
Antiangiogenic Compound Axitinib Demonstrates Low Toxicity and Antitumoral Effects against Medulloblastoma

 , , , , , , and
, , , , , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Lentiviral Infections
2.3. MTT Proliferation Assay, Cell Viability and Cumulative Population Doubling Assays
2.4. Coculture and FACS Experiments
2.5. Spheroid Assays
2.6. Subcutaneous Xenografts
2.7. Intracranial Tumor Xenografts and Metronomic Administration of the Treatments
2.8. Tumor Growth Analysis
2.9. Histopathology and Automated Image Analysis
2.10. Juvenile Rats Toxicity Experiment and Measurement of the Axitinib Concentration in the Brain
2.11. BBB Permeability Assay
2.12. Antibody Array Screenings
2.13. Statistical Analyses
3. Results
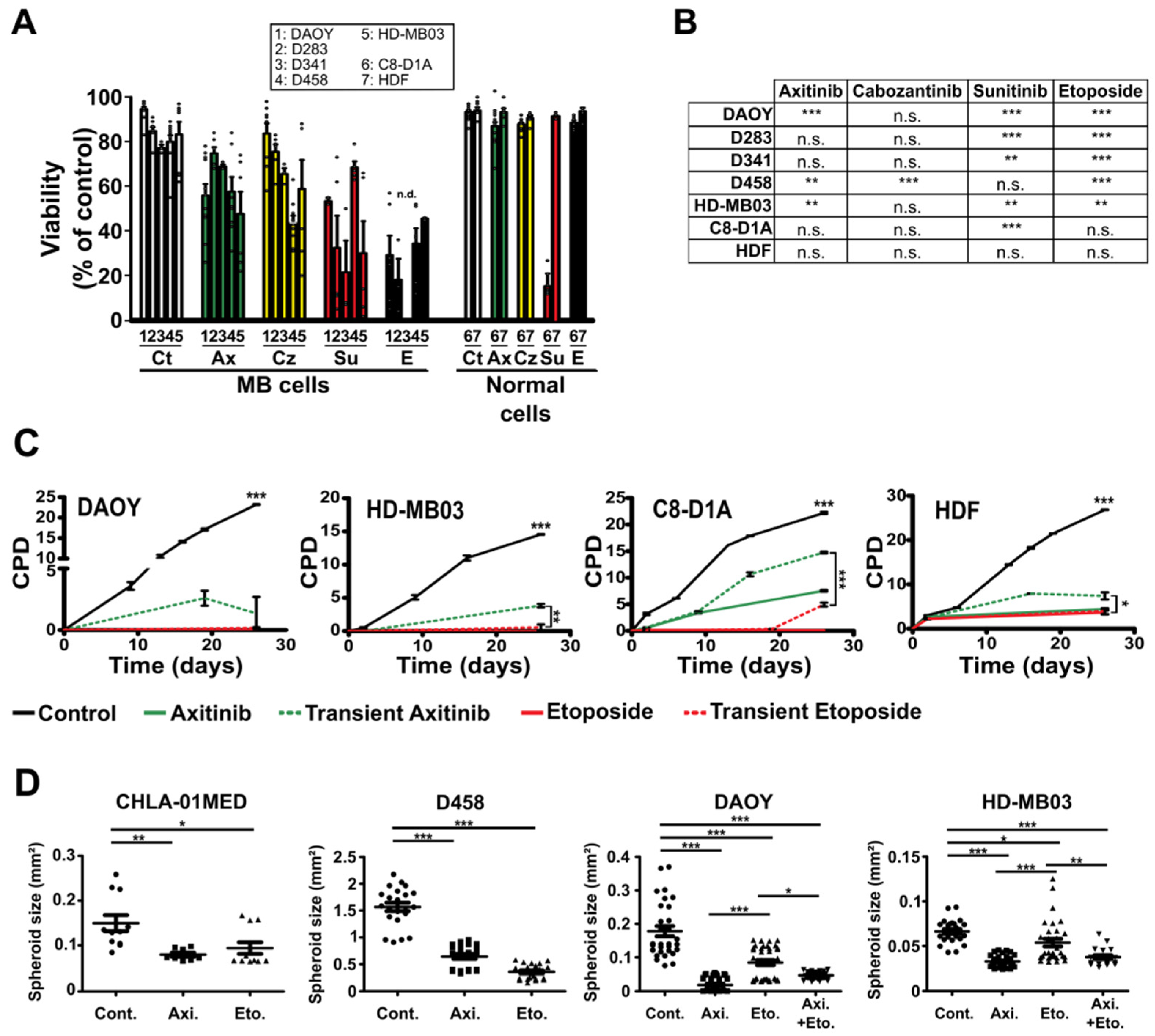
3.1. Antiangiogenic Compounds Are Effective against MB Cells in 2D and 3D Culture Conditions
3.2. Axitinib Leads to Low Toxicity towards Normal Brain Cells
3.3. Axitinib and Axitinib/Etoposide Combination Decrease Tumor Growth
3.4. High Expression of Axitinib Targets Is Linked to Poor Prognosis in Shh Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pritchard-Jones, K.; Pieters, R.; Reaman, G.H.; Hjorth, L.; Downie, P.; Calaminus, G.; Naafs-Wilstra, M.C.; Steliarova-Foucher, E. Sustaining innovation and improvement in the treatment of childhood cancer: Lessons from high-income countries. Lancet Oncol. 2013, 14, e95–e103. [Google Scholar] [CrossRef]
- Northcott, P.A.; Buchhalter, I.; Morrissy, A.S.; Hovestadt, V.; Weischenfeldt, J.; Ehrenberger, T.; Gröbner, S.; Segura-Wang, M.; Zichner, T.; Rudneva, V.A.; et al. The whole-genome landscape of medulloblastoma subtypes. Nature 2017, 547, 311–317. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Kumar, V.; McGuire, T.; Coulter, D.W.; Sharp, J.G.; Mahato, R.I. Challenges and Recent Advances in Medulloblastoma Therapy. Trends Pharmacol. Sci. 2017, 38, 1061–1084. [Google Scholar] [CrossRef]
- Kumar, G.G.; Sarathi, V.S.; Nahm, K.S. Recent advances and challenges in the anode architecture and their modifications for the applications of microbial fuel cells. Biosens. Bioelectron. 2013, 43, 461–475. [Google Scholar] [CrossRef]
