transduct. Target. Ther. 2020, 5, 1–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Ma, L.; Wang, B.; Liu, J.; Wei, W. E3 ubiquitin ligases in cancer and implications for therapies. Cancer Metastasis Rev. 2017, 36, 683–702. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, K.I.; Nakayama, K. Ubiquitin ligases: Cell-cycle control and cancer. Nat. Rev. Cancer 2006, 6, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Senft, D.; Qi, J.; Ronai, Z.A. Ubiquitin ligases in oncogenic transformation and cancer therapy. Nat. Rev. Cancer 2018, 18, 69–88. [Google Scholar] [CrossRef] [PubMed]
- Callis, J. The Ubiquitination Machinery of the Ubiquitin System. Arab. Book 2014, 12, e0174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amm, I.; Sommer, T.; Wolf, D.H. Protein quality control and elimination of protein waste: The role of the ubiquitin–proteasome system. Biochim. et Biophys. Acta (BBA)-Bioenerg. 2014, 1843, 182–196. [Google Scholar] [CrossRef] [Green Version]
- Akutsu, M.; Dikic, I.; Bremm, A. Ubiquitin chain diversity at a glance. J. Cell Sci. 2016, 129, 875–880. [Google Scholar] [CrossRef] [Green Version]
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef] [PubMed]
- Deshaies, R.J.; Joazeiro, C.A. RING Domain E3 Ubiquitin Ligases. Annu. Rev. Biochem. 2009, 78, 399–434. [Google Scholar] [CrossRef]
- Weber, J.; Polo, S.; Maspero, E. HECT E3 Ligases: A Tale With Multiple Facets. Front. Physiol. 2019, 10, 370. [Google Scholar] [CrossRef] [Green Version]
- Reiter, K.H.; Klevit, R.E. Characterization of RING-Between-RING E3 Ubiquitin Transfer Mechanisms. Methods Mol. Biol. 2018, 1844, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Ronai, Z.A. Monoubiquitination in proteasomal degradation. Proc. Natl. Acad. Sci. USA 2016, 113, 8894–8896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hicke, L. Protein regulation by monoubiquitin. Nat. Rev. Mol. Cell Biol. 2001, 2, 195–201. [Google Scholar] [CrossRef]
- Haglund, K.; Sigismund, S.; Polo, S.; Szymkiewicz, I.; Di Fiore, P.P.; Dikic, I. Multiple monoubiquitination of RTKs is sufficient for their endocytosis and degradation. Nat. Cell Biol. 2003, 5, 461–466. [Google Scholar] [CrossRef]
- Ohtake, F.; Tsuchiya, H. The emerging complexity of ubiquitin architecture. J. Biochem. 2016, 161, 125–133. [Google Scholar] [CrossRef] [Green Version]
- Wu-Baer, F.; Lagrazon, K.; Yuan, W.; Baer, R. The BRCA1/BARD1 Heterodimer Assembles Polyubiquitin Chains through an Unconventional Linkage Involving Lysine Residue K6 of Ubiquitin. J. Biol. Chem. 2003, 278, 34743–34746. [Google Scholar] [CrossRef] [Green Version]
- Acquaviva, C.; Pines, J. The anaphase-promoting complex/cyclosome: APC/C. J. Cell Sci. 2006, 119, 2401–2404. [Google Scholar] [CrossRef] [Green Version]
- Gatti, M.; Pinato, S.; Maiolica, A.; Rocchio, F.; Prato, M.G.; Aebersold, R.; Penengo, L. RNF168 Promotes Noncanonical K27 Ubiquitination to Signal DNA Damage. Cell Rep. 2015, 10, 226–238. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.-H.; Chen, Y.-H.; Huang, T.-Y. Ubiquitin-mediated regulation of autophagy. J. Biomed. Sci. 2019, 26, 1–12. [Google Scholar] [CrossRef]
- Yuan, W.-C.; Lee, Y.-R.; Lin, S.-Y.; Chang, L.-Y.; Tan, Y.P.; Hung, C.-C.; Kuo, J.-C.; Liu, C.-H.; Lin, M.-Y.; Xu, M.; et al. K33-Linked Polyubiquitination of Coronin 7 by Cul3-KLHL20 Ubiquitin E3 Ligase Regulates Protein Trafficking. Mol. Cell 2014, 54, 586–600. [Google Scholar] [CrossRef] [Green Version]
