
Computational Screening of Anti-Cancer Drugs Identifies a New BRCA Independent Gene Expression Signature to Predict Breast Cancer Sensitivity to Cisplatin

 , , , , ,
, , , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Datasets
2.2. Deriving Drug Efficacy Signatures
2.3. Scoring Samples Using the Singscore and Stingscore Methods
2.4. Training Prediction Models
2.5. Test Prediction Models
2.6. Gene-Set Enrichment Analysis
2.7. In Vivo Experiment
2.8. Single-Cell Suspension Preparation
2.9. mRNA Extraction and Bulk RNA-Seq
3. Results
3.1. Generation of Drug Efficacy Signatures with Training Data Sets
3.2. Assessing Drug Similarity Based on Observed Response and Prediction
3.3. Computational Validation Using Independent Testing Datasets
3.4. Validation of Response Predictions in Patient-Derived Xenografts
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dawson, S.J.; Rueda, O.M.; Aparicio, S.; Caldas, C. A new genome-driven integrated classification of breast cancer and its implications. EMBO J. 2013, 32, 617–628. [Google Scholar] [CrossRef] [Green Version]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Parker, J.S.; Mullins, M.; Cheang, M.C.; Leung, S.; Voduc, D.; Vickery, T.; Davies, S.; Fauron, C.; He, X.; Hu, Z.; et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009, 27, 1160–1167. [Google Scholar] [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modeling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Millikan, R.C.; Newman, B.; Tse, C.K.; Moorman, P.G.; Conway, K.; Dressler, L.G.; Smith, L.V.; Labbok, M.H.; Geradts, J.; Bensen, J.T.; et al. Epidemiology of basal-like breast cancer. Breast Cancer Res. Treat. 2008, 109, 123–139. [Google Scholar] [CrossRef]
- Cheang, M.C.; Chia, S.K.; Voduc, D.; Gao, D.; Leung, S.; Snider, J.; Watson, M.; Davies, S.; Bernard, P.S.; Parker, J.S.; et al. Ki67 index, HER2 status, and prognosis of patients with luminal B breast cancer. J. Natl. Cancer Inst. 2009, 101, 736–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, T.O.; Parker, J.S.; Leung, S.; Voduc, D.; Ebbert, M.; Vickery, T.; Davies, S.R.; Snider, J.; Stijleman, I.J.; Reed, J.; et al. A comparison of PAM50 intrinsic subtyping with immunohistochemistry and clinical prognostic factors in tamoxifen-treated estrogen receptor-positive breast cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010, 16, 5222–5232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heiser, L.M.; Sadanandam, A.; Kuo, W.L.; Benz, S.C.; Goldstein, T.C.; Ng, S.; Gibb, W.J.; Wang, N.J.; Ziyad, S.; Tong, F.; et al. Subtype and pathway specific responses to anticancer compounds in breast cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 2724–2729. [Google Scholar] [CrossRef] [Green Version]