- Salloum, R.; Chen, Y.; Yasui, Y.; Packer, R.; Leisenring, W.; Wells, E.; King, A.; Howell, R.; Gibson, T.M.; Krull, K.R.; et al. Late Morbidity and Mortality Among Medulloblastoma Survivors Diagnosed Across Three Decades: A Report from the Childhood Cancer Survivor Study. J. Clin. Oncol. 2019, 37, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, K.; Hinkes, B.; Gerber, N.U.; Deinlein, F.; Mittler, U.; Urban, C.; Benesch, M.; Warmuth-Metz, M.; Soerensen, N.; Zwiener, I.; et al. Long-term outcome and clinical prognostic factors in children with medulloblastoma treated in the prospective randomised multicentre trial HIT’91. Eur. J. Cancer 2009, 45, 1209–1217. [Google Scholar] [CrossRef]
- Packer, R.J.; Zhou, T.; Holmes, E.; Vezina, G.; Gajjar, A. Survival and secondary tumors in children with medulloblastoma receiving radiotherapy and adjuvant chemotherapy: Results of Children’s Oncology Group trial A9961. Neuro-Oncology 2012, 15, 97–103. [Google Scholar] [CrossRef] [Green Version]
- Joo, W.D.; Visintin, I.; Mor, G. Targeted cancer therapy—Are the days of systemic chemotherapy numbered? Maturitas 2013, 76, 308–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, E.M.; Ashley, D.; Landi, D. Current medulloblastoma subgroup specific clinical trials. Transl. Pediatr. 2020, 9, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Frappaz, D.; Barritault, M.; Montané, L.; Laigle-Donadey, F.; Chinot, O.; Le Rhun, E.; Bonneville-Levard, A.; Hottinger, A.F.; Meyronnet, D.; Bidaux, A.-S.; et al. MEVITEM—A phase I/II trial of vismodegib + temozolomide vs temozolomide in patients with recurrent/refractory medulloblastoma with Sonic Hedgehog pathway activation. Neuro-Oncology 2021, 23, 1949–1960. [Google Scholar] [CrossRef]
- Yauch, R.L.; Dijkgraaf, G.J.P.; Alicke, B.; Januario, T.; Ahn, C.P.; Holcomb, T.; Pujara, K.; Stinson, J.; Callahan, C.A.; Tang, T.; et al. Smoothened Mutation Confers Resistance to a Hedgehog Pathway Inhibitor in Medulloblastoma. Science 2009, 326, 572–574. [Google Scholar] [CrossRef] [Green Version]
- Thompson, E.M.; Keir, S.T.; Venkatraman, T.; Lascola, C.; Yeom, K.W.; Nixon, A.B.; Liu, Y.; Picard, D.; Remke, M.; Bigner, D.D.; et al. The role of angiogenesis in Group 3 medulloblastoma pathogenesis and survival. Neuro-Oncology 2017, 19, 1217–1227. [Google Scholar] [CrossRef]
- Giuliano, S.; Cormerais, Y.; Dufies, M.; Grépin, R.; Colosetti, P.; Belaid, A.; Parola, J.; Martin, A.R.; Lacas-Gervais, S.; Mazure, N.M.; et al. Resistance to sunitinib in renal clear cell carcinoma results from sequestration in lysosomes and inhibition of the autophagic flux. Autophagy 2015, 11, 1891–1904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeWire, M.; Fouladi, M.; Turner, D.C.; Wetmore, C.; Hawkins, C.; Jacobs, C.; Yuan, Y.; Liu, D.; Goldman, S.; Fisher, P.; et al. An open-label, two-stage, phase II study of bevacizumab and lapatinib in children with recurrent or refractory ependymoma: A collaborative ependymoma research network study (CERN). J. Neuro-Oncol. 