- Spit, M.; Rieser, E.; Walczak, H. Linear ubiquitination at a glance. J. Cell Sci. 2019, 132, jcs208512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.; Gan, W.; Su, S.; Hauenstein, A.V.; Fu, T.-M.; Brasher, B.; Schwerdtfeger, C.; Liang, A.C.; Xu, M.; Wei, W. K63-linked polyubiquitin chains bind to DNA to facilitate DNA damage repair. Sci. Signal. 2018, 11, eaar8133. [Google Scholar] [CrossRef] [Green Version]
- Meng, Y.; Liu, C.; Shen, L.; Zhou, M.; Liu, W.; Kowolik, C.; Campbell, J.L.; Zheng, L.; Shen, B. TRAF6 mediates human DNA2 polyubiquitination and nuclear localization to maintain nuclear genome integrity. Nucleic Acids Res. 2019, 47, 7564–7579. [Google Scholar] [CrossRef]
- Nishi, R. Balancing act: To be, or not to be ubiquitylated. Mutat. Res. Mol. Mech. Mutagen. 2017, 803–805, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Seeman, N.C. DNA in a material world. Nat. Cell Biol. 2003, 421, 427–431. [Google Scholar] [CrossRef] [PubMed]
- Bleichert, F.; Botchan, M.R.; Berger, J.M. Mechanisms for initiating cellular DNA replication. Science 2017, 355, eaah6317. [Google Scholar] [CrossRef] [Green Version]
- Yao, N.Y.; O’Donnell, M.E. Replication fork convergence at termination: A multistep process. Proc. Natl. Acad. Sci. USA 2017, 115, 237–239. [Google Scholar] [CrossRef] [Green Version]
- Tognetti, S.; Riera, A.; Speck, C. Switch on the engine: How the eukaryotic replicative helicase MCM2–7 becomes activated. Chromosoma 2014, 124, 13–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benson, T.W.; Weintraub, N.L.; Kim, H.W. A Single High-Fat Meal Provokes Pathological Erythrocyte Remodeling and Increases Myeloperoxidase Levels: Implications for Acute Coronary Syndrome Tyler. Lab Investig. 2018, 98, 1300–1310. [Google Scholar] [CrossRef] [PubMed]
- Petersen, B.O.; Wagener, C.; Marinoni, F.; Kramer, E.R.; Melixetian, M.; Denchi, E.L.; Gieffers, C.; Matteucci, C.; Peters, J.-M.; Helin, K. Cell cycle- and cell growth-regulated proteolysis of mammalian CDC6 is dependent on APC-CDH1. Genes Dev. 2000, 14, 2330–2343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, S.-M.; Zhang, Y.; Utani, K.; Fu, H.; Redon, C.E.; Marks, A.B.; Smith, O.K.; Redmond, C.J.; Baris, A.M.; Tulchinsky, D.A.; et al. The replication initiation determinant protein (RepID) modulates replication by recruiting CUL4 to chromatin. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clijsters, L.; Wolthuis, R. PIP-box-mediated degradation prohibits re-accumulation of Cdc6 during S phase. J. Cell Sci. 2014, 127, 1336–1345. [Google Scholar] [CrossRef] [Green Version]
- Walter, D.; Hoffmann, S.; Komseli, E.-S.; Rappsilber, J.; Gorgoulis, V.; Sørensen, C.S. SCFCyclin F-dependent degradation of CDC6 suppresses DNA re-replication. Nat. Commun. 2016, 7, 10530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, J.R.; Kow, E.; Nevis, K.R.; Lu, C.K.; Luce, K.S.; Zhong, Q.; Cook, J.G. Cdc6 Stability Is Regulated by the Huwe1 Ubiquitin Ligase after DNA Damage. Mol. Biol. Cell 2007, 18, 3340–3350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fok, K.L.; Bose, R.; Sheng, K.; Chang, C.-W.; Katz-Egorov, M.; Culty, M.; Su, S.; Yang, M.; Ruan, Y.C.; Chan, H.C.; et al. Huwe1 Regulates the Establishment and Maintenance of Spermatogonia by Suppressing DNA Damage Response. Endocrinology 2017, 158, 4000–4016. [Google Scholar] [CrossRef] [PubMed]
