
Approaches of the Innate Immune System to Ameliorate Adaptive Immunotherapy for B-Cell Non-Hodgkin Lymphoma in Their Microenvironment

Simple Summary
Abstract
1. Introduction
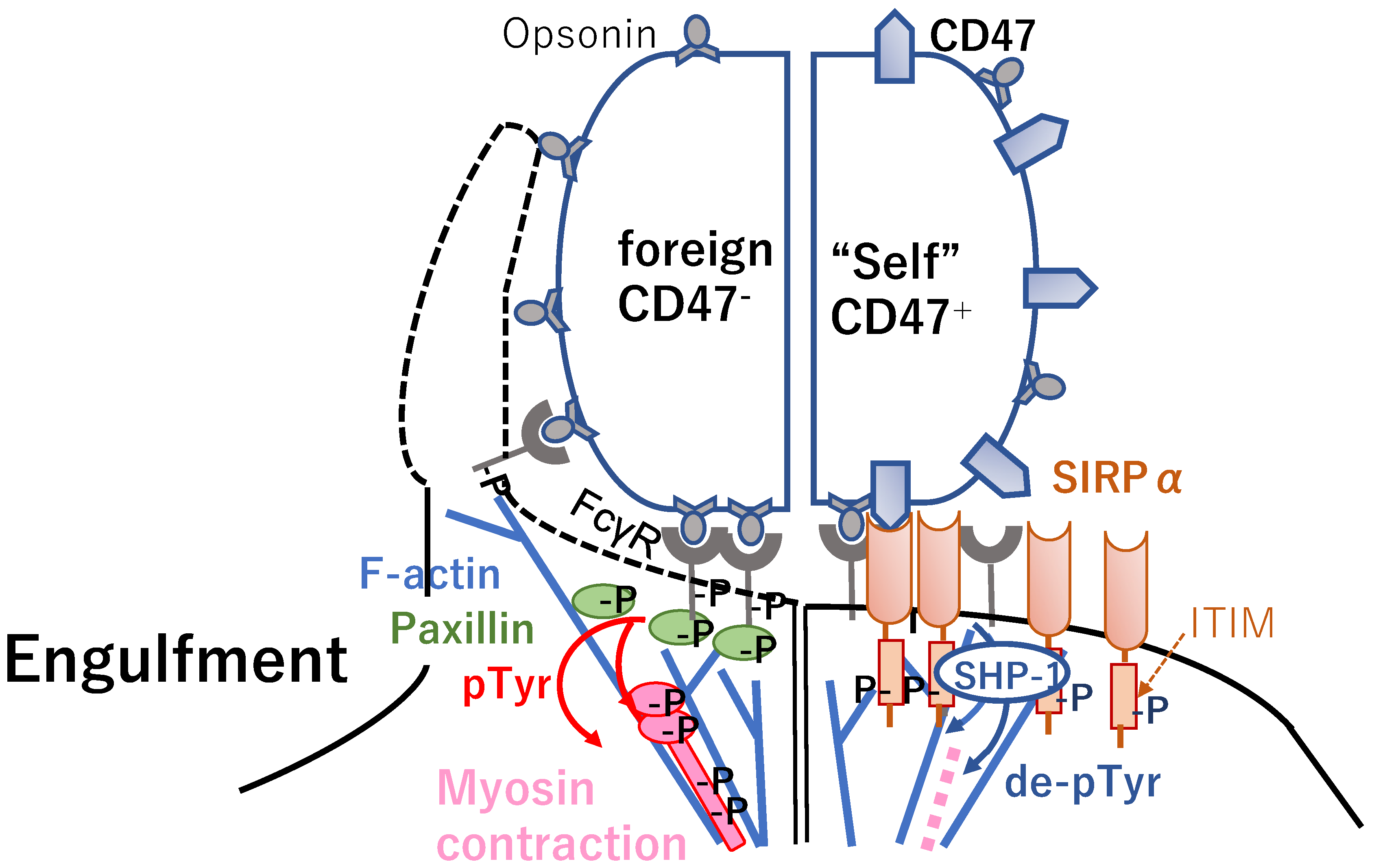
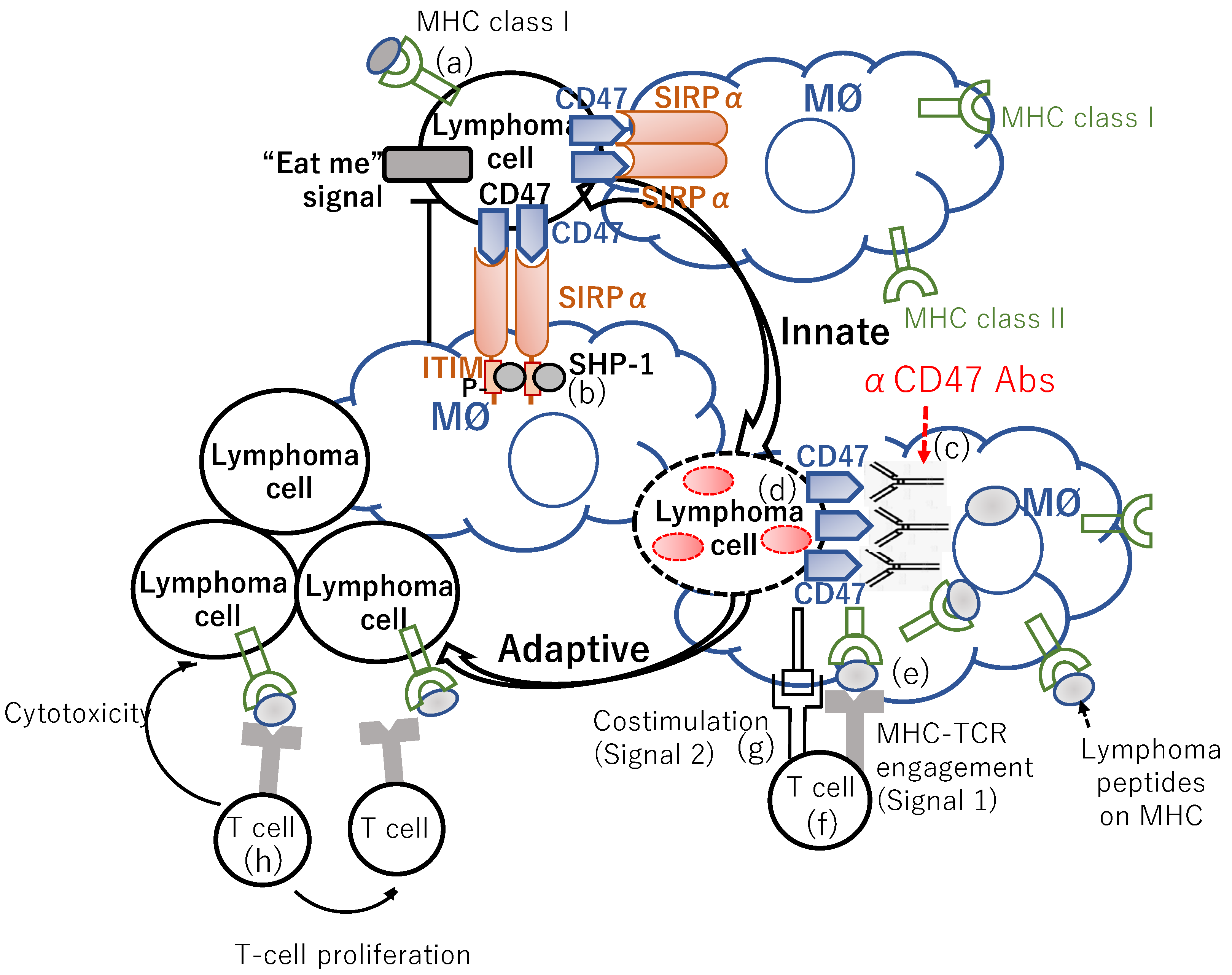
2. “Don’t Eat Me” Signal Blockade Anti-CD47 Antibody Is an Innate Immune Checkpoint Inhibitor
2.1. Anti-CD47 Antibodies
2.2. SIRPα-Fc-CD40 Ligand Agonist Bridging Macrophage-Mediated Tumor Cell Phagocytosis to Antigen-Presenting Cell (APC) Activation and Antigen Presentation
3. Other TAM-Based Therapeutic Approach
3.1. Colony-Stimulating Factor-1 Receptor (CSF-1R) Inhibitor
3.2. Anti-Lymphangiogenic Effect of Lenalidomide in MCL
3.3. Trabectedin and Its Analogue Lurbinetedin
3.4. Artesunate
4. Vaccination
4.1. Idiotype (Id) Vaccination for FL Patients
4.2. Vaccination for MCL Patients
5. Oligodeoxynucleotides Containing Unmethylated CpG Motifs (CpG-ODN)
5.1. CpG-ODN
5.2. CpG-ODN Combined with an Anti-CD20 Antibody Rituximab
5.3. CpG-ODN Combined with Anti-OX40/CD134 and Anti-CTLA4/CD152 MoAbs
5.4. CpG-ODN Combined with BTK Inhibitor Ibrutinib
5.5. CpG-ODN Combined with Low-Dose Radiation
5.6. CpG-ODN Combined with Radioimmunotherapy

{kind=link}
{kind=link}
{kind=link}
| Combination | ODN Class | Name of CpG (Route) | Phase | Object | N | Efficacy (%) | IR (%) | AEs | Authors [Ref] |
|---|---|---|---|---|---|---|---|---|---|
| CpG-ODN alone | B(=Type K) | PF-3512676 [=CPG7909] (IV) | P1 | R/R NHL | 23 | No clinical responses | IgG elevation in 5 pts | Anemia Thrombocytopenia Neutropenia Transient lymphopenia Dyspnea Chills/rigors Nausea/vomiting Fever Hypotension Fatigue Pain Intravenous catheter discomfort ALT increased Hyperglycemia Proteinuria | Link, and Weiner et al. [134] |
| CpG + R | B(=Type K) | PF-3512676 [=CPG7909] (IV or SC) | P1 | Relapsed CD20+ B-NHL | 50 | 12OR (24%) | NA | Systemic flu-like syndromes #1–1, Fatigue or rigors Pyrexia Malaise Injection-site reactions #2–1 Neutropenia Pain in limbs Generalized pruritus Peripheral neuropathy Sjögren’s-like syndrome #3 | Leonard, Link, and Weiner et al. [137] |
| CpG + R | B(=Type K) | 1018 ISS (SC) | P1 | R/R FL | 20 | 6OR [1CRu+5PR] (32%)+ 13SD Median PFS in responding pts: 12 mos | Induction of IFN-α/β—inducible genes | Allergic reaction #4 Headache Fatigue Atypical pneumonia | Friedberg, et al. [138] |
| CpG + R | B(=Type K) | 1018 ISS (SC) | P2 | R/R FL | 23 | 11OR [CR/CRu or PR] (48%) + 6SD (26%) Median PFS: 8.8 mos | T-cell and MØ infiltration in injection sites IFN-γ—secretion increased from T cells in PB (25%) ADCC increased (35%) Increased expression of IFN-α/β-inducible genes (IFIT2, CXCL10, CCL2) (≥60%) Increased CXCL10 expression in PB in responders | Fatigue Rituximab infusion reaction Erythema at the injection sites | Friedberg, et al. [139] |
| CpG + αOX40 + αCTLA4 | B(=Type K) | CpG1826 (IT) | mice | A20 tumor cells | NA | Immune protection from tumor re-challenge over 100 days | IFN-γ-producing, memory T cell increased Treg decreased | NA | Houot and Levy et al. [140] |
| CpG + ibrutinib | B(=Type K) | CpG1826 (IT) | mice | H11 and B3750 tumor cells | NA | Tumor regression in at the distant site | % increase in tumor-specific IFN-γ-producing, memory T cells | NA | Sagiv-Barfi and Levy et al. [141] |
| CpG + LD RT | B(=Type K) | PF-3512676 (IT) | P1/2 | Relapsed low-grade B-NHL | 15 | 1CR, 2PRs | Induced CD137/4-1BB expression and increased intracellular IFN-γ, IL -2, and TNF in memory CD8+ T cells in PB Treg induction in PB and tumor | Injection site reaction #2–2 Flu-like reactions Arthralgia Myalgia | Brody and Levy et al. [32] |
| CpG + LD RT (2 Gy x 2) | C | SD-101 (IT) | P1/2 | Untreated iNHL | 29 | 26 reductions, including 1CR, 7PRs | Increased T cell and decreased Tfh and Treg in injected tumors | Flu-like systemic reactions #1–2 Myalgia Fever Nausea/vomiting Diarrhea Injection-site reaction #2–3 Neutropenia Confusion Decreased appetite Night sweating Thrombocytopenia | Frank and Levy et al. [152] |
| CpG + RIT | B(=Type K) | PF-3512676 [=CPG7909] (IV) | P1 | R/R CD20+B-NHL | 30 | 28OR (93%) 19CRs (63%) Median PFS: 43 mos DOR: 35 mos Median TTP: 40.5 mos | Decrease in IL-10 and TNF-α and increase in IL-1β in serum | Reversible myelosuppression (due to RIT) Pain on bulk neck lymph node (after infusion of CpG 7909) | Witzig and Weiner et al. [158] |
6. CpG-ODN Containing Virus-like Particle (VLP) and CpG ODN Combined with Anti-PD-1 Ab
7. Vaccination with Immunogenic Cell Death Tumors
7.1. Vaccination with Autologous Lymphoma Cell-Loaded DCs
7.2. IFN-α—Conditioned DC (IFN-DC) Vaccination
7.3. Immunomodulatory Drugs Combined with Interferon-α-DC-Based Vaccination
8. STING Agonists (STINGa)
9. Agonist Anti–4-1BB/CD137 Antibody Monotherapy
9.1. Anti–4-1BB/CD137 Antibody Monotherapy
9.2. 4-1BB/CD137 Agonist with Rituximab
10. OX40/CD134 Agonistic MoAb with Rituximab
11. Conclusions
Funding
Conflicts of Interest
References
- Sehn, L.H.; Salles, G. Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2021, 384, 842–858. [Google Scholar] [CrossRef]
- Leonard, J.P.; Trneny, M.; Izutsu, K.; Fowler, N.H.; Hong, X.; Zhu, J.; Zhang, H.; Offner, F.; Scheliga, A.; Nowakowski, G.S.; et al. AUGMENT: A Phase III Study of Lenalidomide Plus Rituximab Versus Placebo Plus Rituximab in Relapsed or Refractory Indolent Lymphoma. J. Clin. Oncol. 2019, 37, 1188–1199. [Google Scholar] [CrossRef] [PubMed]
- Gopal, A.K.; Kahl, B.S.; de Vos, S.; Wagner-Johnston, N.D.; Schuster, S.J.; Jurczak, W.J.; Flinn, I.W.; Flowers, C.R.; Martin, P.; Viardot, A.; et al. PI3Kdelta inhibition by idelalisib in patients with relapsed indolent lymphoma. N. Engl. J. Med. 2014, 370, 1008–1018. [Google Scholar] [CrossRef] [PubMed]
- Dreyling, M.; Jurczak, W.; Jerkeman, M.; Silva, R.S.; Rusconi, C.; Trneny, M.; Offner, F.; Caballero, D.; João, C.; Witzens-Harig, M.; et al. Ibrutinib versus temsirolimus in patients with relapsed or refractory mantle-cell lymphoma: An international, randomised, open-label, phase 3 study. Lancet 2015, 387, 770–778. [Google Scholar] [CrossRef]
- Rolf, J.; Bell, S.E.; Kovesdi, D.; Janas, M.L.; Soond, D.R.; Webb, L.M.C.; Santinelli, S.; Saunders, T.; Hebeis, B.; Killeen, N.; et al. Phosphoinositide 3-Kinase Activity in T Cells Regulates the Magnitude of the Germinal Center Reaction. J. Immunol. 2010, 185, 4042–4052. [Google Scholar] [CrossRef] [PubMed]
- Dubovsky, J.A.; Beckwith, K.A.; Natarajan, G.; Woyach, J.A.; Jaglowski, S.; Zhong, Y.; Hessler, J.D.; Liu, T.-M.; Chang, B.Y.; Larkin, K.M.; et al. Ibrutinib is an irreversible molecular inhibitor of ITK driving a Th1-selective pressure in T lymphocytes. Blood 2013, 122, 2539–2549. [Google Scholar] [CrossRef]
- Casulo, C.; Byrtek, M.; Dawson, K.L.; Zhou, X.; Farber, C.M.; Flowers, C.R.; Hainsworth, J.D.; Maurer, M.J.; Cerhan, J.R.; Link, B.; et al. Early Relapse of Follicular Lymphoma After Rituximab Plus Cyclophosphamide, Doxorubicin, Vincristine, and Prednisone Defines Patients at High Risk for Death: An Analysis from the National LymphoCare Study. J. Clin. Oncol. 2015, 33, 2516–2522. [Google Scholar] [CrossRef]