2015, 123, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Grill, J.; Massimino, M.; Bouffet, E.; Azizi, A.; McCowage, G.; Cañete, A.; Saran, F.; Le Deley, M.-C.; Varlet, P.; Morgan, P.; et al. Phase II, Open-Label, Randomized, Multicenter Trial (HERBY) of Bevacizumab in Pediatric Patients With Newly Diagnosed High-Grade Glioma. J. Clin. Oncol. 2018, 36, 951–958. [Google Scholar] [CrossRef]
- Han, K.; Peyret, T.; Quartino, A.; Gosselin, N.H.; Gururangan, S.; Casanova, M.; Merks, J.H.M.; Massimino, M.; Grill, J.; Daw, N.C.; et al. Bevacizumab dosing strategy in paediatric cancer patients based on population pharmacokinetic analysis with external validation. Br. J. Clin. Pharmacol. 2015, 81, 148–160. [Google Scholar] [CrossRef] [Green Version]
- Venkatramani, R.; Malogolowkin, M.; Davidson, T.B.; May, W.; Sposto, R.; Mascarenhas, L. A Phase I Study of Vincristine, Irinotecan, Temozolomide and Bevacizumab (Vitb) in Pediatric Patients with Relapsed Solid Tumors. PLoS ONE 2013, 8, e68416. [Google Scholar] [CrossRef] [PubMed]
- Venkatramani, R.; Malogolowkin, M.H.; Mascarenhas, L. Treatment of multiply relapsed wilms tumor with vincristine, irinotecan, temozolomide and bevacizumab. Pediatr. Blood Cancer 2013, 61, 756–759. [Google Scholar] [CrossRef] [PubMed]
- Wetmore, C.; Daryani, V.M.; Billups, C.A.; Boyett, J.M.; Leary, S.; Tanos, R.; Goldsmith, K.C.; Stewart, C.F.; Blaney, S.M.; Gajjar, A. Phase II evaluation of sunitinib in the treatment of recurrent or refractory high-grade glioma or ependymoma in children: A children’s Oncology Group Study ACNS1021. Cancer Med. 2016, 5, 1416–1424. [Google Scholar] [CrossRef]
- Geller, J.I.; Fox, E.; Turpin, B.K.; Goldstein, S.L.; Liu, X.; Minard, C.G.; Kudgus, R.A.; Reid, J.M.; Berg, S.L.; Weigel, B.J. A study of axitinib, a VEGF receptor tyrosine kinase inhibitor, in children and adolescents with recurrent or refractory solid tumors: A Children’s Oncology Group phase 1 and pilot consortium trial (ADVL1315). Cancer 2018, 124, 4548–4555. [Google Scholar] [CrossRef] [Green Version]
- Pilot Study of Cabozantinib for Recurrent or Progressive High-Grade Glioma in Children. Available online: https://clinicaltrials.gov/ct2/show/NCT02885324 (accessed on 2 December 2021).
