
Characterization of Temozolomide Resistance Using a Novel Acquired Resistance Model in Glioblastoma Cell Lines

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
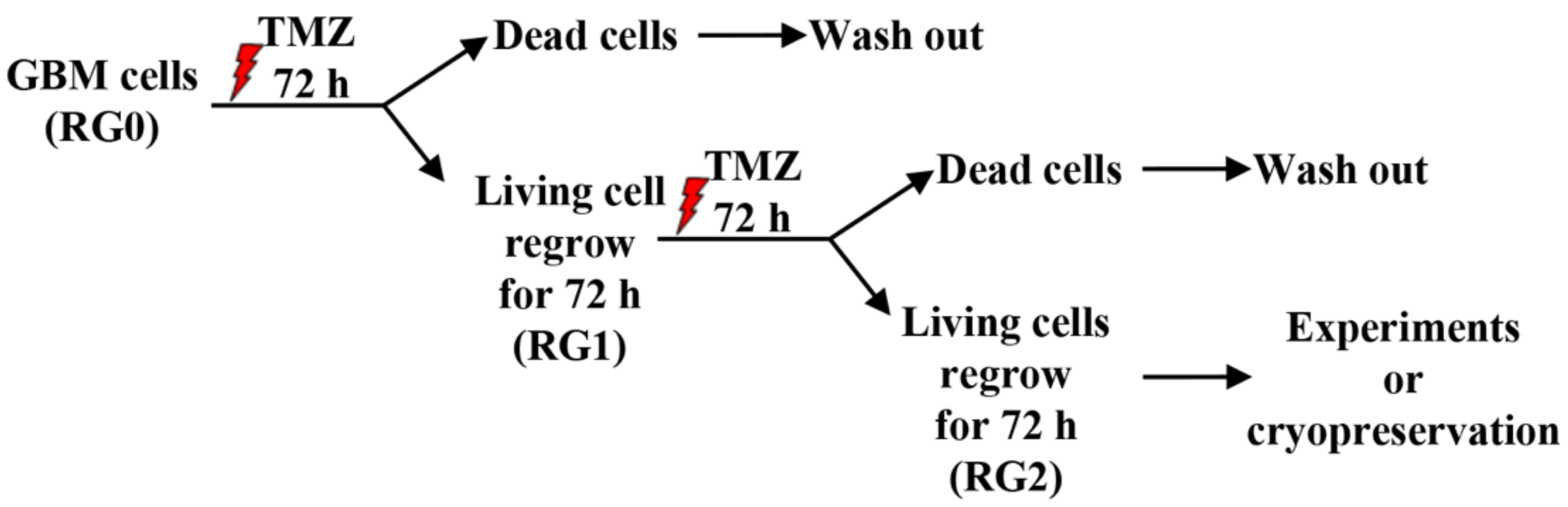
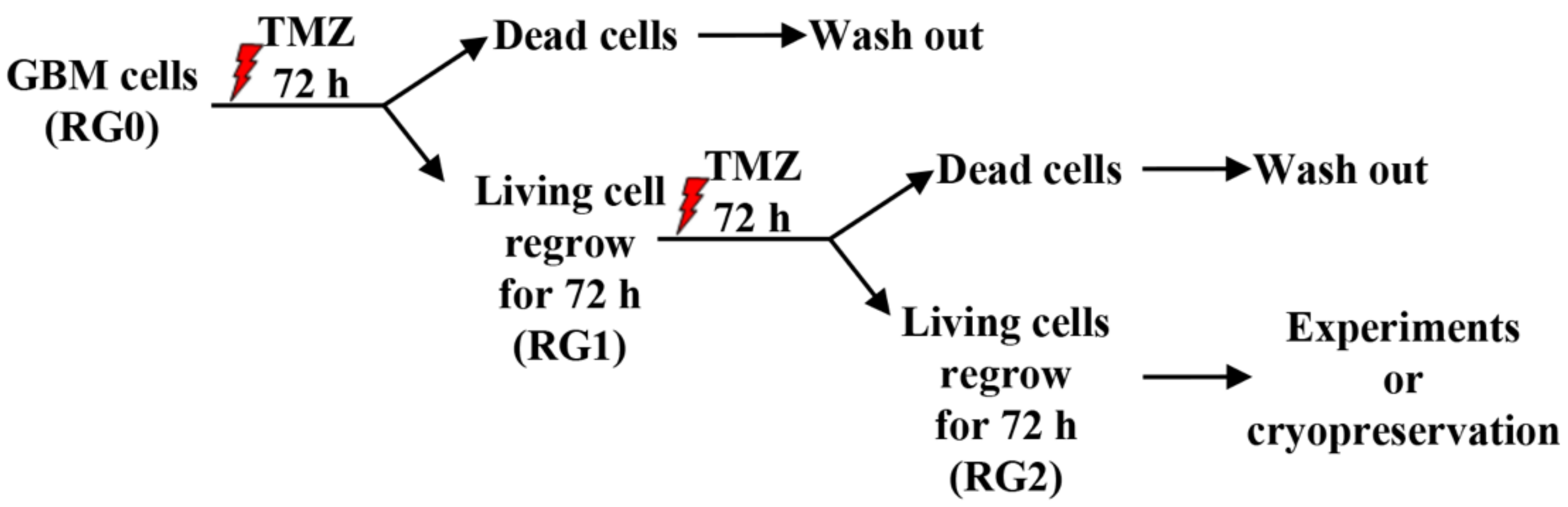
2.1. Cell Culture, Generation of TMZ Resistant GBM Variants and Drug Treatment
2.2. Generation of GBM-RG Cells Expressing a 53BP1-Foci Reporter and Analysis of TMZ Induced Double-Strand Breaks (DSBs)
2.3. MTT Assay
2.4. Flow Cytometry Detection of Cell Cycle and EdU Incorporation
2.5. Apoptosis Assay by Nuclear Staining and Annexin V/PI Detection
2.6. Colony Formation Assay
2.7. Western Blot
2.8. RT2-PCR
2.9. Neurosphere Assay
2.10. Statistics
3. Results
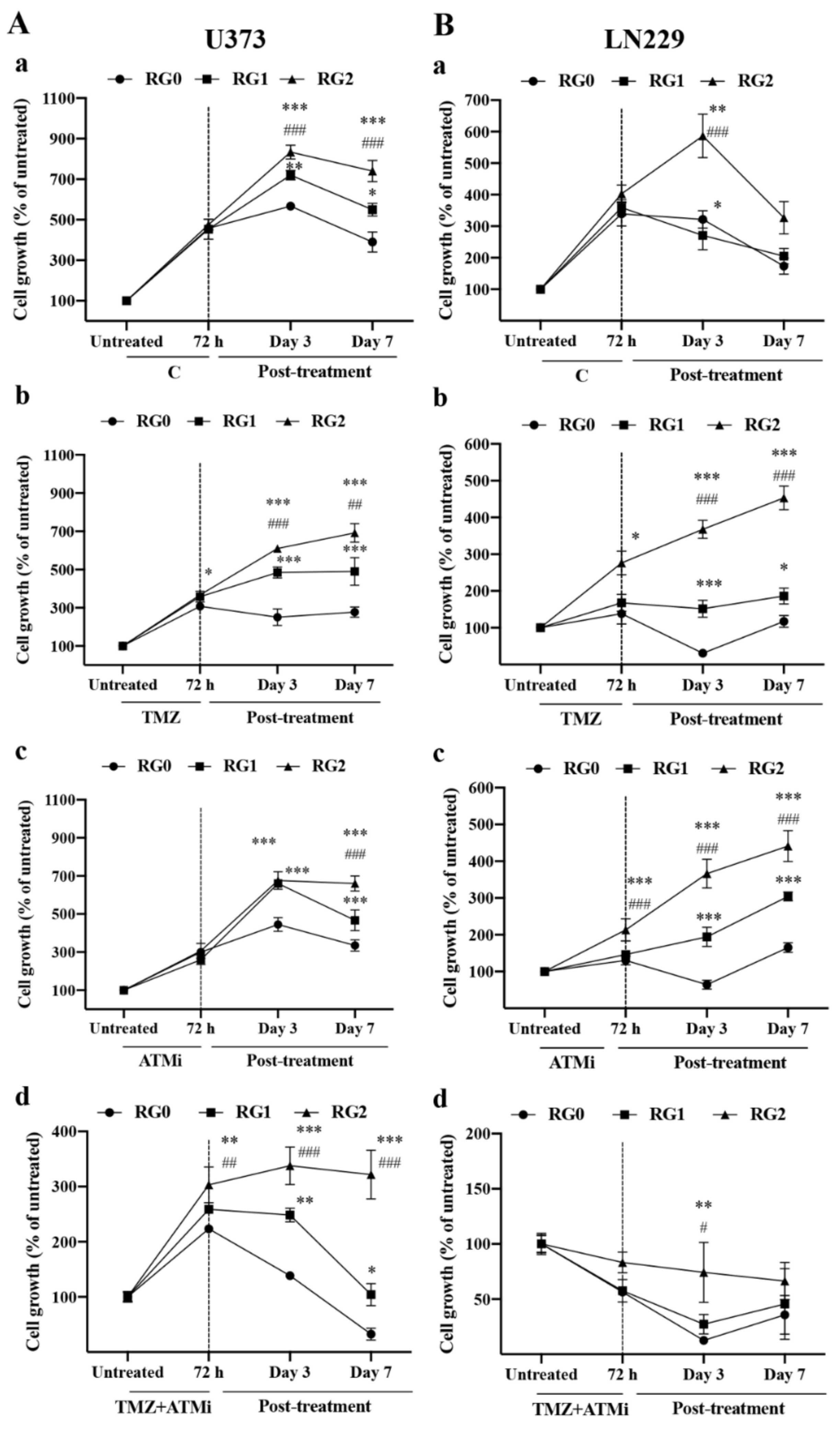
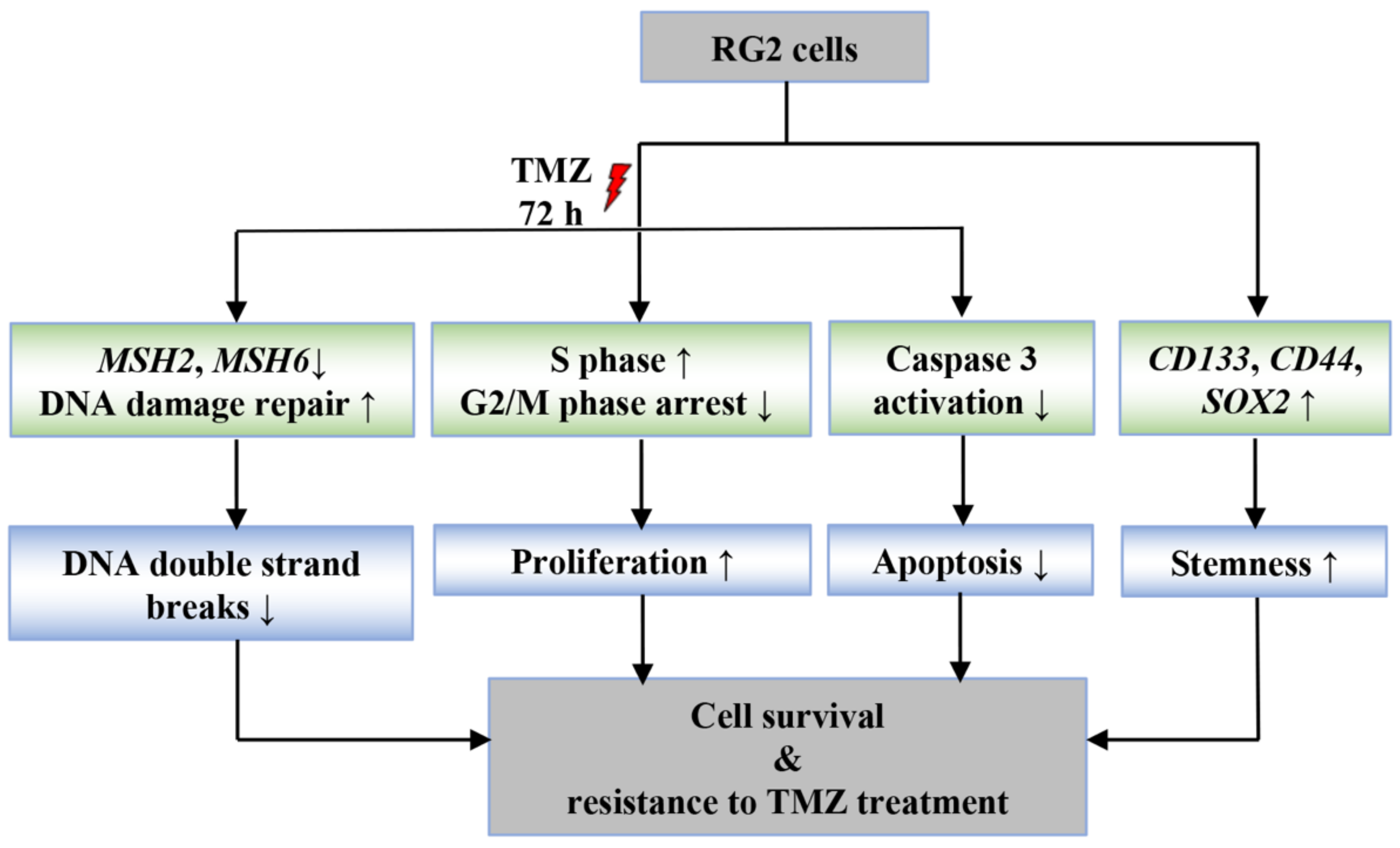
3.1. RG2 Cells Exhibit Resistance to Chemotherapy and Higher Regrowth Capacity in Post-Treatment Phase
3.2. RG2 Cells Exhibit Enhanced Clonogenic Growth Capacity Accompanied by Higher Proliferation Activity in the Post-Treatment Phase
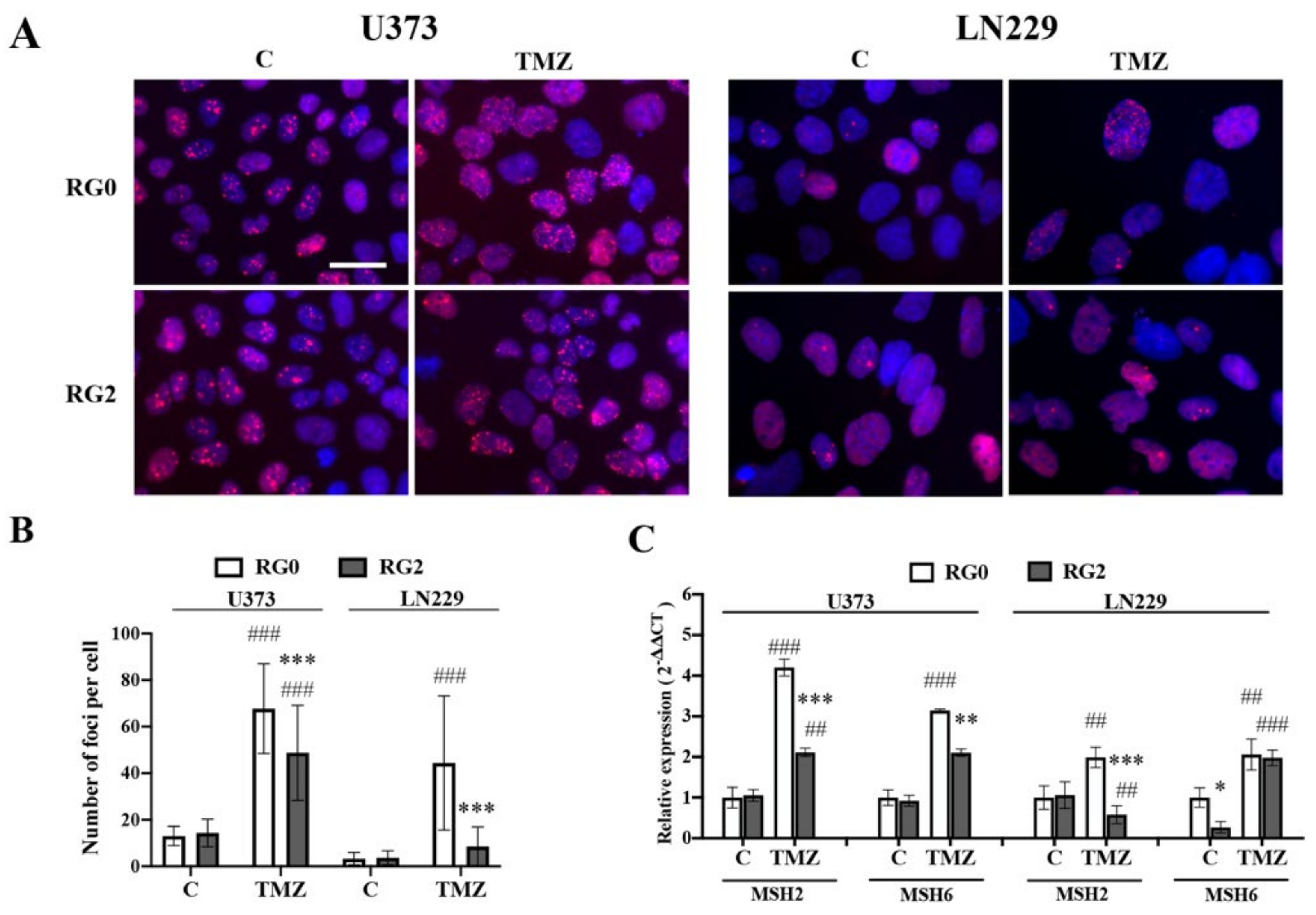
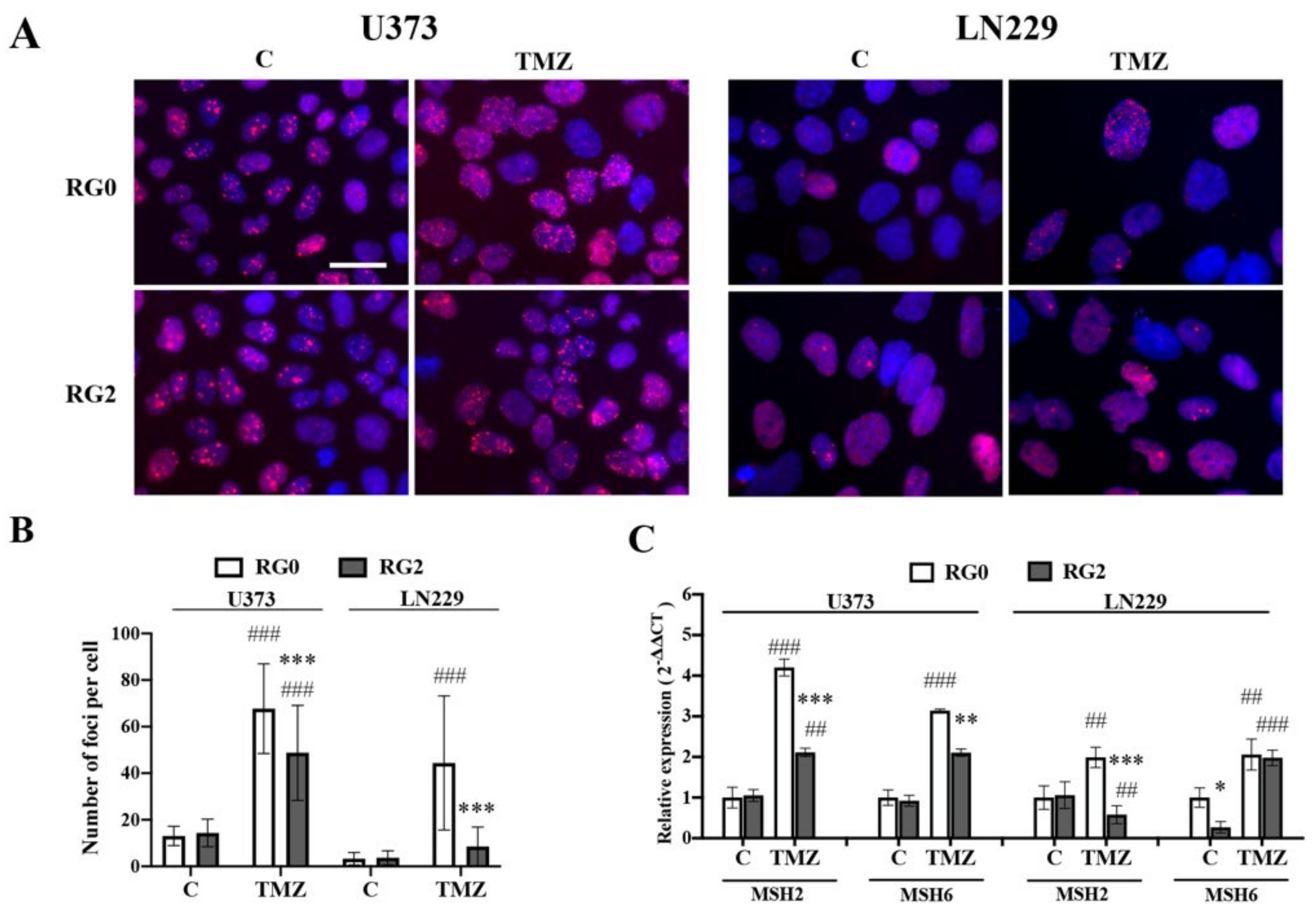
3.3. RG2 Cells Show Less DNA Damage in Individual Cells after TMZ Treatment in Association with the Downregulation of MMR Gene Expression
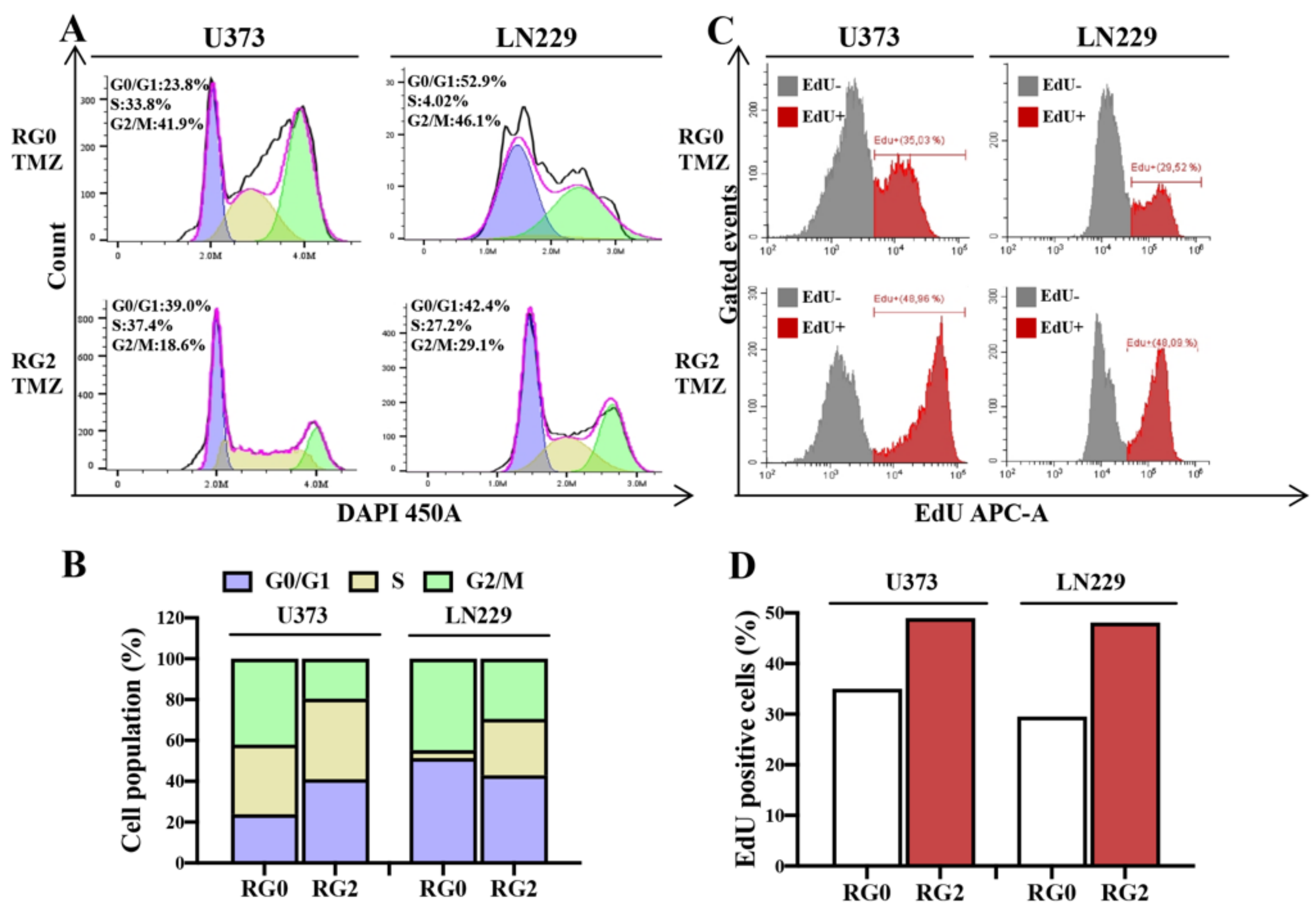
3.4. RG2 Cells Show a Reduction in Cell Cycle Arrest at G2/M Accompanied by an Increase in DNA Replication after TMZ Treatment
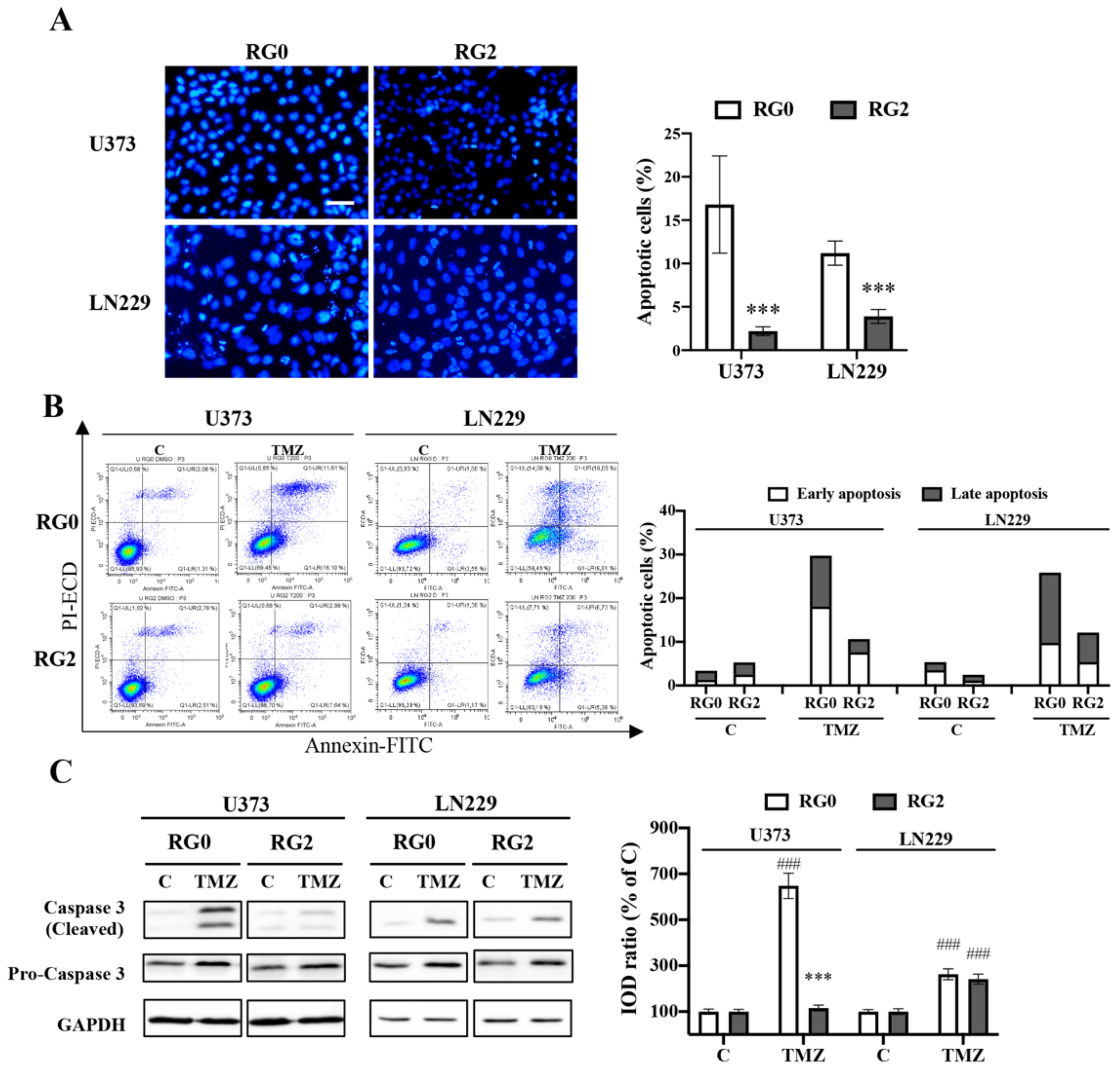
3.5. Apoptosis Is Involved in the Chemoresistance of U373-RG2 and LN229-RG2 Cells via Different Pathways
3.6. U373 RG2 Cells Exhibit Enhanced Self-Renewal Capacity in Association with Upregulation of Stem Cell Markers