- Jurinovic, V.; Kridel, R.; Staiger, A.M.; Szczepanowski, M.; Horn, H.; Dreyling, M.H.; Rosenwald, A.; Ott, G.; Klapper, W.; Zelenetz, A.D.; et al. Clinicogenetic risk models predict early progression of follicular lymphoma after first-line immunochemotherapy. Blood 2016, 128, 1112–1120. [Google Scholar] [CrossRef]
- Casulo, C.; Nastoupil, L.; Fowler, N.H.; Friedberg, J.W.; Flowers, C.R. Unmet needs in the first-line treatment of follicular lymphoma. Ann. Oncol. 2017, 28, 2094–2106. [Google Scholar] [CrossRef]
- Freeman, C.L.; Kridel, R.; Moccia, A.A.; Savage, K.J.; Villa, D.R.; Scott, D.W.; Gerrie, A.S.; Ferguson, D.; Cafferty, F.; Slack, G.W.; et al. Early progression after bendamustine-rituximab is associated with high risk of transformation in advanced stage follicular lymphoma. Blood 2019, 134, 761–764. [Google Scholar] [CrossRef]
- Mozas, P.; Nadeu, F.; Rivas-Delgado, A.; Rivero, A.; Garrote, M.; Balagué, O.; González-Farré, B.; Veloza, L.; Baumann, T.; Giné, E.; et al. Patterns of change in treatment, response, and outcome in patients with follicular lymphoma over the last four decades: A single-center experience. Blood Cancer J. 2020, 10, 31. [Google Scholar] [CrossRef]
- Morschhauser, F.; Tilly, H.; Chaidos, A.; McKay, P.; Phillips, T.; Assouline, S.; Batlevi, C.L.; Campbell, P.; Ribrag, V.; Damaj, G.L.; et al. Tazemetostat for patients with relapsed or refractory follicular lymphoma: An open-label, single-arm, multicentre, phase 2 trial. Lancet Oncol. 2020, 21, 1433–1442. [Google Scholar] [CrossRef]
- Cortelazzo, S.; Ponzoni, M.; Ferreri, A.J.M.; Dreyling, M. Mantle cell lymphoma. Crit. Rev. Oncol. Hematol. 2020, 153, 103038. [Google Scholar] [CrossRef]
- Forstpointner, R.; Unterhalt, M.; Dreyling, M.; Böck, H.-P.; Repp, R.; Wandt, H.; Pott, C.; Seymour, J.F.; Metzner, B.; Hänel, A.; et al. Maintenance therapy with rituximab leads to a significant prolongation of response duration after salvage therapy with a combination of rituximab, fludarabine, cyclophosphamide, and mitoxantrone (R-FCM) in patients with recurring and refractory follicular and mantle cell lymphomas: Results of a prospective randomized study of the German Low Grade Lymphoma Study Group (GLSG). Blood 2006, 108, 4003–4008. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.J.; Svoboda, J.; Chong, E.A.; Nasta, S.D.; Mato, A.R.; Anak, Ö.; Brogdon, J.L.; Pruteanu-Malinici, I.; Bhoj, V.; Landsburg, D.; et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N. Engl. J. Med. 2017, 377, 2545–2554. [Google Scholar] [CrossRef] [PubMed]
- Abramson, J.S.; Palomba, M.L.; Gordon, L.I.; Lunning, M.A.; Wang, M.; Arnason, J.; Mehta, A.; Purev, E.; Maloney, D.G.; Andreadis, C.; et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): A multicentre seamless design study. Lancet 2020, 396, 839–852. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Dudley, M.E.; Kassim, S.H.; Somerville, R.P.; Carpenter, R.O.; Stetler-Stevenson, M.; Yang, J.C.; Phan, G.Q.; Hughes, M.S.; Sherry, R.M.; et al. Chemotherapy-Refractory Diffuse Large B-Cell Lymphoma and Indolent B-Cell Malignancies Can Be Effectively Treated with Autologous T Cells Expressing an Anti-CD19 Chimeric Antigen Receptor. J. Clin. Oncol. 2015, 33, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Kochenderfer, J.N.; Somerville, R.P.; Lu, T.; Shi, V.; Bot, A.; Rossi, J.; Xue, A.; Goff, S.L.; Yang, J.C.; Sherry, R.M.; et al. Lymphoma Remissions Caused by Anti-CD19 Chimeric Antigen Receptor T Cells Are Associated with High Serum Interleukin-15 Levels. J. Clin. Oncol. 2017, 35, 1803–1813. [Google Scholar] [CrossRef] [PubMed]
- Locke, F.L.; Ghobadi, A.; Jacobson, C.A.; Miklos, D.B.; Lekakis, L.J.; Oluwole, O.O.; Lin, Y.; Braunschweig, I.; Hill, B.T.; Timmerman, J.M.; et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): A single-arm, multicentre, phase 1–2 trial. Lancet Oncol. 2019, 20, 31–42. [Google Scholar] [CrossRef]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Cappell, K.M.; Sherry, R.M.; Yang, J.C.; Goff, S.L.; Vanasse, D.A.; McIntyre, L.; Rosenberg, S.A.; Kochenderfer, J.N. Long-Term Follow-Up of Anti-CD19 Chimeric Antigen Receptor T-Cell Therapy. J. Clin. Oncol. 2020, 38, 3805–3815. [Google Scholar] [CrossRef] [PubMed]
- Chong, E.A.; Alanio, C.; Svoboda, J.; Nasta, S.D.; Landsburg, D.J.; Lacey, S.F.; Ruella, M.; Bhattacharyya, S.; Wherry, E.J.; Schuster, S.J. Pembrolizumab for B-cell lymphomas relapsing after or refractory to CD19-directed CAR T-cell therapy. Blood 2021. [Google Scholar] [CrossRef]
- Crump, M.; Neelapu, S.S.; Farooq, U.; Neste, E.V.D.; Kuruvilla, J.; Westin, J.; Link, B.K.; Hay, A.; Cerhan, J.R.; Zhu, L.; et al. Outcomes in refractory diffuse large B-cell lymphoma: Results from the international SCHOLAR-1 study. Blood 2017, 130, 1800–1808. [Google Scholar] [CrossRef]
- Hay, K.A.; Hanafi, L.-A.; Li, D.; Gust, J.; Liles, W.C.; Wurfel, M.M.; López, J.A.; Chen, J.; Chung, D.; Harju-Baker, S.; et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor–modified T-cell therapy. Blood 2017, 130, 2295–2306. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Gutierrez, C.; Locke, F.L.; Komanduri, K.V.; Lin, Y.; Jain, N.; Daver, N.; et al. Chimeric antigen receptor T-cell therapy—Assessment and management of toxicities. Nat. Rev. Clin. Oncol. 2017, 15, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Levy, R.; Ganjoo, K.N.; Leonard, J.P.; Vose, J.M.; Flinn, I.; Ambinder, R.F.; Connors, J.M.; Berinstein, N.L.; Belch, A.R.; Bartlett, N.; et al. Active Idiotypic Vaccination Versus Control Immunotherapy for Follicular Lymphoma. J. Clin. Oncol. 2014, 32, 1797–1803. [Google Scholar] [CrossRef]
- Ruan, J.; Martin, P.; Shah, B.; Schuster, S.J.; Smith, S.M.; Furman, R.R.; Christos, P.J.; Rodriguez, A.; Svoboda, J.; Lewis, J.; et al. Lenalidomide plus Rituximab as Initial Treatment for Mantle-Cell Lymphoma. N. Engl. J. Med. 2015, 373, 1835–1844. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Advani, R.; Flinn, I.; Popplewell, L.; Forero, A.; Bartlett, N.L.; Ghosh, N.; Kline, J.; Roschewski, M.; LaCasce, A.; Collins, G.P.; et al. CD47 Blockade by Hu5F9-G4 and Rituximab in Non-Hodgkin’s Lymphoma. N. Engl. J. Med. 2018, 379, 1711–1721. [Google Scholar] [CrossRef]
- Ansell, S.M.; Maris, M.B.; Lesokhin, A.M.; Chen, R.W.; Flinn, I.W.; Sawas, A.; Minden, M.D.; Villa, D.; Percival, M.-E.M.; Advani, A.S.; et al. Phase I Study of the CD47 Blocker TTI-621 in Patients with Relapsed or Refractory Hematologic Malignancies. Clin. Cancer Res. 2021, 27, 2190–2199. [Google Scholar] [CrossRef] [PubMed]
- Brody, J.D.; Ai, W.Z.; Czerwinski, D.K.; Torchia, J.A.; Levy, M.; Advani, R.H.; Kim, Y.H.; Hoppe, R.T.; Knox, S.J.; Shin, L.K.; et al. In Situ Vaccination with a TLR9 Agonist Induces Systemic Lymphoma Regression: A Phase I/II Study. J. Clin. Oncol. 2010, 28, 4324–4332. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Cencini, E.; Fabbri, A.; Sicuranza, A.; Gozzetti, A.; Bocchia, M. The Role of Tumor-Associated Macrophages in Hematologic Malignancies. Cancers 2021, 13, 3597. [Google Scholar] [CrossRef]