- Waks, A.G.; Winer, E.P. Breast Cancer Treatment: A Review. JAMA 2019, 321, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Loi, S.; Haibe-Kains, B.; Desmedt, C.; Lallemand, F.; Tutt, A.M.; Gillet, C.; Ellis, P.; Harris, A.; Bergh, J.; Foekens, J.A.; et al. Definition of clinically distinct molecular subtypes in estrogen receptor-positive breast carcinomas through genomic grade. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2007, 25, 1239–1246. [Google Scholar] [CrossRef]
- Badve, S.; Dabbs, D.J.; Schnitt, S.J.; Baehner, F.L.; Decker, T.; Eusebi, V.; Fox, S.B.; Ichihara, S.; Jacquemier, J.; Lakhani, S.R.; et al. Basal-like and triple-negative breast cancers: A critical review with an emphasis on the implications for pathologists and oncologists. Mod. Pathol. 2010, 24, 157. [Google Scholar] [CrossRef] [Green Version]
- Arnedos, M.; Vicier, C.; Loi, S.; Lefebvre, C.; Michiels, S.; Bonnefoi, H.; Andre, F. Precision medicine for metastatic breast cancer--limitations and solutions. Nat. Rev. Clin. Oncol. 2015, 12, 693–704. [Google Scholar] [CrossRef]
- Stephens, P.J.; Tarpey, P.S.; Davies, H.; Van Loo, P.; Greenman, C.; Wedge, D.C.; Nik-Zainal, S.; Martin, S.; Varela, I.; Bignell, G.R.; et al. The landscape of cancer genes and mutational processes in breast cancer. Nature 2012, 486, 400–404. [Google Scholar] [CrossRef]
- Tutt, A.; Tovey, H.; Cheang, M.C.U.; Kernaghan, S.; Kilburn, L.; Gazinska, P.; Owen, J.; Abraham, J.; Barrett, S.; Barrett-Lee, P.; et al. Carboplatin in BRCA1/2-mutated and triple-negative breast cancer BRCAness subgroups: The TNT Trial. Nat. Med. 2018, 24, 628–637. [Google Scholar] [CrossRef] [Green Version]
- Malone, E.R.; Oliva, M.; Sabatini, P.J.B.; Stockley, T.L.; Siu, L.L. Molecular profiling for precision cancer therapies. Genome Med. 2020, 12, 8. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Lu, P.; Beeraka, N.M.; Sukocheva, O.A.; Madhunapantula, S.V.; Liu, J.; Sinelnikov, M.Y.; Nikolenko, V.N.; Bulygin, K.V.; Mikhaleva, L.M.; et al. Mitochondrial mutations and mitoepigenetics: Focus on regulation of oxidative stress-induced responses in breast cancers. Semin. Cancer Biol. 2020; in press. [Google Scholar] [CrossRef]
- Su, M.; Zhang, Z.; Zhou, L.; Han, C.; Huang, C.; Nice, E.C. Proteomics, Personalized Medicine and Cancer. Cancers 2021, 13, 2512. [Google Scholar] [CrossRef]
- Castelli, F.A.; Rosati, G.; Moguet, C.; Fuentes, C.; Marrugo-Ramirez, J.; Lefebvre, T.; Volland, H.; Merkoci, A.; Simon, S.; Fenaille, F.; et al. Metabolomics for personalized medicine: The input of analytical chemistry from biomarker discovery to point-of-care tests. Anal. Bioanal. Chem. 2022, 414, 759–789. [Google Scholar] [CrossRef]