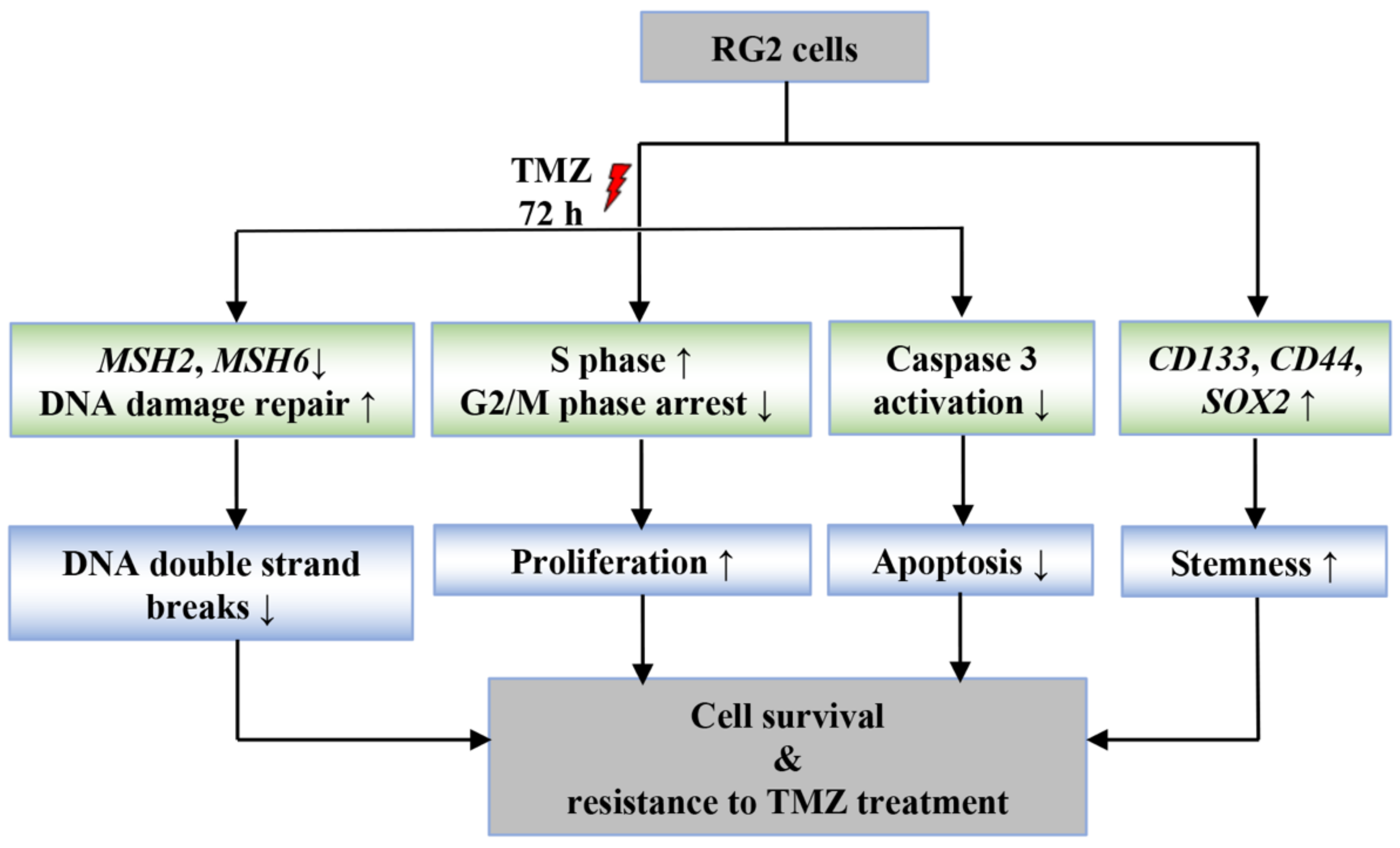
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.B.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Bauchet, L.; Davis, F.G.; Deltour, I.; Fisher, J.L.; Langer, C.E.; Pekmezci, M.; Schwartzbaum, J.A.; Turner, M.C.; Walsh, K.M.; et al. The epidemiology of glioma in adults: A “state of the science” review. Neuro-Oncology 2014, 16, 896–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.Y. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 2016, 3, 198–210. [Google Scholar] [CrossRef] [Green Version]
- Daniel, P.; Sabri, S.; Chaddad, A.; Meehan, B.; Jean-Claude, B.; Rak, J.; Abdulkarim, B.S. Temozolomide Induced Hypermutation in Glioma: Evolutionary Mechanisms and Therapeutic Opportunities. Front. Oncol. 2019, 9, 41. [Google Scholar] [CrossRef] [Green Version]
- Annovazzi, L.; Mellai, M.; Schiffer, D. Chemotherapeutic Drugs: DNA Damage and Repair in Glioblastoma. Cancers 2017, 9, 57. [Google Scholar] [CrossRef] [Green Version]
- Pegg, A.E.; Byers, T.L. Repair of DNA containing O6-alkylguanine. FASEB J. 1992, 6, 2302–2310. [Google Scholar] [CrossRef]
- Rabé, M.; Dumont, S.; Álvarez-Arenas, A.; Janati, H.; Belmonte-Beitia, J.; Calvo, G.F.; Thibault-Carpentier, C.; Séry, Q.; Chauvin, C.; Joalland, N.; et al. Identification of a transient state during the acquisition of temozolomide resistance in glioblastoma. Cell Death Dis. 2020, 11, 19. [Google Scholar] [CrossRef]
- Ferri, A.; Stagni, V.; Barila, D. Targeting the DNA Damage Response to Overcome Cancer Drug Resistance in Glioblastoma. Int. J. Mol. Sci. 2020, 21, 4910. [Google Scholar] [CrossRef]