- Cabozantinib in Combination With 13-cis-Retinoic Acid in Children with Relapsed or Refractory Solid Tumors. Available online: https://clinicaltrials.gov/ct2/show/NCT03611595 (accessed on 2 December 2021).
- European Medicine Agency. Guidelines on the Need for Non-Clinical Testing in Juvenile Animals of Pharmaceuticals for Paediatric Indications. 2008. Available online: https://www.ema.europa.eu/en/need-non-clinical-testing-juvenile-animals-human-pharmaceuticals-paediatric-indications (accessed on 2 December 2021).
- Weksler, B.B.; Subileau, E.A.; Perrière, N.; Charneau, P.; Holloway, C.J.; Leveque, M.; Tricoire-Leignel, H.; Nicotra, A.; Bourdoulous, S.; Turowski, P.; et al. Blood-brain barrier-specific properties of a human adult brain endothelial cell line. FASEB J. 2005, 19, 1872–1874. [Google Scholar] [CrossRef]
- Zhang, X.; Huang, C.T.; Chen, J.; Pankratz, M.T.; Xi, J.; Li, J.; Yang, Y.; LaVaute, T.M.; Li, X.-J.; Ayala, M.; et al. Pax6 Is a Human Neuroectoderm Cell Fate Determinant. Cell Stem Cell 2010, 7, 90–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daubon, T.; Léon, C.; Clarke, K.; Andrique, L.; Salabert, L.; Darbo, E.; Pineau, R.; Guérit, S.; Maitre, M.; Dedieu, S.; et al. Deciphering the complex role of thrombospondin-1 in glioblastoma development. Nat. Commun. 2019, 10, 1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edelstein, A.D.; Amodaj, N.; Hoover, K.H.; Vale, R.D.; Stuurman, N. Computer Control of Microscopes Using µManager. Curr. Protoc. Mol. Biol. 2010, 92, 14.20.1–14.20.17. [Google Scholar] [CrossRef] [Green Version]
- McQuin, C.; Goodman, A.; Chernyshev, V.; Kamentsky, L.; Cimini, B.A.; Karhohs, K.W.; Doan, M.; Ding, L.; Rafelski, S.M.; Thirstrup, D.; et al. CellProfiler 3.0: Next-generation image processing for biology. PLoS Biol. 2018, 16, e2005970. [Google Scholar] [CrossRef] [Green Version]
- Le Guelte, A.; Galan-Moya, E.M.; Dwyer, J.; Treps, L.; Kettler, G.; Hebda, J.K.; Dubois, S.; Auffray, C.; Chneiweiss, H.; Bidere, N.; et al. Semaphorin 3A elevates endothelial cell permeability through PP2A inactivation. J. Cell Sci. 2012, 125, 4137–4146. [Google Scholar] [CrossRef] [Green Version]
- Gustave Roussy, C.C. Study of Sequential High-dose Chemotherapy in Children with High Risk Medulloblastoma (HR MB-5). 2014. Available online: https://clinicaltrials.gov/ct2/show/NCT02025881 (accessed on 2 December 2021).