- Hanna, R.N.; Cekic, C.; Sag, D.; Tacke, R.; Thomas, G.D.; Nowyhed, H.; Herrley, E.; Rasquinha, N.; McArdle, S.; Wu, R.; et al. Patrolling monocytes control tumor metastasis to the lung. Science 2015, 350, 985–990. [Google Scholar] [CrossRef]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Cheng, P.; Condamine, T.; Mony, S.; Languino, L.; McCaffrey, J.C.; Hockstein, N.; Guarino, M.; Masters, G.; Penman, E.; et al. CD45 Phosphatase Inhibits STAT3 Transcription Factor Activity in Myeloid Cells and Promotes Tumor-Associated Macrophage Differentiation. Immunity 2016, 44, 303–315. [Google Scholar] [CrossRef] [PubMed]
- Franklin, R.A.; Liao, W.; Sarkar, A.; Kim, M.V.; Bivona, M.R.; Liu, K.; Pamer, E.G.; Li, M.O. The cellular and molecular origin of tumor-associated macrophages. Science 2014, 344, 921–925. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Xue, J.; Schmidt, S.V.; Sander, J.; Draffehn, A.; Krebs, W.; Quester, I.; De Nardo, D.; Gohel, T.D.; Emde, M.; Schmidleithner, L.; et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 2014, 40, 274–288. [Google Scholar] [CrossRef] [PubMed]
- Mills, C.D. Anatomy of a Discovery: M1 and M2 Macrophages. Front. Immunol. 2015, 6, 212. [Google Scholar] [CrossRef]
- Ricketts, T.D.; Prieto-Dominguez, N.; Gowda, P.S.; Ubil, E. Mechanisms of Macrophage Plasticity in the Tumor Environment: Manipulating Activation State to Improve Outcomes. Front. Immunol. 2021, 12, 642285. [Google Scholar] [CrossRef]
- Khodadoust, M.S.; Olsson, N.; Wagar, L.; Haabeth, O.A.W.; Chen, B.; Swaminathan, K.; Rawson, K.; Liu, C.L.; Steiner, D.; Lund, P.; et al. Antigen presentation profiling reveals recognition of lymphoma immunoglobulin neoantigens. Nature 2017, 543, 723–727. [Google Scholar] [CrossRef]
- Khodadoust, M.S.; Olsson, N.; Chen, B.; Sworder, B.; Shree, T.; Liu, C.L.; Zhang, L.; Czerwinski, D.K.; Davis, M.M.; Levy, R.; et al. B-cell lymphomas present immunoglobulin neoantigens. Blood 2019, 133, 878–881. [Google Scholar] [CrossRef] [PubMed]
- Segal, N.H.; Logan, T.F.; Hodi, F.S.; McDermott, D.; Melero, I.; Hamid, O.; Schmidt, H.; Robert, C.; Sileni, V.C.; Ascierto, P.A.; et al. Results from an Integrated Safety Analysis of Urelumab, an Agonist Anti-CD137 Monoclonal Antibody. Clin. Cancer Res. 2016, 23, 1929–1936. [Google Scholar] [CrossRef] [PubMed]
- Gopal, A.K.; Levy, R.; Houot, R.; Patel, S.P.; Popplewell, L.; Jacobson, C.; Mu, X.J.; Deng, S.; Ching, K.A.; Chen, Y.; et al. First-in-Human Study of Utomilumab, a 4-1BB/CD137 Agonist, in Combination with Rituximab in Patients with Follicular and Other CD20(+) Non-Hodgkin Lymphomas. Clin. Cancer Res. 2020, 26, 2524–2534. [Google Scholar] [CrossRef] [PubMed]
- Turaj, A.H.; Cox, K.L.; Penfold, C.A.; French, R.R.; Mockridge, C.I.; Willoughby, J.E.; Tutt, A.L.; Griffiths, J.; Johnson, P.W.M.; Glennie, M.J.; et al. Augmentation of CD134 (OX40)-dependent NK anti-tumour activity is dependent on antibody cross-linking. Sci. Rep. 2018, 8, 2278. [Google Scholar] [CrossRef] [PubMed]
- Sallets, A.; Robinson, S.; Kardosh, A.; Levy, R. Enhancing immunotherapy of STING agonist for lymphoma in preclinical models. Blood Adv. 2018, 2, 2230–2241. [Google Scholar] [CrossRef] [PubMed]
- Kharitonenkov, A.; Chen, Z.; Sures, I.; Wang, H.; Schilling, J.; Ullrich, A. A family of proteins that inhibit signalling through tyrosine kinase receptors. Nature 1997, 386, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Fujioka, Y.; Matozaki, T.; Noguchi, T.; Iwamatsu, A.; Yamao, T.; Takahashi, N.; Tsuda, M.; Takada, T.; Kasuga, M. A novel membrane glycoprotein, SHPS-1, that binds the SH2-domain-containing protein tyrosine phosphatase SHP-2 in response to mitogens and cell adhesion. Mol. Cell. Biol. 1996, 16, 6887–6899. [Google Scholar] [CrossRef]
- Veillette, A.; Thibaudeau, E.; Latour, S. High Expression of Inhibitory Receptor SHPS-1 and Its Association with Protein-tyrosine Phosphatase SHP-1 in Macrophages. J. Biol. Chem. 1998, 273, 22719–22728. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Lagenaur, C.F.; Narayanan, V. Integrin-associated Protein Is a Ligand for the P84 Neural Adhesion Molecule. J. Biol. Chem. 1999, 274, 559–562. [Google Scholar] [CrossRef] [PubMed]
- Hall, A. Rho GTPases and the Actin Cytoskeleton. Science 1998, 279, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Strzelecka, A.; Pyrzynska, B.; Kwiatkowska, K.; Sobota, A. Syk kinase, tyrosine-phosphorylated proteins and actin filaments accumulate at forming phagosomes during Fc gamma receptor-mediated phagocytosis. Cell Motil. Cytoskel. 1997, 38, 287–296. [Google Scholar] [CrossRef]
- Tsai, R.K.; Discher, D.E. Inhibition of “self” engulfment through deactivation of myosin-II at the phagocytic synapse between human cells. J. Cell Biol. 2008, 180, 989–1003. [Google Scholar] [CrossRef]
- Tsuda, M.; Matozaki, T.; Fukunaga, K.; Fujioka, Y.; Imamoto, A.; Noguchi, T.; Takada, T.; Yamao, T.; Takeda, H.; Ochi, F.; et al. Integrin-mediated Tyrosine Phosphorylation of SHPS-1 and Its Association with SHP-2. J. Biol. Chem. 1998, 273, 13223–13229. [Google Scholar] [CrossRef] [PubMed]
- Vernon-Wilson, E.F.; Kee, W.-J.; Willis, A.C.; Barclay, A.N.; Simmons, D.L.; Brown, M.H. CD47 is a ligand for rat macrophage membrane signal regulatory protein SIRP (OX41) and human SIRPα 1. Eur. J. Immunol. 2000, 30, 2130–2137. [Google Scholar] [CrossRef] [PubMed]
- Kant, A.M.; De, P.; Peng, X.; Yi, T.; Rawlings, D.J.; Kim, J.S.; Durden, D.L. SHP-1 regulates Fcgamma receptor-mediated phagocytosis and the activation of RAC. Blood 2002, 100, 1852–1859. [Google Scholar] [CrossRef]
- Greenberg, S.; Chang, P.; Silverstein, S.C. Tyrosine Phosphorylation of the Gamma-Subunit of Fc(Gamma)-Receptors, P72(Syk), and Paxillin during Fc-Receptor-Mediated Phagocytosis in Macrophages. J. Biol. Chem. 1994, 269, 3897–3902. [Google Scholar] [CrossRef]
- Chao, M.P.; Alizadeh, A.A.; Tang, C.; Myklebust, J.H.; Varghese, B.; Gill, S.; Jan, M.; Cha, A.C.; Chan, C.K.; Tan, B.T.; et al. Anti-CD47 Antibody Synergizes with Rituximab to Promote Phagocytosis and Eradicate Non-Hodgkin Lymphoma. Cell 2010, 142, 699–713. [Google Scholar] [CrossRef]
- Liu, J.; Wang, L.; Zhao, F.; Tseng, S.; Narayanan, C.; Shura, L.; Willingham, S.; Howard, M.; Prohaska, S.; Volkmer, J.; et al. Pre-Clinical Development of a Humanized Anti-CD47 Antibody with Anti-Cancer Therapeutic Potential. PLoS ONE 2015, 10, e0137345. [Google Scholar] [CrossRef]
- Hatherley, D.; Graham, S.; Turner, J.; Harlos, K.; Stuart, D.I.; Barclay, A.N. Paired Receptor Specificity Explained by Structures of Signal Regulatory Proteins Alone and Complexed with CD47. Mol. Cell 2008, 31, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Petrova, P.S.; Viller, N.N.; Wong, M.; Pang, X.; Lin, G.H.; Dodge, K.; Chai, V.; Chen, H.; Lee, V.; House, V.; et al. TTI-621 (SIRPalphaFc): A CD47-Blocking Innate Immune Checkpoint Inhibitor with Broad Antitumor Activity and Minimal Erythrocyte Binding. Clin. Cancer Res. 2017, 23, 1068–1079. [Google Scholar] [CrossRef]
- Bennett, S.R.; Carbone, F.R.; Karamalis, F.; Flavell, R.A.; Miller, J.F.; Heath, W.R. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature 1998, 393, 478–480. [Google Scholar] [CrossRef]
- Schoenberger, S.P.; Toes, R.; Van Der Voort, E.I.H.; Offringa, R.; Melief, C.J.M. T-cell help for cytotoxic T lymphocytes is mediated by CD40–CD40L interactions. Nature 1998, 393, 480–483. [Google Scholar] [CrossRef]