- Yip, S.; Miao, J.; Cahill, D.P.; Iafrate, A.J.; Aldape, K.; Nutt, C.L.; Louis, D.N. MSH6 mutations arise in glioblastomas during temozolomide therapy and mediate temozolomide resistance. Clin. Cancer Res. 2009, 15, 4622–4629. [Google Scholar] [CrossRef] [Green Version]
- Stritzelberger, J.; Distel, L.; Buslei, R.; Fietkau, R.; Putz, F. Acquired temozolomide resistance in human glioblastoma cell line U251 is caused by mismatch repair deficiency and can be overcome by lomustine. Clin. Transl. Oncol. 2018, 20, 508–516. [Google Scholar] [CrossRef] [PubMed]
- Felsberg, J.; Thon, N.; Eigenbrod, S.; Hentschel, B.; Sabel, M.C.; Westphal, M.; Schackert, G.; Kreth, F.W.; Pietsch, T.; Loffler, M.; et al. Promoter methylation and expression of MGMT and the DNA mismatch repair genes MLH1, MSH2, MSH6 and PMS2 in paired primary and recurrent glioblastomas. Int. J. Cancer 2011, 129, 659–670. [Google Scholar] [CrossRef] [PubMed]
- Hunter, C.; Smith, R.; Cahill, D.P.; Stephens, P.; Stevens, C.; Teague, J.; Greenman, C.; Edkins, S.; Bignell, G.; Davies, H.; et al. A hypermutation phenotype and somatic MSH6 mutations in recurrent human malignant gliomas after alkylator chemotherapy. Cancer Res. 2006, 66, 3987–3991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roos, W.P.; Thomas, A.D.; Kaina, B. DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer 2016, 16, 20–33. [Google Scholar] [CrossRef]
- Vidal, S.J.; Rodriguez-Bravo, V.; Galsky, M.; Cordon-Cardo, C.; Domingo-Domenech, J. Targeting cancer stem cells to suppress acquired chemotherapy resistance. Oncogene 2014, 33, 4451–4463. [Google Scholar] [CrossRef] [Green Version]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef] [Green Version]
