
Alterations in Molecular Profiles Affecting Glioblastoma Resistance to Radiochemotherapy: Where Does the Good Go?


Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. The Landscape of Genetic and Genomic Alterations in GBMs
3. Mechanistic Evidence of GBM Resistance to RCT
3.1. Metabolism—Isocitrate Dehydrogenase (IDH) and the Warburg Effect
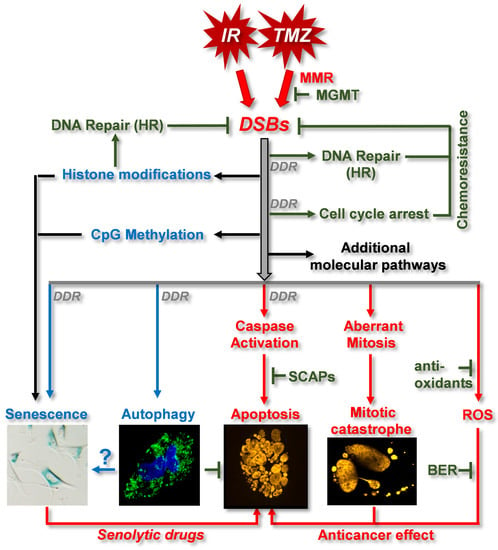
3.2. DNA Damage Response (DDR) and DNA Repair
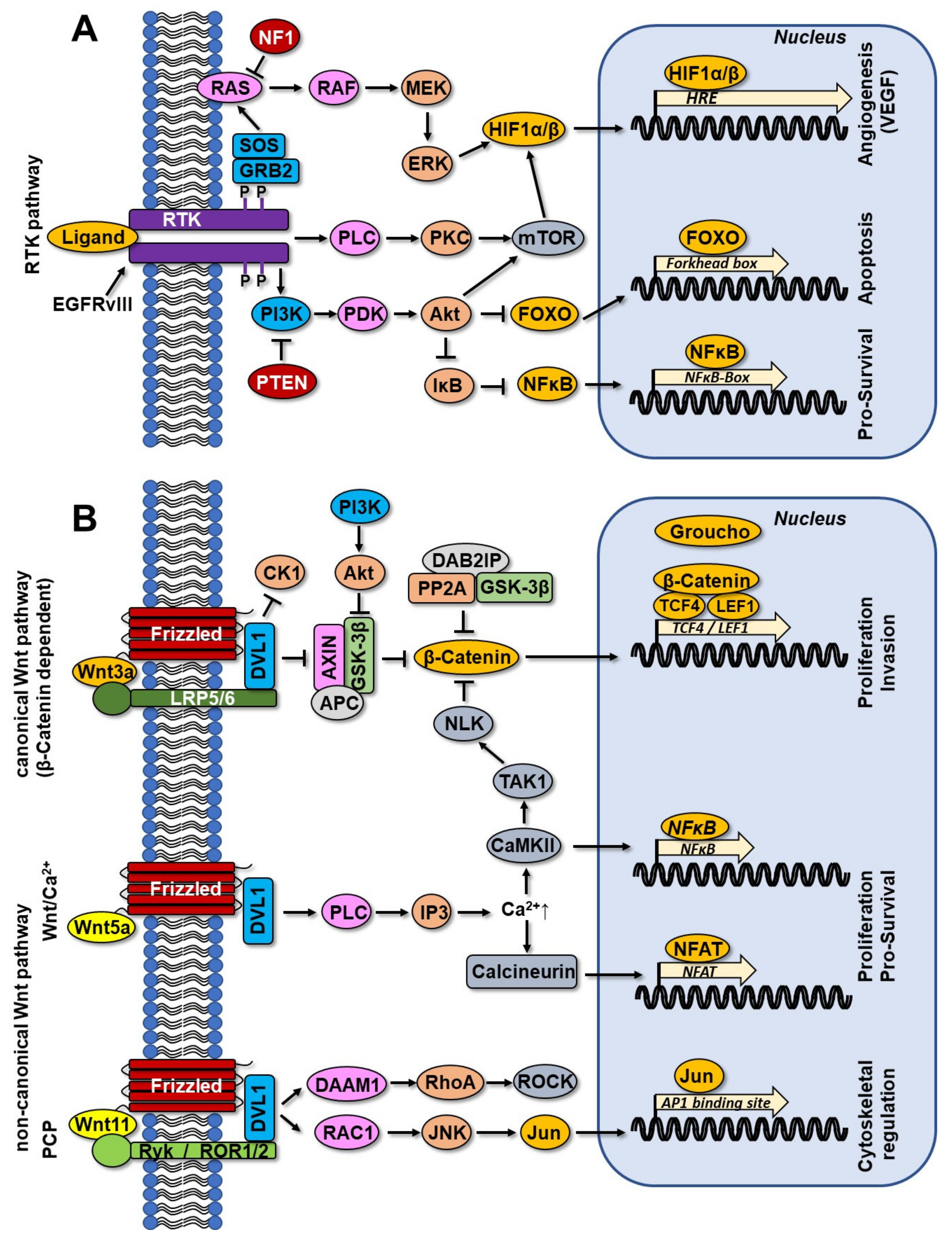
3.3. RTK Pathway and Wnt Signaling
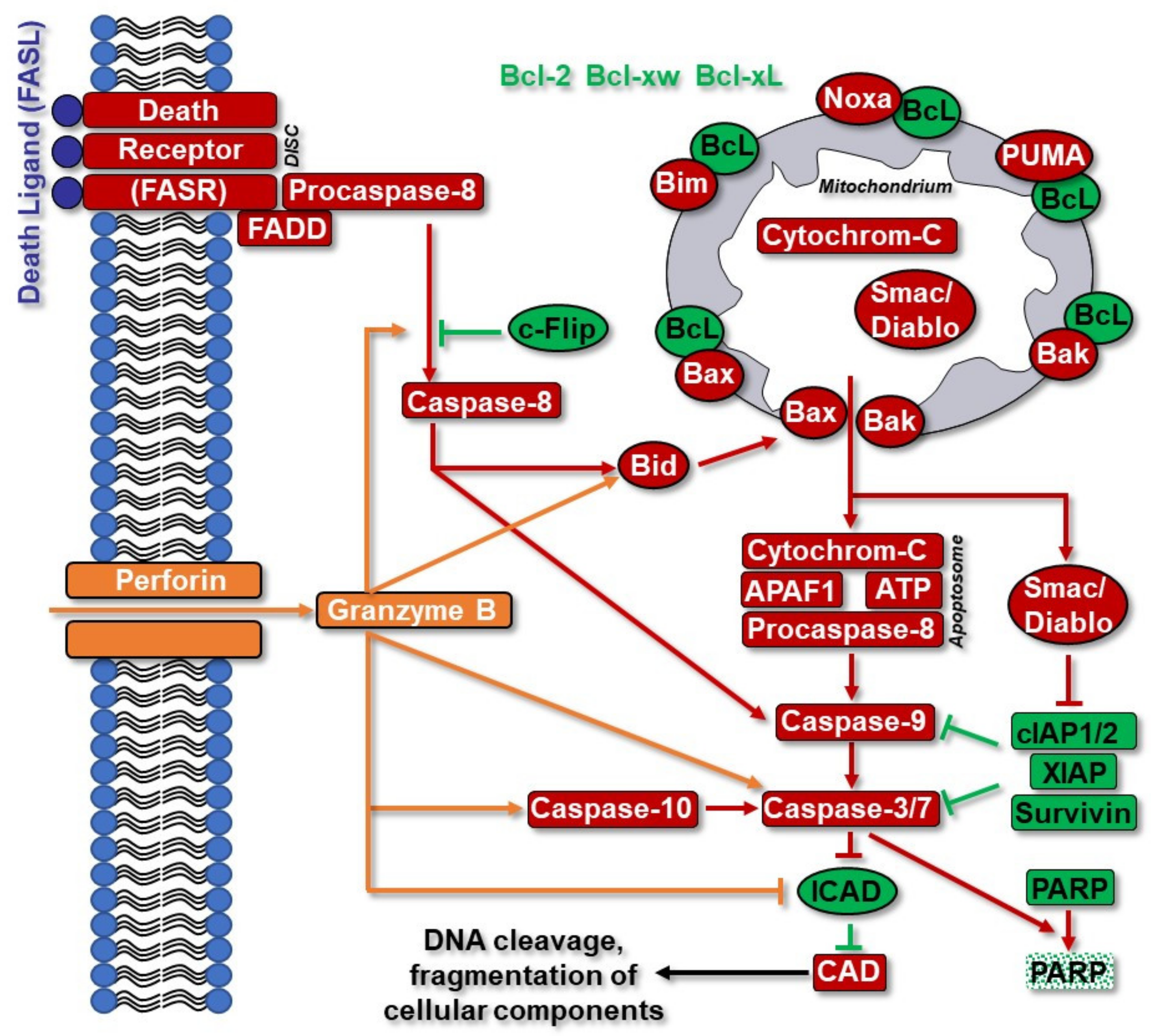
3.4. Apoptotic Pathways
3.5. Senescence
3.6. Stemness
3.7. Autophagy
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cooperman, A.M.; Iskandar, M.E.; Wayne, M.G.; Steele, J.G. Prevention and Early Detection of Pancreatic Cancer. Surg. Clin. N. Am. 2018, 98, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Le Stang, N.; Bouvier, V.; Glehen, O.; Villeneuve, L.; FRANCIM Network; MESOPATH Referent National Center; Galateau-Salle, F.; Clin, B. Incidence and survival of peritoneal malignant mesothelioma between 1989 and 2015: A population-based study. Cancer Epidemiol. 2019, 60, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Patil, N.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2013–2017. Neuro Oncol. 2020, 22, iv1–iv96. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Lin, D.; Wang, M.; Chen, Y.; Gong, J.; Chen, L.; Shi, X.; Lan, F.; Chen, Z.; Xiong, T.; Sun, H. Trends in Intracranial Glioma Incidence and Mortality in the United States, 1975–2018. Front. Oncol. 2021, 11, 748061. [Google Scholar] [CrossRef]
- Bosma, I.; Vos, M.J.; Heimans, J.J.; Taphoorn, M.J.; Aaronson, N.K.; Postma, T.J.; van der Ploeg, H.M.; Muller, M.; Vandertop, W.P.; Slotman, B.J.; et al. The course of neurocognitive functioning in high-grade glioma patients. Neuro-Oncology 2007, 9, 53–62. [Google Scholar] [CrossRef]
- Solanki, C.; Sadana, D.; Arimappamagan, A.; Rao, K.; Rajeswaran, J.; Subbakrishna, D.K.; Santosh, V.; Pandey, P. Impairments in Quality of Life and Cognitive Functions in Long-term Survivors of Glioblastoma. J. Neurosci. Rural Pract. 2017, 8, 228–235. [Google Scholar] [CrossRef]
- Figarella-Branger, D.; Appay, R.; Metais, A.; Tauziede-Espariat, A.; Colin, C.; Rousseau, A.; Varlet, P. The 2021 WHO classification of tumours of the central nervous system. Ann. Pathol. 2021. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.; Pfister, S.M.; Reifenberger, G. The 2021 WHO classification of tumors of the central nervous system: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Banan, R.; Hartmann, C. The new WHO 2016 classification of brain tumors—What neurosurgeons need to know. Acta Neurochir. 2017, 159, 403–418. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; Van Den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Kim, S.-H.; Yoo, H.; Chang, J.H.; Kim, C.-Y.; Chung, D.S.; Kim, S.H.; Park, S.-H.; Lee, Y.S.; Yang, S.H. Procarbazine and CCNU chemotherapy for recurrent glioblastoma with MGMT promoter methylation. J. Korean Med. Sci. 2018, 33, e167. [Google Scholar] [CrossRef] [Green Version]
- Brisman, R.; Housepian, E.M.; Chang, C.; Duffy, P.; Balis, E. Adjuvant nitrosourea therapy for glioblastoma. Arch. Neurol. 1976, 33, 745–750. [Google Scholar] [CrossRef]
- Silvani, A.; Gaviani, P.; Lamperti, E.A.; Eoli, M.; Falcone, C.; DiMeco, F.; Milanesi, I.M.; Erbetta, A.; Boiardi, A.; Fariselli, L. Cisplatinum and BCNU chemotherapy in primary glioblastoma patients. J. Neuro-Oncol. 2009, 94, 57–62. [Google Scholar] [CrossRef]
- Davis, M.E. Glioblastoma: Overview of Disease and Treatment. Clin. J. Oncol. Nurs. 2016, 20, S2–S8. [Google Scholar] [CrossRef] [Green Version]
- Baskar, R.; Lee, K.A.; Yeo, R.; Yeoh, K.W. Cancer and radiation therapy: Current advances and future directions. Int. J. Med. Sci. 2012, 9, 193–199. [Google Scholar] [CrossRef] [Green Version]
- Barani, I.J.; Larson, D.A. Radiation therapy of glioblastoma. Cancer Treat. Res. 2015, 163, 49–73. [Google Scholar] [CrossRef]
- Deloch, L.; Derer, A.; Hartmann, J.; Frey, B.; Fietkau, R.; Gaipl, U.S. Modern Radiotherapy Concepts and the Impact of Radiation on Immune Activation. Front. Oncol. 2016, 6, 141. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.Y.; Jeong, E.K.; Ju, M.K.; Jeon, H.M.; Kim, M.Y.; Kim, C.H.; Park, H.G.; Han, S.I.; Kang, H.S. Induction of metastasis, cancer stem cell phenotype, and oncogenic metabolism in cancer cells by ionizing radiation. Mol. Cancer 2017, 16, 10. [Google Scholar] [CrossRef] [Green Version]
- De Ruysscher, D.; Niedermann, G.; Burnet, N.G.; Siva, S.; Lee, A.W.M.; Hegi-Johnson, F. Radiotherapy toxicity. Nat. Rev. Dis. Primers 2019, 5, 13. [Google Scholar] [CrossRef]
- Riley, P.A. Free radicals in biology: Oxidative stress and the effects of ionizing radiation. Int. J. Radiat. Biol. 1994, 65, 27–33. [Google Scholar] [CrossRef]
- Mortezaee, K.; Goradel, N.H.; Amini, P.; Shabeeb, D.; Musa, A.E.; Najafi, M.; Farhood, B. NADPH Oxidase as a Target for Modulation of Radiation Response; Implications to Carcinogenesis and Radiotherapy. Curr. Mol. Pharm. 2019, 12, 50–60. [Google Scholar] [CrossRef]
- Moody, C.L.; Wheelhouse, R.T. The medicinal chemistry of imidazotetrazine prodrugs. Pharmaceuticals 2014, 7, 797–838. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Stevens, M.F.; Bradshaw, T.D. Temozolomide: Mechanisms of action, repair and resistance. Curr. Mol. Pharm. 2012, 5, 102–114. [Google Scholar] [CrossRef]
- Beranek, D.T. Distribution of methyl and ethyl adducts following alkylation with monofunctional alkylating agents. Mutat. Res. 1990, 231, 11–30. [Google Scholar] [CrossRef]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death: From specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2013, 332, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Johannessen, T.-C.A.; Prestegarden, L.; Grudic, A.; Hegi, M.E.; Tysnes, B.B.; Bjerkvig, R. The DNA repair protein ALKBH2 mediates temozolomide resistance in human glioblastoma cells. Neuro-Oncology 2013, 15, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Hansen, R.J.; Nagasubramanian, R.; Delaney, S.M.; Samson, L.D.; Dolan, M.E. Role of O6-methylguanine-DNA methyltransferase in protecting from alkylating agent-induced toxicity and mutations in mice. Carcinogenesis 2007, 28, 1111–1116. [Google Scholar] [CrossRef] [PubMed]
- Christmann, M.; Verbeek, B.; Roos, W.P.; Kaina, B. O(6)-Methylguanine-DNA methyltransferase (MGMT) in normal tissues and tumors: Enzyme activity, promoter methylation and immunohistochemistry. Biochim. Biophys. Acta BBA Rev. Cancer 2011, 1816, 179–190. [Google Scholar] [CrossRef]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [Green Version]
- Yan, H.; Bigner, D.D.; Velculescu, V.; Parsons, D.W. Mutant metabolic enzymes are at the origin of gliomas. Cancer Res. 2009, 69, 9157–9159. [Google Scholar] [CrossRef] [Green Version]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef]
- Gritsch, S.; Batchelor, T.T.; Gonzalez Castro, L.N. Diagnostic, therapeutic, and prognostic implications of the 2021 World Health Organization classification of tumors of the central nervous system. Cancer 2022, 128, 47–58. [Google Scholar] [CrossRef]
- Stichel, D.; Ebrahimi, A.; Reuss, D.; Schrimpf, D.; Ono, T.; Shirahata, M.; Reifenberger, G.; Weller, M.; Hänggi, D.; Wick, W. Distribution of EGFR amplification, combined chromosome 7 gain and chromosome 10 loss, and TERT promoter mutation in brain tumors and their potential for the reclassification of IDHwt astrocytoma to glioblastoma. Acta Neuropathol. 2018, 136, 793–803. [Google Scholar] [CrossRef] [Green Version]
- Dunn, G.P.; Rinne, M.L.; Wykosky, J.; Genovese, G.; Quayle, S.N.; Dunn, I.F.; Agarwalla, P.K.; Chheda, M.G.; Campos, B.; Wang, A. Emerging insights into the molecular and cellular basis of glioblastoma. Genes Dev. 2012, 26, 756–784. [Google Scholar] [CrossRef] [Green Version]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Yoon, S.J.; Son, H.Y.; Shim, J.K.; Moon, J.H.; Kim, E.H.; Chang, J.H.; Teo, W.Y.; Kim, S.H.; Park, S.W.; Huh, Y.M.; et al. Co-expression of cancer driver genes: IDH-wildtype glioblastoma-derived tumorspheres. J. Transl. Med. 2020, 18, 482. [Google Scholar] [CrossRef]
- Tateishi, K.; Wakimoto, H.; Cahill, D.P. IDH1 Mutation and World Health Organization 2016 Diagnostic Criteria for Adult Diffuse Gliomas: Advances in Surgical Strategy. Neurosurgery 2017, 64, 134–138. [Google Scholar] [CrossRef]
- Reich, T.R.; Switzeny, O.J.; Renovanz, M.; Sommer, C.; Kaina, B.; Christmann, M.; Tomicic, M.T. Epigenetic silencing of XAF1 in high-grade gliomas is associated with IDH1 status and improved clinical outcome. Oncotarget 2017, 8, 15071–15084. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Zhang, J.; Dang, F.; Ren, J.; Wei, W. Biochemical aspects of PD-L1 regulation in cancer immunotherapy. Trends Biochem. Sci. 2018, 43, 1014–1032. [Google Scholar] [CrossRef]
- Ham, S.W.; Jeon, H.Y.; Jin, X.; Kim, E.J.; Kim, J.K.; Shin, Y.J.; Lee, Y.; Kim, S.H.; Lee, S.Y.; Seo, S.; et al. TP53 gain-of-function mutation promotes inflammation in glioblastoma. Cell Death Differ. 2019, 26, 409–425. [Google Scholar] [CrossRef] [Green Version]
- Olafson, L.R.; Gunawardena, M.; Nixdorf, S.; McDonald, K.L.; Rapkins, R.W. The role of TP53 gain-of-function mutation in multifocal glioblastoma. J. Neurooncol. 2020, 147, 37–47. [Google Scholar] [CrossRef] [Green Version]