- Nishitani, H.; Sugimoto, N.; Roukos, V.; Nakanishi, Y.; Saijo, M.; Obuse, C.; Tsurimoto, T.; Nakayama, K.I.; Nakayama, K.; Fujita, M.; et al. Two E3 ubiquitin ligases, SCF-Skp2 and DDB1-Cul4, target human Cdt1 for proteolysis. EMBO J. 2006, 25, 1126–1136. [Google Scholar] [CrossRef] [Green Version]
- Arias, E.E.; Walter, J.C. PCNA functions as a molecular platform to trigger Cdt1 destruction and prevent re-replication. Nat. Cell Biol. 2005, 8, 84–90. [Google Scholar] [CrossRef] [PubMed]
- McGarry, T.J.; Kirschner, M.W. Geminin, an Inhibitor of DNA Replication, Is Degraded during Mitosis. Cell 1998, 93, 1043–1053. [Google Scholar] [CrossRef] [Green Version]
- Méndez, J.; Zou-Yang, X.; Kim, S.-Y.; Hidaka, M.; Tansey, W.P.; Stillman, B. Human Origin Recognition Complex Large Subunit Is Degraded by Ubiquitin-Mediated Proteolysis after Initiation of DNA Replication. Mol. Cell 2002, 9, 481–491. [Google Scholar] [CrossRef]
- Burgers, P.M.; Kunkel, T.A. Eukaryotic DNA Replication Fork. Annu. Rev. Biochem. 2017, 86, 417–438. [Google Scholar] [CrossRef]
- Dewar, J.M.; Walter, J.C. Mechanisms of DNA replication termination. Nat. Rev. Mol. Cell Biol. 2017, 18, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Maric, M.; Maculins, T.; De Piccoli, G.; Labib, K. Cdc48 and a ubiquitin ligase drive disassembly of the CMG helicase at the end of DNA replication. Science 2014, 346, 1253596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno, S.P.; Bailey, R.; Campion, N.; Herron, S.; Gambus, A. Polyubiquitylation drives replisome disassembly at the termination of DNA replication. Science 2014, 346, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Dewar, J.M.; Low, E.; Mann, M.; Räschle, M.; Walter, J.C. CRL2Lrr1promotes unloading of the vertebrate replisome from chromatin during replication termination. Genes Dev. 2017, 31, 275–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonneville, R.; Moreno, S.P.; Knebel, A.; Johnson, C.; Hastie, C.J.; Gartner, A.; Gambus, A.; Labib, K. CUL-2LRR-1 and UBXN-3 drive replisome disassembly during DNA replication termination and mitosis. Nat. Cell Biol. 2017, 19, 468–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dueva, R.; Iliakis, G. Replication protein A: Multifunctional protein with roles in DNA replication, repair and beyond. NAR Cancer 2020, 2, zcaa022. [Google Scholar] [CrossRef]
- Rageul, J.; Park, J.J.; Jo, U.; Weinheimer, A.S.; Vu, T.T.M.; Kim, H. Conditional degradation of SDE2 by the Arg/N-End rule pathway regulates stress response at replication forks. Nucleic Acids Res. 2019, 47, 3996–4010. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-C.; Wang, Y.; Hsu, R.; Giri, S.; Wopat, S.; Arif, M.K.; Chakraborty, A.; Prasanth, K.V.; Prasanth, S.G. PCNA-mediated stabilization of E3 ligase RFWD3 at the replication fork is essential for DNA replication. Proc. Natl. Acad. Sci. USA 2018, 115, 13282–13287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Mason, C.E.; Melnick, A. Genetic and epigenetic heterogeneity in acute myeloid leukemia. Curr. Opin. Genet. Dev. 2016, 36, 100–106. [Google Scholar] [CrossRef] [Green Version]
- Inano, S.; Sato, K.; Katsuki, Y.; Kobayashi, W.; Tanaka, H.; Nakajima, K.; Nakada, S.; Miyoshi, H.; Knies, K.; Takaori-Kondo, A.; et al. RFWD3-Mediated Ubiquitination Promotes Timely Removal of Both RPA and RAD51 from DNA Damage Sites to Facilitate Homologous Recombination. Mol. Cell 2017, 66, 622–634.e8. [Google Scholar] [CrossRef] [Green Version]
