
Importance of T, NK, CAR T and CAR NK Cell Metabolic Fitness for Effective Anti-Cancer Therapy: A Continuous Learning Process Allowing the Optimization of T, NK and CAR-Based Anti-Cancer Therapies


Abstract
:Simple Summary
Abstract
1. T Cell and CAR T Cell Metabolism Plays a Major Role in Anti-Cancer Immunity
1.1. T Cell Metabolism in a “Healthy” Environment
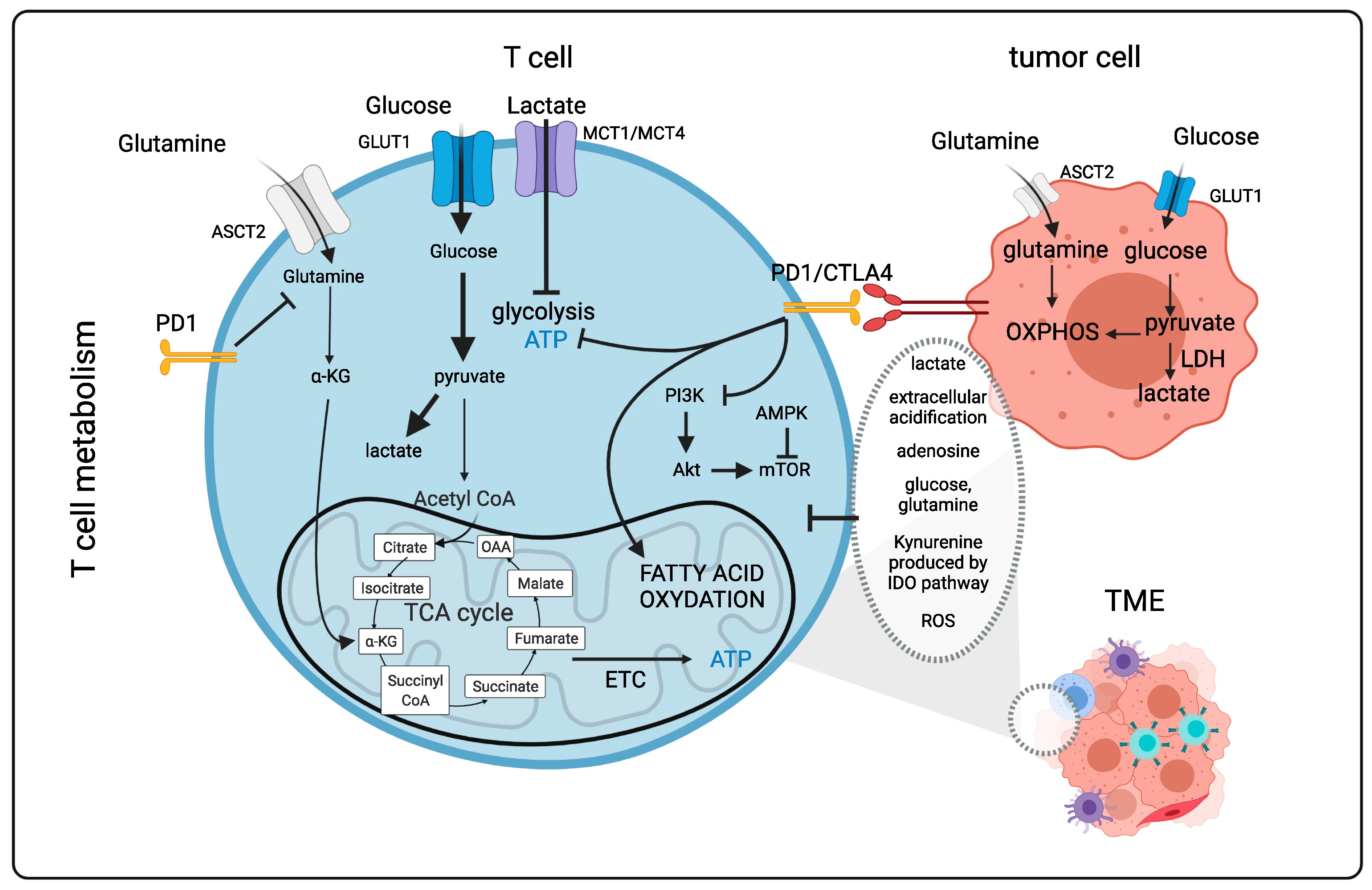
1.2. T Cells Metabolism in the “Tumor” Microenvironment (Figure 1)
1.3. CAR T Cells for Anticancer Treatment: Latest Developments
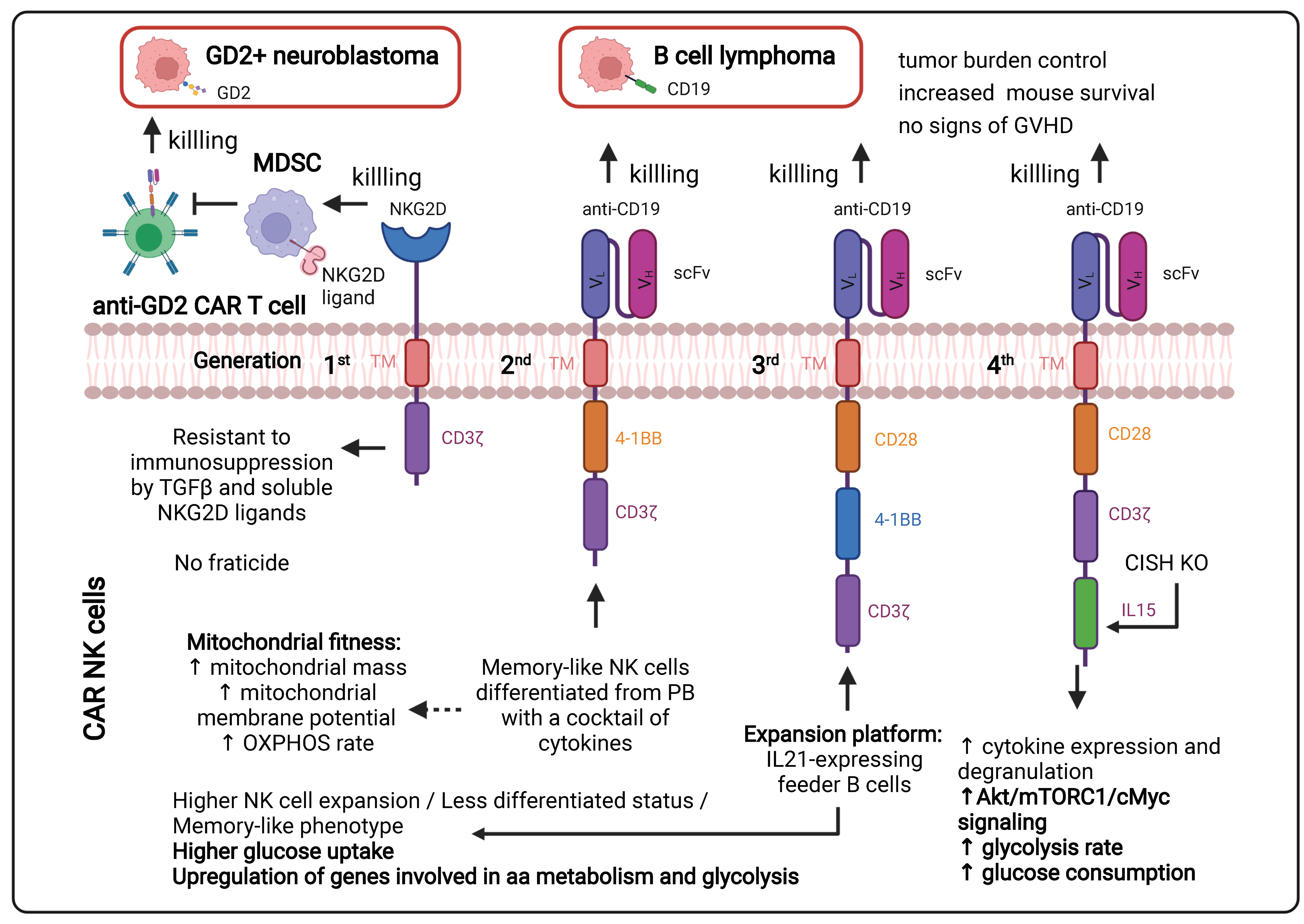
1.3.1. Continuous Improvements in the CAR Design to Stir CAR T Cell Metabolism (Figure 2)

1.3.2. Ex Vivo Expansion of CAR T Cells to Manipulate Their Metabolism
1.4. Importance of Nutrients, Metabolites for T/CAR T Cell Metabolism, Survival and Function (Figure 1)
1.4.1. Glucose Availability in the Tumor Environment
1.4.2. Lactate, a Side Product of Tumor Cell Glycolysis
1.4.3. Limited Amino Acid Availability
1.4.4. Hypoxia in the TME Has Important Effects on TIL Infiltration and Function
1.4.5. Cholesterol
1.4.6. Mitochondria in T Lymphocytes Infiltrating the TME
1.5. Immune Checkpoint Molecules and T Cell Metabolism in the TME (Figure 1)
1.6. Epigenetics Influences the Metabolic Response of T and CAR T Cells (Figure 3)

2. NK and CAR NK Cell-Based Therapies for a First-Line Anti-Cancer Immune Response
2.1. NK Cells Display Unique Anti-Cancer Immunosurveillance Mechanisms (Figure 4)

2.2. The Metabolism of Activated NK Cells in a “Healthy” Environment (Figure 4)
2.3. NK Cells Are Metabolically Challenged within the TME (Figure 5)