- Daemen, A.; Griffith, O.L.; Heiser, L.M.; Wang, N.J.; Enache, O.M.; Sanborn, Z.; Pepin, F.; Durinck, S.; Korkola, J.E.; Griffith, M.; et al. Modeling precision treatment of breast cancer. Genome Biol. 2013, 14, R110. [Google Scholar] [CrossRef] [Green Version]
- Geeleher, P.; Cox, N.J.; Huang, R.S. Clinical drug response can be predicted using baseline gene expression levels and in vitro drug sensitivity in cell lines. Genome Biol. 2014, 15, R47. [Google Scholar] [CrossRef] [Green Version]
- Iorio, F.; Knijnenburg, T.A.; Vis, D.J.; Bignell, G.R.; Menden, M.P.; Schubert, M.; Aben, N.; Goncalves, E.; Barthorpe, S.; Lightfoot, H.; et al. A Landscape of Pharmacogenomic Interactions in Cancer. Cell 2016, 166, 740–754. [Google Scholar] [CrossRef] [Green Version]
- Aben, N.; Vis, D.J.; Michaut, M.; Wessels, L.F. TANDEM: A two-stage approach to maximize interpretability of drug response models based on multiple molecular data types. Bioinformatics 2016, 32, i413–i420. [Google Scholar] [CrossRef] [Green Version]
- Costello, J.C.; Heiser, L.M.; Georgii, E.; Gonen, M.; Menden, M.P.; Wang, N.J.; Bansal, M.; Ammad-ud-din, M.; Hintsanen, P.; Khan, S.A.; et al. A community effort to assess and improve drug sensitivity prediction algorithms. Nat. Biotechnol. 2014, 32, 1202–1212. [Google Scholar] [CrossRef]
- Hess, K.R.; Anderson, K.; Symmans, W.F.; Valero, V.; Ibrahim, N.; Mejia, J.A.; Booser, D.; Theriault, R.L.; Buzdar, A.U.; Dempsey, P.J.; et al. Pharmacogenomic predictor of sensitivity to preoperative chemotherapy with paclitaxel and fluorouracil, doxorubicin, and cyclophosphamide in breast cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2006, 24, 4236–4244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatzis, C.; Pusztai, L.; Valero, V.; Booser, D.J.; Esserman, L.; Lluch, A.; Vidaurre, T.; Holmes, F.; Souchon, E.; Wang, H.; et al. A genomic predictor of response and survival following taxane-anthracycline chemotherapy for invasive breast cancer. JAMA 2011, 305, 1873–1881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGrail, D.J.; Lin, C.C.; Garnett, J.; Liu, Q.; Mo, W.; Dai, H.; Lu, Y.; Yu, Q.; Ju, Z.; Yin, J.; et al. Improved prediction of PARP inhibitor response and identification of synergizing agents through use of a novel gene expression signature generation algorithm. NPJ Syst. Biol. Appl. 2017, 3, 8. [Google Scholar] [CrossRef] [Green Version]