- Fujisawa, H.; Reis, R.M.; Nakamura, M.; Colella, S.; Yonekawa, Y.; Kleihues, P.; Ohgaki, H. Loss of heterozygosity on chromosome 10 is more extensive in primary (de novo) than in secondary glioblastomas. Lab. Investig. 2000, 80, 65–72. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Gines, C.; Cerda-Nicolas, M.; Gil-Benso, R.; Pellin, A.; Lopez-Guerrero, J.; Callaghan, R.; Benito, R.; Roldan, P.; Piquer, J.; Llacer, J. Association of chromosome 7, chromosome 10 and EGFR gene amplification in glioblastoma multiforme. Clin. Neuropathol. 2005, 24, 209–218. [Google Scholar]
- Crespo, I.; Vital, A.L.; Nieto, A.B.; Rebelo, O.; Tão, H.; Lopes, M.C.; Oliveira, C.R.; French, P.J.; Orfao, A.; Tabernero, M.D. Detailed characterization of alterations of chromosomes 7, 9, and 10 in glioblastomas as assessed by single-nucleotide polymorphism arrays. J. Mol. Diagn. 2011, 13, 634–647. [Google Scholar] [CrossRef]
- Killela, P.J.; Pirozzi, C.J.; Healy, P.; Reitman, Z.J.; Lipp, E.; Rasheed, B.A.; Yang, R.; Diplas, B.H.; Wang, Z.; Greer, P.K.; et al. Mutations in IDH1, IDH2, and in the TERT promoter define clinically distinct subgroups of adult malignant gliomas. Oncotarget 2014, 5, 1515–1525. [Google Scholar] [CrossRef] [Green Version]
- Sintupisut, N.; Liu, P.L.; Yeang, C.H. An integrative characterization of recurrent molecular aberrations in glioblastoma genomes. Nucleic Acids Res. 2013, 41, 8803–8821. [Google Scholar] [CrossRef]
- Donehower, L.A.; Soussi, T.; Korkut, A.; Liu, Y.; Schultz, A.; Cardenas, M.; Li, X.; Babur, O.; Hsu, T.K.; Lichtarge, O.; et al. Integrated Analysis of TP53 Gene and Pathway Alterations in The Cancer Genome Atlas. Cell Rep. 2019, 28, 3010. [Google Scholar] [CrossRef]
- Deininger, M.W.; Shah, N.P.; Altman, J.K.; Berman, E.; Bhatia, R.; Bhatnagar, B.; DeAngelo, D.J.; Gotlib, J.; Hobbs, G.; Maness, L.; et al. Chronic Myeloid Leukemia, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Canc. Netw. 2020, 18, 1385–1415. [Google Scholar] [CrossRef]
- Gao, Q.; Liang, W.W.; Foltz, S.M.; Mutharasu, G.; Jayasinghe, R.G.; Cao, S.; Liao, W.W.; Reynolds, S.M.; Wyczalkowski, M.A.; Yao, L.; et al. Driver Fusions and Their Implications in the Development and Treatment of Human Cancers. Cell Rep. 2018, 23, 227–238.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nathansen, J.; Meyer, F.; Muller, L.; Schmitz, M.; Borgmann, K.; Dubrovska, A. Beyond the Double-Strand Breaks: The Role of DNA Repair Proteins in Cancer Stem-Cell Regulation. Cancers 2021, 13, 4818. [Google Scholar] [CrossRef] [PubMed]
- Knijnenburg, T.A.; Wang, L.; Zimmermann, M.T.; Chambwe, N.; Gao, G.F.; Cherniack, A.D.; Fan, H.; Shen, H.; Way, G.P.; Greene, C.S. Genomic and molecular landscape of DNA damage repair deficiency across The Cancer Genome Atlas. Cell Rep. 2018, 23, 239–254.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, S.; Zhang, Y.; Zhou, M.; Li, H.; Jin, W.; Zheng, L.; Yu, X.; Stark, J.M.; Weitzel, J.N.; Shen, B. Functional deficiency of DNA repair gene EXO5 results in androgen-induced genomic instability and prostate tumorigenesis. Oncogene 2020, 39, 1246–1259. [Google Scholar] [CrossRef]
- Hombach-Klonisch, S.; Mehrpour, M.; Shojaei, S.; Harlos, C.; Pitz, M.; Hamai, A.; Siemianowicz, K.; Likus, W.; Wiechec, E.; Toyota, B.D.; et al. Glioblastoma and chemoresistance to alkylating agents: Involvement of apoptosis, autophagy, and unfolded protein response. Pharmacol. Ther. 2018, 184, 13–41. [Google Scholar] [CrossRef]
- Li, X.; He, S.; Ma, B. Autophagy and autophagy-related proteins in cancer. Mol. Cancer 2020, 19, 12. [Google Scholar] [CrossRef]
- Bueno-Martinez, E.; Lara-Almunia, M.; Rodriguez-Arias, C.; Otero-Rodriguez, A.; Garfias-Arjona, S.; Gonzalez-Sarmiento, R. Polymorphisms in autophagy genes are genetic susceptibility factors in glioblastoma development. BMC Cancer 2022, 22, 146. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, W.; Xiao, Z.; Guan, G.; Liu, X.; Zhuang, M. A risk signature with four autophagy-related genes for predicting survival of glioblastoma multiforme. J. Cell Mol. Med. 2020, 24, 3807–3821. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Gao, L.; Guo, X.; Feng, C.; Lian, W.; Deng, K.; Xing, B. Development and validation of a nomogram with an autophagy-related gene signature for predicting survival in patients with glioblastoma. Aging 2019, 11, 12246–12269. [Google Scholar] [CrossRef]
- Gupta, P.B.; Pastushenko, I.; Skibinski, A.; Blanpain, C.; Kuperwasser, C. Phenotypic Plasticity: Driver of Cancer Initiation, Progression, and Therapy Resistance. Cell Stem Cell 2019, 24, 65–78. [Google Scholar] [CrossRef] [Green Version]
- Malta, T.M.; Sokolov, A.; Gentles, A.J.; Burzykowski, T.; Poisson, L.; Weinstein, J.N.; Kaminska, B.; Huelsken, J.; Omberg, L.; Gevaert, O.; et al. Machine Learning Identifies Stemness Features Associated with Oncogenic Dedifferentiation. Cell 2018, 173, 338–354.e315. [Google Scholar] [CrossRef] [Green Version]
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bahr, O.; et al. Effect of Nivolumab vs Bevacizumab in Patients With Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1003–1010. [Google Scholar] [CrossRef]
- Yang, T.; Kong, Z.; Ma, W. PD-1/PD-L1 immune checkpoint inhibitors in glioblastoma: Clinical studies, challenges and potential. Hum. Vaccines Immunother. 2021, 17, 546–553. [Google Scholar] [CrossRef]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018, 174, 1034–1035. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S. Oncogenic signaling pathways in the cancer genome atlas. Cell 2018, 173, 321–337.e310. [Google Scholar] [CrossRef] [Green Version]
- Schaub, F.X.; Dhankani, V.; Berger, A.C.; Trivedi, M.; Richardson, A.B.; Shaw, R.; Zhao, W.; Zhang, X.; Ventura, A.; Liu, Y.; et al. Pan-cancer Alterations of the MYC Oncogene and Its Proximal Network across the Cancer Genome Atlas. Cell Syst. 2018, 6, 282–300.e282. [Google Scholar] [CrossRef] [Green Version]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Shen, R.; Mo, Q.; Schultz, N.; Seshan, V.E.; Olshen, A.B.; Huse, J.; Ladanyi, M.; Sander, C. Integrative subtype discovery in glioblastoma using iCluster. PLoS One 2012, 7, e35236. [Google Scholar] [CrossRef]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; deCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56.e46. [Google Scholar] [CrossRef] [Green Version]
- Couturier, C.P.; Ayyadhury, S.; Le, P.U.; Nadaf, J.; Monlong, J.; Riva, G.; Allache, R.; Baig, S.; Yan, X.; Bourgey, M.; et al. Single-cell RNA-seq reveals that glioblastoma recapitulates a normal neurodevelopmental hierarchy. Nat. Commun. 2020, 11, 3406. [Google Scholar] [CrossRef]
- Brat, D.J.; Aldape, K.; Colman, H.; Holland, E.C.; Louis, D.N.; Jenkins, R.B.; Kleinschmidt-DeMasters, B.; Perry, A.; Reifenberger, G.; Stupp, R. cIMPACT-NOW update 3: Recommended diagnostic criteria for “Diffuse astrocytic glioma, IDH-wildtype, with molecular features of glioblastoma, WHO grade IV”. Acta Neuropathol. 2018, 136, 805–810. [Google Scholar] [CrossRef] [Green Version]
- Weller, M.; van den Bent, M.; Preusser, M.; Le Rhun, E.; Tonn, J.C.; Minniti, G.; Bendszus, M.; Balana, C.; Chinot, O.; Dirven, L. EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nat. Rev. Clin. Oncol. 2021, 18, 170–186. [Google Scholar] [CrossRef]
- Bleeker, F.E.; Atai, N.A.; Lamba, S.; Jonker, A.; Rijkeboer, D.; Bosch, K.S.; Tigchelaar, W.; Troost, D.; Vandertop, W.P.; Bardelli, A. The prognostic IDH1 R132 mutation is associated with reduced NADP+-dependent IDH activity in glioblastoma. Acta Neuropathol. 2010, 119, 487–494. [Google Scholar] [CrossRef] [Green Version]
- Oh, S.; Yeom, J.; Cho, H.J.; Kim, J.-H.; Yoon, S.-J.; Kim, H.; Sa, J.K.; Ju, S.; Lee, H.; Oh, M.J. Integrated pharmaco-proteogenomics defines two subgroups in isocitrate dehydrogenase wild-type glioblastoma with prognostic and therapeutic opportunities. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Calvert, A.E.; Chalastanis, A.; Wu, Y.; Hurley, L.A.; Kouri, F.M.; Bi, Y.; Kachman, M.; May, J.L.; Bartom, E.; Hua, Y. Cancer-associated IDH1 promotes growth and resistance to targeted therapies in the absence of mutation. Cell Rep. 2017, 19, 1858–1873. [Google Scholar] [CrossRef] [Green Version]
- Dang, L.; Jin, S.; Su, S.M. IDH mutations in glioma and acute myeloid leukemia. Trends Mol. Med. 2010, 16, 387–397. [Google Scholar] [CrossRef]
- Turcan, S.; Rohle, D.; Goenka, A.; Walsh, L.A.; Fang, F.; Yilmaz, E.; Campos, C.; Fabius, A.W.; Lu, C.; Ward, P.S. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012, 483, 479–483. [Google Scholar] [CrossRef]
- Hitosugi, T.; Fan, J.; Chung, T.-W.; Lythgoe, K.; Wang, X.; Xie, J.; Ge, Q.; Gu, T.-L.; Polakiewicz, R.D.; Roesel, J.L. Tyrosine phosphorylation of mitochondrial pyruvate dehydrogenase kinase 1 is important for cancer metabolism. Mol. Cell 2011, 44, 864–877. [Google Scholar] [CrossRef] [Green Version]
- Velpula, K.K.; Guda, M.R.; Sahu, K.; Tuszynski, J.; Asuthkar, S.; Bach, S.E.; Lathia, J.D.; Tsung, A.J. Metabolic targeting of EGFRvIII/PDK1 axis in temozolomide resistant glioblastoma. Oncotarget 2017, 8, 35639. [Google Scholar] [CrossRef] [Green Version]
- Velpula, K.K.; Tsung, A.J. PDK1: A new therapeutic target for glioblastoma? CNS Oncol. 2014, 3, 177–179. [Google Scholar] [CrossRef]
- Velpula, K.K.; Bhasin, A.; Asuthkar, S.; Tsung, A.J. Combined targeting of PDK1 and EGFR triggers regression of glioblastoma by reversing the Warburg effect. Cancer Res. 2013, 73, 7277–7289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Switzeny, O.J.; Christmann, M.; Renovanz, M.; Giese, A.; Sommer, C.; Kaina, B. MGMT promoter methylation determined by HRM in comparison to MSP and pyrosequencing for predicting high-grade glioma response. Clin. Epigenetics 2016, 8, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mojas, N.; Lopes, M.; Jiricny, J. Mismatch repair-dependent processing of methylation damage gives rise to persistent single-stranded gaps in newly replicated DNA. Genes Dev. 2007, 21, 3342–3355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caporali, S.; Falcinelli, S.; Starace, G.; Russo, M.T.; Bonmassar, E.; Jiricny, J.; D’Atri, S. DNA damage induced by temozolomide signals to both ATM and ATR: Role of the mismatch repair system. Mol. Pharmacol. 2004, 66, 478–491. [Google Scholar]