2.3.1. The Hypoxic Tumor Environment and NK Cell Function
2.3.2. Effect of the Metabolite Lactate and Other Metabolites on NK Cell Performance
2.3.3. Limited Amino Acid Availability in the TME
2.3.4. Targeting the Metabolism in the TME to Re-Establish NK Effector Function
2.4. How Can CAR NK Cells Persist, Remain Cytotoxic and Metabolically Fit in TME?
3. Perspectives
3.1. “Off the Shelf” Universal CAR T Cells
3.2. Combining CAR T and CAR NK Cells to Increase Their Anti-Tumor Activity
3.3. NKT Cells, the Natural Hybrids of T and NK Cells at the Cutting Edge of CAR Therapies
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shah, K.; Al-Haidari, A.; Sun, J.; Kazi, J.U. T cell receptor (TCR) signaling in health and disease. Signal Transduct. Target. Ther. 2021, 6, 412. [Google Scholar] [CrossRef] [PubMed]
- Palmer, C.S.; Duette, G.A.; Wagner, M.C.E.; Henstridge, D.C.; Saleh, S.; Pereira, C.; Zhou, J.; Simar, D.; Lewin, S.R.; Ostrowski, M.; et al. Metabolically active CD4+ T cells expressing Glut1 and OX40 preferentially harbor HIV during in vitro infection. FEBS Lett. 2017, 591, 3319–3332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, C.S.; Ostrowski, M.; Gouillou, M.; Tsai, L.; Yu, D.; Zhou, J.; Henstridge, D.C.; Maisa, A.; Hearps, A.C.; Lewin, S.R.; et al. Increased glucose metabolic activity is associated with CD4+ T-cell activation and depletion during chronic HIV infection. AIDS 2014, 28, 297–309. [Google Scholar] [CrossRef]
- Kang, S.; Tang, H. HIV-1 Infection and Glucose Metabolism Reprogramming of T Cells: Another Approach Toward Functional Cure and Reservoir Eradication. Front. Immunol. 2020, 11, 572677. [Google Scholar] [CrossRef] [PubMed]
- Andrejeva, G.; Rathmell, J.C. Similarities and Distinctions of Cancer and Immune Metabolism in Inflammation and Tumors. Cell Metab. 2017, 26, 49–70. [Google Scholar] [CrossRef] [Green Version]
- Ma, E.H.; Verway, M.J.; Johnson, R.M.; Roy, D.G.; Steadman, M.; Hayes, S.; Williams, K.S.; Sheldon, R.D.; Samborska, B.; Kosinski, P.A.; et al. Metabolic Profiling Using Stable Isotope Tracing Reveals Distinct Patterns of Glucose Utilization by Physiologically Activated CD8+ T Cells. Immunity 2019, 51, 856–870.e5. [Google Scholar] [CrossRef]
- Menk, A.V.; Scharping, N.E.; Moreci, R.S.; Zeng, X.; Guy, C.; Salvatore, S.; Bae, H.; Xie, J.; Young, H.A.; Wendell, S.G.; et al. Early TCR Signaling Induces Rapid Aerobic Glycolysis Enabling Distinct Acute T Cell Effector Functions. Cell Rep. 2018, 22, 1509–1521. [Google Scholar] [CrossRef] [Green Version]
- Pearce, E.L.; Pearce, E.J. Metabolic Pathways in Immune Cell Activation and Quiescence. Immunity 2013, 38, 633–643. [Google Scholar] [CrossRef] [Green Version]
- Roy, S.; Rizvi, Z.A.; Awasthi, A. Metabolic Checkpoints in Differentiation of Helper T Cells in Tissue Inflammation. Front. Immunol. 2019, 9, 3036. [Google Scholar] [CrossRef]
- Doedens, A.L.; Phan, A.T.; Stradner, M.H.; Fujimoto, J.K.; Nguyen, J.V.; Yang, E.; Johnson, R.S.; Goldrath, A.W. Hypoxia-inducible factors enhance the effector responses of CD8+ T cells to persistent antigen. Nat. Immunol. 2013, 14, 1173–1182. [Google Scholar] [CrossRef] [Green Version]
- Michalek, R.D.; Gerriets, V.A.; Jacobs, S.R.; Macintyre, A.N.; MacIver, N.J.; Mason, E.F.; Sullivan, S.A.; Nichols, A.G.; Rathmell, J.C. Cutting Edge: Distinct Glycolytic and Lipid Oxidative Metabolic Programs Are Essential for Effector and Regulatory CD4+ T Cell Subsets. J. Immunol. 2011, 186, 3299–3303. [Google Scholar] [CrossRef] [Green Version]
- Carr, E.L.; Kelman, A.; Wu, G.S.; Gopaul, R.; Senkevitch, E.; Aghvanyan, A.; Turay, A.M.; Frauwirth, K.A. Glutamine Uptake and Metabolism Are Coordinately Regulated by ERK/MAPK during T Lymphocyte Activation. J. Immunol. 2010, 185, 1037–1044. [Google Scholar] [CrossRef] [Green Version]
- Nakaya, M.; Xiao, Y.; Zhou, X.; Chang, J.-H.; Chang, M.; Cheng, X.; Blonska, M.; Lin, X.; Sun, S.-C. Inflammatory T Cell Responses Rely on Amino Acid Transporter ASCT2 Facilitation of Glutamine Uptake and mTORC1 Kinase Activation. Immunity 2014, 40, 692–705. [Google Scholar] [CrossRef] [Green Version]
- Sinclair, L.V.; Rolf, J.; Emslie, E.; Shi, Y.-B.; Taylor, P.M.; Cantrell, D.A. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat. Immunol. 2013, 14, 500–508. [Google Scholar] [CrossRef] [Green Version]
- Boomer, J.S.; Green, J.M. An Enigmatic Tail of CD28 Signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a002436. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Dillon, C.P.; Shi, L.Z.; Milasta, S.; Carter, R.; Finkelstein, D.; McCormick, L.L.; Fitzgerald, P.; Chi, H.; Munger, J.; et al. The Transcription Factor Myc Controls Metabolic Reprogramming upon T Lymphocyte Activation. Immunity 2011, 35, 871–882. [Google Scholar] [CrossRef] [Green Version]
- Frauwirth, K.A.; Riley, J.L.; Harris, M.H.; Parry, R.V.; Rathmell, J.C.; Plas, D.R.; Elstrom, R.L.; June, C.; Thompson, C.B. The CD28 Signaling Pathway Regulates Glucose Metabolism. Immunity 2002, 16, 769–777. [Google Scholar] [CrossRef] [Green Version]
- West, E.E.; Kolev, M.; Kemper, C. Complement and the Regulation of T Cell Responses. Annu. Rev. Immunol. 2018, 36, 309–338. [Google Scholar] [CrossRef] [PubMed]
- Wieman, H.L.; Wofford, J.A.; Rathmell, J.C. Cytokine Stimulation Promotes Glucose Uptake via Phosphatidylinositol-3 Kinase/Akt Regulation of Glut1 Activity and Trafficking. Mol. Biol. Cell 2007, 18, 1437–1446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, C.J.; Hammerman, P.S.; Thompson, C.B. Fuel feeds function: Energy metabolism and the T-cell response. Nat. Rev. Immunol. 2005, 5, 844–852. [Google Scholar] [CrossRef] [PubMed]
- Wofford, J.A.; Wieman, H.L.; Jacobs, S.R.; Zhao, Y.; Rathmell, J.C. IL-7 promotes Glut1 trafficking and glucose uptake via STAT5-mediated activation of Akt to support T-cell survival. Blood 2008, 111, 2101–2111. [Google Scholar] [CrossRef] [Green Version]
- Macintyre, A.N.; Gerriets, V.A.; Nichols, A.G.; Michalek, R.D.; Rudolph, M.C.; DeOliveira, D.; Anderson, S.M.; Abel, E.D.; Chen, B.J.; Hale, L.P.; et al. The Glucose Transporter Glut1 Is Selectively Essential for CD4 T Cell Activation and Effector Function. Cell Metab. 2014, 20, 61–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobs, S.R.; Herman, C.E.; MacIver, N.J.; Wofford, J.A.; Wieman, H.L.; Hammen, J.J.; Rathmell, J.C. Glucose Uptake Is Limiting in T Cell Activation and Requires CD28-Mediated Akt-Dependent and Independent Pathways. J. Immunol. 2008, 180, 4476–4486. [Google Scholar] [CrossRef] [Green Version]
- Murray, C.M.; Hutchinson, R.; Bantick, J.R.; Belfield, G.P.; Benjamin, A.D.; Brazma, D.; Bundick, R.V.; Cook, I.D.; Craggs, R.I.; Edwards, S.; et al. Monocarboxylate transporter MCT1 is a target for immunosuppression. Nat. Chem. Biol. 2005, 1, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Ryuge, A.; Kosugi, T.; Maeda, K.; Banno, R.; Gou, Y.; Zaitsu, K.; Ito, T.; Sato, Y.; Hirayama, A.; Tsubota, S.; et al. Basigin deficiency prevents anaplerosis and ameliorates insulin resistance and hepatosteatosis. JCI Insight 2021, 6. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-H.; Curtis, J.D.; Maggi, L.B.; Faubert, B.; Villarino, A.V.; O’Sullivan, D.; Huang, S.C.-C.; van der Windt, G.J.W.; Blagih, J.; Qiu, J.; et al. Posttranscriptional Control of T Cell Effector Function by Aerobic Glycolysis. Cell 2013, 153, 1239–1251. [Google Scholar] [CrossRef] [Green Version]
- Blagih, J.; Coulombe, F.; Vincent, E.E.; Dupuy, F.; Galicia-Vazquez, G.; Yurchenko, E.; Raissi, T.C.; van der Windt, G.J.; Viollet, B.; Pearce, E.L.; et al. The Energy Sensor AMPK Regulates T Cell Metabolic Adaptation and Effector Responses in vivo. Immunity 2015, 42, 41–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buck, M.D.; O’Sullivan, D.; Geltink, R.I.K.; Curtis, J.D.; Chang, C.-H.; Sanin, D.E.; Qiu, J.; Kretz, O.; Braas, D.; Van Der Windt, G.J.; et al. Mitochondrial Dynamics Controls T Cell Fate through Metabolic Programming. Cell 2016, 166, 63–76. [Google Scholar] [CrossRef] [Green Version]
- Sena, L.A.; Li, S.; Jairaman, A.; Prakriya, M.; Ezponda, T.; Hildeman, D.A.; Wang, C.-R.; Schumacker, P.T.; Licht, J.D.; Perlman, H.; et al. Mitochondria Are Required for Antigen-Specific T Cell Activation through Reactive Oxygen Species Signaling. Immunity 2013, 38, 225–236. [Google Scholar] [CrossRef] [Green Version]
- Bailis, W.; Shyer, J.A.; Zhao, J.; García-Cañaveras, J.C.; Al Khazal, F.J.; Qu, R.; Steach, H.R.; Bielecki, P.; Khan, O.; Jackson, R.; et al. Distinct modes of mitochondrial metabolism uncouple T cell differentiation and function. Nature 2019, 571, 403–407. [Google Scholar] [CrossRef]
- Ansó, E.; Weinberg, S.E.; Diebold, L.P.; Thompson, B.J.; Malinge, S.; Schumacker, P.T.; Liu, X.; Zhang, Y.; Shao, Z.; Steadman, M.; et al. The mitochondrial respiratory chain is essential for haematopoietic stem cell function. Nat. Cell Biol. 2017, 19, 614–625. [Google Scholar] [CrossRef]
- Balmer, M.L.; Ma, E.H.; Bantug, G.R.; Grählert, J.; Pfister, S.; Glatter, T.; Jauch, A.; Dimeloe, S.; Slack, E.; Dehio, P.; et al. Memory CD8 + T Cells Require Increased Concentrations of Acetate Induced by Stress for Optimal Function. Immunity 2016, 44, 1312–1324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, J.; Villa, M.; Sanin, D.E.; Buck, M.D.; O’Sullivan, D.; Ching, R.; Matsushita, M.; Grzes, K.M.; Winkler, F.; Chang, C.-H.; et al. Acetate Promotes T Cell Effector Function during Glucose Restriction. Cell Rep. 2019, 27, 2063–2074.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Windt, G.J.W.; Everts, B.; Chang, C.-H.; Curtis, J.D.; Freitas, T.C.; Amiel, E.; Pearce, E.J.; Pearce, E.L. Mitochondrial Respiratory Capacity Is a Critical Regulator of CD8+ T Cell Memory Development. Immunity 2012, 36, 68–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gubser, P.M.; Bantug, G.R.; Razik, L.; Fischer, M.; Dimeloe, S.; Hoenger, G.; Müller-Durovic, B.; Jauch, A.; Hess, C. Rapid effector function of memory CD8+ T cells requires an immediate-early glycolytic switch. Nat. Immunol. 2013, 14, 1064–1072. [Google Scholar] [CrossRef] [PubMed]
- Van der Windt, G.J.W.; O’Sullivan, D.; Everts, B.; Huang, S.C.-C.; Buck, M.D.; Curtis, J.D.; Chang, C.-H.; Smith, A.M.; Ai, T.; Faubert, B.; et al. CD8 memory T cells have a bioenergetic advantage that underlies their rapid recall ability. Proc. Natl. Acad. Sci. USA 2013, 110, 14336–14341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef]
- Park, J.H.; Rivière, I.; Gonen, M.; Wang, X.; Sénéchal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef]
- Turtle, C.J.; Hay, K.A.; Hanafi, L.-A.; Li, D.; Cherian, S.; Chen, X.; Wood, B.; Lozanski, A.; Byrd, J.C.; Heimfeld, S.; et al. Durable Molecular Remissions in Chronic Lymphocytic Leukemia Treated With CD19-Specific Chimeric Antigen Receptor–Modified T Cells After Failure of Ibrutinib. J. Clin. Oncol. 2017, 35, 3010–3020. [Google Scholar] [CrossRef]
- Chamoto, K.; Chowdhury, P.S.; Kumar, A.; Sonomura, K.; Matsuda, F.; Fagarasan, S.; Honjo, T. Mitochondrial activation chemicals synergize with surface receptor PD-1 blockade for T cell-dependent antitumor activity. Proc. Natl. Acad. Sci. USA 2017, 114, E761–E770. [Google Scholar] [CrossRef] [Green Version]