- Foroutan, M.; Bhuva, D.D.; Lyu, R.; Horan, K.; Cursons, J.; Davis, M.J. Single sample scoring of molecular phenotypes. BMC Bioinform. 2018, 19, 404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013, 41, D955–D961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basu, A.; Bodycombe, N.E.; Cheah, J.H.; Price, E.V.; Liu, K.; Schaefer, G.I.; Ebright, R.Y.; Stewart, M.L.; Ito, D.; Wang, S.; et al. An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules. Cell 2013, 154, 1151–1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klijn, C.; Durinck, S.; Stawiski, E.W.; Haverty, P.M.; Jiang, Z.; Liu, H.; Degenhardt, J.; Mayba, O.; Gnad, F.; Liu, J.; et al. A comprehensive transcriptional portrait of human cancer cell lines. Nat. Biotechnol. 2015, 33, 306–312. [Google Scholar] [CrossRef]
- Haverty, P.M.; Lin, E.; Tan, J.; Yu, Y.; Lam, B.; Lianoglou, S.; Neve, R.M.; Martin, S.; Settleman, J.; Yauch, R.L.; et al. Reproducible pharmacogenomic profiling of cancer cell line panels. Nature 2016, 533, 333–337. [Google Scholar] [CrossRef]
- Mpindi, J.P.; Yadav, B.; Ostling, P.; Gautam, P.; Malani, D.; Murumagi, A.; Hirasawa, A.; Kangaspeska, S.; Wennerberg, K.; Kallioniemi, O.; et al. Consistency in drug response profiling. Nature 2016, 540, E5–E6. [Google Scholar] [CrossRef]
- Bruna, A.; Rueda, O.M.; Greenwood, W.; Batra, A.S.; Callari, M.; Batra, R.N.; Pogrebniak, K.; Sandoval, J.; Cassidy, J.W.; Tufegdzic-Vidakovic, A.; et al. A Biobank of Breast Cancer Explants with Preserved Intra-tumor Heterogeneity to Screen Anticancer Compounds. Cell 2016, 167, 260–274.e222. [Google Scholar] [CrossRef] [Green Version]
- Rahman, M.; Jackson, L.K.; Johnson, W.E.; Li, D.Y.; Bild, A.H.; Piccolo, S.R. Alternative preprocessing of RNA-Sequencing data in The Cancer Genome Atlas leads to improved analysis results. Bioinformatics 2015, 31, 3666–3672. [Google Scholar] [CrossRef]
- Birkbak, N.J.; Li, Y.; Pathania, S.; Greene-Colozzi, A.; Dreze, M.; Bowman-Colin, C.; Sztupinszki, Z.; Krzystanek, M.; Diossy, M.; Tung, N.; et al. Overexpression of BLM promotes DNA damage and increased sensitivity to platinum salts in triple-negative breast and serous ovarian cancers. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2018, 29, 903–909. [Google Scholar] [CrossRef]
- Ter Brugge, P.; Kristel, P.; Van Der Burg, E.; Boon, U.; De Maaker, M.; Lips, E.; Mulder, L.; De Ruiter, J.; Moutinho, C.; Gevensleben, H.; et al. Mechanisms of Therapy Resistance in Patient-Derived Xenograft Models of BRCA1-Deficient Breast Cancer. J. Natl. Cancer Inst. 2016, 108, djw148. [Google Scholar] [CrossRef]
- Fallahi-Sichani, M.; Honarnejad, S.; Heiser, L.M.; Gray, J.W.; Sorger, P.K. Metrics other than potency reveal systematic variation in responses to cancer drugs. Nat. Chem. Biol. 2013, 9, 708–714. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. The Subread aligner: Fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013, 41, e108. [Google Scholar] [CrossRef]