- O’Connell, P.J.; Morelli, A.E.; Logar, A.J.; Thomson, A.W. Phenotypic and Functional Characterization of Mouse Hepatic CD8α+ Lymphoid-Related Dendritic Cells. J. Immunol. 2000, 165, 795–803. [Google Scholar] [CrossRef]
- Delamarre, L.; Holcombe, H.; Mellman, I. Presentation of Exogenous Antigens on Major Histocompatibility Complex (MHC) Class I and MHC Class II Molecules Is Differentially Regulated during Dendritic Cell Maturation. J. Exp. Med. 2003, 198, 111–122. [Google Scholar] [CrossRef]
- Yasumi, T.; Katamura, K.; Yoshioka, T.; Meguro, T.-A.; Nishikomori, R.; Heike, T.; Inobe, M.; Kon, S.; Uede, T.; Nakahata, T. Differential Requirement for the CD40-CD154 Costimulatory Pathway during Th Cell Priming by CD8α+and CD8α−Murine Dendritic Cell Subsets. J. Immunol. 2004, 172, 4826–4833. [Google Scholar] [CrossRef]
- Byrne, K.T.; Vonderheide, R.H. CD40 Stimulation Obviates Innate Sensors and Drives T Cell Immunity in Cancer. Cell Rep. 2016, 15, 2719–2732. [Google Scholar] [CrossRef] [PubMed]
- de Silva, S.; Fromm, G.; Shuptrine, C.W.; Johannes, K.; Patel, A.; Yoo, K.J.; Huang, K.; Schreiber, T.H. CD40 Enhances Type I Interferon Responses Downstream of CD47 Blockade, Bridging Innate and Adaptive Immunity. Cancer Immunol. Res. 2020, 8, 230–245. [Google Scholar] [CrossRef] [PubMed]
- Valero, J.G.; Matas-Céspedes, A.; Arenas, F.; Rodriguez, V.; Carreras, J.; Serrat, N.; Guerrero-Hernández, M.; Yahiaoui, A.; Balagué, O.; Martin, S.; et al. The receptor of the colony-stimulating factor-1 (CSF-1R) is a novel prognostic factor and therapeutic target in follicular lymphoma. Leukemia 2021, 35, 2635–2649. [Google Scholar] [CrossRef]
- Papin, A.; Tessoulin, B.; Bellanger, C.; Moreau, A.; Le Bris, Y.; Maisonneuve, H.; Moreau, P.; Touzeau, C.; Amiot, M.; Deceunynck, C.; et al. CSF1R and BTK inhibitions as novel strategies to disrupt the dialog between mantle cell lymphoma and macrophages. Leukemia 2019, 33, 2442–2453. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Herzog, B.H.; Sheng, M.; Fu, J.; McDaniel, J.M.; Ruan, J.; Xia, L. Lenalidomide Inhibits Lymphangiogenesis in Preclinical Models of Mantle Cell Lymphoma. Cancer Res. 2013, 73, 7254–7264. [Google Scholar] [CrossRef] [PubMed]
- Schoppmann, S.F.; Birner, P.; Stöckl, J.; Kalt, R.; Ullrich, R.; Caucig, C.; Kriehuber, E.; Nagy, K.; Alitalo, K.; Kerjaschki, D. Tumor-Associated Macrophages Express Lymphatic Endothelial Growth Factors and Are Related to Peritumoral Lymphangiogenesis. Am. J. Pathol. 2002, 161, 947–956. [Google Scholar] [CrossRef]
- Kerjaschki, D. The crucial role of macrophages in lymphangiogenesis. J. Clin. Investig. 2005, 115, 2316–2319. [Google Scholar] [CrossRef]
- Maruyama, K.; Ii, M.; Cursiefen, C.; Jackson, D.G.; Keino, H.; Tomita, M.; Van Rooijen, N.; Takenaka, H.; D’Amore, P.A.; Stein-Streilein, J.; et al. Inflammation-induced lymphangiogenesis in the cornea arises from CD11b-positive macrophages. J. Clin. Investig. 2005, 115, 2363–2372. [Google Scholar] [CrossRef] [PubMed]
- Yona, S.; Kim, K.-W.; Wolf, Y.; Mildner, A.; Varol, D.; Breker, M.; Strauss-Ayali, D.; Viukov, S.; Guilliams, M.; Misharin, A.; et al. Fate Mapping Reveals Origins and Dynamics of Monocytes and Tissue Macrophages under Homeostasis. Immunity 2013, 38, 79–91. [Google Scholar] [CrossRef]
- Banerjee, P.; Zhang, R.; Ivan, C.; Galletti, G.; Clise-Dwyer, K.; Barbaglio, F.; Scarfo’, L.; Aracil, M.; Klein, C.; Wierda, W.; et al. Trabectedin Reveals a Strategy of Immunomodulation in Chronic Lymphocytic Leukemia. Cancer Immunol. Res. 2019, 7, 2036–2051. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Kang, K.; Giannopoulou, E.; Qiao, Y.; Kang, K.; Kim, G.; Park-Min, K.-H.; Ivashkiv, L.B. Type I interferons and the cytokine TNF cooperatively reprogram the macrophage epigenome to promote inflammatory activation. Nat. Immunol. 2017, 18, 1104–1116. [Google Scholar] [CrossRef]
- Majety, M.; Runza, V.; Lehmann, C.; Hoves, S.; Ries, C.H. A drug development perspective on targeting tumor-associated myeloid cells. FEBS J. 2017, 285, 763–776. [Google Scholar] [CrossRef]
- Belgiovine, C.; Bello, E.; Liguori, M.; Craparotta, I.; Mannarino, L.; Paracchini, L.; Beltrame, L.; Marchini, S.; Galmarini, C.M.; Mantovani, A.; et al. Lurbinectedin reduces tumour-associated macrophages and the inflammatory tumour microenvironment in preclinical models. Br. J. Cancer 2017, 117, 628–638. [Google Scholar] [CrossRef]
- Zyad, A.; Tilaoui, M.; Jaafari, A.; Oukerrou, M.A.; Mouse, H.A. More insights into the pharmacological effects of artemisinin. Phytother. Res. 2017, 32, 216–229. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, R.I.; Saad, S.T.O.; Azambuja, H. Artesunate Switches Monocytes to an Inflammatory Phenotype with the Ability to Kill Leukemic Cells. Int. J. Mol. Sci. 2021, 22, 608. [Google Scholar] [CrossRef] [PubMed]
- Timmerman, J.M.; Czerwinski, D.K.; Davis, T.A.; Hsu, F.J.; Benike, C.; Hao, Z.M.; Taidi, B.; Rajapaksa, R.; Caspar, C.B.; Okada, C.Y.; et al. Idiotype-pulsed dendritic cell vaccination for B-cell lymphoma: Clinical and immune responses in 35 patients. Blood 2002, 99, 1517–1526. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.J.; Neelapu, S.S.; Gause, B.L.; Janik, J.E.; Muggia, F.; Gockerman, J.P.; Winter, J.N.; Flowers, C.R.; Nikcevich, D.A.; Sotomayor, E.M.; et al. Vaccination with Patient-Specific Tumor-Derived Antigen in First Remission Improves Disease-Free Survival in Follicular Lymphoma. J. Clin. Oncol. 2011, 29, 2787–2794. [Google Scholar] [CrossRef]
- Cheson, B.D.; Horning, S.J.; Coiffier, B.; Shipp, M.A.; Fisher, R.I.; Connors, J.M.; Lister, T.A.; Vose, J.; Grillo-López, A.; Hagenbeek, A.; et al. Report of an international workshop to standardize response criteria for non-Hodgkin’s lymphomas. NCI Sponsored International Working Group. J. Clin. Oncol. 1999, 17, 1244. [Google Scholar] [CrossRef]
- Jahrsdörfer, B.; Hartmann, G.; Racila, E.; Jackson, W.; Mühlenhoff, L.; Meinhardt, G.; Endres, S.; Link, B.; Krieg, A.M.; Weiner, G. CpG DNA increases primary malignant B cell expression of costimulatory molecules and target antigens. J. Leukoc. Biol. 2001, 69, 81–88. [Google Scholar]
- Frank, M.J.; Khodadoust, M.S.; Czerwinski, D.K.; Haabeth, O.A.; Chu, M.P.; Miklos, D.B.; Advani, R.H.; Alizadeh, A.A.; Gupta, N.K.; Maeda, L.S.; et al. Autologous tumor cell vaccine induces antitumor T cell immune responses in patients with mantle cell lymphoma: A phase I/II trial. J. Exp. Med. 2020, 217, e20191712. [Google Scholar] [CrossRef]
- Kurtz, D.M.; Green, M.R.; Bratman, S.V.; Scherer, F.; Liu, C.L.; Kunder, C.A.; Takahashi, K.; Glover, C.; Keane, C.; Kihira, S.; et al. Noninvasive monitoring of diffuse large B-cell lymphoma by immunoglobulin high-throughput sequencing. Blood 2015, 125, 3679–3687. [Google Scholar] [CrossRef]
- Wolfl, M.; Kuball, J.; Ho, W.Y.; Nguyen, H.; Manley, T.J.; Bleakley, M.; Greenberg, P.D. Activation-induced expression of CD137 permits detection, isolation, and expansion of the full repertoire of CD8+ T cells responding to antigen without requiring knowledge of epitope specificities. Blood 2007, 110, 201–210. [Google Scholar] [CrossRef]