- Scharping, N.E.; Menk, A.V.; Moreci, R.S.; Whetstone, R.D.; Dadey, R.E.; Watkins, S.C.; Ferris, R.L.; Delgoffe, G.M. The Tumor Microenvironment Represses T Cell Mitochondrial Biogenesis to Drive Intratumoral T Cell Metabolic Insufficiency and Dysfunction. Immunity 2016, 45, 701–703. [Google Scholar] [CrossRef] [PubMed]
- Siska, P.J.; Beckermann, K.E.; Mason, F.M.; Andrejeva, G.; Greenplate, A.R.; Sendor, A.B.; Chiang, Y.-C.J.; Corona, A.L.; Gemta, L.F.; Vincent, B.G.; et al. Mitochondrial dysregulation and glycolytic insufficiency functionally impair CD8 T cells infiltrating human renal cell carcinoma. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Chandel, N.S. We need to talk about the Warburg effect. Nat. Metab. 2020, 2, 127–129. [Google Scholar] [CrossRef] [PubMed]
- Fantin, V.R.; St-Pierre, J.; Leder, P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell 2006, 9, 425–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef]
- Patra, K.C.; Wang, Q.; Bhaskar, P.T.; Miller, L.; Wang, Z.; Wheaton, W.; Chandel, N.; Laakso, M.; Muller, W.J.; Allen, E.L.; et al. Hexokinase 2 Is Required for Tumor Initiation and Maintenance and Its Systemic Deletion Is Therapeutic in Mouse Models of Cancer. Cancer Cell 2013, 24, 213–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [Green Version]
- Ho, P.-C.; Bihuniak, J.D.; Macintyre, A.N.; Staron, M.; Liu, X.; Amezquita, R.; Tsui, Y.-C.; Cui, G.; Micevic, G.; Perales, J.C.; et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell 2015, 162, 1217–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, S.Y.C.; Collins, C.C.; Gout, P.W.; Wang, Y. Cancer-generated lactic acid: A regulatory, immunosuppressive metabolite? J. Pathol. 2013, 230, 350–355. [Google Scholar] [CrossRef] [Green Version]
- Helmlinger, G.; Sckell, A.; Dellian, M.; Forbes, N.S.; Jain, R.K. Acid production in glycolysis-impaired tumors provides new insights into tumor metabolism. Clin. Cancer Res. 2002, 8, 1284–1291. [Google Scholar]
- Kato, Y.; Ozawa, S.; Miyamoto, C.; Maehata, Y.; Suzuki, A.; Maeda, T.; Baba, Y. Acidic extracellular microenvironment and cancer. Cancer Cell Int. 2013, 13, 89. [Google Scholar] [CrossRef] [Green Version]
- Brand, A.; Singer, K.; Koehl, G.E.; Kolitzus, M.; Schoenhammer, G.; Thiel, A.; Matos, C.; Bruss, C.; Klobuch, S.; Peter, K.; et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab. 2016, 24, 657–671. [Google Scholar] [CrossRef] [Green Version]
- Cham, C.M.; Driessens, G.; O’Keefe, J.P.; Gajewski, T.F. Glucose deprivation inhibits multiple key gene expression events and effector functions in CD8+ T cells. Eur. J. Immunol. 2008, 38, 2438–2450. [Google Scholar] [CrossRef] [Green Version]
- McNamee, E.N.; Johnson, D.K.; Homann, D.; Clambey, E.T. Hypoxia and hypoxia-inducible factors as regulators of T cell development, differentiation, and function. Immunol. Res. 2012, 55, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Chemnitz, J.M.; Parry, R.V.; Nichols, K.E.; June, C.H.; Riley, J.L. SHP-1 and SHP-2 Associate with Immunoreceptor Tyrosine-Based Switch Motif of Programmed Death 1 upon Primary Human T Cell Stimulation, but Only Receptor Ligation Prevents T Cell Activation. J. Immunol. 2004, 173, 945–954. [Google Scholar] [CrossRef] [PubMed]
- Staron, M.M.; Gray, S.M.; Marshall, H.D.; Parish, I.A.; Chen, J.H.; Perry, C.J.; Cui, G.; Li, M.O.; Kaech, S.M. The Transcription Factor FoxO1 Sustains Expression of the Inhibitory Receptor PD-1 and Survival of Antiviral CD8+ T Cells during Chronic Infection. Immunity 2014, 41, 802–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teft, W.A.; Chau, T.A.; Madrenas, J. Structure-Function analysis of the CTLA-4 interaction with PP2A. BMC Immunol. 2009, 10, 23. [Google Scholar] [CrossRef] [Green Version]
- Wlodarchak, N.; Xing, Y. PP2A as a master regulator of the cell cycle. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 162–184. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.Q.; Tsiaras, W.G.; Araki, T.; Wen, G.; Minichiello, L.; Klein, R.; Neel, B.G. Receptor-Specific Regulation of Phosphatidylinositol 3′-Kinase Activation by the Protein Tyrosine Phosphatase Shp2. Mol. Cell. Biol. 2002, 22, 4062–4072. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.-H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; Van Der Windt, G.J.W.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Su, E.W.; Zhu, C.; Hainline, S.; Phuah, J.; Moroco, J.A.; Smithgall, T.E.; Kuchroo, V.K.; Kane, L.P. Phosphotyrosine-Dependent Coupling of Tim-3 to T-Cell Receptor Signaling Pathways. Mol. Cell. Biol. 2011, 31, 3963–3974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platten, M.; Wick, W.; Van den Eynde, B.J. Tryptophan Catabolism in Cancer: Beyond IDO and Tryptophan Depletion. Cancer Res. 2012, 72, 5435–5440. [Google Scholar] [CrossRef] [Green Version]
- Mastelic-Gavillet, B.; Rodrigo, B.N.; Décombaz, L.; Wang, H.; Ercolano, G.; Ahmed, R.; Lozano, L.E.; Ianaro, A.; Derré, L.; Valerio, M.; et al. Adenosine mediates functional and metabolic suppression of peripheral and tumor-infiltrating CD8+ T cells. J. Immunother. Cancer 2019, 7, 257. [Google Scholar] [CrossRef]
- Fultang, L.; Booth, S.; Yogev, O.; da Costa, B.M.; Tubb, V.; Panetti, S.; Stavrou, V.; Scarpa, U.; Jankevics, A.; Lloyd, G.; et al. Metabolic engineering against the arginine microenvironment enhances CAR-T cell proliferation and therapeutic activity. Blood 2020, 136, 1155–1160. [Google Scholar] [CrossRef]
- Lind, D.S. Arginine and Cancer. J. Nutr. 2004, 134, 2837S–2841S. [Google Scholar] [CrossRef] [Green Version]
- Munder, M. Arginase: An emerging key player in the mammalian immune system. Mov. Disord. 2009, 158, 638–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, R.K.; O’Connor, R.S.; Grupp, S.A.; Barrett, D.M. Lingering effects of chemotherapy on mature T cells impair proliferation. Blood Adv. 2020, 4, 4653–4664. [Google Scholar] [CrossRef]
- Philipson, B.I.; O’Connor, R.S.; May, M.J.; June, C.H.; Albelda, S.M.; Milone, M.C. 4-1BB costimulation promotes CAR T cell survival through noncanonical NF-κB signaling. Sci. Signal. 2020, 13. [Google Scholar] [CrossRef]
- Brentjens, R.J.; Davila, M.L.; Riviere, I.; Park, J.; Wang, X.; Cowell, L.G.; Bartido, S.; Stefanski, J.; Taylor, C.; Olszewska, M.; et al. CD19-Targeted T Cells Rapidly Induce Molecular Remissions in Adults with Chemotherapy-Refractory Acute Lymphoblastic Leukemia. Sci. Transl. Med. 2013, 5, 177ra38. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.W.; Kochenderfer, J.N.; Stetler-Stevenson, M.; Cui, Y.K.; Delbrook, C.; Feldman, S.A.; Fry, T.J.; Orentas, R.; Sabatino, M.; Shah, N.N.; et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose-escalation trial. Lancet 2015, 385, 517–528. [Google Scholar] [CrossRef]
- Long, A.H.; Haso, W.M.; Shern, J.F.; Wanhainen, K.M.; Murgai, M.; Ingaramo, M.; Smith, J.P.; Walker, A.J.; Kohler, M.E.; Venkateshwara, V.R.; et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 2015, 21, 581–590. [Google Scholar] [CrossRef] [Green Version]
- Porter, D.L.; Hwang, W.-T.; Frey, N.V.; Lacey, S.F.; Shaw, P.A.; Loren, A.W.; Bagg, A.; Marcucci, K.T.; Shen, A.; Gonzalez, V.; et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 2015, 7, 303ra139. [Google Scholar] [CrossRef] [Green Version]
- Choi, B.K.; Lee, D.Y.; Lee, D.G.; Kim, Y.H.; Kim, S.-H.; Oh, H.S.; Han, C.; Kwon, B.S. 4-1BB signaling activates glucose and fatty acid metabolism to enhance CD8+ T cell proliferation. Cell. Mol. Immunol. 2016, 14, 748–757. [Google Scholar] [CrossRef]
- Kawalekar, O.U.; O’Connor, R.S.; Fraietta, J.A.; Guo, L.; Mcgettigan, S.E.; Posey, A.D.; Patel, P.R.; Guedan, S.; Scholler, J.; Keith, B.; et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity 2016, 44, 380–390. [Google Scholar] [CrossRef] [Green Version]
- Zeng, H.; Cohen, S.; Guy, C.; Shrestha, S.; Neale, G.; Brown, S.A.; Cloer, C.; Kishton, R.J.; Gao, X.; Youngblood, B.; et al. mTORC1 and mTORC2 Kinase Signaling and Glucose Metabolism Drive Follicular Helper T Cell Differentiation. Immunity 2016, 45, 540–554. [Google Scholar] [CrossRef] [Green Version]
- Petersen, C.T.; Krenciute, G. Next Generation CAR T Cells for the Immunotherapy of High-Grade Glioma. Front. Oncol. 2019, 9, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alizadeh, D.; Wong, R.A.; Yang, X.; Wang, D.; Pecoraro, J.R.; Kuo, C.-F.; Aguilar, B.; Qi, Y.; Ann, D.K.; Starr, R.; et al. IL15 Enhances CAR-T Cell Antitumor Activity by Reducing mTORC1 Activity and Preserving Their Stem Cell Memory Phenotype. Cancer Immunol. Res. 2019, 7, 759–772. [Google Scholar] [CrossRef]
- Kagoya, Y.; Tanaka, S.; Guo, T.; Anczurowski, M.; Wang, C.-H.; Saso, K.; Butler, M.O.; Minden, M.D.; Hirano, N. A novel chimeric antigen receptor containing a JAK–STAT signaling domain mediates superior antitumor effects. Nat. Med. 2018, 24, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Sukumar, M.; Liu, J.; Ji, Y.; Subramanian, M.; Crompton, J.G.; Yu, Z.; Roychoudhuri, R.; Palmer, D.C.; Muranski, P.; Karoly, E.D.; et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J. Clin. Investig. 2013, 123, 4479–4488. [Google Scholar] [CrossRef] [PubMed]
- Ghassemi, S.; Martinez-Becerra, F.J.; Master, A.M.; Richman, S.A.; Heo, D.; Leferovich, J.; Tu, Y.; García-Cañaveras, J.C.; Ayari, A.; Lu, Y.; et al. Enhancing Chimeric Antigen Receptor T Cell Anti-tumor Function through Advanced Media Design. Mol. Ther. Methods Clin. Dev. 2020, 18, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Preston, G.C.; Sinclair, L.V.; Kaskar, A.; Hukelmann, J.L.; Navarro, M.N.; Ferrero, I.; Macdonald, H.R.; Cowling, V.H.; Cantrell, D.A. Single cell tuning of Myc expression by antigen receptor signal strength and interleukin-2 in T lymphocytes. EMBO J. 2015, 34, 2008–2024. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Weidner, T.; Thalheimer, F.B.; Buchholz, C.J. In vivo generated human CAR T cells eradicate tumor cells. OncoImmunology 2019, 8, e1671761. [Google Scholar] [CrossRef] [Green Version]
- Frank, A.M.; Braun, A.H.; Scheib, L.; Agarwal, S.; Schneider, I.C.; Fusil, F.; Perian, S.; Sahin, U.; Thalheimer, F.B.; Verhoeyen, E.; et al. Combining T-cell-specific activation and in vivo gene delivery through CD3-targeted lentiviral vectors. Blood Adv. 2020, 4, 5702–5715. [Google Scholar]
- Mhaidly, R.; Verhoeyen, E. Humanized Mice Are Precious Tools for Preclinical Evaluation of CAR T and CAR NK Cell Therapies. Cancers 2020, 12, 1915. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, A.; Thalheimer, F.B.; Hartmann, S.; Frank, A.M.; Bender, R.R.; Danisch, S.; Costa, C.; Wels, W.S.; Modlich, U.; Stripecke, R.; et al. In vivo generation of human CD 19- CAR T cells results in B-cell depletion and signs of cytokine release syndrome. EMBO Mol. Med. 2018, 10, e9158. [Google Scholar] [CrossRef] [PubMed]
- Weidner, T.; Agarwal, S.; Perian, S.; Fusil, F.; Braun, G.; Hartmann, J.; Verhoeyen, E.; Buchholz, C.J. Genetic in vivo engineering of human T lymphocytes in mouse models. Nat. Protoc. 2021, 16, 3210–3240. [Google Scholar] [CrossRef]