- Foroutan, M.B.; Bhuva, D.D.; Lyu, R. Singscore: Rank-Based Single-Sample Gene Set Scoring Method; R package version 1.0.0; 2018. Available online: https://davislaboratory.github.io/singscore/ (accessed on 10 April 2022).
- Bhuva, D.D.; Cursons, J.; Davis, M.J. Stable gene expression for normalisation and single-sample scoring. Nucleic Acids Res. 2020, 48, e113. [Google Scholar] [CrossRef]
- Cortes, C.V.; Vladimir, N. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdottir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Roswall, P.; Bocci, M.; Bartoschek, M.; Li, H.; Kristiansen, G.; Jansson, S.; Lehn, S.; Sjolund, J.; Reid, S.; Larsson, C.; et al. Microenvironmental control of breast cancer subtype elicited through paracrine platelet-derived growth factor-CC signaling. Nat. Med. 2018, 24, 463–473. [Google Scholar] [CrossRef]
- Berthelet, J.; Wimmer, V.C.; Whitfield, H.J.; Serrano, A.; Boudier, T.; Mangiola, S.; Merdas, M.; El-Saafin, F.; Baloyan, D.; Wilcox, J.; et al. The site of breast cancer metastases dictates their clonal composition and reversible transcriptomic profile. Sci. Adv. 2021, 7, eabf4408. [Google Scholar] [CrossRef]
- Charafe-Jauffret, E.; Ginestier, C.; Bertucci, F.; Cabaud, O.; Wicinski, J.; Finetti, P.; Josselin, E.; Adelaide, J.; Nguyen, T.T.; Monville, F.; et al. ALDH1-positive cancer stem cells predict engraftment of primary breast tumors and are governed by a common stem cell program. Cancer Res. 2013, 73, 7290–7300. [Google Scholar] [CrossRef] [Green Version]
- Ben-Hur, A.; Horn, D.; Siegelmann, H.T.; Vapnik, V. Support vector clustering. J. Mach. Learn. Res. 2001, 2, 125–137. [Google Scholar]
- Haibe-Kains, B.; El-Hachem, N.; Birkbak, N.J.; Jin, A.C.; Beck, A.H.; Aerts, H.; Quackenbush, J. Inconsistency in large pharmacogenomic studies. Nature 2013, 504, 389–393. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Umbach, D.M.; Krahn, J.M.; Shats, I.; Li, X.; Li, L. Predicting tumor response to drugs based on gene-expression biomarkers of sensitivity learned from cancer cell lines. BMC Genom. 2021, 22, 272. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Ma, L.; Paik, H.; Sirota, M.; Wei, W.; Chua, M.S.; So, S.; Butte, A.J. Reversal of cancer gene expression correlates with drug efficacy and reveals therapeutic targets. Nat. Commun. 2017, 8, 16022. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Lv, Q.L.; Yu, J.; Hu, L.; Zhang, L.H.; Cheng, Y.; Zhou, H.H. Correlating transcriptional networks with pathological complete response following neoadjuvant chemotherapy for breast cancer. Breast Cancer Res. Treat. 2015, 151, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Cursons, J.; Pillman, K.A.; Scheer, K.G.; Gregory, P.A.; Foroutan, M.; Hediyeh-Zadeh, S.; Toubia, J.; Crampin, E.J.; Goodall, G.J.; Bracken, C.P.; et al. Combinatorial Targeting by MicroRNAs Co-ordinates Post-transcriptional Control of EMT. Cell Syst. 2018, 7, 77–91.e77. [Google Scholar] [CrossRef]