- Feeney, L.; Muñoz, I.M.; Lachaud, C.; Toth, R.; Appleton, P.L.; Schindler, D.; Rouse, J. RPA-Mediated Recruitment of the E3 Ligase RFWD3 Is Vital for Interstrand Crosslink Repair and Human Health. Mol. Cell 2017, 66, 610–621.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, H.; Mansour, S.; Reed, R.; Gillis, M.K.; Parent, B.; Liu, B.; Sztupinszki, Z.; Birkbak, N.; Szallasi, Z.; Elia, A.E.; et al. E3 ligase RFWD3 is a novel modulator of stalled fork stability in BRCA2-deficient cells. J. Cell Biol. 2020, 219. [Google Scholar] [CrossRef]
- Dubois, J.-C.; Yates, M.; Gaudreau-Lapierre, A.; Clément, G.; Cappadocia, L.; Gaudreau, L.; Zou, L.; Maréchal, A. A phosphorylation-and-ubiquitylation circuitry driving ATR activation and homologous recombination. Nucleic Acids Res. 2017, 45, 8859–8872. [Google Scholar] [CrossRef] [PubMed]
- Maréchal, A.; Li, J.-M.; Ji, X.Y.; Wu, C.-S.; Yazinski, S.A.; Nguyen, H.D.; Liu, S.; Jiménez, A.E.; Jin, J.; Zou, L. PRP19 Transforms into a Sensor of RPA-ssDNA after DNA Damage and Drives ATR Activation via a Ubiquitin-Mediated Circuitry. Mol. Cell 2014, 53, 235–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kannouche, P.L.; Wing, J.; Lehmann, A.R. Interaction of Human DNA Polymerase η with Monoubiquitinated PCNA. Mol. Cell 2004, 14, 491–500. [Google Scholar] [CrossRef]
- Motegi, A.; Liaw, H.-J.; Lee, K.-Y.; Roest, H.; Maas, A.; Wu, X.; Moinova, H.; Markowitz, S.; Ding, H.; Hoeijmakers, J.; et al. Polyubiquitination of proliferating cell nuclear antigen by HLTF and SHPRH prevents genomic instability from stalled replication forks. Proc. Natl. Acad. Sci. USA 2008, 105, 12411–12416. [Google Scholar] [CrossRef] [Green Version]
- Ciccia, A.; Nimonkar, A.V.; Hu, Y.; Hajdu, I.; Achar, Y.J.; Izhar, L.; Petit, S.A.; Adamson, B.; Yoon, J.C.; Kowalczykowski, S.C.; et al. Polyubiquitinated PCNA Recruits the ZRANB3 Translocase to Maintain Genomic Integrity after Replication Stress. Mol. Cell 2012, 47, 396–409. [Google Scholar] [CrossRef] [Green Version]
- Choe, K.N.; Nicolae, C.M.; Constantin, D.; Kawasawa, Y.I.; Delgado-Diaz, M.R.; De, S.; Freire, R.; Smits, V.A.; Moldovan, G. HUWE 1 interacts with PCNA to alleviate replication stress. EMBO Rep. 2016, 17, 874–886. [Google Scholar] [CrossRef] [Green Version]
- Coleman, K.E.; Huang, T.T. HUWE 1 comes to the rescue at stalled replication forks. EMBO Rep. 2016, 17, 781–782. [Google Scholar] [CrossRef]
- Kirchmaier, A.L. Ub-family modifications at the replication fork: Regulating PCNA-interacting components. FEBS Lett. 2011, 585, 2920–2928. [Google Scholar] [CrossRef] [Green Version]
- Wu, R.A.; Semlow, D.R.; Kamimae-Lanning, A.N.; Kochenova, O.V.; Chistol, G.; Hodskinson, M.R.; Amunugama, R.; Sparks, J.L.; Wang, M.; Deng, L.; et al. TRAIP is a master regulator of DNA interstrand crosslink repair. Nat. Cell Biol. 2019, 567, 267–272. [Google Scholar] [CrossRef]
- Larsen, N.B.; Gao, A.O.; Sparks, J.L.; Gallina, I.; Wu, R.A.; Mann, M.; Räschle, M.; Walter, J.C.; Duxin, J.P. Replication-Coupled DNA-Protein Crosslink Repair by SPRTN and the Proteasome in Xenopus Egg Extracts. Mol. Cell 2019, 73, 574–588.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonneville, R.; Bhowmick, R.; Hoffmann, S.; Mailand, N.; Hickson, I.D.; Labib, K. TRAIP drives replisome disassembly and mitotic DNA repair synthesis at sites of incomplete DNA replication. eLife 2019, 8. [Google Scholar] [CrossRef]