- Weber, E.W.; Parker, K.R.; Sotillo, E.; Lynn, R.C.; Anbunathan, H.; Lattin, J.; Good, Z.; Belk, J.A.; Daniel, B.; Klysz, D.; et al. Transient rest restores functionality in exhausted CAR-T cells through epigenetic remodeling. Science 2021, 372. [Google Scholar] [CrossRef]
- Finlay, D.K.; Rosenzweig, E.; Sinclair, L.V.; Carmen, F.C.; Hukelmann, J.L.; Rolf, J.; Panteleyev, A.A.; Okkenhaug, K.; Cantrell, D.A. PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells. J. Exp. Med. 2012, 209, 2441–2453. [Google Scholar] [CrossRef] [Green Version]
- Kedia-Mehta, N.; Finlay, D.K. Competition for nutrients and its role in controlling immune responses. Nat. Commun. 2019, 10, 2123. [Google Scholar] [CrossRef]
- Greiner, E.F.; Guppy, M.; Brand, K. Glucose is essential for proliferation and the glycolytic enzyme induction that provokes a transition to glycolytic energy production. J. Biol. Chem. 1994, 269, 31484–31490. [Google Scholar] [CrossRef]
- Geltink, R.I.K.; Edwards-Hicks, J.; Apostolova, P.; O’Sullivan, D.; Sanin, D.E.; Patterson, A.E.; Puleston, D.J.; Ligthart, N.A.M.; Buescher, J.M.; Grzes, K.M.; et al. Metabolic conditioning of CD8+ effector T cells for adoptive cell therapy. Nat. Metab. 2020, 2, 703–716. [Google Scholar] [CrossRef] [PubMed]
- Díaz, F.E.; Dantas, E.; Geffner, J. Unravelling the Interplay between Extracellular Acidosis and Immune Cells. Mediat. Inflamm. 2018, 2018, 1218297. [Google Scholar] [CrossRef]
- Angelin, A.; Gil-De-Gómez, L.; Dahiya, S.; Jiao, J.; Guo, L.; Levine, M.H.; Wang, Z.; Quinn, W.J., II; Kopinski, P.K.; Wang, L.; et al. Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab. 2017, 25, 1282–1293.e7. [Google Scholar] [CrossRef] [Green Version]
- Colegio, O.R.; Chu, N.-Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef]
- Haas, R.; Smith, J.; Rocher-Ros, V.; Nadkarni, S.; Montero-Melendez, T.; D’Acquisto, F.; Bland, E.J.; Bombardieri, M.; Pitzalis, C.; Perretti, M.; et al. Lactate Regulates Metabolic and Pro-inflammatory Circuits in Control of T Cell Migration and Effector Functions. PLoS Biol. 2015, 13, e1002202. [Google Scholar] [CrossRef]
- Hermans, D.; Gautam, S.; García-Cañaveras, J.C.; Gromer, D.; Mitra, S.; Spolski, R.; Li, P.; Christensen, S.; Nguyen, R.; Lin, J.-X.; et al. Lactate dehydrogenase inhibition synergizes with IL-21 to promote CD8+T cell stemness and antitumor immunity. Proc. Natl. Acad. Sci. USA 2020, 117, 6047–6055. [Google Scholar] [CrossRef]
- Mane, M.M.; Cohen, I.J.; Ackerstaff, E.; Shalaby, K.; Ijoma, J.N.; Ko, M.; Maeda, M.; Albeg, A.S.; Vemuri, K.; Satagopan, J.; et al. Lactate Dehydrogenase A Depletion Alters MyC-CaP Tumor Metabolism, Microenvironment, and CAR T Cell Therapy. Mol. Ther. Oncolytics 2020, 18, 382–395. [Google Scholar] [CrossRef]
- Comito, G.; Iscaro, A.; Bacci, M.; Morandi, A.; Ippolito, L.; Parri, M.; Montagnani, I.; Raspollini, M.R.; Serni, S.; Simeoni, L.; et al. Lactate modulates CD4+ T-cell polarization and induces an immunosuppressive environment, which sustains prostate carcinoma progression via TLR8/miR21 axis. Oncogene 2019, 38, 3681–3695. [Google Scholar] [CrossRef]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory effect of tumor cell–derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef]
- Ping, W.; Senyan, H.; Li, G.; Yan, C.; Long, L. Increased Lactate in Gastric Cancer Tumor-Infiltrating Lymphocytes Is Related to Impaired T Cell Function Due to miR-34a Deregulated Lactate Dehydrogenase A. Cell. Physiol. Biochem. 2018, 49, 828–836. [Google Scholar] [CrossRef] [PubMed]
- Renner, K.; Bruss, C.; Schnell, A.; Koehl, G.; Becker, H.M.; Fante, M.; Menevse, A.-N.; Kauer, N.; Blazquez, R.; Hacker, L.; et al. Restricting Glycolysis Preserves T Cell Effector Functions and Augments Checkpoint Therapy. Cell Rep. 2019, 29, 135–150.e9. [Google Scholar] [CrossRef] [Green Version]
- Newsholme, E.A.; Crabtree, B.; Ardawi, M.S.M. Glutamine metabolism in lymphocytes: Its biochemical, physiological and clinical importance. Q. J. Exp. Physiol. 1985, 70, 473–489. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Hardie, R.A.; Hoy, A.J.; Van Geldermalsen, M.; Gao, D.; Fazli, L.; Sadowski, M.C.; Balaban, S.; Schreuder, M.; Nagarajah, R.; et al. Targeting ASCT2 -mediated glutamine uptake blocks prostate cancer growth and tumour development. J. Pathol. 2015, 236, 278–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammami, I.; Chen, J.; Bronte, V.; DeCrescenzo, G.; Jolicoeur, M. l-glutamine is a key parameter in the immunosuppression phenomenon. Biochem. Biophys. Res. Commun. 2012, 425, 724–729. [Google Scholar] [CrossRef] [PubMed]
- Leone, R.D.; Zhao, L.; Englert, J.M.; Sun, I.-M.; Oh, M.-H.; Arwood, M.L.; Bettencourt, I.A.; Patel, C.H.; Wen, J.; Tam, A.; et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 2019, 366, 1013–1021. [Google Scholar] [CrossRef]
- Nabe, S.; Yamada, T.; Suzuki, J.; Toriyama, K.; Yasuoka, T.; Kuwahara, M.; Shiraishi, A.; Takenaka, K.; Yasukawa, M.; Yamashita, M. Reinforce the antitumor activity of CD 8 + T cells via glutamine restriction. Cancer Sci. 2018, 109, 3737–3750. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Wang, X.; Wang, L.; Ma, X.; Gong, Z.; Zhang, S.; Li, Y. Targeting the IDO1 pathway in cancer: From bench to bedside. J. Hematol. Oncol. 2018, 11, 100. [Google Scholar] [CrossRef] [Green Version]
- Wainwright, D.A.; Balyasnikova, I.V.; Chang, A.L.; Ahmed, A.U.; Moon, K.-S.; Auffinger, B.; Tobias, A.L.; Han, Y.; Lesniak, M.S. IDO Expression in Brain Tumors Increases the Recruitment of Regulatory T Cells and Negatively Impacts Survival. Clin. Cancer Res. 2012, 18, 6110–6121. [Google Scholar] [CrossRef] [Green Version]
- Munn, D.H.; Mellor, A.L. Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol. 2012, 34, 137–143. [Google Scholar] [CrossRef] [Green Version]
- Prendergast, G.C.; Malachowski, W.J.; Mondal, A.; Scherle, P.; Muller, A.J. Indoleamine 2,3-Dioxygenase and Its Therapeutic Inhibition in Cancer. Int. Rev. Cell Mol. Biol. 2018, 336, 175–203. [Google Scholar] [CrossRef]
- Geiger, R.; Rieckmann, J.C.; Wolf, T.; Basso, C.; Feng, Y.; Fuhrer, T.; Kogadeeva, M.; Picotti, P.; Meissner, F.; Mann, M.; et al. L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity. Cell 2016, 167, 829–842.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fletcher, M.; Ramirez, M.E.; Sierra, R.A.; Raber, P.; Thevenot, P.; Al-Khami, A.A.; Sanchez-Pino, D.; Hernandez, C.; Wyczechowska, D.D.; Ochoa, A.C.; et al. l-Arginine Depletion Blunts Antitumor T-cell Responses by Inducing Myeloid-Derived Suppressor Cells. Cancer Res. 2014, 75, 275–283. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, P.C.; Quiceno, D.G.; Zabaleta, J.; Ortiz, B.; Zea, A.H.; Piazuelo, M.B.; Delgado, A.; Correa, P.; Brayer, J.; Sotomayor, E.M.; et al. Arginase I Production in the Tumor Microenvironment by Mature Myeloid Cells Inhibits T-Cell Receptor Expression and Antigen-Specific T-Cell Responses. Cancer Res. 2004, 64, 5839–5849. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, P.C.; Zea, A.H.; DeSalvo, J.; Culotta, K.S.; Zabaleta, J.; Quiceno, D.G.; Ochoa, J.B.; Ochoa, A.C. l-Arginine Consumption by Macrophages Modulates the Expression of CD3ζ Chain in T Lymphocytes. J. Immunol. 2003, 171, 1232–1239. [Google Scholar] [CrossRef] [Green Version]
- Hatfield, S.M.; Kjaergaard, J.; Lukashev, D.; Schreiber, T.H.; Belikoff, B.; Abbott, R.; Sethumadhavan, S.; Philbrook, P.; Ko, K.; Cannici, R.; et al. Immunological mechanisms of the antitumor effects of supplemental oxygenation. Sci. Transl. Med. 2015, 7, 277ra30. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.Z.; Wang, R.; Huang, G.; Vogel, P.; Neale, G.; Green, D.R.; Chi, H. HIF1α–dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J. Exp. Med. 2011, 208, 1367–1376. [Google Scholar] [CrossRef] [Green Version]
- Riera-Domingo, C.; Audigé, A.; Granja, S.; Cheng, W.-C.; Ho, P.-C.; Baltazar, F.; Stockmann, C.; Mazzone, M. Immunity, Hypoxia, and Metabolism–the Ménage à Trois of Cancer: Implications for Immunotherapy. Physiol. Rev. 2020, 100, 1–102. [Google Scholar] [CrossRef] [PubMed]
- Scharping, N.E.; Rivadeneira, D.B.; Menk, A.V.; Vignali, P.D.A.; Ford, B.R.; Rittenhouse, N.L.; Peralta, R.; Wang, Y.; Wang, Y.; DePeaux, K.; et al. Mitochondrial stress induced by continuous stimulation under hypoxia rapidly drives T cell exhaustion. Nat. Immunol. 2021, 22, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Kosti, P.; Opzoomer, J.W.; Larios-Martinez, K.I.; Henley-Smith, R.; Scudamore, C.L.; Okesola, M.; Taher, M.Y.M.; Davies, D.M.; Muliaditan, T.; Larcombe-Young, D.; et al. Hypoxia-sensing CAR T cells provide safety and efficacy in treating solid tumors. Cell Rep. Med. 2021, 2, 100227. [Google Scholar] [CrossRef] [PubMed]
- Ohta, A.; Gorelik, E.; Prasad, S.J.; Ronchese, F.; Lukashev, D.; Wong, M.K.K.; Huang, X.; Caldwell, S.; Liu, K.; Smith, P.; et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc. Natl. Acad. Sci. USA 2006, 103, 13132–13137. [Google Scholar] [CrossRef] [Green Version]
- Sitkovsky, M.V. Lessons from the A2A Adenosine Receptor Antagonist–Enabled Tumor Regression and Survival in Patients with Treatment-Refractory Renal Cell Cancer. Cancer Discov. 2020, 10, 16–19. [Google Scholar] [CrossRef]
- Giuffrida, L.; Sek, K.; Henderson, M.A.; Lai, J.; Chen, A.X.Y.; Meyran, D.; Todd, K.L.; Petley, E.V.; Mardiana, S.; Mølck, C.; et al. CRISPR/Cas9 mediated deletion of the adenosine A2A receptor enhances CAR T cell efficacy. Nat. Commun. 2021, 12, 3236. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Bi, E.; Lu, Y.; Su, P.; Huang, C.; Liu, L.; Wang, Q.; Yang, M.; Kalady, M.F.; Qian, J.; et al. Cholesterol Induces CD8+ T Cell Exhaustion in the Tumor Microenvironment. Cell Metab. 2019, 30, 143–156.e5. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Bai, Y.; Xiong, Y.; Zhang, J.; Chen, S.; Zheng, X.; Meng, X.; Li, L.; Wang, J.; Xu, C.; et al. Potentiating the antitumour response of CD8+ T cells by modulating cholesterol metabolism. Nature 2016, 531, 651–655. [Google Scholar] [CrossRef] [Green Version]
- Bengsch, B.; Johnson, A.L.; Kurachi, M.; Odorizzi, P.M.; Pauken, K.E.; Attanasio, J.; Stelekati, E.; McLane, L.M.; Paley, M.A.; Delgoffe, G.M.; et al. Bioenergetic Insufficiencies Due to Metabolic Alterations Regulated by the Inhibitory Receptor PD-1 Are an Early Driver of CD8 + T Cell Exhaustion. Immunity 2016, 45, 358–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geltink, R.K.; O’Sullivan, D.; Corrado, M.; Bremser, A.; Buck, M.D.; Buescher, J.M.; Firat, E.; Zhu, X.; Niedermann, G.; Caputa, G.; et al. Mitochondrial Priming by CD28. Cell 2017, 171, 385–397.e11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parry, R.V.; Chemnitz, J.M.; Frauwirth, K.A.; Lanfranco, A.R.; Braunstein, I.; Kobayashi, S.V.; Linsley, P.S.; Thompson, C.B.; Riley, J.L. CTLA-4 and PD-1 Receptors Inhibit T-Cell Activation by Distinct Mechanisms. Mol. Cell. Biol. 2005, 25, 9543–9553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patsoukis, N.; Bardhan, K.; Chatterjee, P.; Sari, D.; Liu, B.; Bell, L.N.; Karoly, E.D.; Freeman, G.J.; Petkova, V.; Seth, P.; et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 2015, 6, 6692. [Google Scholar] [CrossRef] [Green Version]