- Foroutan, M.; Cursons, J.; Hediyeh-Zadeh, S.; Thompson, E.W.; Davis, M.J. A Transcriptional Program for Detecting TGFbeta-Induced EMT in Cancer. Mol. Cancer Res. MCR 2017, 15, 619–631. [Google Scholar] [CrossRef] [Green Version]
- Bhuva, D.D.; Foroutan, M.; Xie, Y.; Lyu, R.; Cursons, J.; Davis, M.J. Using singscore to predict mutation status in acute myeloid leukemia from transcriptomic signatures. F1000Res 2019, 8, 776. [Google Scholar] [CrossRef]
- Cursons, J.; Souza-Fonseca-Guimaraes, F.; Foroutan, M.; Anderson, A.; Hollande, F.; Hediyeh-Zadeh, S.; Behren, A.; Huntington, N.D.; Davis, M.J. A Gene Signature Predicting Natural Killer Cell Infiltration and Improved Survival in Melanoma Patients. Cancer Immunol. Res. 2019, 7, 1162–1174. [Google Scholar] [CrossRef] [Green Version]
- Foroutan, M.; Molania, R.; Pfefferle, A.; Behrenbruch, C.; Scheer, S.; Kallies, A.; Speed, T.P.; Cursons, J.; Huntington, N.D. The Ratio of Exhausted to Resident Infiltrating Lymphocytes Is Prognostic for Colorectal Cancer Patient Outcome. Cancer Immunol. Res. 2021, 9, 1125–1140. [Google Scholar] [CrossRef]
- Jaeger, S.; Duran-Frigola, M.; Aloy, P. Drug sensitivity in cancer cell lines is not tissue-specific. Mol. Cancer 2015, 14, 40. [Google Scholar] [CrossRef] [Green Version]
- Yao, F.; Madani Tonekaboni, S.A.; Safikhani, Z.; Smirnov, P.; El-Hachem, N.; Freeman, M.; Manem, V.S.K.; Haibe-Kains, B. Tissue specificity of in vitro drug sensitivity. J. Am. Med. Inform. Assoc. 2018, 25, 158–166. [Google Scholar] [CrossRef]
- Khirade, M.F.; Lal, G.; Bapat, S.A. Derivation of a fifteen gene prognostic panel for six cancers. Sci. Rep. 2015, 5, 13248. [Google Scholar] [CrossRef] [Green Version]
- Cheng, W.Y.; Ou Yang, T.H.; Anastassiou, D. Biomolecular events in cancer revealed by attractor metagenes. PLoS Comput. Biol. 2013, 9, e1002920. [Google Scholar] [CrossRef] [Green Version]
- Koberle, B.; Schoch, S. Platinum Complexes in Colorectal Cancer and Other Solid Tumors. Cancers 2021, 13, 2073. [Google Scholar] [CrossRef]
- Sluyser, M.; Mester, J. Oncogenes homologous to steroid receptors? Nature 1985, 315, 546. [Google Scholar] [CrossRef]
- Alluri, P.G.; Speers, C.; Chinnaiyan, A.M. Estrogen receptor mutations and their role in breast cancer progression. Breast Cancer Res. 2014, 16, 494. [Google Scholar] [CrossRef] [Green Version]