- Hoster, E.; Dreyling, M.; Klapper, W.; Gisselbrecht, C.; Van Hoof, A.; Kluin-Nelemans, J.C.; Pfreundschuh, M.; Reiser, M.; Metzner, B.; Einsele, H.; et al. A new prognostic index (MIPI) for patients with advanced-stage mantle cell lymphoma. Blood 2008, 111, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Finn, O.J. Human Tumor Antigens Yesterday, Today, and Tomorrow. Cancer Immunol. Res. 2017, 5, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.D.; Pope, B.; Murray, A.; Esdale, W.; Sze, D.M.; Gibson, J.; Ho, P.J.; Hart, D.; Joshua, D. Dendritic cells from patients with myeloma are numerically normal but functionally defective as they fail to up-regulate CD80 (B7-1) expression after huCD40LT stimulation because of inhibition by transforming growth factor-beta1 and interleukin-10. Blood 2001, 98, 2992–2998. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, G.; Weiner, G.J.; Krieg, A.M. CpG DNA: A potent signal for growth, activation, and maturation of human dendritic cells. Proc. Natl. Acad. Sci. USA 1999, 96, 9305–9310. [Google Scholar] [CrossRef]
- Kadowaki, N.; Antonenko, S.; Liu, Y.J. Distinct CpG DNA and polyinosinic-polycytidylic acid double-stranded RNA, respectively, stimulate CD11c- type 2 dendritic cell precursors and CD11c+ dendritic cells to produce type I IFN. J. Immunol. 2001, 166, 2291–2295. [Google Scholar] [CrossRef]
- Krug, A.; Towarowski, A.; Britsch, S.; Rothenfusser, S.; Hornung, V.; Bals, R.; Giese, T.; Engelmann, H.; Endres, S.; Krieg, A.M.; et al. Toll-like receptor expression reveals CpG DNA as a unique microbial stimulus for plasmacytoid dendritic cells which synergizes with CD40 ligand to induce high amounts of IL-12. Eur. J. Immunol. 2001, 31, 3026–3037. [Google Scholar] [CrossRef]
- Kadowaki, N.; Ho, S.; Antonenko, S.; de Waal Malefyt, R.; Kastelein, R.A.; Bazan, F.; Liu, Y.-J. Subsets of Human Dendritic Cell Precursors Express Different Toll-like Receptors and Respond to Different Microbial Antigens. J. Exp. Med. 2001, 194, 863–870. [Google Scholar] [CrossRef]
- Krieg, A.M.; Yi, A.-K.; Matson, S.; Waldschmidt, T.J.; Bishop, G.A.; Teasdale, R.; Koretzky, G.A.; Klinman, D.M. CpG motifs in bacterial DNA trigger direct B-cell activation. Nature 1995, 374, 546–549. [Google Scholar] [CrossRef] [PubMed]
- Davis, H.L.; Weeratna, R.; Waldschmidt, T.J.; Tygrett, L.; Schorr, J.; Krieg, A.M. CpG DNA is a potent enhancer of specific immunity in mice immunized with recombinant hepatitis B surface antigen. J. Immunol. 1998, 160, 870–876. [Google Scholar] [PubMed]
- Kobayashi, H.; Horner, A.A.; Takabayashi, K.; Nguyen, M.D.; Huang, E.; Cinman, N.; Raz, E. Immunostimulatory DNA pre-priming: A novel approach for prolonged Th1-biased immunity. Cell Immunol. 1999, 198, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, G.; Krieg, A.M. Mechanism and Function of a Newly Identified CpG DNA Motif in Human Primary B Cells. J. Immunol. 2000, 164, 944–953. [Google Scholar] [CrossRef] [PubMed]
- Krieg, A.M. CpG DNA: A pathogenic factor in systemic lupus erythematosus? J. Clin. Immunol. 1995, 15, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Jakob, T.; Walker, P.S.; Krieg, A.M.; Udey, M.C.; Vogel, J.C. Activation of cutaneous dendritic cells by CpG-containing oligodeoxynucleotides: A role for dendritic cells in the augmentation of Th1 responses by immunostimulatory DNA. J. Immunol. 1998, 161, 3042–3049. [Google Scholar]
- Sparwasser, T.; Koch, E.S.; Vabulas, R.M.; Heeg, K.; Lipford, G.B.; Ellwart, J.W.; Wagner, H. Bacterial DNA and immunostimulatory CpG oligonucleotides trigger maturation and activation of murine dendritic cells. Eur. J. Immunol. 1998, 28, 2045–2054. [Google Scholar] [CrossRef]
- Krug, A.; Rothenfusser, S.; Hornung, V.; Jahrsdorfer, B.; Blackwell, S.; Ballas, Z.K.; Endres, S.; Krieg, A.M.; Hartmann, G. Identification of CpG oligonucleotide sequences with high induction of IFN-alpha/beta in plasmacytoid dendritic cells. Eur. J. Immunol. 2001, 31, 2154–2163. [Google Scholar] [CrossRef]
- Scheiermann, J.; Klinman, D.M. Clinical evaluation of CpG oligonucleotides as adjuvants for vaccines targeting infectious diseases and cancer. Vaccine 2014, 32, 6377–6389. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Rothenfusser, S.; Britsch, S.; Krug, A.; Jahrsdörfer, B.; Giese, T.; Endres, S.; Hartmann, G. Quantitative Expression of Toll-Like Receptor 1–10 mRNA in Cellular Subsets of Human Peripheral Blood Mononuclear Cells and Sensitivity to CpG Oligodeoxynucleotides. J. Immunol. 2002, 168, 4531–4537. [Google Scholar] [CrossRef] [PubMed]
- Bernasconi, N.L.; Onai, N.; Lanzavecchia, A.; Cotta, C.V.; Zhang, Z.; Kim, H.-G.; Klug, C.A. A role for Toll-like receptors in acquired immunity: Up-regulation of TLR9 by BCR triggering in naive B cells and constitutive expression in memory B cells. Blood 2003, 101, 4500–4504. [Google Scholar] [CrossRef]
- Suzuki, Y.; Wakita, D.; Chamoto, K.; Narita, Y.; Tsuji, T.; Takeshima, T.; Gyobu, H.; Kawarada, Y.; Kondo, S.; Akira, S.; et al. Liposome-Encapsulated CpG Oligodeoxynucleotides as a Potent Adjuvant for Inducing Type 1 Innate Immunity. Cancer Res. 2004, 64, 8754–8760. [Google Scholar] [CrossRef]
- Bauer, S.; Kirschning, C.J.; Hacker, H.; Redecke, V.; Hausmann, S.; Akira, S.; Wagner, H.; Lipford, G.B. Human TLR9 confers responsiveness to bacterial DNA via species-specific CpG motif recognition. Proc. Natl. Acad. Sci. USA 2001, 98, 9237–9242. [Google Scholar] [CrossRef]
- Chu, R.S.; Targoni, O.S.; Krieg, A.M.; Lehmann, P.V.; Harding, C.V. CpG Oligodeoxynucleotides Act as Adjuvants that Switch on T Helper 1 (Th1) Immunity. J. Exp. Med. 1997, 186, 1623–1631. [Google Scholar] [CrossRef]
- Roman, M.; Martin-Orozco, E.; Goodman, J.S.; Nguyen, M.-D.; Sato, Y.; Ronaghy, A.; Kornbluth, R.; Richman, D.D.; Carson, D.A.; Raz, E. Immunostimulatory DNA sequences function as T helper-1-promoting adjuvants. Nat. Med. 1997, 3, 849–854. [Google Scholar] [CrossRef] [PubMed]
- Brazolot Millan, C.L.; Weeratna, R.; Krieg, A.M.; Siegrist, C.A.; Davis, H.L. CpG DNA can induce strong Th1 humoral and cell-mediated immune responses against hepatitis B surface antigen in young mice. Proc. Natl. Acad. Sci. USA 1998, 95, 15553–15558. [Google Scholar] [CrossRef]
- Klinman, D.M.; Yi, A.K.; Beaucage, S.L.; Conover, J.; Krieg, A.M. CpG motifs present in bacteria DNA rapidly induce lymphocytes to secrete interleukin 6, interleukin 12, and interferon gamma. Proc. Natl. Acad. Sci. USA 1996, 93, 2879–2883. [Google Scholar] [CrossRef] [PubMed]
- Ballas, Z.K.; Rasmussen, W.L.; Krieg, A.M. Induction of NK activity in murine and human cells by CpG motifs in oligodeoxynucleotides and bacterial DNA. J. Immunol. 1996, 157, 1840–1845. [Google Scholar] [PubMed]
- Cowdery, J.S.; Chace, J.H.; Yi, A.K.; Krieg, A.M. Bacterial DNA induces NK cells to produce IFN-gamma in vivo and increases the toxicity of lipopolysaccharides. J. Immunol. 1996, 156, 4570–4575. [Google Scholar] [PubMed]
- Cowdery, J.S.; Boerth, N.J.; Norian, L.; Myung, P.S.; Koretzky, G.A. Differential regulation of the IL-12 p40 promoter and of p40 secretion by CpG DNA and lipopolysaccharide. J. Immunol. 1999, 162, 6770–6775. [Google Scholar] [PubMed]