- Waickman, A.T.; Powell, J.D. mTOR, metabolism, and the regulation of T-cell differentiation and function. Immunol. Rev. 2012, 249, 43–58. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.; Liu, X.; Fang, C.; Jiang, S.; June, C.H.; Zhao, Y. Multiplex Genome Editing to Generate Universal CAR T Cells Resistant to PD1 Inhibition. Clin. Cancer Res. 2016, 23, 2255–2266. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Shi, L.; Zhang, W.; Han, J.; Zhang, S.; Fu, Z.; Cai, J. CRISPR knock out of programmed cell death protein 1 enhances anti-tumor activity of cytotoxic T lymphocytes. Oncotarget 2017, 9, 5208–5215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rupp, L.J.; Schumann, K.; Roybal, K.T.; Gate, R.E.; Chun, J.Y.; Lim, W.A.; Marson, A. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci. Rep. 2017, 7, 737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Jiang, H.; Shi, B.; Zhou, M.; Zhang, H.; Shi, Z.; Du, G.; Luo, H.; Wu, X.; Wang, Y.; et al. Disruption of PD-1 Enhanced the Anti-tumor Activity of Chimeric Antigen Receptor T Cells Against Hepatocellular Carcinoma. Front. Pharmacol. 2018, 9, 1118. [Google Scholar] [CrossRef]
- Pomeroy, E.J.; Hunzeker, J.T.; Kluesner, M.G.; Lahr, W.S.; Smeester, B.A.; Crosby, M.R.; Lonetree, C.-L.; Yamamoto, K.; Bendzick, L.; Miller, J.S.; et al. A Genetically Engineered Primary Human Natural Killer Cell Platform for Cancer Immunotherapy. Mol. Ther. 2019, 28, 52–63. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, X.; Cheng, C.; Mu, W.; Liu, X.; Li, N.; Wei, X.; Liu, X.; Xia, C.; Wang, H. CRISPR-Cas9 mediated LAG-3 disruption in CAR-T cells. Front. Med. 2017, 11, 554–562. [Google Scholar] [CrossRef]
- Suarez, E.R.; de Chang, K.; Sun, J.; Sui, J.; Freeman, G.J.; Signoretti, S.; Zhu, Q.; Marasco, W.A. Chimeric antigen receptor T cells secreting anti-PD-L1 antibodies more effectively regress renal cell carcinoma in a humanized mouse model. Oncotarget 2016, 7, 34341–34355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeku, O.O.; Purdon, T.J.; Koneru, M.; Spriggs, D.; Brentjens, R.J. Armored CAR T cells enhance antitumor efficacy and overcome the tumor microenvironment. Sci. Rep. 2017, 7, 10541. [Google Scholar] [CrossRef] [Green Version]
- Chmielewski, M.; Kopecky, C.; Hombach, A.A.; Abken, H. IL-12 Release by Engineered T Cells Expressing Chimeric Antigen Receptors Can Effectively Muster an Antigen-Independent Macrophage Response on Tumor Cells That Have Shut Down Tumor Antigen Expression. Cancer Res. 2011, 71, 5697–5706. [Google Scholar] [CrossRef] [Green Version]
- Kerkar, S.P.; Muranski, P.; Kaiser, A.; Boni, A.; Sanchez-Perez, L.; Yu, Z.; Palmer, D.C.; Reger, R.N.; Borman, Z.A.; Zhang, L.; et al. Tumor-Specific CD8+ T Cells Expressing Interleukin-12 Eradicate Established Cancers in Lymphodepleted Hosts. Cancer Res. 2010, 70, 6725–6734. [Google Scholar] [CrossRef] [Green Version]
- Pegram, H.J.; Lee, J.C.; Hayman, E.G.; Imperato, G.H.; Tedder, T.F.; Sadelain, M.; Brentjens, R.J. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood 2012, 119, 4133–4141. [Google Scholar] [CrossRef] [PubMed]
- Sachdeva, M.; Busser, B.W.; Temburni, S.; Jahangiri, B.; Gautron, A.-S.; Maréchal, A.; Juillerat, A.; Williams, A.; Depil, S.; Duchateau, P.; et al. Repurposing endogenous immune pathways to tailor and control chimeric antigen receptor T cell functionality. Nat. Commun. 2019, 10, 5100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chmielewski, M.; Abken, H. CAR T cells transform to trucks: Chimeric antigen receptor–redirected T cells engineered to deliver inducible IL-12 modulate the tumour stroma to combat cancer. Cancer Immunol. Immunother. 2012, 61, 1269–1277. [Google Scholar] [CrossRef]
- McCoy, K.D.; Le Gros, G. The role of CTLA-4 in the regulation of T cell immune responses. Immunol. Cell Biol. 1999, 77, 1–10. [Google Scholar] [CrossRef]
- Lee, M.J.; Yun, S.J.; Lee, B.; Jeong, E.; Yoon, G.; Kim, K.; Park, S. Association of TIM-3 expression with glucose metabolism in Jurkat T cells. BMC Immunol. 2020, 21, 48. [Google Scholar] [CrossRef] [PubMed]
- Previte, D.M.; Martins, C.P.; O’Connor, E.C.; Marre, M.L.; Coudriet, G.M.; Beck, N.W.; Menk, A.V.; Wright, R.H.; Tse, H.M.; Delgoffe, G.M.; et al. Lymphocyte Activation Gene-3 Maintains Mitochondrial and Metabolic Quiescence in Naive CD4+ T Cells. Cell Rep. 2019, 27, 129–141.e4. [Google Scholar] [CrossRef] [Green Version]
- Andrews, L.P.; Marciscano, A.E.; Drake, C.G.; Vignali, D.A.A. LAG3 (CD223) as a cancer immunotherapy target. Immunol. Rev. 2017, 276, 80–96. [Google Scholar] [CrossRef]
- Cai, G.; Anumanthan, A.; Brown, J.A.; Greenfield, E.A.; Zhu, B.; Freeman, G.J. CD160 inhibits activation of human CD4+ T cells through interaction with herpesvirus entry mediator. Nat. Immunol. 2008, 9, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Pauken, K.E.; Wherry, E.J. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. 2015, 36, 265–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phan, A.T.; Goldrath, A.W.; Glass, C.K. Metabolic and Epigenetic Coordination of T Cell and Macrophage Immunity. Immunity 2017, 46, 714–729. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y.; Wang, J.; Hu, Q.; Hochu, G.M.; Xin, H.; Wang, C.; Gu, Z. Synergistic Transcutaneous Immunotherapy Enhances Antitumor Immune Responses through Delivery of Checkpoint Inhibitors. ACS Nano 2016, 10, 8956–8963. [Google Scholar] [CrossRef] [PubMed]
- Tabana, Y.; Moon, T.C.; Siraki, A.; Elahi, S.; Barakat, K. Reversing T-cell exhaustion in immunotherapy: A review on current approaches and limitations. Expert Opin. Ther. Targets 2021, 25, 347–363. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, R.; Beura, L.K.; Quarnstrom, C.F.; Ghoneim, H.E.; Fan, Y.; Zebley, C.C.; Scott, M.C.; Fares-Frederickson, N.J.; Wijeyesinghe, S.; Thompson, E.A.; et al. Developmental plasticity allows outside-in immune responses by resident memory T cells. Nat. Immunol. 2020, 21, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Zebley, C.C.; Gottschalk, S.; Youngblood, B. Rewriting History: Epigenetic Reprogramming of CD8+ T Cell Differentiation to Enhance Immunotherapy. Trends Immunol. 2020, 41, 665–675. [Google Scholar] [CrossRef]
- Ghoneim, H.E.; Fan, Y.; Moustaki, A.; Abdelsamed, H.A.; Dash, P.; Dogra, P.; Carter, R.; Awad, W.; Neale, G.; Thomas, P.G.; et al. De Novo Epigenetic Programs Inhibit PD-1 Blockade-Mediated T Cell Rejuvenation. Cell 2017, 170, 142–157.e19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koss, B.; Shields, B.D.; Taylor, E.M.; Storey, A.J.; Byrum, S.D.; Gies, A.J.; Washam, C.L.; Choudhury, S.R.; Ahn, J.H.; Uryu, H.; et al. Epigenetic Control of Cdkn2a.Arf Protects Tumor-Infiltrating Lymphocytes from Metabolic Exhaustion. Cancer Res. 2020, 80, 4707–4719. [Google Scholar] [CrossRef]
- Zhao, E.; Maj, T.; Kryczek, I.; Li, W.; Wu, K.; Zhao, L.; Wei, S.; Crespo, J.; Wan, S.; Vatan, L.; et al. Cancer mediates effector T cell dysfunction by targeting microRNAs and EZH2 via glycolysis restriction. Nat. Immunol. 2015, 17, 95–103. [Google Scholar] [CrossRef]
- Bian, Y.; Li, W.; Kremer, D.M.; Sajjakulnukit, P.; Li, S.; Crespo, J.; Nwosu, Z.C.; Zhang, L.; Czerwonka, A.; Pawłowska, A.; et al. Cancer SLC43A2 alters T cell methionine metabolism and histone methylation. Nature 2020, 585, 277–282. [Google Scholar] [CrossRef]
- Roy, D.G.; Chen, J.; Mamane, V.; Ma, E.H.; Muhire, B.M.; Sheldon, R.D.; Shorstova, T.; Koning, R.; Johnson, R.M.; Esaulova, E.; et al. Methionine Metabolism Shapes T Helper Cell Responses through Regulation of Epigenetic Reprogramming. Cell Metab. 2020, 31, 250–266.e9. [Google Scholar] [CrossRef]
- Schvartzman, J.M.; Thompson, C.B.; Finley, L.W.S. Metabolic regulation of chromatin modifications and gene expression. J. Cell Biol. 2018, 217, 2247–2259. [Google Scholar] [CrossRef]
- Johnson, M.O.; Wolf, M.M.; Madden, M.Z.; Andrejeva, G.; Sugiura, A.; Contreras, D.C.; Maseda, D.; Liberti, M.V.; Paz, K.; Kishton, R.J.; et al. Distinct Regulation of Th17 and Th1 Cell Differentiation by Glutaminase-Dependent Metabolism. Cell 2018, 175, 1780–1795.e19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, M.; Yin, N.; Chhangawala, S.; Xu, K.; Leslie, C.S.; Li, M.O. Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science 2016, 354, 481–484. [Google Scholar] [CrossRef] [PubMed]
- McDonnell, E.; Crown, S.B.; Fox, D.B.; Kitir, B.; Ilkayeva, O.R.; Olsen, C.A.; Grimsrud, P.A.; Hirschey, M.D. Lipids Reprogram Metabolism to Become a Major Carbon Source for Histone Acetylation. Cell Rep. 2016, 17, 1463–1472. [Google Scholar] [CrossRef] [Green Version]
- Navas, L.E.; Carnero, A. NAD+ metabolism, stemness, the immune response, and cancer. Signal Transduct. Target. Ther. 2021, 6, 2. [Google Scholar] [CrossRef]
- Jeng, M.Y.; Hull, P.A.; Fei, M.; Kwon, H.-S.; Tsou, C.-L.; Kasler, H.; Ng, C.-P.; Gordon, D.E.; Johnson, J.; Krogan, N.; et al. Metabolic reprogramming of human CD8+ memory T cells through loss of SIRT1. J. Exp. Med. 2017, 215, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.-S.; Brent, M.M.; Getachew, R.; Jayakumar, P.; Chen, L.-F.; Schnolzer, M.; McBurney, M.W.; Marmorstein, R.; Greene, W.C.; Ott, M. Human Immunodeficiency Virus Type 1 Tat Protein Inhibits the SIRT1 Deacetylase and Induces T Cell Hyperactivation. Cell Host Microbe 2008, 3, 158–167. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Tang, Z.; Huang, H.; Zhou, G.; Cui, C.; Weng, Y.; Liu, W.; Kim, S.; Lee, S.; Perez-Neut, M.; et al. Metabolic regulation of gene expression by histone lactylation. Nature 2019, 574, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Van Acker, H.H.; Ma, S.; Scolaro, T.; Kaech, S.M.; Mazzone, M. How metabolism bridles cytotoxic CD8+ T cells through epigenetic modifications. Trends Immunol. 2021, 42, 401–417. [Google Scholar] [CrossRef]
- Vivier, E.; Nunès, J.A.; Vély, F. Natural Killer Cell Signaling Pathways. Science 2004, 306, 1517–1519. [Google Scholar] [CrossRef]
- Fang, F.; Xiao, W.; Tian, Z. NK cell-based immunotherapy for cancer. Semin. Immunol. 2017, 31, 37–54. [Google Scholar] [CrossRef]
- Gardiner, C.M.; Finlay, D.K. What Fuels Natural Killers? Metabolism and NK Cell Responses. Front. Immunol. 2017, 8, 367. [Google Scholar] [CrossRef] [Green Version]
- Myers, J.A.; Miller, J.S. Exploring the NK cell platform for cancer immunotherapy. Nat. Rev. Clin. Oncol. 2020, 18, 85–100. [Google Scholar] [CrossRef]
- Sivori, S.; Pende, D.; Quatrini, L.; Pietra, G.; Della Chiesa, M.; Vacca, P.; Tumino, N.; Moretta, F.; Mingari, M.C.; Locatelli, F.; et al. NK cells and ILCs in tumor immunotherapy. Mol. Asp. Med. 2020, 80, 100870. [Google Scholar] [CrossRef]
- Ljunggren, H.-G.; Kärre, K. In search of the ‘missing self’: MHC molecules and NK cell recognition. Immunol. Today 1990, 11, 237–244. [Google Scholar] [CrossRef]
- Morandi, F.; Pistoia, V. Interactions between HLA-G and HLA-E in Physiological and Pathological Conditions. Front. Immunol. 2014, 5, 394. [Google Scholar] [CrossRef] [Green Version]
- Bollino, D.; Webb, T.J. Chimeric antigen receptor–engineered natural killer and natural killer T cells for cancer immunotherapy. Transl. Res. 2017, 187, 32–43. [Google Scholar] [CrossRef]
- Wu, S.-Y.; Fu, T.; Jiang, Y.-Z.; Shao, Z.-M. Natural killer cells in cancer biology and therapy. Mol. Cancer 2020, 19, 120. [Google Scholar] [CrossRef]