- Skourti-Stathaki, K.; Proudfoot, N.J. A double-edged sword: R loops as threats to genome integrity and powerful regulators of gene expression. Genes Dev. 2014, 28, 1384–1396. [Google Scholar] [CrossRef] [Green Version]
- Sahtoe, D.D.; Van Dijk, W.J.; Ekkebus, R.; Ovaa, H.; Sixma, T.K. BAP1/ASXL1 recruitment and activation for H2A deubiquitination. Nat. Commun. 2016, 7, 10292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klusmann, I.; Wohlberedt, K.; Magerhans, A.; Teloni, F.; Korbel, J.O.; Altmeyer, M.; Dobbelstein, M. Chromatin modifiers Mdm2 and RNF2 prevent RNA:DNA hybrids that impair DNA replication. Proc. Natl. Acad. Sci. USA 2018, 115, E11311–E11320. [Google Scholar] [CrossRef] [Green Version]
- Latif, C.; Harvey, S.H.; O’Connell, M.J. Ensuring the Stability of the Genome: DNA Damage Checkpoints. Sci. World J. 2001, 1, 684–702. [Google Scholar] [CrossRef] [Green Version]
- Maréchal, A.; Zou, L. DNA Damage Sensing by the ATM and ATR Kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef]
- Giglia-Mari, G.; Zotter, A.; Vermeulen, W. DNA Damage Response. Cold Spring Harb. Perspect. Biol. 2010, 3, a000745. [Google Scholar] [CrossRef]
- Donzelli, M.; Squatrito, M.; Ganoth, D.; Hershko, A.; Pagano, M.; Draetta, G.F. Dual mode of degradation of Cdc25 A phosphatase. EMBO J. 2002, 21, 4875–4884. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez, G.J.; Ronai, Z. Ubiquitin and SUMO systems in the regulation of mitotic checkpoints. Trends Biochem. Sci. 2006, 31, 324–332. [Google Scholar] [CrossRef] [Green Version]
- Eyers, P.A.; Keeshan, K.; Kannan, N. Tribbles in the 21st Century: The Evolving Roles of Tribbles Pseudokinases in Biology and Disease. Trends Cell Biol. 2017, 27, 284–298. [Google Scholar] [CrossRef] [Green Version]
- Frescas, D.; Pagano, M. Deregulated proteolysis by the F-box proteins SKP2 and β-TrCP: Tipping the scales of cancer. Nat. Rev. Cancer 2008, 8, 438–449. [Google Scholar] [CrossRef] [Green Version]
- Paull, T.T. Mechanisms of ATM Activation. Annu. Rev. Biochem. 2015, 84, 711–738. [Google Scholar] [CrossRef]
- Syed, A.; Tainer, J.A. The MRE11–RAD50–NBS1 Complex Conducts the Orchestration of Damage Signaling and Outcomes to Stress in DNA Replication and Repair. Annu. Rev. Biochem. 2018, 87, 263–294. [Google Scholar] [CrossRef]
- Uziel, T.; Lerenthal, Y.; Moyal, L.; Andegeko, Y.; Mittelman, L.; Shiloh, Y. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J. 2003, 22, 5612–5621. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Q.; Chen, J. Mechanism of p53 stabilization by ATM after DNA damage. Cell Cycle 2010, 9, 472–478. [Google Scholar] [CrossRef] [Green Version]
- Williams, A.B.; Schumacher, B. p53 in the DNA-Damage-Repair Process. Cold Spring Harb. Perspect. Med. 2016, 6, a026070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coster, G.; Goldberg, M. The cellular response to DNA damage: A focus on MDC1 and its interacting proteins. Nucleus 2010, 1, 166–178. [Google Scholar] [CrossRef]
- Bartocci, C.; Denchi, E.L. Put a RING on it: Regulation and inhibition of RNF8 and RNF168 RING finger E3 ligases at DNA damage sites. Front. Genet. 2013, 4, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakada, S. Opposing roles of RNF8/RNF168 and deubiquitinating enzymes in ubiquitination-dependent DNA double-strand break response signaling and DNA-repair pathway choice. J. Radiat. Res. 2016, 57, i33–i40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Halaby, M.-J.; Hakem, A.; Cardoso, R.; El Ghamrasni, S.; Harding, S.; Chan, N.; Bristow, R.; Sanchez, O.; Durocher, D.; et al. Rnf8 deficiency impairs class switch recombination, spermatogenesis, and genomic integrity and predisposes for cancer. J. Exp. Med. 2010, 207, 983–997. [Google Scholar] [CrossRef] [Green Version]