- Yan, S.; Sorrell, M.; Berman, Z. Functional interplay between ATM/ATR-mediated DNA damage response and DNA repair pathways in oxidative stress. Cell. Mol. Life Sci. 2014, 71, 3951–3967. [Google Scholar] [CrossRef] [Green Version]
- Eich, M.; Roos, W.P.; Nikolova, T.; Kaina, B. Contribution of ATM and ATR to the resistance of glioblastoma and malignant melanoma cells to the methylating anticancer drug temozolomide. Mol. Cancer Ther. 2013, 12, 2529–2540. [Google Scholar] [CrossRef] [Green Version]
- Ito, M.; Ohba, S.; Gaensler, K.; Ronen, S.M.; Mukherjee, J.; Pieper, R.O. Early Chk1 phosphorylation is driven by temozolomide-induced, DNA double strand break-and mismatch repair-independent DNA damage. PLoS ONE 2013, 8, e62351. [Google Scholar] [CrossRef]
- Aasland, D.; Götzinger, L.; Hauck, L.; Berte, N.; Meyer, J.; Effenberger, M.; Schneider, S.; Reuber, E.E.; Roos, W.P.; Tomicic, M.T. Temozolomide induces senescence and repression of DNA repair pathways in glioblastoma cells via activation of ATR–CHK1, p21, and NF-κB. Cancer Res. 2019, 79, 99–113. [Google Scholar] [CrossRef] [Green Version]
- Squatrito, M.; Brennan, C.W.; Helmy, K.; Huse, J.T.; Petrini, J.H.; Holland, E.C. Loss of ATM/Chk2/p53 pathway components accelerates tumor development and contributes to radiation resistance in gliomas. Cancer Cell 2010, 18, 619–629. [Google Scholar] [CrossRef] [Green Version]
- Tomicic, M.T.; Meise, R.; Aasland, D.; Berte, N.; Kitzinger, R.; Krämer, O.H.; Kaina, B.; Christmann, M. Apoptosis induced by temozolomide and nimustine in glioblastoma cells is supported by JNK/c-Jun-mediated induction of the BH3-only protein BIM. Oncotarget 2015, 6, 33755. [Google Scholar] [CrossRef] [Green Version]
- Rocha, C.R.R.; Rocha, A.R.; Silva, M.M.; Gomes, L.R.; Latancia, M.T.; Andrade-Tomaz, M.; de Souza, I.; Monteiro, L.K.S.; Menck, C.F.M. Revealing temozolomide resistance mechanisms via genome-wide CRISPR libraries. Cells 2020, 9, 2573. [Google Scholar] [CrossRef]
- Quiros, S.; Roos, W.P.; Kaina, B. Processing of O6-methylguanine into DNA double-strand breaks requires two rounds of replication whereas apoptosis is also induced in subsequent cell cycles. Cell Cycle 2010, 9, 168–178. [Google Scholar] [CrossRef] [Green Version]
- Roos, W.P.; Frohnapfel, L.; Quiros, S.; Ringel, F.; Kaina, B. XRCC3 contributes to temozolomide resistance of glioblastoma cells by promoting DNA double-strand break repair. Cancer Lett. 2018, 424, 119–126. [Google Scholar] [CrossRef]
- Quiros, S.; Roos, W.P.; Kaina, B. Rad51 and BRCA2-New molecular targets for sensitizing glioma cells to alkylating anticancer drugs. PLoS ONE 2011, 6, e27183. [Google Scholar] [CrossRef] [Green Version]
- McFaline-Figueroa, J.L.; Braun, C.J.; Stanciu, M.; Nagel, Z.D.; Mazzucato, P.; Sangaraju, D.; Cerniauskas, E.; Barford, K.; Vargas, A.; Chen, Y. Minor changes in expression of the mismatch repair protein MSH2 exert a major impact on glioblastoma response to temozolomide. Cancer Res. 2015, 75, 3127–3138. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Shao, Z.; Jiang, W.; Lee, B.J.; Zha, S. PAXX promotes KU accumulation at DNA breaks and is essential for end-joining in XLF-deficient mice. Nat. Commun. 2017, 8, 13816. [Google Scholar] [CrossRef]
- Yang, B.; Fu, X.; Hao, J.; Sun, J.; Li, Z.; Li, H.; Xu, H. PAXX participates in base excision repair via interacting with Pol β and Contributes to TMZ resistance in glioma cells. J. Mol. Neurosci. 2018, 66, 214–221. [Google Scholar] [CrossRef] [Green Version]
- Wyatt, M.D.; Pittman, D.L. Methylating agents and DNA repair responses: Methylated bases and sources of strand breaks. Chem. Res. Toxicol. 2006, 19, 1580–1594. [Google Scholar] [CrossRef] [Green Version]
- Erasimus, H.; Gobin, M.; Niclou, S.; Van Dyck, E. DNA repair mechanisms and their clinical impact in glioblastoma. Mutat. Res./Rev. Mutat. Res. 2016, 769, 19–35. [Google Scholar] [CrossRef]
- Serrano-Heras, G.; Castro-Robles, B.; Romero-Sánchez, C.M.; Carrión, B.; Barbella-Aponte, R.; Sandoval, H.; Segura, T. Involvement of N-methylpurine DNA glycosylase in resistance to temozolomide in patient-derived glioma cells. Sci. Rep. 2020, 10, 22185. [Google Scholar] [CrossRef]
- Madhusudan, S.; Hickson, I.D. DNA repair inhibition: A selective tumour targeting strategy. Trends Mol. Med. 2005, 11, 503–511. [Google Scholar] [CrossRef]
- Tang, J.-B.; Svilar, D.; Trivedi, R.N.; Wang, X.-H.; Goellner, E.M.; Moore, B.; Hamilton, R.L.; Banze, L.A.; Brown, A.R.; Sobol, R.W. N-methylpurine DNA glycosylase and DNA polymerase β modulate BER inhibitor potentiation of glioma cells to temozolomide. Neuro-Oncology 2011, 13, 471–486. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Gerson, S.L. Therapeutic impact of methoxyamine: Blocking repair of abasic sites in the base excision repair pathway. Curr. Opin. Investig. Drugs 2004, 5, 623–627. [Google Scholar]
- Zandarashvili, L.; Langelier, M.-F.; Velagapudi, U.K.; Hancock, M.A.; Steffen, J.D.; Billur, R.; Hannan, Z.M.; Wicks, A.J.; Krastev, D.B.; Pettitt, S.J. Structural basis for allosteric PARP-1 retention on DNA breaks. Science 2020, 368, eaax6367. [Google Scholar] [CrossRef]
- Pandey, N.; Black, B.E. Rapid detection and signaling of DNA damage by PARP-1. Trends Biochem. Sci. 2021, 46, 744–757. [Google Scholar] [CrossRef]
- Tentori, L.; Ricci-Vitiani, L.; Muzi, A.; Ciccarone, F.; Pelacchi, F.; Calabrese, R.; Runci, D.; Pallini, R.; Caiafa, P.; Graziani, G. Pharmacological inhibition of poly (ADP-ribose) polymerase-1 modulates resistance of human glioblastoma stem cells to temozolomide. BMC Cancer 2014, 14, 151. [Google Scholar] [CrossRef] [Green Version]
- Montaldi, A.P.; Lima, S.C.; Godoy, P.R.; Xavier, D.J.; Sakamoto-Hojo, E.T. PARP-1 inhibition sensitizes temozolomide-treated glioblastoma cell lines and decreases drug resistance independent of MGMT activity and PTEN proficiency. Oncol. Rep. 2020, 44, 2275–2287. [Google Scholar] [CrossRef]
- Higuchi, F.; Nagashima, H.; Ning, J.; Koerner, M.V.; Wakimoto, H.; Cahill, D.P. Restoration of temozolomide sensitivity by PARP inhibitors in mismatch repair deficient glioblastoma is independent of base excision repair. Clin. Cancer Res. 2020, 26, 1690–1699. [Google Scholar] [CrossRef]
- Ko, H.L.; Ren, E.C. Functional aspects of PARP1 in DNA repair and transcription. Biomolecules 2012, 2, 524–548. [Google Scholar] [CrossRef] [Green Version]
- Hedglin, M.; Aitha, M.; Benkovic, S.J. Monitoring the retention of human proliferating cell nuclear antigen at primer/template junctions by proteins that bind single-stranded DNA. Biochemistry 2017, 56, 3415–3421. [Google Scholar] [CrossRef] [Green Version]
- Sasatani, M.; Xu, Y.; Kawai, H.; Cao, L.; Tateishi, S.; Shimura, T.; Li, J.; Iizuka, D.; Noda, A.; Hamasaki, K. RAD18 activates the G2/M checkpoint through DNA damage signaling to maintain genome integrity after ionizing radiation exposure. PLoS ONE 2015, 10, e0117845. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Wang, H.; Cheng, H.; Li, J.; Wang, Z.; Yue, W. RAD18 mediates resistance to ionizing radiation in human glioma cells. Biochem. Biophys. Res. Commun. 2014, 445, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Hanisch, D.; Krumm, A.; Diehl, T.; Stork, C.M.; Dejung, M.; Butter, F.; Kim, E.; Brenner, W.; Fritz, G.; Hofmann, T.G. Class I HDAC overexpression promotes temozolomide resistance in glioma cells by regulating RAD18 expression. Cell Death Dis. 2022, 13, 293. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Wang, H.; Zhang, L.; Sun, C.; Li, H.; Jiang, C.; Liu, X. High expression of RAD18 in glioma induces radiotherapy resistance via down-regulating P53 expression. Biomed. Pharmacother. 2019, 112, 108555. [Google Scholar] [CrossRef]
- Choi, J.-S.; Kim, C.S.; Berdis, A. Inhibition of translesion DNA synthesis as a novel therapeutic strategy to treat brain cancer. Cancer Res. 2018, 78, 1083–1096. [Google Scholar] [CrossRef] [Green Version]
- Yamanaka, K.; Dorjsuren, D.; Eoff, R.L.; Egli, M.; Maloney, D.J.; Jadhav, A.; Simeonov, A.; Lloyd, R.S. A comprehensive strategy to discover inhibitors of the translesion synthesis DNA polymerase κ. PLoS ONE 2012, 7, e45032. [Google Scholar] [CrossRef] [Green Version]
- Ketkar, A.; Maddukuri, L.; Penthala, N.R.; Reed, M.R.; Zafar, M.K.; Crooks, P.A.; Eoff, R.L. Inhibition of human DNA polymerases eta and kappa by indole-derived molecules occurs through distinct mechanisms. ACS Chem. Biol. 2019, 14, 1337–1351. [Google Scholar] [CrossRef]
- Menck, C.F.; Munford, V. DNA repair diseases: What do they tell us about cancer and aging? Genet. Mol. Biol. 2014, 37, 220–233. [Google Scholar] [CrossRef] [Green Version]
- Tomicic, M.T.; Aasland, D.; Naumann, S.C.; Meise, R.; Barckhausen, C.; Kaina, B.; Christmann, M. Translesion polymerase η is upregulated by cancer therapeutics and confers anticancer drug resistance. Cancer Res. 2014, 74, 5585–5596. [Google Scholar] [CrossRef] [Green Version]
- Stern, H.R.; Sefcikova, J.; Chaparro, V.E.; Beuning, P.J. Mammalian DNA polymerase kappa activity and specificity. Molecules 2019, 24, 2805. [Google Scholar] [CrossRef] [Green Version]
- Peng, C.; Chen, Z.; Wang, S.; Wang, H.-W.; Qiu, W.; Zhao, L.; Xu, R.; Luo, H.; Chen, Y.; Chen, D. The error-prone DNA polymerase κ promotes temozolomide resistance in glioblastoma through Rad17-dependent activation of ATR-Chk1 signaling. Cancer Res. 2016, 76, 2340–2353. [Google Scholar] [CrossRef] [Green Version]