- Auffinger, B.; Tobias, A.L.; Han, Y.; Lee, G.; Guo, D.; Dey, M.; Lesniak, M.S.; Ahmed, A.U. Conversion of differentiated cancer cells into cancer stem-like cells in a glioblastoma model after primary chemotherapy. Cell Death Differ. 2014, 21, 1119–1131. [Google Scholar] [CrossRef]
- Oliva, C.R.; Nozell, S.E.; Diers, A.; McClugage, S.G., 3rd; Sarkaria, J.N.; Markert, J.M.; Darley-Usmar, V.M.; Bailey, S.M.; Gillespie, G.Y.; Landar, A.; et al. Acquisition of temozolomide chemoresistance in gliomas leads to remodeling of mitochondrial electron transport chain. J. Biol. Chem. 2010, 285, 39759–39767. [Google Scholar] [CrossRef] [Green Version]
- Tiek, D.M.; Rone, J.D.; Graham, G.T.; Pannkuk, E.L.; Haddad, B.R.; Riggins, R.B. Alterations in Cell Motility, Proliferation, and Metabolism in Novel Models of Acquired Temozolomide Resistant Glioblastoma. Sci. Rep. 2018, 8, 7222. [Google Scholar] [CrossRef]
- Perazzoli, G.; Prados, J.; Ortiz, R.; Caba, O.; Cabeza, L.; Berdasco, M.; Gonzalez, B.; Melguizo, C. Temozolomide Resistance in Glioblastoma Cell Lines: Implication of MGMT, MMR, P-Glycoprotein and CD133 Expression. PLoS ONE 2015, 10, e0140131. [Google Scholar] [CrossRef] [Green Version]
- Nickel, A.-C.; Wan, X.-Y.; Saban, D.-V.; Weng, Y.-L.; Zhang, S.; Keyvani, K.; Sure, U.; Zhu, Y. Loss of programmed cell death 10 activates tumor cells and leads to temozolomide-resistance in glioblastoma. J. Neuro-Oncol. 2019, 141, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Karanam, K.; Kafri, R.; Loewer, A.; Lahav, G. Quantitative live cell imaging reveals a gradual shift between DNA repair mechanisms and a maximal use of HR in mid S phase. Mol. Cell 2012, 47, 