- Hacker, G.; Redecke, V.; Hacker, H. Activation of the immune system by bacterial CpG-DNA. Immunology 2002, 105, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Vezys, V.; Yates, A.; Casey, K.A.; Lanier, G.; Ahmed, R.; Antia, R.; Masopust, D. Memory CD8 T-cell compartment grows in size with immunological experience. Nature 2008, 457, 196–199. [Google Scholar] [CrossRef] [PubMed]
- Guimond, M.; Veenstra, R.G.; Grindler, D.J.; Zhang, H.; Cui, Y.; Murphy, R.D.; Kim, S.Y.; Na, R.; Hennighausen, L.; Kurtulus, S.; et al. Interleukin 7 signaling in dendritic cells regulates the homeostatic proliferation and niche size of CD4+ T cells. Nat. Immunol. 2009, 10, 149–157. [Google Scholar] [CrossRef]
- Ahlers, J.D.; Belyakov, I.M. Memories that last forever: Strategies for optimizing vaccine T-cell memory. Blood 2010, 115, 1678–1689. [Google Scholar] [CrossRef] [PubMed]
- Cockburn, I.; Chen, Y.-C.; Overstreet, M.; Lees, J.; Van Rooijen, N.; Farber, D.; Zavala, F. Prolonged Antigen Presentation Is Required for Optimal CD8+ T Cell Responses against Malaria Liver Stage Parasites. PLoS Pathog. 2010, 6, e1000877. [Google Scholar] [CrossRef] [PubMed]
- Messina, J.P.; Gilkeson, G.S.; Pisetsky, D.S. Stimulation of in vitro murine lymphocyte proliferation by bacterial DNA. J. Immunol. 1991, 147, 1759–1764. [Google Scholar]
- Yamamoto, S.; Yamamoto, T.; Shimada, S.; Kuramoto, E.; Yano, O.; Kataoka, T.; Tokunaga, T. DNA from Bacteria, but Not from Vertebrates, Induces Interferons, Activates Natural Killer Cells and Inhibits Tumor Growth. Microbiol. Immunol. 1992, 36, 983–997. [Google Scholar] [CrossRef]
- Stacey, K.J.; Sweet, M.; Hume, D.A. Macrophages ingest and are activated by bacterial DNA. J. Immunol. 1996, 157, 2116–2122. [Google Scholar] [PubMed]
- Weiner, G.J.; Liu, H.-M.; Wooldridge, J.E.; Dahle, C.E.; Krieg, A.M. Immunostimulatory oligodeoxynucleotides containing the CpG motif are effective as immune adjuvants in tumor antigen immunization. Proc. Natl. Acad. Sci. USA 1997, 94, 10833–10837. [Google Scholar] [CrossRef]
- Ahmad-Nejad, P.; Hacker, H.; Rutz, M.; Bauer, S.; Vabulas, R.M.; Wagner, H. Bacterial CpG-DNA and lipopolysaccharides activate Toll-like receptors at distinct cellular compartments. Eur. J. Immunol. 2002, 32, 1958–1968. [Google Scholar] [CrossRef]
- Askew, D.; Chu, R.S.; Krieg, A.M.; Harding, C.V. CpG DNA Induces Maturation of Dendritic Cells with Distinct Effects on Nascent and Recycling MHC-II Antigen-Processing Mechanisms. J. Immunol. 2000, 165, 6889–6895. [Google Scholar] [CrossRef] [PubMed]
- Krieg, A.M. CpG Motifs in Bacterial DNA and Their Immune Effects. Annu. Rev. Immunol. 2002, 20, 709–760. [Google Scholar] [CrossRef]
- van Ojik, H.H.; Bevaart, L.; Dahle, C.E.; Bakker, A.; Jansen, M.J.; van Vugt, M.J.; van de Winkel, J.G.; Weiner, G.J. CpG-A and B oligodeoxynucleotides enhance the efficacy of antibody therapy by activating different effector cell populations. Cancer Res. 2003, 63, 5595–5600. [Google Scholar]
- Krieg, A.M. CpG motifs: The active ingredient in bacterial extracts? Nat. Med. 2003, 9, 831–835. [Google Scholar] [CrossRef]
- Sester, D.P.; Naik, S.; Beasley, S.; Hume, D.; Stacey, K. Phosphorothioate Backbone Modification Modulates Macrophage Activation by CpG DNA. J. Immunol. 2000, 165, 4165–4173. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Song, W.; Czerwinski, D.K.; Varghese, B.; Uematsu, S.; Akira, S.; Krieg, A.M.; Levy, R. Lymphoma Immunotherapy with CpG Oligodeoxynucleotides Requires TLR9 Either in the Host or in the Tumor Itself. J. Immunol. 2007, 179, 2493–2500. [Google Scholar] [CrossRef]
- Link, B.K.; Ballas, Z.K.; Weisdorf, D.; Wooldridge, J.E.; Bossler, A.D.; Shannon, M.; Rasmussen, W.L.; Krieg, A.M.; Weiner, G.J. Oligodeoxynucleotide CpG 7909 Delivered as Intravenous Infusion Demonstrates Immunologic Modulation in Patients with Previously Treated Non-Hodgkin Lymphoma. J. Immunother. 2006, 29, 558–568. [Google Scholar] [CrossRef]
- Avalos, A.M.; Latz, E.; Mousseau, B.; Christensen, S.R.; Shlomchik, M.J.; Lund, F.; Marshak-Rothstein, A. Differential cytokine production and bystander activation of autoreactive B cells in response to CpG-A and CpG-B oligonucleotides. J. Immunol. 2009, 183, 6262–6268. [Google Scholar] [CrossRef]
- Wooldridge, J.E.; Ballas, Z.; Krieg, A.M.; Weiner, G.J. Immunostimulatory oligodeoxynucleotides containing CpG motifs enhance the efficacy of monoclonal antibody therapy of lymphoma. Blood 1997, 89, 2994–2998. [Google Scholar] [CrossRef] [PubMed]
- Leonard, J.P.; Link, B.K.; Emmanouilides, C.; Gregory, S.A.; Weisdorf, D.; Andrey, J.; Hainsworth, J.; Sparano, J.A.; Tsai, D.E.; Horning, S.; et al. Phase I trial of toll-like receptor 9 agonist PF-3512676 with and following rituximab in patients with recurrent indolent and aggressive non Hodgkin’s lymphoma. Clin. Cancer Res. 2007, 13, 6168–6174. [Google Scholar] [CrossRef]
- Friedberg, J.W.; Kim, H.; McCauley, M.; Hessel, E.M.; Sims, P.; Fisher, D.C.; Nadler, L.M.; Coffman, R.L.; Freedman, A.S. Combination immunotherapy with a CpG oligonucleotide (1018 ISS) and rituximab in patients with non-Hodgkin lymphoma: Increased interferon-alpha/beta-inducible gene expression, without significant toxicity. Blood 2005, 105, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Friedberg, J.W.; Kelly, J.L.; Neuberg, D.; Peterson, D.R.; Kutok, J.L.; Salloum, R.; Brenn, T.; Fisher, D.C.; Ronan, E.; Dalton, V.; et al. Phase II study of a TLR-9 agonist (1018 ISS) with rituximab in patients with relapsed or refractory follicular lymphoma. Br. J. Haematol. 2009, 146, 282–291. [Google Scholar] [CrossRef]
- Houot, R.; Levy, R. T-cell modulation combined with intratumoral CpG cures lymphoma in a mouse model without the need for chemotherapy. Blood 2009, 113, 3546–3552. [Google Scholar] [CrossRef]
- Sagiv-Barfi, I.; Kohrt, H.E.; Burckhardt, L.; Czerwinski, D.K.; Levy, R. Ibrutinib enhances the antitumor immune response induced by intratumoral injection of a TLR9 ligand in mouse lymphoma. Blood 2015, 125, 2079–2086. [Google Scholar] [CrossRef] [PubMed]
- Grignano, E.; Deau-Fischer, B.; Loganadane, G.; Breton, M.; Burroni, B.; Bouscary, D.; Kirova, Y. Radiotherapy of relapse-refractory follicular lymphoma. Cancer Radiother. 2018, 22, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T. The Tumor Microenvironment in Follicular Lymphoma: Its Pro-Malignancy Role with Therapeutic Potential. Int. J. Mol. Sci. 2021, 22, 5352. [Google Scholar] [CrossRef]
- Reits, E.A.; Hodge, J.W.; Herberts, C.A.; Groothuis, T.; Chakraborty, M.; Wansley, E.; Camphausen, K.; Luiten, R.; De Ru, A.H.; Neijssen, J.; et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J. Exp. Med. 2006, 203, 1259–1271. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Bowerman, N.A.; Salama, J.K.; Schmidt, H.; Spiotto, M.T.; Schietinger, A.; Yu, P.; Fu, Y.-X.; Weichselbaum, R.R.; Rowley, D.A.; et al. Induced sensitization of tumor stroma leads to eradication of established cancer by T cells. J. Exp. Med. 2007, 204, 49–55. [Google Scholar] [CrossRef]