- Luo, H.; Chen, Z.; Wang, S.; Zhang, R.; Qiu, W.; Zhao, L.; Peng, C.; Xu, R.; Chen, W.; Wang, H.-W. c-Myc–miR-29c–REV3L signalling pathway drives the acquisition of temozolomide resistance in glioblastoma. Brain 2015, 138, 3654–3672. [Google Scholar] [CrossRef] [Green Version]
- Roos, W.P.; Tsaalbi-Shtylik, A.; Tsaryk, R.; Güvercin, F.; de Wind, N.; Kaina, B. The translesion polymerase Rev3L in the tolerance of alkylating anticancer drugs. Mol. Pharmacol. 2009, 76, 927–934. [Google Scholar] [CrossRef]
- Pearson, J.R.; Regad, T. Targeting cellular pathways in glioblastoma multiforme. Signal. Transduct. Target. Ther. 2017, 2, 17040. [Google Scholar] [CrossRef] [Green Version]
- Gan, H.K.; Cvrljevic, A.N.; Johns, T.G. The epidermal growth factor receptor variant III (EGFRvIII): Where wild things are altered. FEBS J. 2013, 280, 5350–5370. [Google Scholar] [CrossRef]
- Montano, N.; Cenci, T.; Martini, M.; D’Alessandris, Q.G.; Pelacchi, F.; Ricci-Vitiani, L.; Maira, G.; De Maria, R.; Larocca, L.M.; Pallini, R. Expression of EGFRvIII in glioblastoma: Prognostic significance revisited. Neoplasia 2011, 13, 1113–1121. [Google Scholar] [CrossRef]
- Heimberger, A.; Suki, D.; Yang, D.; Shi, W.; Aldape, K. The natural history of EGFR and EGFRvIII in glioblastoma patients. J Transl. Med. 2005, 3, 38. [Google Scholar] [CrossRef] [Green Version]
- Struve, N.; Binder, Z.A.; Stead, L.F.; Brend, T.; Bagley, S.J.; Faulkner, C.; Ott, L.; Müller-Goebel, J.; Weik, A.-S.; Hoffer, K. EGFRvIII upregulates DNA mismatch repair resulting in increased temozolomide sensitivity of MGMT promoter methylated glioblastoma. Oncogene 2020, 39, 3041–3055. [Google Scholar] [CrossRef] [Green Version]
- Kolch, W. Meaningful relationships: The regulation of the Ras/Raf/Mek/Erk signal transduction pathway. Biochem. J. 2000, 351, 289–305. [Google Scholar] [CrossRef]
- Lim, J.-H.; Lee, E.-S.; You, H.-J.; Lee, J.W.; Park, J.-W.; Chun, Y.-S. Ras-dependent induction of HIF-1α785 via the Raf/MEK/ERK pathway: A novel mechanism of Ras-mediated tumor promotion. Oncogene 2004, 23, 9427–9431. [Google Scholar] [CrossRef] [Green Version]
- Yunoue, S.; Tokuo, H.; Fukunaga, K.; Feng, L.; Ozawa, T.; Nishi, T.; Kikuchi, A.; Hattori, S.; Kuratsu, J.; Saya, H. Neurofibromatosis type I tumor suppressor neurofibromin regulates neuronal differentiation via its GTPase-activating protein function toward Ras. J. Biol. Chem. 2003, 278, 26958–26969. [Google Scholar] [CrossRef] [Green Version]
- LoPiccolo, J.; Blumenthal, G.M.; Bernstein, W.B.; Dennis, P.A. Targeting the PI3K/Akt/mTOR pathway: Effective combinations and clinical considerations. Drug Resist. Updates 2008, 11, 32–50. [Google Scholar] [CrossRef] [Green Version]
- Carracedo, A.; Pandolfi, P. The PTEN–PI3K pathway: Of feedbacks and cross-talks. Oncogene 2008, 27, 5527–5541. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.I.; Puc, J.; Li, J.; Bruce, J.N.; Cairns, P.; Sidransky, D.; Parsons, R. Somatic mutations of PTEN in glioblastoma multiforme. Cancer Res. 1997, 57, 4183–4186. [Google Scholar]
- Wang, G.; Wang, J.-J.; Fu, X.-L.; Guang, R.; To, S.-S.T. Advances in the targeting of HIF-1α and future therapeutic strategies for glioblastoma multiforme. Oncol. Rep. 2017, 37, 657–670. [Google Scholar] [CrossRef] [Green Version]
- Fang, X.; Yu, S.X.; Lu, Y.; Bast, R.C.; Woodgett, J.R.; Mills, G.B. Phosphorylation and inactivation of glycogen synthase kinase 3 by protein kinase A. Proc. Natl. Acad. Sci. USA 2000, 97, 11960–11965. [Google Scholar] [CrossRef] [Green Version]
- Oeckinghaus, A.; Ghosh, S. The NF-κB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef]
- Tompa, M.; Kalovits, F.; Nagy, A.; Kalman, B. Contribution of the Wnt pathway to defining biology of glioblastoma. Neuromolecular Med. 2018, 20, 437–451. [Google Scholar] [CrossRef]
- Han, W.; Shi, J.; Cao, J.; Dong, B.; Guan, W. Current advances of long non-coding RNAs mediated by wnt signaling in glioma. Pathol. Res. Pract. 2020, 216, 153008. [Google Scholar] [CrossRef]
- Rezaei, O.; Tamizkar, K.H.; Sharifi, G.; Taheri, M.; Ghafouri-Fard, S. Emerging role of long non-coding RNAs in the pathobiology of glioblastoma. Front. Oncol. 2021, 10, 3381. [Google Scholar] [CrossRef]
- Gareev, I.; Beylerli, O.; Liang, Y.; Xiang, H.; Liu, C.; Xu, X.; Yuan, C.; Ahmad, A.; Yang, G. The role of MicroRNAs in therapeutic resistance of malignant primary brain tumors. Front. Cell Dev. Biol. 2021, 9, 2810. [Google Scholar] [CrossRef] [PubMed]
- Movahedpour, A.; Khatami, S.H.; Khorsand, M.; Salehi, M.; Savardashtaki, A.; Mirmajidi, S.H.; Negahdari, B.; Khanjani, N.; Naeli, P.; Vakili, O. Exosomal noncoding RNAs: Key players in glioblastoma drug resistance. Mol. Cell. Biochem. 2021, 476, 4081–4092. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Miner, A.; Hennis, L.; Mittal, S. Mechanisms of temozolomide resistance in glioblastoma—A comprehensive review. Cancer Drug Resist. 2021, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.S.; Al Mamun, A.; Alghamdi, B.S.; Tewari, D.; Jeandet, P.; Sarwar, M.S.; Ashraf, G.M. Epigenetics of glioblastoma multiforme: From molecular mechanisms to therapeutic approaches. Semin. Cancer Biol. 2020. [Google Scholar] [CrossRef]
- Ahmed, S.; Hasan, M.M.; Aschner, M.; Mirzaei, H.; Alam, W.; Shah, S.M.M.; Khan, H. Therapeutic potential of marine peptides in glioblastoma: Mechanistic insights. Cell. Signal. 2021, 87, 110142. [Google Scholar] [CrossRef]
- Setlai, B.P.; Hull, R.; Reis, R.M.; Agbor, C.; Ambele, M.A.; Mulaudzi, T.V.; Dlamini, Z. MicroRNA Interrelated Epithelial Mesenchymal Transition (EMT) in Glioblastoma. Genes 2022, 13, 244. [Google Scholar] [CrossRef]
- Sabbagh, Q.; Andre-Gregoire, G.; Guevel, L.; Gavard, J. Vesiclemia: Counting on extracellular vesicles for glioblastoma patients. Oncogene 2020, 39, 6043–6052. [Google Scholar] [CrossRef]
- Yekula, A.; Yekula, A.; Muralidharan, K.; Kang, K.; Carter, B.S.; Balaj, L. Extracellular vesicles in glioblastoma tumor microenvironment. Front. Immunol. 2020, 10, 3137. [Google Scholar] [CrossRef]
- Broekman, M.L.; Maas, S.L.; Abels, E.R.; Mempel, T.R.; Krichevsky, A.M.; Breakefield, X.O. Multidimensional communication in the microenvirons of glioblastoma. Nat. Rev. Neurol. 2018, 14, 482–495. [Google Scholar] [CrossRef]
- Mondal, A.A.; Kumari Singh, K.; Panda, S.; Shiras, A. Extracellular Vesicles as Modulators of Tumor Microenvironment and Disease Progression in Glioma (Invited review). Front. Oncol. 2017, 7, 144. [Google Scholar] [CrossRef]
- Colardo, M.; Segatto, M.; Di Bartolomeo, S. Targeting RTK-PI3K-mTOR axis in gliomas: An update. Int. J. Mol. Sci. 2021, 22, 4899. [Google Scholar] [CrossRef]
- Qin, A.; Musket, A.; Musich, P.R.; Schweitzer, J.B.; Xie, Q. Receptor tyrosine kinases as druggable targets in glioblastoma: Do signaling pathways matter? Neuro-Oncol. Adv. 2021, 3, vdab133. [Google Scholar] [CrossRef]
- Humphreys, L.M.; Smith, P.; Chen, Z.; Fouad, S.; D’Angiolella, V. The role of E3 ubiquitin ligases in the development and progression of glioblastoma. Cell Death Differ. 2021, 28, 522–537. [Google Scholar] [CrossRef]
- Tilak, M.; Holborn, J.; New, L.A.; Lalonde, J.; Jones, N. Receptor tyrosine kinase signaling and targeting in glioblastoma multiforme. Int. J. Mol. Sci. 2021, 22, 1831. [Google Scholar] [CrossRef]
- Chakravarti, A.; Chakladar, A.; Delaney, M.A.; Latham, D.E.; Loeffler, J.S. The epidermal growth factor receptor pathway mediates resistance to sequential administration of radiation and chemotherapy in primary human glioblastoma cells in a RAS-dependent manner. Cancer Res. 2002, 62, 4307–4315. [Google Scholar]
- Osuka, S.; Sampetrean, O.; Shimizu, T.; Saga, I.; Onishi, N.; Sugihara, E.; Okubo, J.; Fujita, S.; Takano, S.; Matsumura, A. IGF1 receptor signaling regulates adaptive radioprotection in glioma stem cells. Stem Cells 2013, 31, 627–640. [Google Scholar] [CrossRef]
- Osuka, S.; Zhu, D.; Zhang, Z.; Li, C.; Stackhouse, C.T.; Sampetrean, O.; Olson, J.J.; Gillespie, G.Y.; Saya, H.; Willey, C.D. N-cadherin upregulation mediates adaptive radioresistance in glioblastoma. J. Clin. Investig. 2021, 131, e136098. [Google Scholar] [CrossRef]
- Huang, M.; Zhang, D.; Wu, J.Y.; Xing, K.; Yeo, E.; Li, C.; Zhang, L.; Holland, E.; Yao, L.; Qin, L. Wnt-mediated endothelial transformation into mesenchymal stem cell–like cells induces chemoresistance in glioblastoma. Sci. Transl. Med. 2020, 12, eaay7522. [Google Scholar] [CrossRef]
- Gao, X.-Y.; Zang, J.; Zheng, M.-H.; Zhang, Y.-F.; Yue, K.-Y.; Cao, X.-L.; Cao, Y.; Li, X.-X.; Han, H.; Jiang, X.-F. Temozolomide treatment induces HMGB1 to promote the formation of glioma stem cells via the TLR2/NEAT1/Wnt pathway in glioblastoma. Front. Cell Dev. Biol. 2021, 9, 76. [Google Scholar] [CrossRef]
- Casili, G.; Caffo, M.; Campolo, M.; Barresi, V.; Caruso, G.; Cardali, S.M.; Lanza, M.; Mallamace, R.; Filippone, A.; Conti, A. TLR-4/Wnt modulation as new therapeutic strategy in the treatment of glioblastomas. Oncotarget 2018, 9, 37564. [Google Scholar] [CrossRef]
- Ma, C.; Nguyen, H.P.; Jones, J.J.; Stylli, S.S.; Whitehead, C.A.; Paradiso, L.; Luwor, R.B.; Areeb, Z.; Hanssen, E.; Cho, E. Extracellular Vesicles Secreted by Glioma Stem Cells Are Involved in Radiation Resistance and Glioma Progression. Int. J. Mol. Sci. 2022, 23, 2770. [Google Scholar] [CrossRef]
- Ma, Z.; Cai, S.; Xiong, Q.; Liu, W.; Xia, H.; Zhu, Z.; Huang, Z.; Yan, X.; Wang, Q. WNT signaling modulates chemoresistance to temozolomide in p53-mutant glioblastoma multiforme. Apoptosis 2022, 27, 80–89. [Google Scholar] [CrossRef]