- Caligiuri, M.A. Human natural killer cells. Blood 2008, 112, 461–469. [Google Scholar] [CrossRef]
- Clynes, R.A.; Towers, T.L.; Presta, L.G.; Ravetch, J.V. Inhibitory Fc receptors modulate in vivo cytoxicity against tumor targets. Nat. Med. 2000, 6, 443–446. [Google Scholar] [CrossRef]
- Conlon, K.C.; Lugli, E.; Welles, H.C.; Rosenberg, S.A.; Fojo, A.T.; Morris, J.C.; Fleisher, T.A.; Dubois, S.P.; Perera, L.P.; Stewart, D.M.; et al. Redistribution, Hyperproliferation, Activation of Natural Killer Cells and CD8 T Cells, and Cytokine Production During First-in-Human Clinical Trial of Recombinant Human Interleukin-15 in Patients With Cancer. J. Clin. Oncol. 2015, 33, 74–82. [Google Scholar] [CrossRef]
- Fallon, J.; Tighe, R.; Kradjian, G.; Guzman, W.; Bernhardt, A.; Neuteboom, B.; Lan, Y.; Sabzevari, H.; Schlom, J.; Greiner, J.W. The immunocytokine NHS-IL12 as a potential cancer therapeutic. Oncotarget 2014, 5, 1869–1884. [Google Scholar] [CrossRef] [Green Version]
- Floros, T.; Tarhini, A.A. Anticancer Cytokines: Biology and Clinical Effects of Interferon-α2, Interleukin (IL)-2, IL-15, IL-21, and IL-12. Semin. Oncol. 2015, 42, 539–548. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, S.; Pelloso, D.; Feng, H.; Voiles, L.; Lewis, D.; Haskova, Z.; Whitacre, M.; Trulli, S.; Chen, Y.-J.; Toso, J.; et al. Effects of interleukin-18 on natural killer cells: Costimulation of activation through Fc receptors for immunoglobulin. Cancer Immunol. Immunother. 2013, 62, 1073–1082. [Google Scholar] [CrossRef] [Green Version]
- Koerner, S.P.; André, M.C.; Leibold, J.S.; Kousis, P.C.; Kübler, A.; Pal, M.; Haen, S.; Bühring, H.-J.; Grosse-Hovest, L.; Jung, G.; et al. An Fc-optimized CD133 antibody for induction of NK cell reactivity against myeloid leukemia. Leukemia 2016, 31, 459–469. [Google Scholar] [CrossRef]
- Kohrt, H.E.; Houot, R.; Weiskopf, K.; Goldstein, M.J.; Scheeren, F.; Czerwinski, D.; Colevas, A.D.; Weng, W.-K.; Clarke, M.F.; Carlson, R.W.; et al. Stimulation of natural killer cells with a CD137-specific antibody enhances trastuzumab efficacy in xenotransplant models of breast cancer. J. Clin. Investig. 2012, 122, 1066–1075. [Google Scholar] [CrossRef] [Green Version]
- Romagné, F.; André, P.; Spee, P.; Zahn, S.; Anfossi, N.; Gauthier, L.; Capanni, M.; Ruggeri, L.; Benson, D.M., Jr.; Blaser, B.W.; et al. Preclinical characterization of 1-7F9, a novel human anti–KIR receptor therapeutic antibody that augments natural killer–mediated killing of tumor cells. Blood 2009, 114, 2667–2677. [Google Scholar] [CrossRef] [PubMed]
- Ruggeri, L.; Urbani, E.; André, P.; Mancusi, A.; Tosti, A.; Topini, F.; Bléry, M.; Animobono, L.; Romagné, F.; Wagtmann, N.; et al. Effects of anti-NKG2A antibody administration on leukemia and normal hematopoietic cells. Haematologica 2015, 101, 626–633. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Fu, J.; Zhang, M.; Liu, D. AFM13: A first-in-class tetravalent bispecific anti-CD30/CD16A antibody for NK cell-mediated immunotherapy. J. Hematol. Oncol. 2015, 8, 96. [Google Scholar] [CrossRef] [Green Version]
- Bachanova, V.; Burns, L.J.; McKenna, D.H.; Curtsinger, J.; Panoskaltsis-Mortari, A.; Lindgren, B.R.; Cooley, S.; Weisdorf, D.; Miller, J.S. Allogeneic natural killer cells for refractory lymphoma. Cancer Immunol. Immunother. 2010, 59, 1739–1744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klingemann, H.; Grodman, C.; Cutler, E.; Duque, M.; Kadidlo, D.; Klein, A.K.; Sprague, K.A.; Miller, K.B.; Comenzo, R.L.; Kewalramani, T.; et al. Autologous stem cell transplant recipients tolerate haploidentical related-donor natural killer cell-enriched infusions. Transfusion 2012, 53, 412–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romee, R.; Rosario, M.; Berrien-Elliott, M.M.; Wagner, J.A.; Jewell, B.A.; Schappe, T.; Leong, J.W.; Abdel-Latif, S.; Schneider, S.E.; Willey, S.; et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci. Transl. Med. 2016, 8, 357ra123. [Google Scholar] [CrossRef] [Green Version]
- Rubnitz, J.E.; Inaba, H.; Ribeiro, R.C.; Pounds, S.; Rooney, B.; Bell, T.; Pui, C.-H.; Leung, W. NKAML: A Pilot Study to Determine the Safety and Feasibility of Haploidentical Natural Killer Cell Transplantation in Childhood Acute Myeloid Leukemia. J. Clin. Oncol. 2010, 28, 955–959. [Google Scholar] [CrossRef] [Green Version]
- Geller, M.A.; Cooley, S.; Judson, P.L.; Ghebre, R.; Carson, L.F.; Argenta, P.A.; Jonson, A.L.; Panoskaltsis-Mortari, A.; Curtsinger, J.; McKenna, D.; et al. A phase II study of allogeneic natural killer cell therapy to treat patients with recurrent ovarian and breast cancer. Cytotherapy 2011, 13, 98–107. [Google Scholar] [CrossRef] [Green Version]
- Iliopoulou, E.G.; Kountourakis, P.; Karamouzis, M.V.; Doufexis, D.; Ardavanis, A.; Baxevanis, C.N.; Rigatos, G.; Papamichail, M.; Perez, S.A. A phase I trial of adoptive transfer of allogeneic natural killer cells in patients with advanced non-small cell lung cancer. Cancer Immunol. Immunother. 2010, 59, 1781–1789. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, N.; Ishikawa, T.; Kokura, S.; Okayama, T.; Oka, K.; Ideno, M.; Sakai, F.; Kato, A.; Tanabe, M.; Enoki, T.; et al. Phase I clinical trial of autologous NK cell therapy using novel expansion method in patients with advanced digestive cancer. J. Transl. Med. 2015, 13, 277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parkhurst, M.R.; Riley, J.P.; Dudley, M.E.; Rosenberg, S.A. Adoptive Transfer of Autologous Natural Killer Cells Leads to High Levels of Circulating Natural Killer Cells but Does Not Mediate Tumor Regression. Clin. Cancer Res. 2011, 17, 6287–6297. [Google Scholar] [CrossRef] [Green Version]
- Terrén, I.; Orrantia, A.; Vitallé, J.; Zenarruzabeitia, O.; Borrego, F. NK Cell Metabolism and Tumor Microenvironment. Front. Immunol. 2019, 10, 2278. [Google Scholar] [CrossRef] [PubMed]
- Dolstra, H.; Roeven, M.W.H.; Spanholtz, J.; Hangalapura, B.N.; Tordoir, M.; Maas, F.; Leenders, M.; Bohme, F.; Kok, N.; Trilsbeek, C.; et al. Successful Transfer of Umbilical Cord Blood CD34+ Hematopoietic Stem and Progenitor-derived NK Cells in Older Acute Myeloid Leukemia Patients. Clin. Cancer Res. 2017, 23, 4107–4118. [Google Scholar] [CrossRef] [Green Version]
- Colamartino, A.B.L.; Lemieux, W.; Bifsha, P.; Nicoletti, S.; Chakravarti, N.; Sanz, J.; Roméro, H.; Selleri, S.; Béland, K.; Guiot, M.; et al. Efficient and Robust NK-Cell Transduction With Baboon Envelope Pseudotyped Lentivector. Front. Immunol. 2019, 10, 2873. [Google Scholar] [CrossRef]
- Jochems, C.; Hodge, J.W.; Fantini, M.; Fujii, R.; Maurice, Y.M., II; Greiner, J.W.; Padget, M.R.; Tritsch, S.R.; Tsang, K.Y.; Campbell, K.S.; et al. An NK cell line (haNK) expressing high levels of granzyme and engineered to express the high affinity CD16 allele. Oncotarget 2016, 7, 86359–86373. [Google Scholar] [CrossRef] [Green Version]
- Sahm, C.; Schönfeld, K.; Wels, W.S. Expression of IL-15 in NK cells results in rapid enrichment and selective cytotoxicity of gene-modified effectors that carry a tumor-specific antigen receptor. Cancer Immunol. Immunother. 2012, 61, 1451–1461. [Google Scholar] [CrossRef]
- Schirrmann, T.; Pecher, G. Human natural killer cell line modified with a chimeric immunoglobulin T-cell receptor gene leads to tumor growth inhibition in vivo. Cancer Gene Ther. 2002, 9, 390–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uherek, C.; Tonn, T.; Uherek, B.; Becker, S.; Schnierle, B.; Klingemann, H.-G.; Wels, W. Retargeting of natural killer–cell cytolytic activity to ErbB2-expressing cancer cells results in efficient and selective tumor cell destruction. Blood 2002, 100, 1265–1273. [Google Scholar] [CrossRef] [PubMed]
- Müller, T.; Uherek, C.; Maki, G.; Chow, K.U.; Schimpf, A.; Klingemann, H.-G.; Tonn, T.; Wels, W.S. Expression of a CD20-specific chimeric antigen receptor enhances cytotoxic activity of NK cells and overcomes NK-resistance of lymphoma and leukemia cells. Cancer Immunol. Immunother. 2007, 57, 411–423. [Google Scholar] [CrossRef]
- Shimasaki, N.; Fujisaki, H.; Cho, D.; Masselli, M.; Lockey, T.; Eldridge, P.; Leung, W.; Campana, D. A clinically adaptable method to enhance the cytotoxicity of natural killer cells against B-cell malignancies. Cytotherapy 2012, 14, 830–840. [Google Scholar] [CrossRef]
- Tang, X.; Yang, L.; Li, Z.; Nalin, A.P.; Dai, H.; Xu, T.; Yin, J.; You, F.; Zhu, M.; Shen, W.; et al. First-in-man clinical trial of CAR NK-92 cells: Safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am. J. Cancer Res. 2018, 8, 1083–1089. [Google Scholar] [PubMed]
- Jiang, L.; Fang, X.; Wang, H.; Li, D.; Wang, X. Ovarian Cancer-Intrinsic Fatty Acid Synthase Prevents Anti-tumor Immunity by Disrupting Tumor-Infiltrating Dendritic Cells. Front. Immunol. 2018, 9, 2927. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Liu, Q.; Zhong, M.; Wang, Z.; Chen, Z.; Zhang, Y.; Xing, H.; Tian, Z.; Tang, K.; Liao, X.; et al. 2B4 costimulatory domain enhancing cytotoxic ability of anti-CD5 chimeric antigen receptor engineered natural killer cells against T cell malignancies. J. Hematol. Oncol. 2019, 12, 491. [Google Scholar] [CrossRef] [PubMed]
- Altvater, B.; Landmeier, S.; Pscherer, S.; Temme, J.; Schweer, K.; Kailayangiri, S.; Campana, D.; Juergens, H.; Pule, M.; Rossig, C. 2B4 (CD244) Signaling by Recombinant Antigen-specific Chimeric Receptors Costimulates Natural Killer Cell Activation to Leukemia and Neuroblastoma Cells. Clin. Cancer Res. 2009, 15, 4857–4866. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Liu, R.; Zhu, X.; Wang, L.; Ma, J.; Han, H.; Wang, X.; Zhang, G.; He, W.; Wang, W.; et al. Retargeting NK-92 for anti-melanoma activity by a TCR-like single-domain antibody. Immunol. Cell Biol. 2013, 91, 615–624. [Google Scholar] [CrossRef]
- Han, J.; Chu, J.; Chan, W.K.; Zhang, J.; Wang, Y.; Cohen, J.B.; Victor, A.; Meisen, W.H.; Kim, S.-H.; Grandi, P.; et al. CAR-Engineered NK Cells Targeting Wild-Type EGFR and EGFRvIII Enhance Killing of Glioblastoma and Patient-Derived Glioblastoma Stem Cells. Sci. Rep. 2015, 5, 11483. [Google Scholar] [CrossRef]
- Kruschinski, A.; Moosmann, A.; Poschke, I.; Norell, H.; Chmielewski, M.; Seliger, B.; Kiessling, R.; Blankenstein, T.; Abken, H.; Charo, J. Engineering antigen-specific primary human NK cells against HER-2 positive carcinomas. Proc. Natl. Acad. Sci. USA 2008, 105, 17481–17486. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Yang, N.; Li, H.; Wang, Z. Robo1-specific chimeric antigen receptor natural killer cell therapy for pancreatic ductal adenocarcinoma with liver metastasis. J. Cancer Res. Ther. 2020, 16, 393–396. [Google Scholar] [CrossRef]
- Wrona, E.; Borowiec, M.; Potemski, P. CAR-NK Cells in the Treatment of Solid Tumors. Int. J. Mol. Sci. 2021, 22, 5899. [Google Scholar] [CrossRef]
- O’Brien, K.L.; Finlay, D.K. Immunometabolism and natural killer cell responses. Nat. Rev. Immunol. 2019, 19, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Keating, S.E.; Bittencourt, V.Z.; Loftus, R.M.; Keane, C.; Brennan, K.; Finlay, D.K.; Gardiner, C.M. Metabolic Reprogramming Supports IFN-γ Production by CD56bright NK Cells. J. Immunol. 2016, 196, 2552–2560. [Google Scholar] [CrossRef] [Green Version]