- Ivanov, D.P.; Coyle, B.; Walker, D.A.; Grabowska, A.M. In Vitro models of medulloblastoma: Choosing the right tool for the job. J. Biotechnol. 2016, 236, 10–25. [Google Scholar] [CrossRef] [PubMed]
- Badisa, R.B.; Darling-Reed, S.F.; Joseph, P.; Cooperwood, J.S.; Latinwo, L.M.; Goodman, C.B. Selective cytotoxic activities of two novel synthetic drugs on human breast carcinoma MCF-7 cells. Anticancer Res. 2009, 29, 2993–2996. [Google Scholar] [PubMed]
- Ehrhardt, M.; Craveiro, R.B.; Velz, J.; Olschewski, M.; Casati, A.; Schönberger, S.; Pietsch, T.; Dilloo, D. The FDA approved PI 3K inhibitor GDC -0941 enhances in vitro the anti-neoplastic efficacy of Axitinib against c-myc-amplified high-risk medulloblastoma. J. Cell. Mol. Med. 2018, 22, 2153–2161. [Google Scholar] [CrossRef] [Green Version]
- Schwinn, S.; Mokhtari, Z.; Thusek, S.; Schneider, T.; Sirén, A.-L.; Tiemeyer, N.; Caruana, I.; Miele, E.; Schlegel, P.G.; Beilhack, A.; et al. Cytotoxic effects and tolerability of gemcitabine and axitinib in a xenograft model for c-myc amplified medulloblastoma. Sci. Rep. 2021, 11, 14062. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, P. The Laboratory Rat: Relating Its Age with Human’s. Int. J. Prev. Med. 2013, 4, 624–630. [Google Scholar]
- Poller, B.; Iusuf, D.; Sparidans, R.W.; Wagenaar, E.; Beijnen, J.H.; Schinkel, A.H. Differential Impact of P-Glycoprotein (ABCB1) and Breast Cancer Resistance Protein (ABCG2) on Axitinib Brain Accumulation and Oral Plasma Pharmacokinetics. Drug Metab. Dispos. 2011, 39, 729–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slobbe, P.; Poot, A.J.; Haumann, R.; Schuit, R.C.; Windhorst, A.D.; van Dongen, G.A. Two anti-angiogenic TKI-PET tracers, [11 C]axitinib and [11 C]nintedanib: Radiosynthesis, in vivo metabolism and initial biodistribution studies in rodents. Nucl. Med. Biol. 2016, 43, 612–624. [Google Scholar] [CrossRef]
- Panigrahy, D.; Kaipainen, A.; Butterfield, C.E.; Chaponis, D.M.; Laforme, A.M.; Folkman, J.; Kieran, M.W. Inhibition of tumor angiogenesis by oral etoposide. Exp. Ther. Med. 2010, 1, 739–746. [Google Scholar] [CrossRef] [Green Version]
- André, N.; Tsai, K.; Carré, M.; Pasquier, E. Metronomic Chemotherapy: Direct Targeting of Cancer Cells after all? Trends Cancer 2017, 3, 319–325. [Google Scholar] [CrossRef]
- Cavalli, F.M.; Remke, M.; Rampasek, L.; Peacock, J.; Shih, D.J.H.; Luu, B.; Garzia, L.; Torchia, J.; Nor, C.; Morrissy, S.; et al. Intertumoral Heterogeneity within Medulloblastoma Subgroups. Cancer Cell 2017, 31, 737–754.e6. [Google Scholar] [CrossRef] [Green Version]