- Yi, G.-z.; Liu, Y.-w.; Xiang, W.; Wang, H.; Chen, Z.-y.; Qi, S.-t. Akt and β-catenin contribute to TMZ resistance and EMT of MGMT negative malignant glioma cell line. J. Neurol. Sci. 2016, 367, 101–106. [Google Scholar] [CrossRef]
- Xu, K.; Zhang, Z.; Pei, H.; Wang, H.; Li, L.; Xia, Q. FoxO3a induces temozolomide resistance in glioblastoma cells via the regulation of β-catenin nuclear accumulation Corrigendum in/10.3892/or. 2020.7842. Oncol. Rep. 2017, 37, 2391–2397. [Google Scholar] [CrossRef] [Green Version]
- Tomar, V.S.; Patil, V.; Somasundaram, K. Temozolomide induces activation of Wnt/β-catenin signaling in glioma cells via PI3K/Akt pathway: Implications in glioma therapy. Cell Biol. Toxicol. 2020, 36, 273–278. [Google Scholar] [CrossRef]
- Yun, E.-J.; Kim, S.; Hsieh, J.-T.; Baek, S.T. Wnt/β-catenin signaling pathway induces autophagy-mediated temozolomide-resistance in human glioblastoma. Cell Death Dis. 2020, 11, 771. [Google Scholar] [CrossRef]
- Lu, C.; Cui, C.; Liu, B.; Zou, S.; Song, H.; Tian, H.; Zhao, J.; Li, Y. FERMT3 contributes to glioblastoma cell proliferation and chemoresistance to temozolomide through integrin mediated Wnt signaling. Neurosci. Lett. 2017, 657, 77–83. [Google Scholar] [CrossRef]
- Christmann, M.; Diesler, K.; Majhen, D.; Steigerwald, C.; Berte, N.; Freund, H.; Stojanović, N.; Kaina, B.; Osmak, M.; Ambriović-Ristov, A. Integrin αVβ3 silencing sensitizes malignant glioma cells to temozolomide by suppression of homologous recombination repair. Oncotarget 2017, 8, 27754. [Google Scholar] [CrossRef] [Green Version]
- Tsai, C.-Y.; Ko, H.-J.; Chiou, S.-J.; Lai, Y.-L.; Hou, C.-C.; Javaria, T.; Huang, Z.-Y.; Cheng, T.-S.; Hsu, T.-I.; Chuang, J.-Y. Nbm-bmx, an hdac8 inhibitor, overcomes temozolomide resistance in glioblastoma multiforme by downregulating the β-catenin/c-myc/sox2 pathway and upregulating p53-mediated mgmt inhibition. Int. J. Mol. Sci. 2021, 22, 5907. [Google Scholar] [CrossRef]
- Wickström, M.; Dyberg, C.; Milosevic, J.; Einvik, C.; Calero, R.; Sveinbjörnsson, B.; Sandén, E.; Darabi, A.; Siesjö, P.; Kool, M. Wnt/β-catenin pathway regulates MGMT gene expression in cancer and inhibition of Wnt signalling prevents chemoresistance. Nat. Commun. 2015, 6, 8904. [Google Scholar] [CrossRef]
- Bhuvanalakshmi, G.; Gamit, N.; Patil, M.; Arfuso, F.; Sethi, G.; Dharmarajan, A.; Prem Kumar, A.; Warrier, S. Stemness, pluripotentiality, and Wnt antagonism: sFRP4, a Wnt antagonist mediates pluripotency and stemness in glioblastoma. Cancers 2018, 11, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elmore, S. A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Trapani, J.A.; Sutton, V.R. Granzyme B: Pro-apoptotic, antiviral and antitumor functions. Curr. Opin. Immunol. 2003, 15, 533–543. [Google Scholar] [CrossRef]
- Beaudouin, J.; Liesche, C.; Aschenbrenner, S.; Hörner, M.; Eils, R. Caspase-8 cleaves its substrates from the plasma membrane upon CD95-induced apoptosis. Cell Death Differ. 2013, 20, 599–610. [Google Scholar] [CrossRef] [Green Version]
- Hill, M.M.; Adrain, C.; Duriez, P.J.; Creagh, E.M.; Martin, S.J. Analysis of the composition, assembly kinetics and activity of native Apaf-1 apoptosomes. EMBO J. 2004, 23, 2134–2145. [Google Scholar] [CrossRef] [Green Version]
- Pistritto, G.; Trisciuoglio, D.; Ceci, C.; Garufi, A.; D’Orazi, G. Apoptosis as anticancer mechanism: Function and dysfunction of its modulators and targeted therapeutic strategies. Aging 2016, 8, 603. [Google Scholar] [CrossRef] [Green Version]
- Valdés-Rives, S.A.; Casique-Aguirre, D.; Germán-Castelán, L.; Velasco-Velázquez, M.A.; González-Arenas, A. Apoptotic signaling pathways in glioblastoma and therapeutic implications. BioMed Res. Int. 2017, 2017, 7403747. [Google Scholar] [CrossRef] [Green Version]
- Saito, R.; Bringas, J.R.; Panner, A.; Tamas, M.; Pieper, R.O.; Berger, M.S.; Bankiewicz, K.S. Convection-enhanced delivery of tumor necrosis factor-related apoptosis-inducing ligand with systemic administration of temozolomide prolongs survival in an intracranial glioblastoma xenograft model. Cancer Res. 2004, 64, 6858–6862. [Google Scholar] [CrossRef] [Green Version]
- Deng, L.; Zhai, X.; Liang, P.; Cui, H. Overcoming TRAIL resistance for glioblastoma treatment. Biomolecules 2021, 11, 572. [Google Scholar] [CrossRef]
- Gratas, C.; Tohma, Y.; Meir, E.G.V.; Klein, M.; Tenan, M.; Ishii, N.; Tachibana, O.; Kleihues, P.; Ohgaki, H. Fas ligand expression in glioblastoma cell lines and primary astrocytic brain tumors. Brain Pathol. 1997, 7, 863–869. [Google Scholar] [CrossRef]
- Ho, I.A.; Ng, W.H.; Lam, P.Y. FasL and FADD delivery by a glioma-specific and cell cycle-dependent HSV-1 amplicon virus enhanced apoptosis in primary human brain tumors. Mol. Cancer 2010, 9, 270. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.-B.; Li, T.; Ma, D.-Z.; Ji, Y.-X.; Zhi, H. Overexpression of FADD and Caspase-8 inhibits proliferation and promotes apoptosis of human glioblastoma cells. Biomed. Pharmacother. 2017, 93, 1–7. [Google Scholar] [CrossRef]
- Kiraz, Y.; Adan, A.; Kartal Yandim, M.; Baran, Y. Major apoptotic mechanisms and genes involved in apoptosis. Tumor Biol. 2016, 37, 8471–8486. [Google Scholar] [CrossRef] [Green Version]
- Ho, T.; Tan, B.X.; Lane, D. How the other half lives: What p53 does when it is not being a transcription factor. Int. J. Mol. Sci. 2019, 21, 13. [Google Scholar] [CrossRef] [Green Version]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death by apoptosis. Trends Mol. Med. 2006, 12, 440–450. [Google Scholar] [CrossRef]
- He, Y.; Roos, W.P.; Wu, Q.; Hofmann, T.G.; Kaina, B. The SIAH1–HIPK2–p53ser46 damage response pathway is involved in temozolomide-induced glioblastoma cell death. Mol. Cancer Res. 2019, 17, 1129–1141. [Google Scholar] [CrossRef] [Green Version]
- Batista, L.F.; Roos, W.P.; Christmann, M.; Menck, C.F.; Kaina, B. Differential sensitivity of malignant glioma cells to methylating and chloroethylating anticancer drugs: p53 determines the switch by regulating xpc, ddb2, and DNA double-strand breaks. Cancer Res. 2007, 67, 11886–11895. [Google Scholar] [CrossRef] [Green Version]
- Rubner, Y.; Muth, C.; Strnad, A.; Derer, A.; Sieber, R.; Buslei, R.; Frey, B.; Fietkau, R.; Gaipl, U.S. Fractionated radiotherapy is the main stimulus for the induction of cell death and of Hsp70 release of p53 mutated glioblastoma cell lines. Radiat. Oncol. 2014, 9, 89. [Google Scholar] [CrossRef] [Green Version]
- Bocangel, D.B.; Finkelstein, S.; Schold, S.C.; Bhakat, K.K.; Mitra, S.; Kokkinakis, D.M. Multifaceted resistance of gliomas to temozolomide. Clin. Cancer Res. 2002, 8, 2725–2734. [Google Scholar]
- Hirose, Y.; Berger, M.S.; Pieper, R.O. p53 effects both the duration of G2/M arrest and the fate of temozolomide-treated human glioblastoma cells. Cancer Res. 2001, 61, 1957–1963. [Google Scholar]
- Xu, G.W.; Mymryk, J.S.; Cairncross, J.G. Inactivation of p53 sensitizes astrocytic glioma cells to BCNU and temozolomide, but not cisplatin. J. Neuro-Oncol. 2005, 74, 141–149. [Google Scholar] [CrossRef]
- Dinca, E.B.; Lu, K.V.; Sarkaria, J.N.; Pieper, R.O.; Prados, M.D.; Haas-Kogan, D.A.; VandenBerg, S.R.; Berger, M.S.; James, C.D. p53 Small-molecule inhibitor enhances temozolomide cytotoxic activity against intracranial glioblastoma xenografts. Cancer Res. 2008, 68, 10034–10039. [Google Scholar] [CrossRef] [Green Version]
- Reich, T.R.; Schwarzenbach, C.; Vilar, J.B.; Unger, S.; Mühlhäusler, F.; Nikolova, T.; Poplawski, A.; Baymaz, H.; Beli, P.; Christmann, M. Localization matters: Nuclear-trapped Survivin sensitizes glioblastoma cells to temozolomide by elevating cellular senescence and impairing homologous recombination. Cell. Mol. Life Sci. 2021, 78, 5587–5604. [Google Scholar] [CrossRef]
- Blough, M.D.; Beauchamp, D.C.; Westgate, M.R.; Kelly, J.J.; Cairncross, J.G. Effect of aberrant p53 function on temozolomide sensitivity of glioma cell lines and brain tumor initiating cells from glioblastoma. J. Neuro-Oncol. 2011, 102, 1–7. [Google Scholar] [CrossRef]
- Birner, P.; Piribauer, M.; Fischer, I.; Gatterbauer, B.; Marosi, C.; Ungersbock, K.; Rossler, K.; Budka, H.; Hainfellner, J.A. Prognostic relevance of p53 protein expression in glioblastoma. Oncol. Rep. 2002, 9, 703–707. [Google Scholar] [CrossRef]
- Kraus, J.A.; Glesmann, N.; Beck, M.; Krex, D.; Klockgether, T.; Schackert, G.; Schlegel, U. Molecular analysis of the PTEN, TP53 and CDKN2A tumor suppressor genes in long-term survivors of glioblastoma multiforme. J. Neuro-Oncol. 2000, 48, 89–94. [Google Scholar] [CrossRef]
- Kraus, A.; Gross, M.W.; Knuechel, R.; Münkel, K.; Neff, F.; Schlegel, J. Aberrant p21 regulation in radioresistant primary glioblastoma multiforme cells bearing wild-type p53. J. Neurosurg. 2000, 93, 863–872. [Google Scholar] [CrossRef]
- Rich, J.N.; Hans, C.; Jones, B.; Iversen, E.S.; McLendon, R.E.; Rasheed, B.A.; Dobra, A.; Dressman, H.K.; Bigner, D.D.; Nevins, J.R. Gene expression profiling and genetic markers in glioblastoma survival. Cancer Res. 2005, 65, 4051–4058. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Li, C.; Pazgier, M.; Li, C.; Mao, Y.; Lv, Y.; Gu, B.; Wei, G.; Yuan, W.; Zhan, C. D-peptide inhibitors of the p53–MDM2 interaction for targeted molecular therapy of malignant neoplasms. Proc. Natl. Acad. Sci. USA 2010, 107, 14321–14326. [Google Scholar] [CrossRef] [Green Version]
- Villalonga-Planells, R.; Coll-Mulet, L.; Martínez-Soler, F.; Castaño, E.; Acebes, J.-J.; Giménez-Bonafé, P.; Gil, J.; Tortosa, A. Activation of p53 by nutlin-3a induces apoptosis and cellular senescence in human glioblastoma multiforme. PLoS ONE 2011, 6, e18588. [Google Scholar]