- Bohgaki, T.; Bohgaki, M.; Cardoso, R.; Panier, S.; Zeegers, D.; Li, L.; Stewart, G.S.; Sanchez, O.; Hande, M.P.; Durocher, D.; et al. Genomic Instability, Defective Spermatogenesis, Immunodeficiency, and Cancer in a Mouse Model of the RIDDLE Syndrome. PLoS Genet. 2011, 7, e1001381. [Google Scholar] [CrossRef]
- Luo, K.; Zhang, H.; Wang, L.; Yuan, J.; Lou, Z. Sumoylation of MDC1 is important for proper DNA damage response. EMBO J. 2012, 31, 3008–3019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, C.J.; Gibb, B.; Kwon, Y.; Sung, P.; Greene, E.C. Protein dynamics of human RPA and RAD51 on ssDNA during assembly and disassembly of the RAD51 filament. Nucleic Acids Res. 2017, 45, 749–761. [Google Scholar] [CrossRef] [Green Version]
- Galanty, Y.; Belotserkovskaya, R.; Coates, J.; Jackson, S.P. RNF4, a SUMO-targeted ubiquitin E3 ligase, promotes DNA double-strand break repair. Genes Dev. 2012, 26, 1179–1195. [Google Scholar] [CrossRef] [Green Version]
- Rigakos, G.; Razis, E. BRCAness: Finding the Achilles Heel in Ovarian Cancer. Oncologist 2012, 17, 956–962. [Google Scholar] [CrossRef] [Green Version]
- Trenner, A.; Sartori, A.A. Harnessing DNA Double-Strand Break Repair for Cancer Treatment. Front. Oncol. 2019, 9, 1388. [Google Scholar] [CrossRef] [PubMed]
- Kane, R.C.; Farrell, A.T.; Sridhara, R.; Pazdur, R. United States Food and Drug Administration Approval Summary: Bortezomib for the Treatment of Progressive Multiple Myeloma after One Prior Therapy. Clin. Cancer Res. 2006, 12, 2955–2960. [Google Scholar] [CrossRef] [Green Version]
- Kane, R.C.; Dagher, R.; Farrell, A.; Ko, C.-W.; Sridhara, R.; Justice, R.; Pazdur, R. Bortezomib for the Treatment of Mantle Cell Lymphoma. Clin. Cancer Res. 2007, 13, 5291–5294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herndon, T.M.; Deisseroth, A.; Kaminskas, E.; Kane, R.C.; Koti, K.M.; Rothmann, M.D.; Habtemariam, B.; Bullock, J.; Bray, J.D.; Hawes, J.; et al. U.S. Food and Drug Administration Approval: Carfilzomib for the Treatment of Multiple Myeloma. Clin. Cancer Res. 2013, 19, 4559–4563. [Google Scholar] [CrossRef] [Green Version]
- Shirley, M. Ixazomib: First Global Approval. Drugs 2016, 76, 405–411. [Google Scholar] [CrossRef]
- Ghobrial, I.M.; Vij, R.; Siegel, D.S.; Badros, A.Z.; Kaufman, J.L.; Raje, N.S.; Jakubowiak, A.J.; Savona, M.R.; Obreja, M.; Berdeja, J.G. A Phase Ib/II Study of Oprozomib in Patients with Advanced Multiple Myeloma and Waldenström Macroglobulinemia. Clin. Cancer Res. 2019, 25, 4907–4916. [Google Scholar] [CrossRef]
- Spencer, A.; Harrison, S.; Zonder, J.; Badros, A.; Laubach, J.; Bergin, K.; Khot, A.; Zimmerman, T.; Chauhan, D.; Levin, N.; et al. A phase 1 clinical trial evaluating marizomib, pomalidomide and low-dose dexamethasone in relapsed and refractory multiple myeloma (NPI-0052-107): Final study results. Br. J. Haematol. 2018, 180, 41–51. [Google Scholar] [CrossRef] [Green Version]