- Suri, A.; Bailey, A.W.; Tavares, M.T.; Gunosewoyo, H.; Dyer, C.P.; Grupenmacher, A.T.; Piper, D.R.; Horton, R.A.; Tomita, T.; Kozikowski, A.P.; et al. Evaluation of Protein Kinase Inhibitors with PLK4 Cross-Over Potential in a Pre-Clinical Model of Cancer. Int. J. Mol. Sci. 2019, 20, 2112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, Y.; Gharbi, N.; Yuan, X.; Olsen, J.R.; Blicher, P.; Dalhus, B.; Brokstad, K.A.; Lin, B.; Øyan, A.M.; Zhang, W.; et al. Axitinib blocks Wnt/β-catenin signaling and directs asymmetric cell division in cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 9339–9344. [Google Scholar] [CrossRef] [Green Version]
- Davson, H.; Spaziani, E. The blood-brain barrier and the extracellular space of brain. J. Physiol. 1959, 149, 135–143. [Google Scholar] [CrossRef] [Green Version]
- Davson, H. Review lecture. The blood-brain barrier. J. Physiol. 1976, 255, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Reyner, E.L.; Sevidal, S.; West, M.A.; Clouser-Roche, A.; Freiwald, S.; Fenner, K.; Ullah, M.; Lee, C.A.; Smith, B.J. In Vitro Characterization of Axitinib Interactions with Human Efflux and Hepatic Uptake Transporters: Implications for Disposition and Drug Interactions. Drug Metab. Dispos. 2013, 41, 1575–1583. [Google Scholar] [CrossRef] [Green Version]
- Lu, L.; Saha, D.; Martuza, R.L.; Rabkin, S.; Wakimoto, H. Single agent efficacy of the VEGFR kinase inhibitor axitinib in preclinical models of glioblastoma. J. Neuro-Oncol. 2014, 121, 91–100. [Google Scholar] [CrossRef] [Green Version]
- Liebner, S.; Corada, M.; Bangsow, T.; Babbage, J.; Taddei, A.; Czupalla, C.J.; Reis, M.; Felici, A.; Wolburg, H.; Fruttiger, M.; et al. Wnt/β-catenin signaling controls development of the blood–brain barrier. J. Cell Biol. 2008, 183, 409–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stenman, J.M.; Rajagopal, J.; Carroll, T.J.; Ishibashi, M.; McMahon, J.; McMahon, A.P. Canonical Wnt Signaling Regulates Organ-Specific Assembly and Differentiation of CNS Vasculature. Science 2008, 322, 1247–1250. [Google Scholar] [CrossRef]
- Corada, M.; Orsenigo, F.; Bhat, G.P.; Conze, L.L.; Breviario, F.; Cunha, S.I.; Claesson-Welsh, L.; Beznoussenko, G.V.; Mironov, A.A.; Bacigaluppi, M.; et al. Fine-Tuning of Sox17 and Canonical Wnt Coordinates the Permeability Properties of the Blood-Brain Barrier. Circ. Res. 2019, 124, 511–525. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Normalization of Tumor Vasculature: An Emerging Concept in Antiangiogenic Therapy. Science 2005, 307, 58–62. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Normal Cells | Medulloblastoma Cells | |||||
|---|---|---|---|---|---|---|
| IC50 (µM) | HDF | C8D1A | DAOY | HD-MB03 | D458 | CHLA-01-Med |