- Mah-Som, A.Y.; Rashidi, A.; Keppel, M.P.; Saucier, N.; Moore, E.K.; Alinger, J.B.; Tripathy, S.K.; Agarwal, S.K.; Jeng, E.K.; Wong, H.C.; et al. Glycolytic requirement for NK cell cytotoxicity and cytomegalovirus control. JCI Insight 2017, 2, e95128. [Google Scholar] [CrossRef]
- Loftus, R.M.; Assmann, N.; Kedia-Mehta, N.; O’Brien, K.L.; Garcia, A.; Gillespie, C.; Hukelmann, J.L.; Oefner, P.J.; Lamond, A.I.; Gardiner, C.M.; et al. Amino acid-dependent cMyc expression is essential for NK cell metabolic and functional responses in mice. Nat. Commun. 2018, 9, 2341. [Google Scholar] [CrossRef]
- Donnelly, R.P.; Loftus, R.M.; Keating, S.E.; Liou, K.T.; Biron, C.A.; Gardiner, C.M.; Finlay, D.K. mTORC1-Dependent Metabolic Reprogramming Is a Prerequisite for NK Cell Effector Function. J. Immunol. 2014, 193, 4477–4484. [Google Scholar] [CrossRef] [Green Version]
- Marçais, A.; Cherfils-Vicini, J.; Viant, C.; Degouve, S.; Viel, S.; Fenis, A.; Rabilloud, J.; Mayol, K.; Tavares, A.; Bienvenu, J.; et al. The metabolic checkpoint kinase mTOR is essential for IL-15 signaling during the development and activation of NK cells. Nat. Immunol. 2014, 15, 749–757. [Google Scholar] [CrossRef]
- Sun, J.C.; Beilke, J.N.; Lanier, L.L. Adaptive immune features of natural killer cells. Nature 2009, 457, 557–561. [Google Scholar] [CrossRef]
- Assmann, N.; O’Brien, K.L.; Donnelly, R.P.; Dyck, L.; Zaiatz-Bittencourt, V.; Loftus, R.M.; Heinrich, P.; Oefner, P.J.; Lynch, L.; Gardiner, C.M.; et al. Srebp-controlled glucose metabolism is essential for NK cell functional responses. Nat. Immunol. 2017, 18, 1197–1206. [Google Scholar] [CrossRef]
- O’Brien, K.L.; Assmann, N.; O’Connor, E.; Keane, C.; Walls, J.; Choi, C.; Oefner, P.J.; Gardiner, C.M.; Dettmer, K.; Finlay, D.K. De novo polyamine synthesis supports metabolic and functional responses in activated murine NK cells. Eur. J. Immunol. 2020, 51, 91–102. [Google Scholar] [CrossRef]
- Saetersmoen, M.L.; Hammer, Q.; Valamehr, B.; Kaufman, D.S.; Malmberg, K.-J. Off-the-shelf cell therapy with induced pluripotent stem cell-derived natural killer cells. Semin. Immunopathol. 2018, 41, 59–68. [Google Scholar] [CrossRef] [Green Version]
- Viel, S.; Marçais, A.; Guimaraes, F.S.-F.; Loftus, R.; Rabilloud, J.; Grau, M.; Degouve, S.; Djebali, S.; Sanlaville, A.; Charrier, E.; et al. TGF-β inhibits the activation and functions of NK cells by repressing the mTOR pathway. Sci. Signal. 2016, 9, ra19. [Google Scholar] [CrossRef]
- Chambers, A.M.; Wang, J.; Lupo, K.B.; Yu, H.; Lanman, N.M.A.; Matosevic, S. Adenosinergic Signaling Alters Natural Killer Cell Functional Responses. Front. Immunol. 2018, 9, 2533. [Google Scholar] [CrossRef]
- Abarca-Rojano, E.; Muñiz-Hernández, S.; Moreno-Altamirano, M.M.B.; Mondragón-Flores, R.; Enriquez-Rincón, F.; Sánchez-García, F.J. Re-organization of mitochondria at the NK cell immune synapse. Immunol. Lett. 2009, 122, 18–25. [Google Scholar] [CrossRef]
- Netter, P.; Anft, M.; Watzl, C. Termination of the Activating NK Cell Immunological Synapse Is an Active and Regulated Process. J. Immunol. 2017, 199, 2528–2535. [Google Scholar] [CrossRef]
- Cong, J.; Wang, X.; Zheng, X.; Wang, D.; Fu, B.; Sun, R.; Tian, Z.; Wei, H. Dysfunction of Natural Killer Cells by FBP1-Induced Inhibition of Glycolysis during Lung Cancer Progression. Cell Metab. 2018, 28, 243–255.e5. [Google Scholar] [CrossRef] [Green Version]
- Stiff, A.; Trikha, P.; Mundy-Bosse, B.L.; McMichael, E.L.; Mace, T.A.; Benner, B.; Kendra, K.; Campbell, A.; Gautam, S.; Abood, D.; et al. Nitric Oxide Production by Myeloid-Derived Suppressor Cells Plays a Role in Impairing Fc Receptor–Mediated Natural Killer Cell Function. Clin. Cancer Res. 2018, 24, 1891–1904. [Google Scholar] [CrossRef] [Green Version]
- Della Chiesa, M.; Carlomagno, S.; Frumento, G.; Balsamo, M.; Cantoni, C.; Conte, R.; Moretta, L.; Moretta, A.; Vitale, M. The tryptophan catabolite l-kynurenine inhibits the surface expression of NKp46- and NKG2D-activating receptors and regulates NK-cell function. Blood 2006, 108, 4118–4125. [Google Scholar] [CrossRef]
- Melaiu, O.; Lucarini, V.; Cifaldi, L.; Fruci, D. Influence of the Tumor Microenvironment on NK Cell Function in Solid Tumors. Front. Immunol. 2020, 10, 3038. [Google Scholar] [CrossRef]
- Bailey, C.P.; Figueroa, M.; Gangadharan, A.; Lee, D.A.; Chandra, J. Scaffolding LSD1 Inhibitors Impair NK Cell Metabolism and Cytotoxic Function Through Depletion of Glutathione. Front. Immunol. 2020, 11, 2196. [Google Scholar] [CrossRef]
- Bader, J.E.; Voss, K.; Rathmell, J.C. Targeting Metabolism to Improve the Tumor Microenvironment for Cancer Immunotherapy. Mol. Cell 2020, 78, 1019–1033. [Google Scholar] [CrossRef]
- Xie, G.; Dong, H.; Liang, Y.; Ham, J.D.; Rizwan, R.; Chen, J. CAR-NK cells: A promising cellular immunotherapy for cancer. EBioMedicine 2020, 59, 102975. [Google Scholar] [CrossRef]
- Gang, M.; Marin, N.D.; Wong, P.; Neal, C.C.; Marsala, L.; Foster, M.; Schappe, T.; Meng, W.; Tran, J.; Schaettler, M.; et al. CAR-modified memory-like NK cells exhibit potent responses to NK-resistant lymphomas. Blood 2020, 136, 2308–2318. [Google Scholar] [CrossRef]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Kerbauy, L.N.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef]
- Koltai, T. Cancer: Fundamentals behind pH targeting and the double-edged approach. OncoTargets Ther. 2016, 9, 6343–6360. [Google Scholar] [CrossRef] [Green Version]
- Kouidhi, S.; Ayed, F.B.; Elgaaied, A.B. Targeting Tumor Metabolism: A New Challenge to Improve Immunotherapy. Front. Immunol. 2018, 9, 353. [Google Scholar] [CrossRef] [Green Version]
- Faubert, B.; Li, K.Y.; Cai, L.; Hensley, C.T.; Kim, J.; Zacharias, L.G.; Yang, C.; Do, Q.N.; Doucette, S.; Burguete, D.; et al. Lactate Metabolism in Human Lung Tumors. Cell 2017, 171, 358–371.e9. [Google Scholar] [CrossRef] [Green Version]
- Feichtinger, R.G.; Lang, R. Targeting L-Lactate Metabolism to Overcome Resistance to Immune Therapy of Melanoma and Other Tumor Entities. J. Oncol. 2019, 2019, 2084195. [Google Scholar] [CrossRef]
- Yeung, C.; Gibson, A.E.; Issaq, S.H.; Oshima, N.; Baumgart, J.T.; Edessa, L.D.; Rai, G.; Urban, D.J.; Johnson, M.S.; Benavides, G.A.; et al. Targeting Glycolysis through Inhibition of Lactate Dehydrogenase Impairs Tumor Growth in Preclinical Models of Ewing Sarcoma. Cancer Res. 2019, 79, 5060–5073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Li, J.; Wang, F.; Hu, J.; Wang, S.; Sun, Y. 2-Deoxy-D-glucose targeting of glucose metabolism in cancer cells as a potential therapy. Cancer Lett. 2014, 355, 176–183. [Google Scholar] [CrossRef]
- Benjamin, D.; Robay, D.; Hindupur, S.K.; Pohlmann, J.; Colombi, M.; El-Shemerly, M.Y.; Maira, S.-M.; Moroni, C.; Lane, H.A.; Hall, M.N. Dual Inhibition of the Lactate Transporters MCT1 and MCT4 Is Synthetic Lethal with Metformin due to NAD+ Depletion in Cancer Cells. Cell Rep. 2018, 25, 3047–3058.e4. [Google Scholar] [CrossRef] [Green Version]
- Chirasani, S.R.; Leukel, P.; Gottfried, E.; Hochrein, J.; Stadler, K.; Neumann, B.; Oefner, P.J.; Gronwald, W.; Bogdahn, U.; Hau, P.; et al. Diclofenac inhibits lactate formation and efficiently counteracts local immune suppression in a murine glioma model. Int. J. Cancer 2012, 132, 843–853. [Google Scholar] [CrossRef] [PubMed]
- Oh, M.-H.; Sun, I.-H.; Zhao, L.; Leone, R.D.; Sun, I.M.; Xu, W.; Collins, S.L.; Tam, A.J.; Blosser, R.L.; Patel, C.H.; et al. Targeting glutamine metabolism enhances tumor-specific immunity by modulating suppressive myeloid cells. J. Clin. Investig. 2020, 130, 3865–3884. [Google Scholar] [CrossRef]
- Miret, J.J.; Kirschmeier, P.; Koyama, S.; Zhu, M.; Li, Y.Y.; Naito, Y.; Wu, M.; Malladi, V.S.; Huang, W.; Walker, W.; et al. Suppression of Myeloid Cell Arginase Activity leads to Therapeutic Response in a NSCLC Mouse Model by Activating Anti-Tumor Immunity. J. Immunother. Cancer 2019, 7, 32. [Google Scholar] [CrossRef]
- Labadie, B.W.; Bao, R.; Luke, J.J. Reimagining IDO Pathway Inhibition in Cancer Immunotherapy via Downstream Focus on the Tryptophan–Kynurenine–Aryl Hydrocarbon Axis. Clin. Cancer Res. 2018, 25, 1462–1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herber, D.L.; Cao, W.; Nefedova, Y.; Novitskiy, S.V.; Nagaraj, S.; Tyurin, V.A.; Corzo, A.; Cho, H.-I.; Celis, E.; Lennox, B.; et al. Lipid accumulation and dendritic cell dysfunction in cancer. Nat. Med. 2010, 16, 880–886. [Google Scholar] [CrossRef] [Green Version]
- Chaoul, N.; Fayolle, C.; Desrues, B.; Oberkampf, M.; Tang, A.; Ladant, D.; Leclerc, C. Rapamycin Impairs Antitumor CD8+ T-cell Responses and Vaccine-Induced Tumor Eradication. Cancer Res. 2015, 75, 3279–3291. [Google Scholar] [CrossRef] [Green Version]
- Mao, Y.; van Hoef, V.; Zhang, X.; Wennerberg, E.; Lorent, J.; Witt, K.; Masvidal, L.; Liang, S.; Murray, S.; Larsson, O.; et al. IL-15 activates mTOR and primes stress-activated gene expression leading to prolonged antitumor capacity of NK cells. Blood 2016, 128, 1475–1489. [Google Scholar] [CrossRef] [Green Version]
- Eikawa, S.; Nishida, M.; Mizukami, S.; Yamazaki, C.; Nakayama, E.; Udono, H. Immune-mediated antitumor effect by type 2 diabetes drug, metformin. Proc. Natl. Acad. Sci. USA 2015, 112, 1809–1814. [Google Scholar] [CrossRef] [Green Version]
- Chang, D.-K.; Moniz, R.J.; Xu, Z.; Sun, J.; Signoretti, S.; Zhu, Q.; Marasco, W.A. Human anti-CAIX antibodies mediate immune cell inhibition of renal cell carcinoma in vitro and in a humanized mouse model in vivo. Mol. Cancer 2015, 14, 119. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Zhang, Q.; Song, Q.; Wang, H.; Dmitriev, P.; Sun, M.Y.; Cao, X.; Wang, Y.; Guo, L.; Indig, I.H.; et al. Targeting hypoxia downstream signaling protein, CAIX, for CAR T-cell therapy against glioblastoma. Neuro-Oncology 2019, 21, 1436–1446. [Google Scholar] [CrossRef]
- Bernadin, O.; Amirache, F.; Girard-Gagnepain, A.; Moirangthem, R.D.; Lévy, C.; Ma, K.; Costa, C.; Negre, D.; Reimann, C.; Fenard, D.; et al. Baboon envelope LVs efficiently transduced human adult, fetal, and progenitor T cells and corrected SCID-X1 T-cell deficiency. Blood Adv. 2019, 3, 461–475. [Google Scholar] [CrossRef]
- Girard-Gagnepain, A.; Amirache, F.; Costa, C.; Lévy, C.; Frecha, C.; Fusil, F.; Negre, D.; Lavillette, D.; Cosset, F.-L.; Verhoeyen, E. Baboon envelope pseudotyped LVs outperform VSV-G-LVs for gene transfer into early-cytokine-stimulated and resting HSCs. Blood 2014, 124, 1221–1231. [Google Scholar] [CrossRef] [Green Version]
- Bari, R.; Granzin, M.; Tsang, K.S.; Roy, A.; Krueger, W.; Orentas, R.; Schneider, D.; Pfeifer, R.; Moeker, N.; Verhoeyen, E.; et al. A Distinct Subset of Highly Proliferative and Lentiviral Vector (LV)-Transducible NK Cells Define a Readily Engineered Subset for Adoptive Cellular Therapy. Front. Immunol. 2019, 10, 2001. [Google Scholar] [CrossRef] [Green Version]
- Müller, S.; Bexte, T.; Gebel, V.; Kalensee, F.; Stolzenberg, E.; Hartmann, J.; Koehl, U.; Schambach, A.; Wels, W.S.; Modlich, U.; et al. High Cytotoxic Efficiency of Lentivirally and Alpharetrovirally Engineered CD19-Specific Chimeric Antigen Receptor Natural Killer Cells Against Acute Lymphoblastic Leukemia. Front. Immunol. 2020, 10, 3123. [Google Scholar] [CrossRef]