320–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chehrehasa, F.; Meedeniya, A.C.; Dwyer, P.; Abrahamsen, G.; Mackay-Sim, A. EdU, a new thymidine analogue for labelling proliferating cells in the nervous system. J. Neurosci. Methods 2009, 177, 122–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Hindy, N.; Keyvani, K.; Pagenstecher, A.; Dammann, P.; Sandalcioglu, I.E.; Sure, U.; Zhu, Y. Implications of Dll4-Notch signaling activation in primary glioblastoma multiforme. Neuro-Oncology 2013, 15, 1366–1378. [Google Scholar] [CrossRef] [Green Version]
- Lou, J.; Priest, D.G.; Solano, A.; Kerjouan, A.; Hinde, E. Spatiotemporal dynamics of 53BP1 dimer recruitment to a DNA double strand break. Nat. Commun. 2020, 11, 5776. [Google Scholar] [CrossRef]
- Margison, G.P.; Santibáñez Koref, M.F.; Povey, A.C. Mechanisms of carcinogenicity/chemotherapy by O6-methylguanine. Mutagenesis 2002, 17, 483–487. [Google Scholar] [CrossRef] [Green Version]
- Hermisson, M.; Klumpp, A.; Wick, W.; Wischhusen, J.; Nagel, G.; Roos, W.; Kaina, B.; Weller, M. O6-methylguanine DNA methyltransferase and p53 status predict temozolomide sensitivity in human malignant glioma cells. J. Neurochem. 2006, 96, 766–776. [Google Scholar] [CrossRef]
- Helleday, T.; Petermann, E.; Lundin, C.; Hodgson, B.; Sharma, R.A. DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer 2008, 8, 193–204. [Google Scholar] [CrossRef]
- Nadkarni, A.; Shrivastav, M.; Mladek, A.C.; Schwingler, P.M.; Grogan, P.T.; Chen, J.; Sarkaria, J.N. ATM inhibitor KU-55933 increases the TMZ responsiveness of only inherently TMZ sensitive GBM cells. J. Neuro-Oncol. 2012, 110, 349–357. [Google Scholar] [CrossRef] [Green Version]