| Sunitinib | 1.8 ± 0.04 | 5.7 ± 2.2 | 7.9 ± 5.5 | 4.6 ± 0.3 | 4.2 ± 0.3 | 7.5 ± 0.8 |
| Cabozantinib | 4.1 ± 1.9 | 4 ± 2.2 | 8.3 ± 1.2 | 0.78 ± 0.2 | 5.1 ± 1.6 | 11.4 ± 2.1 |
| Carbo/Eto | 1.6 ± 1.1 | 3 ± 0.8 | 1 ± 0.4 | 0.16 ± 0.07 | 0.56 ± 0.7 | 0.11 ± 0.08 |
| Axitinib | >100 | >100 | 2.3 ± 1.5 | 0.47 ± 0.2 | 0.49 ± 0.4 | 3.9 ± 0.23 |
| Specificity Index | Relative to HDF | Relative to C8D1A | ||||||
|---|---|---|---|---|---|---|---|---|
| DAOY | HD-MB03 | D458 | CHLA-01-Med | DAOY | HDMB | D458 | CHLA-01-Med | |
| Sunitinib | 0.23 | 0.39 | 0.43 | 0.24 | 0.72 | 1.24 | 1.36 | 0.76 |
| Cabozantinib | 0.49 | 5.26 | 0.80 | 0.36 | 0.48 | 5.13 | 0.78 | 0.35 |
| Carbo/Eto | 1.60 | 10.00 | 2.86 | 14.3 | 3.00 | 18.75 | 5.36 | 26.8 |
| Axitinib | >40 | >200 | >200 | >25 | >40 | >200 | >200 | >25 |
| IC50 (µM) | DAOY | HD-MB03 | D458 | HDF | C8-D1A |
|---|---|---|---|---|---|
| Etoposide | 0.2 | 0.09 ± 0.006 | 0.52 ± 0.15 | 0.06 ± 2 | 2.2 ± 0.6 |
| Carboplatin | >20 | >20 | >20 | >20 | >20 |
| Etoposide/carboplatin | 1 ± 0.41 | 0.17 ± 0.05 | 0.58 ± 0.51 | 2 ± 1.1 | 3 ± 0.8 |
| Gene/Best Cutoff | Group 3 (113 Patients) | Group 4 (264 Patients) | Shh (172 Patients) |
|---|---|---|---|
| cKit | p = 0.04 | p = 0.083 | p = 8.7 × 10−5 |
| PDGFRA | p = 0.012 | p = 0.061 | p = 6.7 × 10−5 |
| PDGFRB | p = 0.017 | p = 0.04 | p = 5 × 10−8 |
| VEGFR1 | p = 0.041 | p = 5.5 × 10−3 | p = 4.8 × 10−3 |
| VEGFR2 | p = 0.325 | p = 0.043 | p = 5.2 × 10−3 |
| VEGFR3 | p = 0.075 | p = 0.1 | p = 0.015 |
| Score | 0 | −2 | −5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pagnuzzi-Boncompagni, M.; Picco, V.; Vial, V.; Planas-Bielsa, V.; Vandenberghe, A.; Daubon, T.; Derieppe, M.-A.; Montemagno, C.; Durivault, J.; Grépin, R.; et al. Antiangiogenic Compound Axitinib Demonstrates Low Toxicity and Antitumoral Effects against Medulloblastoma. Cancers 2022, 14, 70. https://doi.org/10.3390/cancers14010070
Pagnuzzi-Boncompagni M, Picco V, Vial V, Planas-Bielsa V, Vandenberghe A, Daubon T, Derieppe M-A, Montemagno C, Durivault J, Grépin R, et al. Antiangiogenic Compound Axitinib Demonstrates Low Toxicity and Antitumoral Effects against Medulloblastoma. Cancers. 2022; 14(1):70. https://doi.org/10.3390/cancers14010070
Chicago/Turabian StylePagnuzzi-Boncompagni, Marina, Vincent Picco, Valérie Vial, Victor Planas-Bielsa, Ashaina Vandenberghe, Thomas Daubon, Marie-Alix Derieppe, Christopher Montemagno, Jérôme Durivault, Renaud Grépin, and et al. 2022. "Antiangiogenic Compound Axitinib Demonstrates Low Toxicity and Antitumoral Effects against Medulloblastoma" Cancers 14, no. 1: 70. https://doi.org/10.3390/cancers14010070