- Recupero, D.; Daniele, L.; Marchio, C.; Molinaro, L.; Castellano, I.; Cassoni, P.; Righi, A.; Montemurro, F.; Sismondi, P.; Biglia, N.; et al. Spontaneous and pronase-induced HER2 truncation increases the trastuzumab binding capacity of breast cancer tissues and cell lines. J. Pathol. 2013, 229, 390–399. [Google Scholar] [CrossRef] [Green Version]
- Silver, D.P.; Richardson, A.L.; Eklund, A.C.; Wang, Z.C.; Szallasi, Z.; Li, Q.; Juul, N.; Leong, C.O.; Calogrias, D.; Buraimoh, A.; et al. Efficacy of Neoadjuvant Cisplatin in Triple-Negative Breast Cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 1145–1153. [Google Scholar] [CrossRef]
- Salehi, S.; Kabeer, F.; Ceglia, N.; Andronescu, M.; Williams, M.J.; Campbell, K.R.; Masud, T.; Wang, B.; Biele, J.; Brimhall, J.; et al. Clonal fitness inferred from time-series modelling of single-cell cancer genomes. Nature 2021, 595, 585–590. [Google Scholar] [CrossRef]
- Merino, D.; Weber, T.S.; Serrano, A.; Vaillant, F.; Liu, K.; Pal, B.; Di Stefano, L.; Schreuder, J.; Lin, D.; Chen, Y.; et al. Barcoding reveals complex clonal behavior in patient-derived xenografts of metastatic triple negative breast cancer. Nat. Commun. 2019, 10, 766. [Google Scholar] [CrossRef] [Green Version]
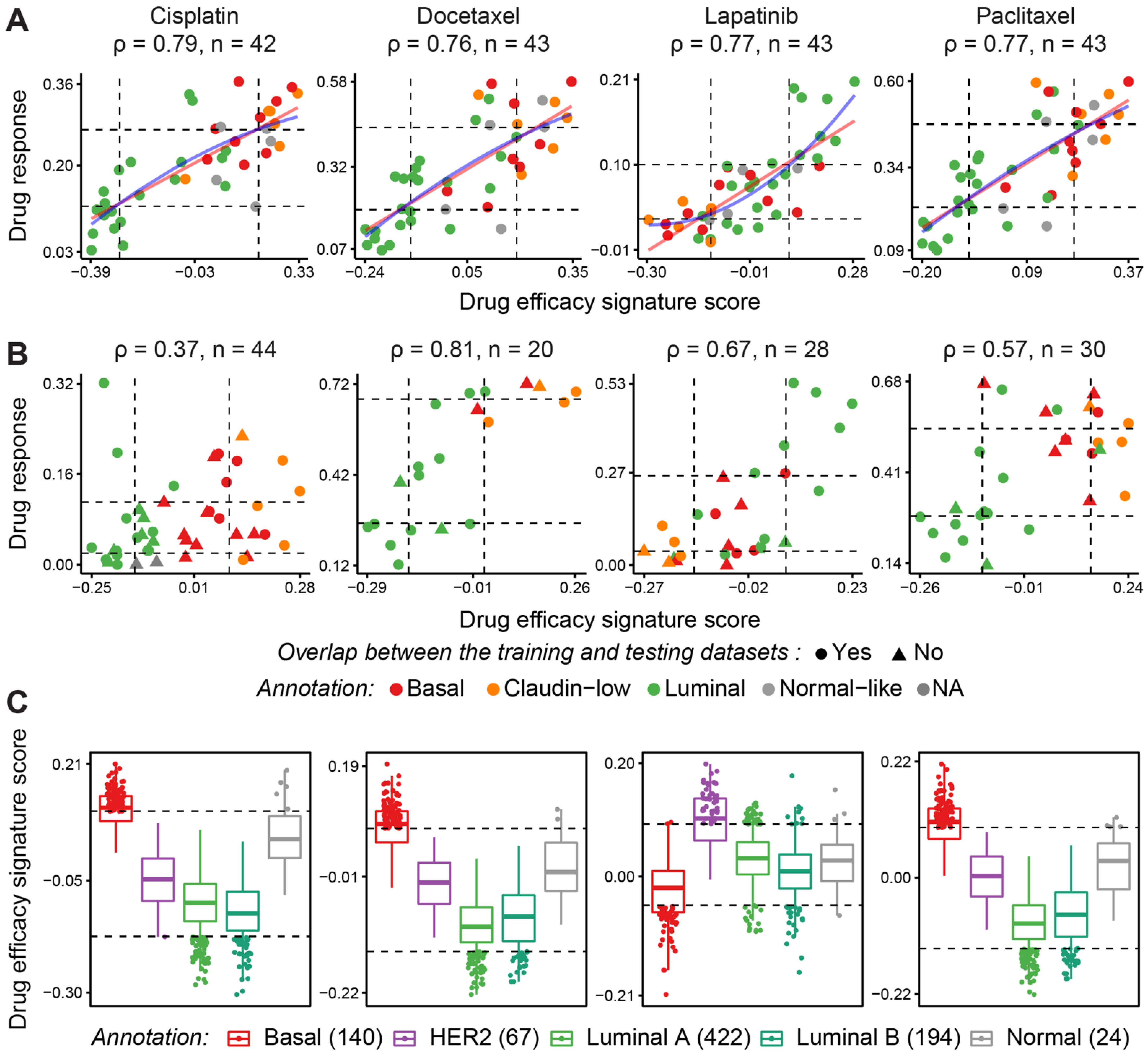




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Set | NSamples | Type of Sample | NDrugs | RNASeq | Microarray | Response Metrics | Use | Ref |
|---|---|---|---|---|---|---|---|---|
| Gray | 84 | Cell line | 90 | Yes | Yes | AUC | Train | [20] |
| CCLE | 60 | Cell line | 24 | Yes | Yes | AUC | Test | [4] |
| GDSC1000 | 50 | Cell line | 251 | No | Yes | AUC | Test | [29] |
| CTRPv2 | 40 | Cell line | 545 | From CCLE | No | AUC | Test | [30] |
| gCSI | 30 | Cell line | 16 | Yes | No | AUC | Test | [31,32] |
| FIMM | 21 | Cell line | 52 | From CCLE | No | AUC | [33] | |
| Caldas | 20 | PDTX-PDTC | 104 | No | Yes | AUC | Test | [34] |
| TCGA | 1102 | Patient | - | Yes | Yes | - | Test | GSE62944 [35] |
| GSE100925 | 50 | Patient | - | Yes | - | - | Test | GSE100925 |
| GSE103668 (cisplatin) | 21 TN | Clinical trial | 1 | No | Yes | Miller-Payne and RCB | Test | [36] |
| ONJCRI-PDX | 4 | PDX | 1 | Yes | No | NTR | Test | In-house |
| Jonkers-PDX | 3 | PDX | 1 | Yes | No | Proportion of remissions and resistance | Test | [37] |
| Drugs | GRAY | CCLE | CTRPv2 | FIMM | gCSI | GDSC1000 | Caldas | Confidence |
|---|---|---|---|---|---|---|---|---|
| AZD6244 | 0.72 | 0.42 | 0.42 | 0.28 | - | - | - | High |
| Bortezomib | 0.68 | - | 0.29 | 0.43 | 0.4 | 0.41 | 0.5 | High |
| Docetaxel | 0.76 | - | 0.8 | - | 0.48 | 0.43 | 0.43 | High |
| Doxorubicin | 0.78 | - | - | 0.28 | 0.6 | −0.05 | - | High |