- Brazdova, M.; Quante, T.; Togel, L.; Walter, K.; Loscher, C.; Tichy, V.; Cincarova, L.; Deppert, W.; Tolstonog, G.V. Modulation of gene expression in U251 glioblastoma cells by binding of mutant p53 R273H to intronic and intergenic sequences. Nucleic Acids Res. 2009, 37, 1486–1500. [Google Scholar] [CrossRef] [PubMed]
- Ellert-Miklaszewska, A.; Ciechomska, I.A.; Kaminska, B. Synthetic Cannabinoids Induce Autophagy and Mitochondrial Apoptotic Pathways in Human Glioblastoma Cells Independently of Deficiency in or Tumor Suppressors. Cancers 2021, 13, 419. [Google Scholar] [CrossRef] [PubMed]
- Pyrko, P.; Soriano, N.; Kardosh, A.; Liu, Y.-T.; Uddin, J.; Petasis, N.A.; Hofman, F.M.; Chen, C.-S.; Chen, T.C.; Schönthal, A.H. Downregulation of survivin expression and concomitant induction of apoptosis by celecoxib and its non-cyclooxygenase-2-inhibitory analog, dimethyl-celecoxib (DMC), in tumor cells in vitro and in vivo. Mol. Cancer 2006, 5, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fink, D.; Aebi, S.; Howell, S.B. The role of DNA mismatch repair in drug resistance. Clin. Cancer Res. 1998, 4, 1. [Google Scholar] [PubMed]
- Choi, S.; Yu, Y.; Grimmer, M.R.; Wahl, M.; Chang, S.M.; Costello, J.F. Temozolomide-associated hypermutation in gliomas. Neuro-Oncology 2018, 20, 1300–1309. [Google Scholar] [CrossRef] [Green Version]
- Kat, A.; Thilly, W.G.; Fang, W.H.; Longley, M.J.; Li, G.M.; Modrich, P. An alkylation-tolerant, mutator human cell line is deficient in strand-specific mismatch repair. Proc. Natl. Acad. Sci. USA 1993, 90, 6424–6428. [Google Scholar] [CrossRef] [Green Version]