- Dewan, M.Z.; Galloway, A.E.; Kawashima, N.; Dewyngaert, J.K.; Babb, J.S.; Formenti, S.C.; Demaria, S. Fractionated but Not Single-Dose Radiotherapy Induces an Immune-Mediated Abscopal Effect when Combined with Anti–CTLA-4 Antibody. Clin. Cancer Res. 2009, 15, 5379–5388. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Auh, S.L.; Wang, Y.; Burnette, B.; Meng, Y.; Beckett, M.; Sharma, R.; Chin, R.; Tu, T.; Weichselbaum, R.R.; et al. Therapeutic effects of ablative radiation on local tumor require CD8+ T cells: Changing strategies for cancer treatment. Blood 2009, 114, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Burnette, B.C.; Liang, H.; Lee, Y.; Chlewicki, L.; Khodarev, N.N.; Weichselbaum, R.R.; Fu, Y.-X.; Auh, S.L. The Efficacy of Radiotherapy Relies upon Induction of Type I Interferon–Dependent Innate and Adaptive Immunity. Cancer Res. 2011, 71, 2488–2496. [Google Scholar] [CrossRef] [PubMed]
- Postow, M.A.; Callahan, M.K.; Barker, C.A.; Yamada, Y.; Yuan, J.; Kitano, S.; Mu, Z.; Rasalan, T.; Adamow, M.; Ritter, E.; et al. Immunologic Correlates of the Abscopal Effect in a Patient with Melanoma. N. Engl. J. Med. 2012, 366, 925–931. [Google Scholar] [CrossRef]
- Klug, F.; Prakash, H.; Huber, P.E.; Seibel, T.; Bender, N.; Halama, N.; Pfirschke, C.; Voss, R.H.; Timke, C.; Umansky, L.; et al. Low-dose irradiation programs macrophage differentiation to an iNOS(+)/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell 2013, 24, 589–602. [Google Scholar] [CrossRef]
- Reynders, K.; Illidge, T.; Siva, S.; Chang, J.Y.; De Ruysscher, D. The abscopal effect of local radiotherapy: Using immunotherapy to make a rare event clinically relevant. Cancer Treat. Rev. 2015, 41, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Frank, M.J.; Reagan, P.M.; Bartlett, N.; Gordon, L.I.; Friedberg, J.W.; Czerwinski, D.K.; Long, S.R.; Hoppe, R.T.; Janssen, R.S.; Candia, A.F.; et al. In Situ Vaccination with a TLR9 Agonist and Local Low-Dose Radiation Induces Systemic Responses in Untreated Indolent Lymphoma. Cancer Discov. 2018, 8, 1258–1269. [Google Scholar] [CrossRef]
- Cheson, B.D.; Pfistner, B.; Juweid, M.E.; Gascoyne, R.D.; Specht, L.; Horning, S.J.; Coiffier, B.; Fisher, R.I.; Hagenbeek, A.; Zucca, E.; et al. Revised Response Criteria for Malignant Lymphoma. J. Clin. Oncol. 2007, 25, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Sagiv-Barfi, I.; Czerwinski, D.K.; Levy, S.; Alam, I.S.; Mayer, A.T.; Gambhir, S.S.; Levy, R. Eradication of spontaneous malignancy by local immunotherapy. Sci. Transl. Med. 2018, 10, eaan4488. [Google Scholar] [CrossRef] [PubMed]
- Green, M.R.; Kihira, S.; Liu, C.L.; Nair, R.V.; Salari, R.; Gentles, A.J.; Irish, J.; Stehr, H.; Vicente-Dueñas, C.; Romero-Camarero, I.; et al. Mutations in early follicular lymphoma progenitors are associated with suppressed antigen presentation. Proc. Natl. Acad. Sci. USA 2015, 112, E1116–E1125. [Google Scholar] [CrossRef]
- Lackraj, T.; Goswami, R.; Kridel, R. Pathogenesis of follicular lymphoma. Best Pract. Res. Clin. Haematol. 2018, 31, 2–14. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.C.J.; Tadros, S.; Bouska, A.; Heavican, T.; Yang, H.; Deng, Q.; Moore, D.; Akhter, A.; Hartert, K.; Jain, N.; et al. Subtype-specific and co-occurring genetic alterations in B-cell non-Hodgkin lymphoma. Haematologica 2021. [Google Scholar] [CrossRef]
- Witzig, T.E.; Wiseman, G.A.; Maurer, M.; Habermann, T.M.; Micallef, I.N.; Nowakowski, G.S.; Ansell, S.M.; Colgan, J.P.; Inwards, D.J.; Porrata, L.F.; et al. A phase I trial of immunostimulatory CpG 7909 oligodeoxynucleotide and 90 yttrium ibritumomab tiuxetan radioimmunotherapy for relapsed B-cell non-Hodgkin lymphoma. Am. J. Hematol. 2013, 88, 589–593. [Google Scholar] [CrossRef]
- Sands, H.; Goreyferet, L.J.; Cocuzza, A.J.; Hobbs, F.W.; Chidester, D.; Trainor, G.L. Biodistribution and Metabolism of Internally H-3 Labeled Oligonucleotides.1. Comparison of a Phosphodiester and a Phosphorothioate. Mol. Pharmacol. 1994, 45, 932–943. [Google Scholar]
- Lemke-Miltner, C.D.; Blackwell, S.E.; Yin, C.; Krug, A.E.; Morris, A.J.; Krieg, A.M.; Weiner, G.J. Antibody Opsonization of a TLR9 Agonist–Containing Virus-like Particle Enhances In Situ Immunization. J. Immunol. 2020, 204, 1386–1394. [Google Scholar] [CrossRef]
- Tang, H.; Liang, Y.; Anders, R.A.; Taube, J.M.; Qiu, X.; Mulgaonkar, A.; Liu, X.; Harrington, S.M.; Guo, J.; Xin, Y.; et al. PD-L1 on host cells is essential for PD-L1 blockade–mediated tumor regression. J. Clin. Investig. 2018, 128, 580–588. [Google Scholar] [CrossRef]
- Fourcade, J.; Sun, Z.; Pagliano, O.; Chauvin, J.-M.; Sander, C.; Janjic, B.; Tarhini, A.A.; Tawbi, H.A.; Kirkwood, J.M.; Moschos, S.; et al. PD-1 and Tim-3 Regulate the Expansion of Tumor Antigen–Specific CD8+ T Cells Induced by Melanoma Vaccines. Cancer Res. 2013, 74, 1045–1055. [Google Scholar] [CrossRef]
- Di Nicola, M.; Zappasodi, R.; Carlo-Stella, C.; Mortarini, R.; Pupa, S.M.; Magni, M.; Devizzi, L.; Matteucci, P.; Baldassari, P.; Ravagnani, F.; et al. Vaccination with autologous tumor-loaded dendritic cells induces clinical and immunologic responses in indolent B-cell lymphoma patients with relapsed and measurable disease: A pilot study. Blood 2009, 113, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Kepp, O.; Chan, F.K.-M.; Kroemer, G. Necroptosis: Mechanisms and Relevance to Disease. Annu. Rev. Pathol. Mech. Dis. 2017, 12, 103–130. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Legrand, A.; Konstantinou, M.; Goode, E.F.; Meier, P. The Diversification of Cell Death and Immunity: Memento Mori. Mol. Cell 2019, 76, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.; Tait, S.W. Targeting immunogenic cell death in cancer. Mol. Oncol. 2020, 14, 2994–3006. [Google Scholar] [CrossRef]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Warren, S.; Adjemian, S.; Agostinis, P.; Martinez, A.B.; Chan, T.A.; Coukos, G.; Demaria, S.; Deutsch, E.; et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J. Immunother. Cancer 2020, 8, e000337. [Google Scholar] [CrossRef] [PubMed]
- Fucikova, J.; Kepp, O.; Kasikova, L.; Petroni, G.; Yamazaki, T.; Liu, P.; Zhao, L.; Spisek, R.; Kroemer, G.; Galluzzi, L. Detection of immunogenic cell death and its relevance for cancer therapy. Cell Death Dis. 2020, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zappasodi, R.; Pupa, S.; Ghedini, G.C.; Bongarzone, I.; Magni, M.; Cabras, A.D.; Colombo, M.P.; Carlo-Stella, C.; Gianni, A.M.; Di Nicola, M. Improved Clinical Outcome in Indolent B-Cell Lymphoma Patients Vaccinated with Autologous Tumor Cells Experiencing Immunogenic Death. Cancer Res. 2010, 70, 9062–9072. [Google Scholar] [CrossRef]
- Montico, B.; Lapenta, C.; Ravo, M.; Martorelli, D.; Muraro, E.; Zeng, B.; Comaro, E.; Spada, M.; Donati, S.; Santini, S.M.; et al. Exploiting a new strategy to induce immunogenic cell death to improve dendritic cell-based vaccines for lymphoma immunotherapy. OncoImmunology 2017, 6, e1356964. [Google Scholar] [CrossRef]