- Chen, X.; Tai, L.; Gao, J.; Qian, J.; Zhang, M.; Li, B.; Xie, C.; Lu, L.; Lu, W.; Lu, W. A stapled peptide antagonist of MDM2 carried by polymeric micelles sensitizes glioblastoma to temozolomide treatment through p53 activation. J. Control. Release 2015, 218, 29–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verreault, M.; Schmitt, C.; Goldwirt, L.; Pelton, K.; Haidar, S.; Levasseur, C.; Guehennec, J.; Knoff, D.; Labussiere, M.; Marie, Y. Preclinical efficacy of the MDM2 inhibitor RG7112 in MDM2-amplified and TP53 wild-type glioblastomas. Clin. Cancer Res. 2016, 22, 1185–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Cai, S.; Bailey, B.J.; Saadatzadeh, M.R.; Ding, J.; Tonsing-Carter, E.; Georgiadis, T.M.; Gunter, T.Z.; Long, E.C.; Minto, R.E. Combination therapy in a xenograft model of glioblastoma: Enhancement of the antitumor activity of temozolomide by an MDM2 antagonist. J. Neurosurg. 2017, 126, 446–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Punganuru, S.R.; Arutla, V.; Zhao, W.; Rajaei, M.; Deokar, H.; Zhang, R.; Buolamwini, J.K.; Srivenugopal, K.S.; Wang, W. Targeted brain tumor therapy by inhibiting the MDM2 oncogene: In vitro and in vivo antitumor activity and mechanism of action. Cells 2020, 9, 1592. [Google Scholar] [CrossRef]
- Daniele, S.; La Pietra, V.; Piccarducci, R.; Pietrobono, D.; Cavallini, C.; D’Amore, V.M.; Cerofolini, L.; Giuntini, S.; Russomanno, P.; Puxeddu, M. CXCR4 antagonism sensitizes cancer cells to novel indole-based MDM2/4 inhibitors in glioblastoma multiforme. Eur. J. Pharmacol. 2021, 897, 173936. [Google Scholar] [CrossRef]
- Miles, X.; Vandevoorde, C.; Hunter, A.; Bolcaen, J. MDM2/X Inhibitors as Radiosensitizers for Glioblastoma Targeted Therapy. Front. Oncol. 2021, 11, 2688. [Google Scholar] [CrossRef]
- Haas-Kogan, D.A.; Kogan, S.S.; Yount, G.; Hsu, J.; Haas, M.; Deen, D.F.; Israel, M.A. p53 function influences the effect of fractionated radiotherapy on glioblastoma tumors. Int. J. Radiat. Oncol.* Biol.* Phys. 1999, 43, 399–403. [Google Scholar] [CrossRef]
- Riley, T.; Sontag, E.; Chen, P.; Levine, A. Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 2008, 9, 402–412. [Google Scholar] [CrossRef]
- Ochs, K.; Kaina, B. Apoptosis induced by DNA Damage O-Methylguanine is Bcl-2 and Caspase-9/3 regulated and Fas/Caspase-8 independent. Cancer Res. 2000, 60, 5815–5824. [Google Scholar]
- Jiang, Z.; Zheng, X.; Rich, K.M. Down-regulation of Bcl-2 and Bcl-xL expression with bispecific antisense treatment in glioblastoma cell lines induce cell death. J. Neurochem. 2003, 84, 273–281. [Google Scholar] [CrossRef]
- Hermisson, M.; Klumpp, A.; Wick, W.; Wischhusen, J.; Nagel, G.; Roos, W.; Kaina, B.; Weller, M. O6-methylguanine DNA methyltransferase and p53 status predict temozolomide sensitivity in human malignant glioma cells. J. Neurochem. 2006, 96, 766–776. [Google Scholar] [CrossRef]
- Schwarzenbach, C.; Tatsch, L.; Vilar, J.B.; Rasenberger, B.; Beltzig, L.; Kaina, B.; Tomicic, M.T.; Christmann, M. Targeting c-IAP1, c-IAP2, and Bcl-2 Eliminates Senescent Glioblastoma Cells Following Temozolomide Treatment. Cancers 2021, 13, 3585. [Google Scholar] [CrossRef]
- Glaser, T.; Weller, M. Caspase-dependent chemotherapy-induced death of glioma cells requires mitochondrial cytochrome c release. Biochem. Biophys. Res. Commun. 2001, 281, 322–327. [Google Scholar] [CrossRef]
- Decker, P.; Muller, S. Modulating poly (ADP-ribose) polymerase activity: Potential for the prevention and therapy of pathogenic situations involving DNA damage and oxidative stress. Curr. Pharm. Biotechnol. 2002, 3, 275–283. [Google Scholar] [CrossRef]
- Jo, G.H.; Bögler, O.; Chwae, Y.-J.; Yoo, H.; Lee, S.H.; Park, J.B.; Kim, Y.-J.; Kim, J.H.; Gwak, H.-S. Radiation-induced autophagy contributes to cell death and induces apoptosis partly in malignant glioma cells. Cancer Res. Treat. 2015, 47, 221. [Google Scholar] [CrossRef]
- Golden, E.B.; Cho, H.-Y.; Jahanian, A.; Hofman, F.M.; Louie, S.G.; Schönthal, A.H.; Chen, T.C. Chloroquine enhances temozolomide cytotoxicity in malignant gliomas by blocking autophagy. Neurosurg. Focus 2014, 37, E12. [Google Scholar] [CrossRef] [Green Version]
- Woo, S.R.; Ham, Y.; Kang, W.; Yang, H.; Kim, S.; Jin, J.; Joo, K.M.; Nam, D.-H. KML001, a telomere-targeting drug, sensitizes glioblastoma cells to temozolomide chemotherapy and radiotherapy through DNA damage and apoptosis. BioMed Res. Int. 2014, 2014, 747415. [Google Scholar] [CrossRef]
- Ohba, S.; Yamashiro, K.; Hirose, Y. Inhibition of dna repair in combination with temozolomide or dianhydrogalactiol overcomes temozolomide-resistant glioma cells. Cancers 2021, 13, 2570. [Google Scholar] [CrossRef]
- Kim, G.W.; Lee, D.H.; Yeon, S.-K.; Jeon, Y.H.; Yoo, J.; Lee, S.W.; Kwon, S.H. Temozolomide-resistant glioblastoma depends on HDAC6 activity through regulation of DNA mismatch repair. Anticancer Res. 2019, 39, 6731–6741. [Google Scholar] [CrossRef]
- De Salvo, M.; Maresca, G.; D’agnano, I.; Marchese, R.; Stigliano, A.; Gagliassi, R.; Brunetti, E.; Raza, G.H.; De Paula, U.; Bucci, B. Temozolomide induced c-Myc-mediated apoptosis via Akt signalling in MGMT expressing glioblastoma cells. Int. J. Radiat. Biol. 2011, 87, 518–533. [Google Scholar] [CrossRef]
- Larsen, B.D.; Sørensen, C.S. The caspase-activated DN ase: Apoptosis and beyond. FEBS J. 2017, 284, 1160–1170. [Google Scholar] [CrossRef]
- Masuoka, J.; Shiraishi, T.; Ichinose, M.; Mineta, T.; Tabuchi, K. Expression of ICAD-L and ICAD-S in Human Brain Tumor and Its Cleavage upon Activation of Apoptosis by Anti-Fas Antibody. Jpn. J. Cancer Res. 2001, 92, 806–812. [Google Scholar] [CrossRef]
- Samejima, K.; Earnshaw, W.C. Trashing the genome: The role of nucleases during apoptosis. Nat. Rev. Mol. Cell Biol. 2005, 6, 677–688. [Google Scholar] [CrossRef]
- Nagase, H.; Fukuyama, H.; Tanaka, M.; Kawane, K.; Nagata, S. Mutually regulated expression of caspase-activated DNase and its inhibitor for apoptotic DNA fragmentation. Cell Death Differ. 2003, 10, 142–143. [Google Scholar] [CrossRef]
- Kumar, R.; Gont, A.; Perkins, T.J.; Hanson, J.E.; Lorimer, I.A. Induction of senescence in primary glioblastoma cells by serum and TGFβ. Sci. Rep. 2017, 7, 2156. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Kohli, J.; Demaria, M. Senescent cells in cancer therapy: Friends or foes? Trends Cancer 2020, 6, 838–857. [Google Scholar] [CrossRef]
- Sharpless, N.E.; Sherr, C.J. Forging a signature of in vivo senescence. Nat. Rev. Cancer 2015, 15, 397–408. [Google Scholar] [CrossRef]
- Park, C.; Lee, I.; Kang, W.K. E2F-1 is a critical modulator of cellular senescence in human cancer. Int. J. Mol. Med. 2006, 17, 715–720. [Google Scholar] [CrossRef] [Green Version]
- Kumari, R.; Jat, P. Mechanisms of cellular senescence: Cell cycle arrest and senescence associated secretory phenotype. Front. Cell Dev. Biol. 2021, 9, 485. [Google Scholar] [CrossRef]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 99–118. [Google Scholar] [CrossRef] [Green Version]
- Jeon, H.-Y.; Kim, J.-K.; Ham, S.W.; Oh, S.-Y.; Kim, J.; Park, J.-B.; Lee, J.-Y.; Kim, S.-C.; Kim, H. Irradiation induces glioblastoma cell senescence and senescence-associated secretory phenotype. Tumor Biol. 2016, 37, 5857–5867. [Google Scholar] [CrossRef] [PubMed]
- Fletcher-Sananikone, E.; Kanji, S.; Tomimatsu, N.; Di Cristofaro, L.F.M.; Kollipara, R.K.; Saha, D.; Floyd, J.R.; Sung, P.; Hromas, R.; Burns, T.C. Elimination of radiation-induced senescence in the brain tumor microenvironment attenuates glioblastoma recurrence. Cancer Res. 2021, 81, 5935–5947. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.; Olson, I.; Mansour, M.; Carlstrom, L.P.; Sutiwisesak, R.; Saber, R.; Rajani, K.; Warrington, A.E.; Howard, A.; Schroeder, M. Selective Vulnerability of Senescent Glioblastoma Cells to BCL-XL InhibitionBCL-XL Inhibitors Ablate Senescent GBM Cells. Mol. Cancer Res. 2022, OF1–OF11. [Google Scholar]
- Espinosa-Sánchez, A.; Suárez-Martínez, E.; Sánchez-Díaz, L.; Carnero, A. Therapeutic targeting of signaling pathways related to cancer stemness. Front. Oncol. 2020, 1533. [Google Scholar] [CrossRef] [PubMed]
- Gürsel, D.B.; Shin, B.J.; Burkhardt, J.-K.; Kesavabhotla, K.; Schlaff, C.D.; Boockvar, J.A. Glioblastoma stem-like cells—Biology and therapeutic implications. Cancers 2011, 3, 2655–2666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raso, A.; Vecchio, D.; Cappelli, E.; Ropolo, M.; Poggi, A.; Nozza, P.; Biassoni, R.; Mascelli, S.; Capra, V.; Kalfas, F. Characterization of glioma stem cells through multiple stem cell markers and their specific sensitization to double-strand break-inducing agents by pharmacological inhibition of ataxia telangiectasia mutated protein. Brain Pathol. 2012, 22, 677–688. [Google Scholar] [CrossRef]
- Zhou, W.; Sun, M.; Li, G.-H.; Wu, Y.-Z.; Wang, Y.; Jin, F.; Zhang, Y.-Y.; Yang, L.; Wang, D.-L. Activation of the phosphorylation of ATM contributes to radioresistance of glioma stem cells. Oncol. Rep. 2013, 30, 1793–1801. [Google Scholar] [CrossRef] [Green Version]
- Lim, Y.C.; Roberts, T.L.; Day, B.W.; Stringer, B.W.; Kozlov, S.; Fazry, S.; Bruce, Z.C.; Ensbey, K.S.; Walker, D.G.; Boyd, A.W. Increased sensitivity to ionizing radiation by targeting the homologous recombination pathway in glioma initiating cells. Mol. Oncol. 2014, 8, 1603–1615. [Google Scholar] [CrossRef]
- Lim, Y.C.; Roberts, T.L.; Day, B.W.; Harding, A.; Kozlov, S.; Kijas, A.W.; Ensbey, K.S.; Walker, D.G.; Lavin, M.F. A role for homologous recombination and abnormal cell-cycle progression in radioresistance of glioma-initiating cells. Mol. Cancer Ther. 2012, 11, 1863–1872. [Google Scholar] [CrossRef] [Green Version]