- MacLeod, G.; Bozek, D.A.; Rajakulendran, N.; Monteiro, V.; Ahmadi, M.; Steinhart, Z.; Kushida, M.M.; Yu, H.; Coutinho, F.J.; Cavalli, F.M.G.; et al. Genome-Wide CRISPR-Cas9 Screens Expose Genetic Vulnerabilities and Mechanisms of Temozolomide Sensitivity in Glioblastoma Stem Cells. Cell Rep. 2019, 27, 971–986.e979. [Google Scholar] [CrossRef] [Green Version]
- Gaspar, N.; Marshall, L.; Perryman, L.; Bax, D.A.; Little, S.E.; Viana-Pereira, M.; Sharp, S.Y.; Vassal, G.; Pearson, A.D.J.; Reis, R.M.; et al. MGMT-independent temozolomide resistance in pediatric glioblastoma cells associated with a PI3-kinase-mediated HOX/stem cell gene signature. Cancer Res. 2010, 70, 9243–9252. [Google Scholar] [CrossRef] [Green Version]
- Oldrini, B.; Vaquero-Siguero, N.; Mu, Q.; Kroon, P.; Zhang, Y.; Galan-Ganga, M.; Bao, Z.; Wang, Z.; Liu, H.; Sa, J.K.; et al. MGMT genomic rearrangements contribute to chemotherapy resistance in gliomas. Nat. Commun. 2020, 11, 3883. [Google Scholar] [CrossRef]
- Ryu, C.H.; Yoon, W.S.; Park, K.Y.; Kim, S.M.; Lim, J.Y.; Woo, J.S.; Jeong, C.H.; Hou, Y.; Jeun, S.-S. Valproic acid downregulates the expression of MGMT and sensitizes temozolomide-resistant glioma cells. J. Biomed. Biotechnol. 2012, 2012, 987495. [Google Scholar] [CrossRef] [Green Version]
- Barciszewska, A.-M.; Gurda, D.; Głodowicz, P.; Nowak, S.; Naskręt-Barciszewska, M.Z. A New Epigenetic Mechanism of Temozolomide Action in Glioma Cells. PLoS ONE 2015, 10, e0136669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirose, Y.; Berger, M.S.; Pieper, R.O. p53 effects both the duration of G2/M arrest and the fate of temozolomide-treated human glioblastoma cells. Cancer Res. 2001, 61, 1957–1963. [Google Scholar] [PubMed]