- Lapenta, C.; Donati, S.; Spadaro, F.; Castaldo, P.; Belardelli, F.; Cox, M.C.; Santini, S.M. NK Cell Activation in the Antitumor Response Induced by IFN-α Dendritic Cells Loaded with Apoptotic Cells from Follicular Lymphoma Patients. J. Immunol. 2016, 197, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Morschhauser, F.; Fowler, N.H.; Feugier, P.; Bouabdallah, R.; Tilly, H.; Palomba, M.L.; Fruchart, C.; Libby, E.N.; Casasnovas, R.-O.; Flinn, I.W.; et al. Rituximab plus Lenalidomide in Advanced Untreated Follicular Lymphoma. N. Engl. J. Med. 2018, 379, 934–947. [Google Scholar] [CrossRef] [PubMed]
- Ladetto, M.; Cortelazzo, S.; Ferrero, S.; Evangelista, A.; Mian, M.; Tavarozzi, R.; Zanni, M.; Cavallo, F.; Di Rocco, A.; Stefoni, V.; et al. Lenalidomide maintenance after autologous haematopoietic stem-cell transplantation in mantle cell lymphoma: Results of a Fondazione Italiana Linfomi (FIL) multicentre, randomised, phase 3 trial. Lancet Haematol. 2021, 8, e34–e44. [Google Scholar] [CrossRef]
- Lapenta, C.; Donati, S.; Spadaro, F.; Lattanzi, L.; Urbani, F.; Macchia, I.; Sestili, P.; Spada, M.; Cox, M.C.; Belardelli, F.; et al. Lenalidomide improves the therapeutic effect of an interferon-α-dendritic cell-based lymphoma vaccine. Cancer Immunol. Immunother. 2019, 68, 1791–1804. [Google Scholar] [CrossRef]
- Corrales, L.; Glickman, L.H.; McWhirter, S.M.; Kanne, D.B.; Sivick, K.E.; Katibah, G.E.; Woo, S.-R.; Lemmens, E.; Banda, T.; Leong, J.J.; et al. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep. 2015, 11, 1018–1030. [Google Scholar] [CrossRef] [PubMed]
- Danilchanka, O.; Mekalanos, J.J. Cyclic Dinucleotides and the Innate Immune Response. Cell 2013, 154, 962–970. [Google Scholar] [CrossRef]
- Demaria, O.; De Gassart, A.; Coso, S.; Gestermann, N.; Di Domizio, J.; Flatz, L.; Gaide, O.; Michielin, O.; Hwu, P.; Petrova, T.V.; et al. STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proc. Natl. Acad. Sci. USA 2015, 112, 15408–15413. [Google Scholar] [CrossRef]
- Temizoz, B.; Kuroda, E.; Ohata, K.; Jounai, N.; Ozasa, K.; Kobiyama, K.; Aoshi, T.; Ishii, K.J. TLR9 and STING agonists synergistically induce innate and adaptive type-II IFN. Eur. J. Immunol. 2014, 45, 1159–1169. [Google Scholar] [CrossRef] [PubMed]
- Baird, J.R.; Friedman, D.; Cottam, B.; Dubensky, T.W.; Kanne, D.B., Jr.; Bambina, S.; Bahjat, K.; Crittenden, M.R.; Gough, M.J. Radiotherapy Combined with Novel STING-Targeting Oligonucleotides Results in Regression of Established Tumors. Cancer Res. 2016, 76, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Melero, I.; Shuford, W.W.; Newby, S.A.; Aruffo, A.; Ledbetter, J.A.; Hellström, K.E.; Mittler, R.S.; Chen, L. Monoclonal antibodies against the 4-1BB T-cell activation molecule eradicate established tumors. Nat. Med. 1997, 3, 682–685. [Google Scholar] [CrossRef]
- Melero, I.; Gangadhar, T.C.; Kohrt, H.E.; Segal, N.H.; Logan, T.; Urba, W.J.; Hodi, F.S.; Ott, P.A.; Perez-Gracia, J.L.; Wolchok, J.D.; et al. A phase I study of the safety, tolerability, pharmacokinetics, and immunoregulatory activity of urelumab (BMS-663513) in subjects with advanced and/or metastatic solid tumors and relapsed/refractory B-cell non-Hodgkin’s lymphoma (B-NHL). J. Clin. Oncol. 2013, 31. [Google Scholar] [CrossRef]
- Fisher, T.S.; Kamperschroer, C.; Oliphant, T.; Love, V.A.; Lira, P.D.; Doyonnas, R.; Bergqvist, S.; Baxi, S.; Rohner, A.; Shen, A.C.; et al. Targeting of 4-1BB by monoclonal antibody PF-05082566 enhances T-cell function and promotes anti-tumor activity. Cancer Immunol. Immunother. 2012, 61, 1721–1733. [Google Scholar] [CrossRef]
- Croft, M. Control of Immunity by the TNFR-Related Molecule OX40 (CD134). Annu. Rev. Immunol. 2010, 28, 57–78. [Google Scholar] [CrossRef]
- Willoughby, J.; Griffiths, J.; Tews, I.; Cragg, M.S. OX40: Structure and function—What questions remain? Mol. Immunol. 2017, 83, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, S.; Hori, T.; Imura, A.; Takaori-Kondo, A.; Uchiyama, T. Activation of OX40 signal transduction pathways leads to tumor necrosis factor receptor-associated factor (TRAF) 2- and TRAF5-mediated NF-kappaB activation. J. Biol. Chem. 1998, 273, 5808–5814. [Google Scholar] [CrossRef] [PubMed]
- Arch, R.H.; Thompson, C.B. 4-1BB and Ox40 are members of a tumor necrosis factor (TNF)-nerve growth factor receptor subfamily that bind TNF receptor-associated factors and activate nuclear factor kappaB. Mol. Cell. Biol. 1998, 18, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Salek-Ardakani, S.; Rogers, P.R.; Cheng, M.; Van Parijs, L.; Croft, M. The costimulation-regulated duration of PKB activation controls T cell longevity. Nat. Immunol. 2004, 5, 150–158. [Google Scholar] [CrossRef] [PubMed]
- So, T.; Song, J.; Sugie, K.; Altman, A.; Croft, M. Signals from OX40 regulate nuclear factor of activated T cells c1 and T cell helper 2 lineage commitment. Proc. Natl. Acad. Sci. USA 2006, 103, 3740–3745. [Google Scholar] [CrossRef] [PubMed]
- Marabelle, A.; Kohrt, H.; Sagiv-Barfi, I.; Ajami, B.; Axtell, R.C.; Zhou, G.; Rajapaksa, R.; Green, M.R.; Torchia, J.; Brody, J.; et al. Depleting tumor-specific Tregs at a single site eradicates disseminated tumors. J. Clin. Investig. 2013, 123, 2447–2463. [Google Scholar] [CrossRef] [PubMed]
- Piconese, S.; Valzasina, B.; Colombo, M.P. OX40 triggering blocks suppression by regulatory T cells and facilitates tumor rejection. J. Exp. Med. 2008, 205, 825–839. [Google Scholar] [CrossRef]
- Kjaergaard, J.; Tanaka, J.; Kim, J.A.; Rothchild, K.; Weinberg, A.; Shu, S. Therapeutic efficacy of OX-40 receptor antibody depends on tumor immunogenicity and anatomic site of tumor growth. Cancer Res. 2000, 60, 5514–5521. [Google Scholar]
- Gough, M.; Ruby, C.E.; Redmond, W.; Dhungel, B.; Brown, A.; Weinberg, A.D. OX40 Agonist Therapy Enhances CD8 Infiltration and Decreases Immune Suppression in the Tumor. Cancer Res. 2008, 68, 5206–5215. [Google Scholar] [CrossRef]
- Linch, S.; Kasiewicz, M.J.; McNamara, M.J.; Hilgart-Martiszus, I.F.; Farhad, M.; Redmond, W.L. Combination OX40 agonism/CTLA-4 blockade with HER2 vaccination reverses T-cell anergy and promotes survival in tumor-bearing mice. Proc. Natl. Acad. Sci. USA 2016, 113, E319–E327. [Google Scholar] [CrossRef] [PubMed]
- Curti, B.D.; Kovacsovics-Bankowski, M.; Morris, N.; Walker, E.; Chisholm, L.; Floyd, K.; Walker, J.; Gonzalez, I.; Meeuwsen, T.; Fox, B.A.; et al. OX40 Is a Potent Immune-Stimulating Target in Late-Stage Cancer Patients. Cancer Res. 2013, 73, 7189–7198. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Watanabe, T. Approaches of the Innate Immune System to Ameliorate Adaptive Immunotherapy for B-Cell Non-Hodgkin Lymphoma in Their Microenvironment. Cancers 2022, 14, 141. https://doi.org/10.3390/cancers14010141
Watanabe T. Approaches of the Innate Immune System to Ameliorate Adaptive Immunotherapy for B-Cell Non-Hodgkin Lymphoma in Their Microenvironment. Cancers. 2022; 14(1):141. https://doi.org/10.3390/cancers14010141
Chicago/Turabian StyleWatanabe, Takashi. 2022. "Approaches of the Innate Immune System to Ameliorate Adaptive Immunotherapy for B-Cell Non-Hodgkin Lymphoma in Their Microenvironment" Cancers 14, no. 1: 141. https://doi.org/10.3390/cancers14010141
APA StyleWatanabe, T. (2022). Approaches of the Innate Immune System to Ameliorate Adaptive Immunotherapy for B-Cell Non-Hodgkin Lymphoma in Their Microenvironment. Cancers, 14(1), 141. https://doi.org/10.3390/cancers14010141