- Firat, E.; Gaedicke, S.; Tsurumi, C.; Esser, N.; Weyerbrock, A.; Niedermann, G. Delayed cell death associated with mitotic catastrophe in γ-irradiated stem-like glioma cells. Radiat. Oncol. 2011, 6, 71. [Google Scholar] [CrossRef] [Green Version]
- Cappelli, E.; Vecchio, D.; Frosina, G. Delayed formation of FancD2 foci in glioma stem cells treated with ionizing radiation. J. Cancer Res. Clin. Oncol. 2012, 138, 897–899. [Google Scholar] [CrossRef]
- Tachon, G.; Cortes, U.; Guichet, P.-O.; Rivet, P.; Balbous, A.; Masliantsev, K.; Berger, A.; Boissonnade, O.; Wager, M.; Karayan-Tapon, L. Cell cycle changes after glioblastoma stem cell irradiation: The major role of RAD51. Int. J. Mol. Sci. 2018, 19, 3018. [Google Scholar] [CrossRef] [Green Version]
- Lau, J.; Ilkhanizadeh, S.; Wang, S.; Miroshnikova, Y.A.; Salvatierra, N.A.; Wong, R.A.; Schmidt, C.; Weaver, V.M.; Weiss, W.A.; Persson, A.I. STAT3 blockade inhibits radiation-induced malignant progression in glioma. Cancer Res. 2015, 75, 4302–4311. [Google Scholar] [CrossRef] [Green Version]
- Minata, M.; Audia, A.; Shi, J.; Lu, S.; Bernstock, J.; Pavlyukov, M.S.; Das, A.; Kim, S.-H.; Shin, Y.J.; Lee, Y. Phenotypic plasticity of invasive edge glioma stem-like cells in response to ionizing radiation. Cell Rep. 2019, 26, 1893–1905.e1897. [Google Scholar] [CrossRef] [Green Version]
- Gauthier, L.R.; Saati, M.; Bensalah-Pigeon, H.; Ben M’Barek, K.; Gitton-Quent, O.; Bertrand, R.; Busso, D.; Mouthon, M.-A.; Collura, A.; Junier, M.-P. The HIF1α/JMY pathway promotes glioblastoma stem-like cell invasiveness after irradiation. Sci. Rep. 2020, 10, 18742. [Google Scholar] [CrossRef]
- Dahan, P.; Martinez Gala, J.; Delmas, C.; Monferran, S.; Malric, L.; Zentkowski, D.; Lubrano, V.; Toulas, C.; Cohen-Jonathan Moyal, E.; Lemarie, A. Ionizing radiations sustain glioblastoma cell dedifferentiation to a stem-like phenotype through survivin: Possible involvement in radioresistance. Cell Death Dis. 2014, 5, e1543. [Google Scholar] [CrossRef] [Green Version]
- Berg, T.J.; Marques, C.; Pantazopoulou, V.; Johansson, E.; von Stedingk, K.; Lindgren, D.; Pietras, E.J.; Bergström, T.; Swartling, F.J.; Governa, V. The irradiated brain microenvironment supports glioma stemness and survival via astrocyte-derived Transglutaminase 2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Ong, D.S.T.; Hu, B.; Ho, Y.W.; Sauvé, C.-E.G.; Bristow, C.A.; Wang, Q.; Multani, A.S.; Chen, P.; Nezi, L.; Jiang, S. PAF promotes stemness and radioresistance of glioma stem cells. Proc. Natl. Acad. Sci. USA 2017, 114, E9086–E9095. [Google Scholar] [CrossRef] [Green Version]
- Günther, W.; Pawlak, E.; Damasceno, R.; Arnold, H.; Terzis, A. Temozolomide induces apoptosis and senescence in glioma cells cultured as multicellular spheroids. Br. J. Cancer 2003, 88, 463–469. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Zhao, L.; Gong, S.; Xiong, S.; Wang, J.; Zou, D.; Pan, J.; Deng, Y.; Yan, Q.; Wu, N.; et al. HIF1alpha/HIF2alpha-Sox2/Klf4 promotes the malignant progression of glioblastoma via the EGFR-PI3K/AKT signalling pathway with positive feedback under hypoxia. Cell Death Dis. 2021, 12, 312. [Google Scholar] [CrossRef]
- Auffinger, B.; Tobias, A.; Han, Y.; Lee, G.; Guo, D.; Dey, M.; Lesniak, M.; Ahmed, A. Conversion of differentiated cancer cells into cancer stem-like cells in a glioblastoma model after primary chemotherapy. Cell Death Differ. 2014, 21, 1119–1131. [Google Scholar] [CrossRef]
- Clement, V.; Sanchez, P.; De Tribolet, N.; Radovanovic, I.; i Altaba, A.R. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr. Biol. 2007, 17, 165–172. [Google Scholar] [CrossRef]
- Zhang, M.; Kleber, S.; Röhrich, M.; Timke, C.; Han, N.; Tuettenberg, J.; Martin-Villalba, A.; Debus, J.; Peschke, P.; Wirkner, U. Blockade of TGF-β signaling by the TGFβR-I kinase inhibitor LY2109761 enhances radiation response and prolongs survival in glioblastoma. Cancer Res. 2011, 71, 7155–7167. [Google Scholar] [CrossRef] [Green Version]
- Colamaio, M.; Tosti, N.; Puca, F.; Mari, A.; Gattordo, R.; Kuzay, Y.; Federico, A.; Pepe, A.; Sarnataro, D.; Ragozzino, E. HMGA1 silencing reduces stemness and temozolomide resistance in glioblastoma stem cells. Expert Opin. Ther. Targets 2016, 20, 1169–1179. [Google Scholar] [CrossRef]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Saha, S.; Panigrahi, D.P.; Patil, S.; Bhutia, S.K. Autophagy in health and disease: A comprehensive review. Biomed. Pharmacother. 2018, 104, 485–495. [Google Scholar] [CrossRef]
- Sun, K.; Deng, W.; Zhang, S.; Cai, N.; Jiao, S.; Song, J.; Wei, L. Paradoxical roles of autophagy in different stages of tumorigenesis: Protector for normal or cancer cells. Cell Biosci. 2013, 3, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondo, Y.; Kanzawa, T.; Sawaya, R.; Kondo, S. The role of autophagy in cancer development and response to therapy. Nat. Rev. Cancer 2005, 5, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Tam, S.Y.; Wu, V.W.C.; Law, H.K.W. Influence of autophagy on the efficacy of radiotherapy. Radiat. Oncol. 2017, 12, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul-Samojedny, M.; Pudełko, A.; Kowalczyk, M.; Fila-Daniłow, A.; Suchanek-Raif, R.; Borkowska, P.; Kowalski, J. Combination therapy with AKT3 and PI3KCA siRNA enhances the antitumor effect of temozolomide and carmustine in T98G glioblastoma multiforme cells. BioDrugs 2016, 30, 129–144. [Google Scholar] [CrossRef]
- Guo, J.Y.; Chen, H.-Y.; Mathew, R.; Fan, J.; Strohecker, A.M.; Karsli-Uzunbas, G.; Kamphorst, J.J.; Chen, G.; Lemons, J.M.; Karantza, V. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011, 25, 460–470. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.Y.; Teng, X.; Laddha, S.V.; Ma, S.; Van Nostrand, S.C.; Yang, Y.; Khor, S.; Chan, C.S.; Rabinowitz, J.D.; White, E. Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells. Genes Dev. 2016, 30, 1704–1717. [Google Scholar] [CrossRef] [Green Version]
- Papa, S.; Choy, P.M.; Bubici, C. The ERK and JNK pathways in the regulation of metabolic reprogramming. Oncogene 2019, 38, 2223–2240. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.; Dong, X.; Yap, J.; Hu, J. The MAPK and AMPK signalings: Interplay and implication in targeted cancer therapy. J. Hematol. Oncol. 2020, 13, 113. [Google Scholar] [CrossRef]
- Lo, H.-W. Targeting Ras-RAF-ERK and its interactive pathways as a novel therapy for malignant gliomas. Curr. Cancer Drug Targets 2010, 10, 840–848. [Google Scholar] [CrossRef]
- Ito, H.; Daido, S.; Kanzawa, T.; Kondo, S.; Kondo, Y. Radiation-induced autophagy is associated with LC3 and its inhibition sensitizes malignant glioma cells. Int. J. Oncol. 2005, 26, 1401–1410. [Google Scholar] [CrossRef]
- Ye, X.; Zhou, X.-J.; Zhang, H. Exploring the role of autophagy-related gene 5 (ATG5) yields important insights into autophagy in autoimmune/autoinflammatory diseases. Front. Immunol. 2018, 9, 2334. [Google Scholar] [CrossRef]
- Kang, R.; Zeh, H.; Lotze, M.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011, 18, 571–580. [Google Scholar] [CrossRef]
- Lomonaco, S.L.; Finniss, S.; Xiang, C.; DeCarvalho, A.; Umansky, F.; Kalkanis, S.N.; Mikkelsen, T.; Brodie, C. The induction of autophagy by γ-radiation contributes to the radioresistance of glioma stem cells. Int. J. Cancer 2009, 125, 717–722. [Google Scholar] [CrossRef]
- Liu, C.; He, W.; Jin, M.; Li, H.; Xu, H.; Liu, H.; Yang, K.; Zhang, T.; Wu, G.; Ren, J. Blockage of autophagy in C6 glioma cells enhanced radiosensitivity possibly by attenuating DNA-PK-dependent DSB due to limited Ku nuclear translocation and DNA binding. Curr. Mol. Med. 2015, 15, 663–673. [Google Scholar] [CrossRef]
- Menon, M.B.; Dhamija, S. Beclin 1 phosphorylation—At the center of autophagy regulation. Front. Cell Dev. Biol. 2018, 6, 137. [Google Scholar] [CrossRef] [Green Version]
- Daido, S.; Yamamoto, A.; Fujiwara, K.; Sawaya, R.; Kondo, S.; Kondo, Y. Inhibition of the DNA-dependent protein kinase catalytic subunit radiosensitizes malignant glioma cells by inducing autophagy. Cancer Res. 2005, 65, 4368–4375. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, W.; Li, B.; Long, L.; Chen, L.; Huang, Q.; Liang, Z.-Q. Knockdown of the DNA-dependent protein kinase catalytic subunit radiosensitizes glioma-initiating cells by inducing autophagy. Brain Res. 2011, 1371, 7–15. [Google Scholar] [CrossRef]
- Palumbo, S.; Tini, P.; Toscano, M.; Allavena, G.; Angeletti, F.; Manai, F.; Miracco, C.; Comincini, S.; Pirtoli, L. Combined EGFR and autophagy modulation impairs cell migration and enhances radiosensitivity in human glioblastoma cells. J. Cell. Physiol. 2014, 229, 1863–1873. [Google Scholar] [CrossRef]
- Huang, T.; Kim, C.K.; Alvarez, A.A.; Pangeni, R.P.; Wan, X.; Song, X.; Shi, T.; Yang, Y.; Sastry, N.; Horbinski, C.M. MST4 phosphorylation of ATG4B regulates autophagic activity, tumorigenicity, and radioresistance in glioblastoma. Cancer Cell 2017, 32, 840–855.e848. [Google Scholar] [CrossRef] [Green Version]
- Cho, Y.H.; Lee, S.-H.; Lee, S.-J.; Kim, H.N.; Koh, J.-Y. A role of metallothionein-3 in radiation-induced autophagy in glioma cells. Sci. Rep. 2020, 10, 2015. [Google Scholar] [CrossRef]
- Zheng, W.; Chen, Q.; Wang, C.; Yao, D.; Zhu, L.; Pan, Y.; Zhang, J.; Bai, Y.; Shao, C. Inhibition of Cathepsin D (CTSD) enhances radiosensitivity of glioblastoma cells by attenuating autophagy. Mol. Carcinog. 2020, 59, 651–660. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. An emerging role of mTOR in lipid biosynthesis. Curr. Biol. 2009, 19, R1046–R1052. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.C.; Guan, K.-L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, W.; Li, B.; Long, L.; Chen, L.; Huang, Q.; Liang, Z. Induction of autophagy promotes differentiation of glioma-initiating cells and their radiosensitivity. Int. J. Cancer 2011, 129, 2720–2731. [Google Scholar] [CrossRef] [PubMed]