- Michelet, X.; Dyck, L.; Hogan, A.; Loftus, R.M.; Duquette, D.; Wei, K.; Beyaz, S.; Tavakkoli, A.; Foley, C.; Donnelly, R.; et al. Metabolic reprogramming of natural killer cells in obesity limits antitumor responses. Nat. Immunol. 2018, 19, 1330–1340. [Google Scholar] [CrossRef]
- Pawelec, G.; Verschoor, C.P.; Ostrand-Rosenberg, S. Myeloid-Derived Suppressor Cells: Not Only in Tumor Immunity. Front. Immunol. 2019, 10, 1099. [Google Scholar] [CrossRef]
- Parihar, R.; Rivas, C.; Huynh, M.; Omer, B.; Lapteva, N.; Metelitsa, L.S.; Gottschalk, S.M.; Rooney, C.M. NK Cells Expressing a Chimeric Activating Receptor Eliminate MDSCs and Rescue Impaired CAR-T Cell Activity against Solid Tumors. Cancer Immunol. Res. 2019, 7, 363–375. [Google Scholar] [CrossRef]
- Yang, Y.; Badeti, S.; Tseng, H.-C.; Ma, M.T.; Liu, T.; Jiang, J.-G.; Liu, C.; Liu, D. Superior Expansion and Cytotoxicity of Human Primary NK and CAR-NK Cells from Various Sources via Enriched Metabolic Pathways. Mol. Ther.-Methods Clin. Dev. 2020, 18, 428–445. [Google Scholar] [CrossRef]
- Delconte, R.B.; Guittard, G.; Goh, W.; Hediyeh-Zadeh, S.; Hennessy, R.J.; Rautela, J.; Davis, M.J.; Souza-Fonseca-Guimaraes, F.; Nunès, J.A.; Huntington, N.D. NK Cell Priming From Endogenous Homeostatic Signals Is Modulated by CIS. Front. Immunol. 2020, 11, 75. [Google Scholar] [CrossRef] [PubMed]
- Daher, M.; Basar, R.; Gokdemir, E.; Baran, N.; Uprety, N.; Cortes, A.K.N.; Mendt, M.; Kerbauy, L.N.; Banerjee, P.P.; Shanley, M.; et al. Targeting a cytokine checkpoint enhances the fitness of armored cord blood CAR-NK cells. Blood 2021, 137, 624–636. [Google Scholar] [CrossRef] [PubMed]
- Mhaidly, R.; Verhoeyen, E. The Future: In Vivo CAR T Cell Gene Therapy. Mol. Ther. 2019, 27, 707–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brendel, C.; Rio, P.; Verhoeyen, E. Humanized mice are precious tools for evaluation of hematopoietic gene therapies and preclinical modeling to move towards a clinical trial. Biochem. Pharmacol. 2019, 174, 113711. [Google Scholar] [CrossRef]
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; van der Stegen, S.J.C.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gönen, M.; Sadelain, M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017, 543, 113–117. [Google Scholar] [CrossRef] [Green Version]
- Qasim, W.; Zhan, H.; Samarasinghe, S.; Adams, S.; Amrolia, P.; Stafford, S.; Butler, K.; Rivat, C.; Wright, G.; Somana, K.; et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci. Transl. Med. 2017, 9, eaaj2013. [Google Scholar] [CrossRef]
- Torikai, H.; Reik, A.; Liu, P.-Q.; Zhou, Y.; Zhang, L.; Maiti, S.; Huls, H.; Miller, J.C.; Kebriaei, P.; Rabinovitch, B.; et al. A foundation for universal T-cell based immunotherapy: T cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR. Blood 2012, 119, 5697–5705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, M.A.; Büning, H.; Sauer, M.; Schambach, A. Use of Cell and Genome Modification Technologies to Generate Improved “Off-the-Shelf” CAR T and CAR NK Cells. Front. Immunol. 2020, 11, 1965. [Google Scholar] [CrossRef]
- Mo, F.; Watanabe, N.; McKenna, M.K.; Hicks, M.J.; Srinivasan, M.; Gomes-Silva, D.; Atilla, E.; Smith, T.; Atilla, P.A.; Ma, R.; et al. Engineered off-the-shelf therapeutic T cells resist host immune rejection. Nat. Biotechnol. 2020, 39, 56–63. [Google Scholar] [CrossRef]
- Nezhad, M.S.; Abdollahpour-Alitappeh, M.; Rezaei, B.; Yazdanifar, M.; Seifalian, A.M. Induced Pluripotent Stem Cells (iPSCs) Provide a Potentially Unlimited T Cell Source for CAR-T Cell Development and Off-the-Shelf Products. Pharm. Res. 2021, 38, 931–945. [Google Scholar] [CrossRef] [PubMed]
- Krijgsman, D.; Hokland, M.; Kuppen, P.J.K. The role of natural killer T cells in cancer—A phenotypical and functional approach. Front. Immunol. 2018, 9, 367. [Google Scholar] [CrossRef]
- Heczey, A.; Liu, D.; Tian, G.; Courtney, A.N.; Wei, J.; Marinova, E.; Gao, X.; Guo, L.; Yvon, E.; Hicks, J.; et al. Invariant NKT cells with chimeric antigen receptor provide a novel platform for safe and effective cancer immunotherapy. Blood 2014, 124, 2824–2833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Huang, W.; Heczey, A.; Liu, D.; Guo, L.; Wood, M.; Jin, J.; Courtney, A.N.; Liu, B.; Di Pierro, E.J.; et al. NKT Cells Coexpressing a GD2-Specific Chimeric Antigen Receptor and IL15 Show Enhanced In Vivo Persistence and Antitumor Activity against Neuroblastoma. Clin. Cancer Res. 2019, 25, 7126–7138. [Google Scholar] [CrossRef] [Green Version]
- Cichocki, F.; Valamehr, B.; Bjordahl, R.; Zhang, B.; Rezner, B.; Rogers, P.; Gaidarova, S.; Moreno, S.; Tuininga, K.; Dougherty, P.; et al. GSK3 Inhibition Drives Maturation of NK Cells and Enhances Their Antitumor Activity. Cancer Res. 2017, 77, 5664–5675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangal, J.L.; Handlos, J.L.; Esrafili, A.; Inamdar, S.; Mcmillian, S.; Wankhede, M.; Gottardi, R.; Acharya, A.P. Engineering Metabolism of Chimeric Antigen Receptor (CAR) Cells for Developing Efficient Immunotherapies. Cancers 2021, 13, 1123. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Tong, Y.; Dotti, G.; Shaim, H.; Savoldo, B.; Mukherjee, M.; Orange, J.; Wan, X.; Lu, X.; Reynolds, A.; et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia 2017, 32, 520–531. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Features | Active Effector T Cell | Active Effector NK Cell |
|---|---|---|
| Immune cell activation mechanisms | TCR engagement via recognition of peptides onto MHC-I in target cells [7] | No requirement of MHC-I on target cells, activation through stimulatory receptors [172] |
| Energetic metabolism | aerobic glycolysis and OXPHOS via the TCA cycle [7,20]. | aerobic glycolysis and OXPHOS via the CMS [215] |
| Metabolic phenotype | glycolytic [7] | glycolytic [215] |
| Energetic sources | glucose and glutamine [20] | glucose [215] |
| Metabolic regulators | PI3K/Akt/mTORC1 pathway cMyc, HIF-1α, glutamine [7] | mTORC1 dependent on and independent of PI3K/Akt pathway cMyc, SREBP, glutamine [215] |
| Metabolism of memory (-like) phenotype | OXPHOS [34] | OXPHOS [215] |
| Metabolic approaches to enhance immune cell metabolism, effector functions and persistence upon adoptive cell transfer | In vivo inhibition of the lactate transporters MCT1 and MCT4 by diclofenac in a melanoma mouse model renders tumors sensible to PD1 blockade [102] | Pharmacological inhibition of SREBPs in a melanoma mouse model controls tumor burden [222] |
| Glucose restriction for expansion of CD8 T+ cells prior to adoptive transfer into a lymphoma mouse model drives better tumor burden control [92] | Ex vivo pharmacological inhibition of fructose-1,6-biphosphatase in infiltrating NK cells from lung tumors in mice enhances glycolysis in vitro and in vivo tumor control upon adoptive cell transfer [229] | |
| In vitro and ex vivo administration of acetate in glucose-restricted CD8+ T cells and exhausted T cells, respectively, increases cytokine expression. Silencing of the acetyl-CoA synthetase controls better the tumor burden of a lymphoma mouse model [33] | ||
| Overexpression of PEP carboxykinase 1 [49] and PGC1α [42] in T cells transferred into melanoma-bearing mice lead to higher tumor cytotoxicity. | Pharmacological inhibition of GSK3 in NK cells from PB expanded with IL15 increases maturation and tumor cytotoxicity in mouse model of ovarian cancer [276] | |
| Oral bicarbonate in tumor-bearing mice controls tumor growth upon PD1 and/or CTL4 blockade and upon adoptive T cell transfer in melanoma-bearing mice [234] | ||
| Advantages of adoptive CAR-expressing cell transfer as a therapy | Commercial approval of several CAR T cell therapies by the FDA [277] T cells are more suitable for bioengineering by classical viral vector transduction [257] | No need for cells of autologous origin [172] Less prone to GVHD [172] |
| CAR designs and metabolic fitness | 4-1BB-containing CAR: OXPHOS metabolism [75] and longer in vivo persistence [72] CD28-containing CAR: glycolytic metabolism [75] and shorter in vivo persistence [72] | NKG2D-expressing CAR resistant to the immune and metabolic suppressor TGFβ drives MDSCs clearance and better tumor burden control of CAR T cells targeting neuroblastoma in mice [261]. |
| Hypoxia-inducible CAR expression for better tumor control in mouse models of ovarian cancer and neck and head cancer [120] | IL15-expressing CAR increases in vivo persistence and survival of a lymphoma mouse model [278] | |
| Metabolic strategies to improve fitness of CAR-expressing cells in the TME | IL15 stimulation of CAR T cells reduces glycolysis, increases OXPHOS and FAO genes and leads to a stem cell memory phenotype, high proliferation, longer in vivo persistence, tumor burden control and survival of a lymphoma model [78] | Cytokine-induced memory-like (ML) NK cells modified with a CAR displayed better tumor burden control in lymphoma mouse models as compared to conventional CAR NK cells and ML NK cells [236] |
| LDH depletion in prostate tumors improved cancer growth control by CAR T cells [98] | Genetic deletion of the IL15 immune checkpoint in IL15-expressing CAR NK cells increases mTOR and cMyc pathways, glycolytic rates and survival of a lymphoma model [264] | |
| A2AR deficiency in mouse and human CAR T cells improved tumor burden control in breast and ovarian cancer mouse models, respectively [123] | NK cell expansion with IL21-expressing feeder cells increases the expression of several metabolic genes, glucose uptake and promotes a less differentiated phenotype while enhancing tumor cytotoxicity in lymphoma mouse models [262] | |
| PD1 silencing and expression of IL12 in PD1 deficient CAR T cells increased survival of a lymphoma xenograft mouse model [142] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krug, A.; Martinez-Turtos, A.; Verhoeyen, E. Importance of T, NK, CAR T and CAR NK Cell Metabolic Fitness for Effective Anti-Cancer Therapy: A Continuous Learning Process Allowing the Optimization of T, NK and CAR-Based Anti-Cancer Therapies. Cancers 2022, 14, 183. https://doi.org/10.3390/cancers14010183
Krug A, Martinez-Turtos A, Verhoeyen E. Importance of T, NK, CAR T and CAR NK Cell Metabolic Fitness for Effective Anti-Cancer Therapy: A Continuous Learning Process Allowing the Optimization of T, NK and CAR-Based Anti-Cancer Therapies. Cancers. 2022; 14(1):183. https://doi.org/10.3390/cancers14010183
Chicago/Turabian StyleKrug, Adrien, Adriana Martinez-Turtos, and Els Verhoeyen. 2022. "Importance of T, NK, CAR T and CAR NK Cell Metabolic Fitness for Effective Anti-Cancer Therapy: A Continuous Learning Process Allowing the Optimization of T, NK and CAR-Based Anti-Cancer Therapies" Cancers 14, no. 1: 183. https://doi.org/10.3390/cancers14010183
APA StyleKrug, A., Martinez-Turtos, A., & Verhoeyen, E. (2022). Importance of T, NK, CAR T and CAR NK Cell Metabolic Fitness for Effective Anti-Cancer Therapy: A Continuous Learning Process Allowing the Optimization of T, NK and CAR-Based Anti-Cancer Therapies. Cancers, 14(1), 183. https://doi.org/10.3390/cancers14010183