- Besse, A.; Besse, L.; Kraus, M.; Mendez-Lopez, M.; Bader, J.; Xin, B.-T.; De Bruin, G.; Maurits, E.; Overkleeft, H.S.; Driessen, C. Proteasome Inhibition in Multiple Myeloma: Head-to-Head Comparison of Currently Available Proteasome Inhibitors. Cell Chem. Biol. 2019, 26, 340–351.e3. [Google Scholar] [CrossRef] [PubMed]
- Neri, P.; Ren, L.; Gratton, K.; Stebner, E.; Johnson, J.; Klimowicz, A.; Duggan, P.; Tassone, P.; Mansoor, A.; Stewart, U.A.; et al. Bortezomib-induced “BRCAness” sensitizes multiple myeloma cells to PARP inhibitors. Blood 2011, 118, 6368–6379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivlin, N.; Brosh, R.; Oren, M.; Rotter, V. Mutations in the p53 Tumor Suppressor Gene: Important Milestones at the Various Steps of Tumorigenesis. Genes Cancer 2011, 2, 466–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.W.; Seong, M.W.; Jeon, Y.J.; Chung, C.H. Ubiquitin E3 ligases controlling p53 stability. Anim. Cells Syst. 2012, 16, 173–182. [Google Scholar] [CrossRef]
- Vassilev, L.T. Small-molecule antagonists of p53-MDM2 binding: Research tools and potential therapeutics. Cell Cycle 2004, 3, 417–419. [Google Scholar] [CrossRef] [Green Version]
- Popowicz, G.M.; Dömling, A.; Holak, T.A. The Structure-Based Design of Mdm2/Mdmx–p53 Inhibitors Gets Serious. Angew. Chem. Int. Ed. 2011, 50, 2680–2688. [Google Scholar] [CrossRef] [PubMed]
- Roxburgh, P.; Hock, A.K.; Dickens, M.P.; Mezna, M.; Fischer, P.M.; Vousden, K.H. Small molecules that bind the Mdm2 RING stabilize and activate p53. Carcinogenesis 2012, 33, 791–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konopleva, M.; Martinelli, G.; Daver, N.; Papayannidis, C.; Wei, A.; Higgins, B.; Ott, M.; Mascarenhas, J.; Andreeff, M. MDM2 inhibition: An important step forward in cancer therapy. Leukemia 2020, 34, 2858–2874. [Google Scholar] [CrossRef]
- Harper, J.W.; Burton, J.L.; Solomon, M.J. The anaphase-promoting complex: It’s not just for mitosis any more. Genes Dev. 2002, 16, 2179–2206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sackton, K.L.; Dimova, N.; Zeng, X.; Tian, W.; Zhang, M.; Sackton, T.B.; Meaders, J.; Pfaff, K.L.; Sigoillot, F.; Yu, H.; et al. Synergistic blockade of mitotic exit by two chemical inhibitors of the APC/C. Nat. Cell Biol. 2014, 514, 646–649. [Google Scholar] [CrossRef] [Green Version]
- Swords, R.T.; Kelly, K.R.; Smith, P.G.; Garnsey, J.J.; Mahalingam, D.; Medina, E.; Oberheu, K.; Padmanabhan, S.; O’Dwyer, M.; Nawrocki, S.T.; et al. Inhibition of NEDD8-activating enzyme: A novel approach for the treatment of acute myeloid leukemia. Blood 2010, 115, 3796–3800. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.J.; Milhollen, M.A.; Smith, P.G.; Narayanan, U.; Dutta, A. NEDD8-Targeting Drug MLN4924 Elicits DNA Rereplication by Stabilizing Cdt1 in S Phase, Triggering Checkpoint Activation, Apoptosis, and Senescence in Cancer Cells. Cancer Res. 2010, 70, 10310–10320. [Google Scholar] [CrossRef] [Green Version]
- Gstaiger, M.; Jordan, R.; Lim, M.; Catzavelos, C.; Mestan, J.; Slingerland, J.; Krek, W. Skp2 is oncogenic and overexpressed in human cancers. Proc. Natl. Acad. Sci. USA 2001, 98, 5043–5048. [Google Scholar] [CrossRef] [Green Version]
- Hershko, D.D. Oncogenic properties and prognostic implications of the ubiquitin ligase Skp2 in cancer. Cancer 2008, 112, 1415–1424. [Google Scholar] [CrossRef]