- Nam, H.Y.; Han, M.W.; Chang, H.W.; Lee, Y.S.; Lee, M.; Lee, H.J.; Lee, B.W.; Lee, H.J.; Lee, K.E.; Jung, M.K. Radioresistant cancer cells can be conditioned to enter senescence by mTOR inhibition. Cancer Res. 2013, 73, 4267–4277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vessoni, A.T.; Quinet, A.; de Andrade-Lima, L.C.; Martins, D.J.; Garcia, C.C.M.; Rocha, C.R.R.; Vieira, D.B.; Menck, C.F.M. Chloroquine-induced glioma cells death is associated with mitochondrial membrane potential loss, but not oxidative stress. Free Radic. Biol. Med. 2016, 90, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Chen, M.; Cao, F.; Huang, H.; Zhan, R.; Zheng, X. Chloroquine, an autophagy inhibitor, potentiates the radiosensitivity of glioma initiating cells by inhibiting autophagy and activating apoptosis. BMC Neurol. 2016, 16, 178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natsumeda, M.; Aoki, H.; Miyahara, H.; Yajima, N.; Uzuka, T.; Toyoshima, Y.; Kakita, A.; Takahashi, H.; Fujii, Y. Induction of autophagy in temozolomide treated malignant gliomas. Neuropathology 2011, 31, 486–493. [Google Scholar] [CrossRef] [PubMed]
- Abbas, S.; Singh, S.K.; Saxena, A.K.; Tiwari, S.; Sharma, L.K.; Tiwari, M. Role of autophagy in regulation of glioma stem cells population during therapeutic stress. J. Stem Cells Regen. Med. 2020, 16, 80. [Google Scholar] [PubMed]
- Knizhnik, A.V.; Roos, W.P.; Nikolova, T.; Quiros, S.; Tomaszowski, K.-H.; Christmann, M.; Kaina, B. Survival and death strategies in glioma cells: Autophagy, senescence and apoptosis triggered by a single type of temozolomide-induced DNA damage. PLoS One 2013, 8, e55665. [Google Scholar] [CrossRef] [Green Version]
- Kanzawa, T.; Germano, I.; Komata, T.; Ito, H.; Kondo, Y.; Kondo, S. Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ. 2004, 11, 448–457. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.W.; Kim, H.-K.; Lee, N.-H.; Yi, H.-Y.; Kim, H.-S.; Hong, S.H.; Hong, Y.-K.; Joe, Y.A. The synergistic effect of combination temozolomide and chloroquine treatment is dependent on autophagy formation and p53 status in glioma cells. Cancer Lett. 2015, 360, 195–204. [Google Scholar] [CrossRef]
- Wen, Z.-p.; Zeng, W.-j.; Chen, Y.-h.; Li, H.; Wang, J.-y.; Cheng, Q.; Yu, J.; Zhou, H.-h.; Liu, Z.-z.; Xiao, J. Knockdown ATG4C inhibits gliomas progression and promotes temozolomide chemosensitivity by suppressing autophagic flux. J. Exp. Clin. Cancer Res. 2019, 38, 298. [Google Scholar] [CrossRef]
- Knizhnik, A.; Quiros, S.; Barckhausen, C.; Roos, W.; Kaina, B. DNA Damaging Drugs in the Treatment of Glioblastoma: HR, Apoptosis, Autophagy and Senescence. Klin. Pädiatrie 2012, 224, A8. [Google Scholar] [CrossRef]
- Filippi-Chiela, E.C.; Bueno e Silva, M.M.; Thome, M.P.; Lenz, G. Single-cell analysis challenges the connection between autophagy and senescence induced by DNA damage. Autophagy 2015, 11, 1099–1113. [Google Scholar] [CrossRef] [Green Version]
- Zou, Y.; Wang, Q.; Li, B.; Xie, B.; Wang, W. Temozolomide induces autophagy via ATM-AMPK-ULK1 pathways in glioma. Mol. Med. Rep. 2014, 10, 411–416. [Google Scholar] [CrossRef] [Green Version]
- Galavotti, S.; Bartesaghi, S.; Faccenda, D.; Shaked-Rabi, M.; Sanzone, S.; McEvoy, A.; Dinsdale, D.; Condorelli, F.; Brandner, S.; Campanella, M. The autophagy-associated factors DRAM1 and p62 regulate cell migration and invasion in glioblastoma stem cells. Oncogene 2013, 32, 699–712. [Google Scholar] [CrossRef] [Green Version]
- Hori, Y.S.; Hosoda, R.; Akiyama, Y.; Sebori, R.; Wanibuchi, M.; Mikami, T.; Sugino, T.; Suzuki, K.; Maruyama, M.; Tsukamoto, M. Chloroquine potentiates temozolomide cytotoxicity by inhibiting mitochondrial autophagy in glioma cells. J. Neuro-Oncol. 2015, 122, 11–20. [Google Scholar] [CrossRef]
- Buccarelli, M.; Marconi, M.; Pacioni, S.; De Pascalis, I.; D’Alessandris, Q.G.; Martini, M.; Ascione, B.; Malorni, W.; Larocca, L.M.; Pallini, R. Inhibition of autophagy increases susceptibility of glioblastoma stem cells to temozolomide by igniting ferroptosis. Cell Death Dis. 2018, 9, 841. [Google Scholar] [CrossRef]
- Sato, A.; Okada, M.; Shibuya, K.; Watanabe, E.; Seino, S.; Narita, Y.; Shibui, S.; Kayama, T.; Kitanaka, C. Pivotal role for ROS activation of p38 MAPK in the control of differentiation and tumor-initiating capacity of glioma-initiating cells. Stem Cell Res. 2014, 12, 119–131. [Google Scholar] [CrossRef] [Green Version]
- Rocha, C.R.R.; Kajitani, G.S.; Quinet, A.; Fortunato, R.S.; Menck, C.F.M. NRF2 and glutathione are key resistance mediators to temozolomide in glioma and melanoma cells. Oncotarget 2016, 7, 48081. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Wang, H.-D.; Zhu, L.; Cong, Z.-X.; Li, N.; Ji, X.-J.; Pan, H.; Wang, J.-W.; Li, W.-C. Knockdown of Nrf2 enhances autophagy induced by temozolomide in U251 human glioma cell line. Oncol. Rep. 2013, 29, 394–400. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Hu, X.; Liu, Y.; Dong, S.; Wen, Z.; He, W.; Zhang, S.; Huang, Q.; Shi, M. ROS signaling under metabolic stress: Cross-talk between AMPK and AKT pathway. Mol. Cancer 2017, 16, 79. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.J.; Lee, C.C.; Shih, Y.L.; Lin, T.Y.; Wang, S.H.; Lin, Y.F.; Shih, C.M. Resveratrol enhances the therapeutic effect of temozolomide against malignant glioma in vitro and in vivo by inhibiting autophagy. Free Radic Biol Med. 2012, 52, 377–391. [Google Scholar] [CrossRef]
- Rosenfeld, M.R.; Ye, X.; Supko, J.G.; Desideri, S.; Grossman, S.A.; Brem, S.; Mikkelson, T.; Wang, D.; Chang, Y.C.; Hu, J. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy 2014, 10, 1359–1368. [Google Scholar] [CrossRef]
- Urra, H.; Dufey, E.; Avril, T.; Chevet, E.; Hetz, C. Endoplasmic reticulum stress and the hallmarks of cancer. Trends Cancer 2016, 2, 252–262. [Google Scholar] [CrossRef]
- Dastghaib, S.; Shojaei, S.; Mostafavi-Pour, Z.; Sharma, P.; Patterson, J.B.; Samali, A.; Mokarram, P.; Ghavami, S. Simvastatin induces unfolded protein response and enhances temozolomide-induced cell death in glioblastoma cells. Cells 2020, 9, 2339. [Google Scholar] [CrossRef]
- Lin, C.J.; Lee, C.C.; Shih, Y.L.; Lin, C.H.; Wang, S.H.; Chen, T.H.; Shih, C.M. Inhibition of mitochondria- and endoplasmic reticulum stress-mediated autophagy augments temozolomide-induced apoptosis in glioma cells. PLoS One 2012, 7, e38706. [Google Scholar] [CrossRef] [Green Version]
- Kouroku, Y.; Fujita, E.; Tanida, I.; Ueno, T.; Isoai, A.; Kumagai, H.; Ogawa, S.; Kaufman, R.; Kominami, E.; Momoi, T. ER stress (PERK/eIF2α phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ. 2007, 14, 230–239. [Google Scholar] [CrossRef] [Green Version]
- Bernales, S.; McDonald, K.L.; Walter, P. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol. 2006, 4, e423. [Google Scholar] [CrossRef] [Green Version]
- Ou, A.; Ott, M.; Fang, D.; Heimberger, A.B. The Role and Therapeutic Targeting of JAK/STAT Signaling in Glioblastoma. Cancers 2021, 13, 437. [Google Scholar] [CrossRef]
- Lo, H.W.; Cao, X.; Zhu, H.; Ali-Osman, F. Constitutively activated STAT3 frequently coexpresses with epidermal growth factor receptor in high-grade gliomas and targeting STAT3 sensitizes them to Iressa and alkylators. Clin. Cancer Res. 2008, 14, 6042–6054. [Google Scholar] [CrossRef] [Green Version]
- Kohsaka, S.; Wang, L.; Yachi, K.; Mahabir, R.; Narita, T.; Itoh, T.; Tanino, M.; Kimura, T.; Nishihara, H.; Tanaka, S. STAT3 inhibition overcomes temozolomide resistance in glioblastoma by downregulating MGMT expression. Mol. Cancer 2012, 11, 1289–1299. [Google Scholar] [CrossRef] [Green Version]
- Smith-Cohn, M.A.; Celiku, O.; Gilbert, M.R. Molecularly Targeted Clinical Trials. Neurosurg. Clin. 2021, 32, 191–210. [Google Scholar] [CrossRef] [PubMed]
- Ellison-Hughes, G.M. First evidence that senolytics are effective at decreasing senescent cells in humans. EBioMedicine 2020, 56, 102473. [Google Scholar] [CrossRef] [PubMed]
- Wissler Gerdes, E.O.; Zhu, Y.; Tchkonia, T.; Kirkland, J.L. Discovery, development, and future application of senolytics: Theories and predictions. FEBS J. 2020, 287, 2418–2427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, P.R.; Young, K.H.; Kumar, D.; Jain, N. RNA-mediated immunotherapy regulating tumor immune microenvironment: Next wave of cancer therapeutics. Mol. Cancer 2022, 21, 58. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Bano, S.; Kapse, P.; Kundu, G.C. CRISPR based therapeutics: A new paradigm in cancer precision medicine. Mol. Cancer 2022, 21, 85. [Google Scholar] [CrossRef]


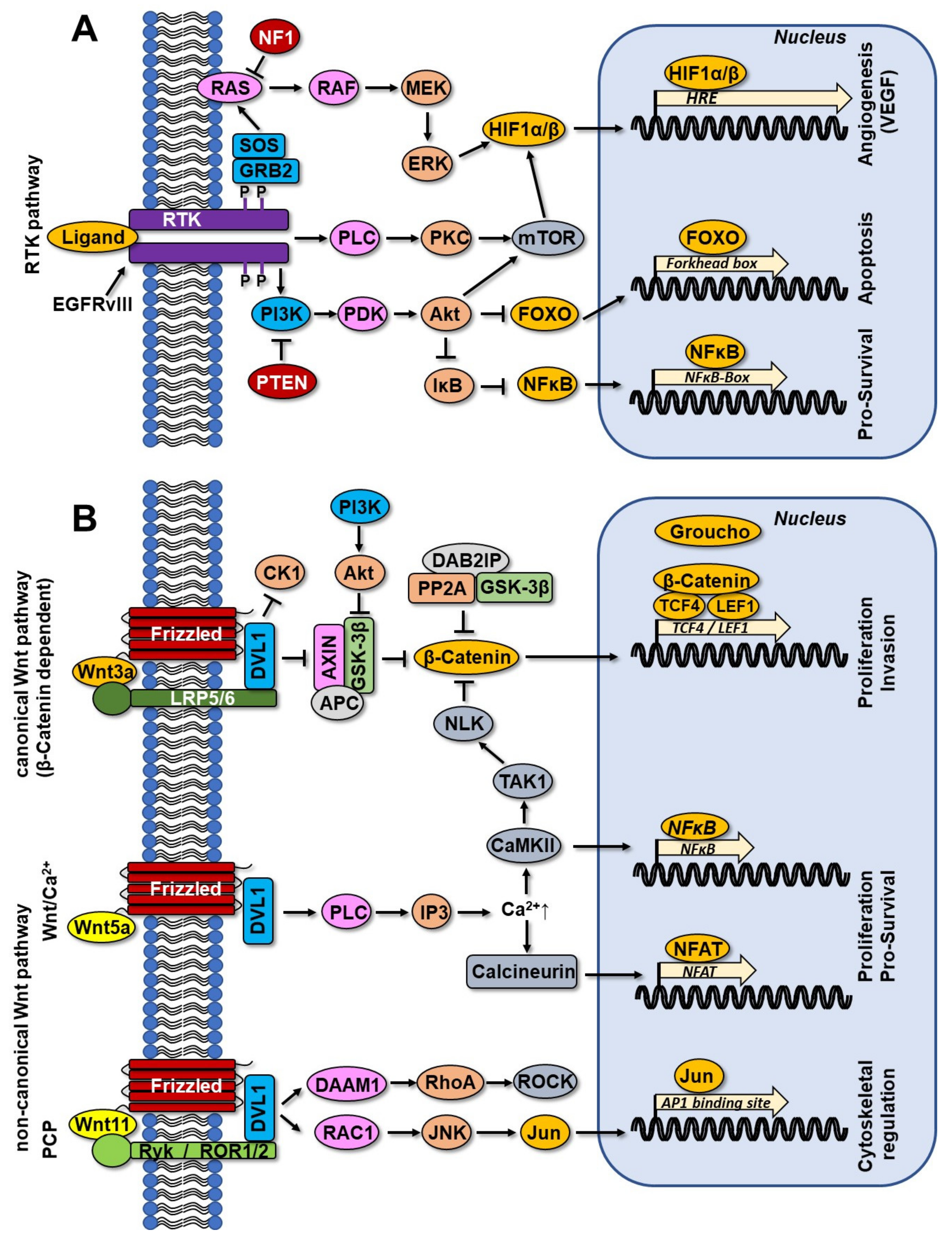

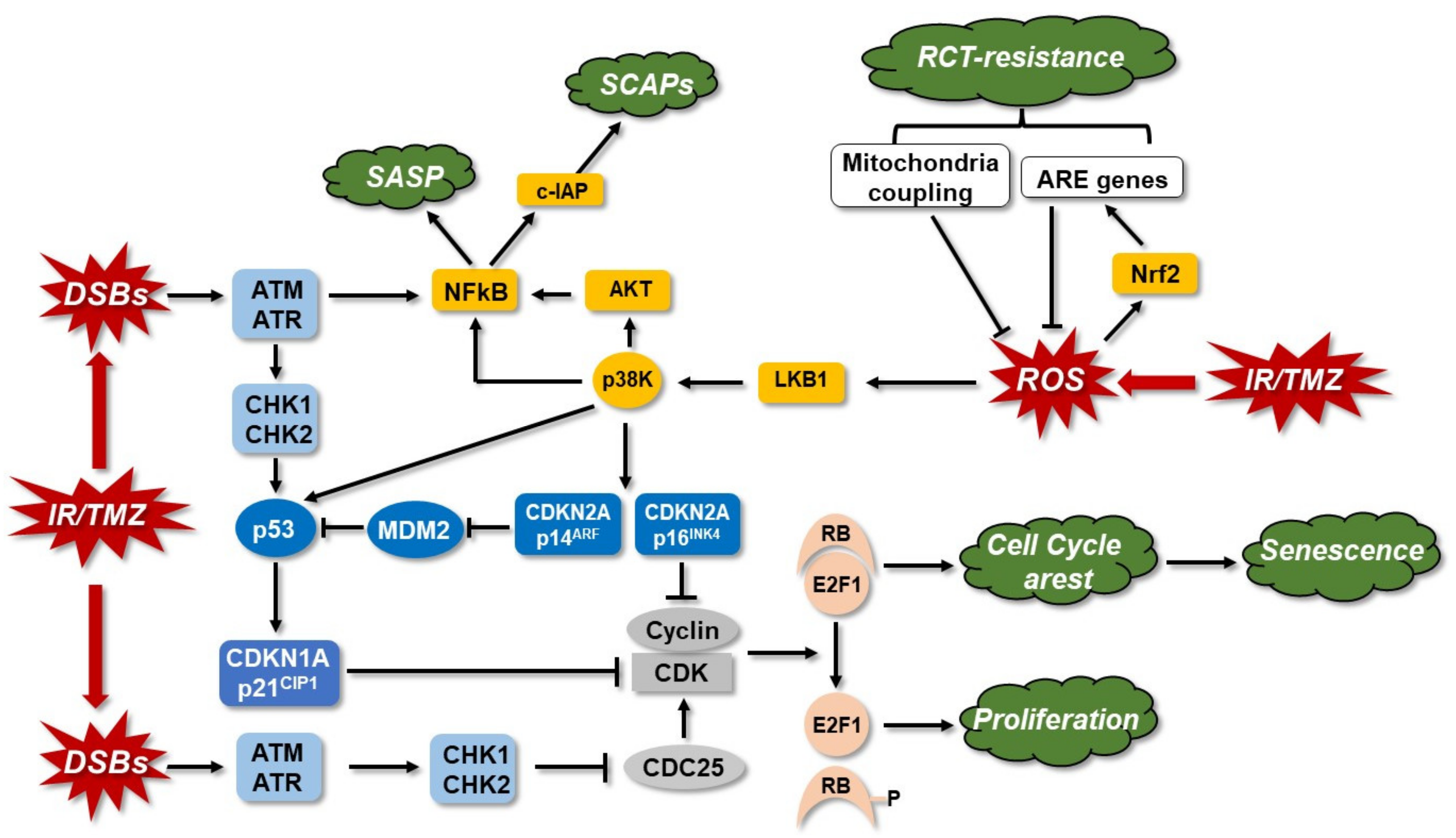

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vilar, J.B.; Christmann, M.; Tomicic, M.T. Alterations in Molecular Profiles Affecting Glioblastoma Resistance to Radiochemotherapy: Where Does the Good Go? Cancers 2022, 14, 2416. https://doi.org/10.3390/cancers14102416
Vilar JB, Christmann M, Tomicic MT. Alterations in Molecular Profiles Affecting Glioblastoma Resistance to Radiochemotherapy: Where Does the Good Go? Cancers. 2022; 14(10):2416. https://doi.org/10.3390/cancers14102416
Chicago/Turabian StyleVilar, Juliana B., Markus Christmann, and Maja T. Tomicic. 2022. "Alterations in Molecular Profiles Affecting Glioblastoma Resistance to Radiochemotherapy: Where Does the Good Go?" Cancers 14, no. 10: 2416. https://doi.org/10.3390/cancers14102416