| Erlotinib | 0.76 | 0.41 | 0.31 | −0.09 | 0.57 | 0.3 | −0.13 | High |
| Gefitinib | 0.74 | - | 0.37 | 0.22 | - | 0.33 | 0.47 | High |
| Gemcitabine | 0.75 | - | 0.35 | - | 0.48 | 0.19 | 0.3 | High |
| GSK1059615 | 0.73 | - | 0.49 | - | - | - | - | High |
| GSK1120212 | 0.78 | - | 0.54 | - | - | - | 0.19 | High |
| GSK461364 | 0.78 | - | 0.72 | - | - | - | - | High |
| Irinotecan | 0.8 | 0.13 | - | −0.13 | 0.57 | - | - | High |
| Lapatinib | 0.77 | 0.68 | 0.54 | 0.5 | 0.34 | 0.26 | 0.9 | High |
| MG-132 | 0.76 | - | 0.47 | - | - | 0.31 | - | High |
| Nutlin-3 | 0.74 | 0.22 | 0.42 | - | - | - | - | High |
| Paclitaxel | 0.77 | 0.36 | 0.61 | 0.37 | 0.42 | 0.12 | −0.1 | High |
| Panobinostat | 0.78 | 0.76 | 0.6 | 0.72 | - | - | - | High |
| Rapamycin | 0.7 | - | 0.44 | - | −0.17 | −0.05 | - | High |
| Topotecan | 0.76 | 0.6 | 0.36 | 0.15 | - | - | - | High |
| VX-680 | 0.69 | - | 0.52 | - | - | 0.21 | - | High |
| ZM-447439 | 0.7 | - | - | - | - | 0.28 | 0.46 | High |
| 5-FU | 0.77 | - | 0.31 | - | - | - | - | Medium |
| BIBW2992 | 0.79 | - | 0.4 | 0.39 | - | - | 0.37 | Medium |
| Cisplatin | 0.79 | - | - | - | - | 0.37 | 0.29 | Medium |
| Crizotinib | 0.75 | 0.21 | 0.36 | −0.04 | 0.39 | 0.03 | - | Medium |
| Etoposide | 0.75 | - | 0.38 | - | - | 0.21 | - | Medium |
| GSK2126458 | 0.73 | - | - | - | - | 0.33 | - | Medium |
| Methotrexate | 0.74 | - | 0.18 | 0.09 | - | 0.39 | - | Medium |
| Temsirolimus | 0.72 | - | - | 0.4 | - | 0.1 | - | Medium |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berthelet, J.; Foroutan, M.; Bhuva, D.D.; Whitfield, H.J.; El-Saafin, F.; Cursons, J.; Serrano, A.; Merdas, M.; Lim, E.; Charafe-Jauffret, E.; et al. Computational Screening of Anti-Cancer Drugs Identifies a New BRCA Independent Gene Expression Signature to Predict Breast Cancer Sensitivity to Cisplatin. Cancers 2022, 14, 2404. https://doi.org/10.3390/cancers14102404
Berthelet J, Foroutan M, Bhuva DD, Whitfield HJ, El-Saafin F, Cursons J, Serrano A, Merdas M, Lim E, Charafe-Jauffret E, et al. Computational Screening of Anti-Cancer Drugs Identifies a New BRCA Independent Gene Expression Signature to Predict Breast Cancer Sensitivity to Cisplatin. Cancers. 2022; 14(10):2404. https://doi.org/10.3390/cancers14102404
Chicago/Turabian StyleBerthelet, Jean, Momeneh Foroutan, Dharmesh D. Bhuva, Holly J. Whitfield, Farrah El-Saafin, Joseph Cursons, Antonin Serrano, Michal Merdas, Elgene Lim, Emmanuelle Charafe-Jauffret, and et al. 2022. "Computational Screening of Anti-Cancer Drugs Identifies a New BRCA Independent Gene Expression Signature to Predict Breast Cancer Sensitivity to Cisplatin" Cancers 14, no. 10: 2404. https://doi.org/10.3390/cancers14102404