- Chan, C.-H.; Morrow, J.K.; Li, C.-F.; Gao, Y.; Jin, G.; Moten, A.; Stagg, L.J.; Ladbury, J.E.; Cai, Z.; Xu, D.; et al. Pharmacological Inactivation of Skp2 SCF Ubiquitin Ligase Restricts Cancer Stem Cell Traits and Cancer Progression. Cell 2013, 154, 556–568. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Du, L.; Ren, Y.; Liu, X.; Jiao, Q.; Cui, D.; Wen, M.; Wang, C.; Wei, G.; Wang, Y.; et al. SKP2 promotes breast cancer tumorigenesis and radiation tolerance through PDCD4 ubiquitination. J. Exp. Clin. Cancer Res. 2019, 38, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, S.; Lee, B.-H. Chemically Induced Cellular Proteolysis: An Emerging Therapeutic Strategy for Undruggable Targets. Mol. Cells 2018, 41, 933–942. [Google Scholar] [PubMed]
- Sun, X.; Gao, H.; Yang, Y.; He, M.; Wu, Y.; Song, Y.; Tong, Y.; Rao, Y. PROTACs: Great opportunities for academia and industry. Signal Transduct. Target. Ther. 2019, 4, 1–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girardini, M.; Maniaci, C.; Hughes, S.J.; Testa, A.; Ciulli, A. Cereblon versus VHL: Hijacking E3 ligases against each other using PROTACs. Bioorganic Med. Chem. 2019, 27, 2466–2479. [Google Scholar] [CrossRef] [PubMed]
- Hines, J.; Lartigue, S.; Dong, H.; Qian, Y.; Crews, C.M. MDM2-Recruiting PROTAC Offers Superior, Synergistic Antiproliferative Activity via Simultaneous Degradation of BRD4 and Stabilization of p53. Cancer Res. 2019, 79, 251–262. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.; Khoo, R.; Peh, K.M.; Teo, J.; Chang, S.C.; Ng, S.; Beilhartz, G.L.; Melnyk, R.A.; Johannes, C.W.; Brown, C.J.; et al. bioPROTACs as versatile modulators of intracellular therapeutic targets including proliferating cell nuclear antigen (PCNA). Proc. Natl. Acad. Sci. USA 2020, 117, 5791–5800. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Inhibitor | Cancer Type | Clinical Stage | Reference/ Trial ID |
|---|---|---|---|---|
| 20S Proteasome (Beta-5 subunit) | Bortezomib | MM, MCL | Approved | [93,94] |
| Carfilzomib | MM | Approved | [95] | |
| Ixazomib | MM | Approved | [96] | |
| Oprozomib | MM | Phase Ib/II | [97] | |
| Marizomib | MM | Phase I | [98] | |
| DIPG | Phase I | NCT04341311 | ||
| MDM2 | Idasanutlin | Glioblastoma | Phase III | NCT03345095 |
| Breast Cancer | Phase I/II | NCT03566485 | ||
| AML/ALL | Phase I/II | NCT04029688 | ||
| AMG-232 | Sarcoma | Phase Ib | NCT03217266 | |
| Lymphoma | Phase I | NCT04502394 | ||
| MM | Phase I | NCT03031730 | ||
| AML | Phase Ib | NCT04190550 | ||
| HDM201 | CRC | Phase I | NCT03714955 | |
| APG-115 | AML | Phase Ib | NCT04275518 | |
| CRL | MLN4924 (Pevonedistat) | MM | Phase I | NCT03770260 |
| AML | Phase Ib | NCT01814826 | ||
| AML | Phase I/II | NCT03862157 | ||
| AML/MDS | Phase I | NCT03772925 | ||
| NSCLC | Phase II | NCT03965689 | ||
| Lymphoma | Phase I | NCT03323034 | ||
| ALL/NHL | Phase I | NCT03349281 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morgan, J.J.; Crawford, L.J. The Ubiquitin Proteasome System in Genome Stability and Cancer. Cancers 2021, 13, 2235. https://doi.org/10.3390/cancers13092235
Morgan JJ, Crawford LJ. The Ubiquitin Proteasome System in Genome Stability and Cancer. Cancers. 2021; 13(9):2235. https://doi.org/10.3390/cancers13092235
Chicago/Turabian StyleMorgan, Jonathan J., and Lisa J. Crawford. 2021. "The Ubiquitin Proteasome System in Genome Stability and Cancer" Cancers 13, no. 9: 2235. https://doi.org/10.3390/cancers13092235
APA StyleMorgan, J. J., & Crawford, L. J. (2021). The Ubiquitin Proteasome System in Genome Stability and Cancer. Cancers, 13(9), 2235. https://doi.org/10.3390/cancers13092235