- Knizhnik, A.V.; Roos, W.P.; Nikolova, T.; Quiros, S.; Tomaszowski, K.H.; Christmann, M.; Kaina, B. Survival and death strategies in glioma cells: Autophagy, senescence and apoptosis triggered by a single type of temozolomide-induced DNA damage. PLoS ONE 2013, 8, e55665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlatkovic, T.; Veldwijk, M.R.; Giordano, F.A.; Herskind, C. Targeting Cell Cycle Checkpoint Kinases to Overcome Intrinsic Radioresistance in Brain Tumor Cells. Cancers 2022, 14, 701. [Google Scholar] [CrossRef]
- Pucci, B.; Kasten, M.; Giordano, A. Cell cycle and apoptosis. Neoplasia 2000, 2, 291–299. [Google Scholar] [CrossRef] [Green Version]
- Roos, W.P.; Batista, L.F.; Naumann, S.C.; Wick, W.; Weller, M.; Menck, C.F.; Kaina, B. Apoptosis in malignant glioma cells triggered by the temozolomide-induced DNA lesion O6-methylguanine. Oncogene 2007, 26, 186–197. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Li, Y.; Yu, T.-S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Zheng, S.; Amini, S.S.; Virk, S.M.; Mikkelsen, T.; Brat, D.J.; Grimsby, J.; Sougnez, C.; Muller, F.; Hu, J.; et al. Whole-genome and multisector exome sequencing of primary and post-treatment glioblastoma reveals patterns of tumor evolution. Genome Res. 2015, 25, 316–327. [Google Scholar] [CrossRef] [Green Version]
- Campos, B.; Olsen, L.R.; Urup, T.; Poulsen, H.S. A comprehensive profile of recurrent glioblastoma. Oncogene 2016, 35, 5819–5825. [Google Scholar] [CrossRef]
- Heddleston, J.M.; Li, Z.; McLendon, R.E.; Hjelmeland, A.B.; Rich, J.N. The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype. Cell Cycle 2009, 8, 3274–3284. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Yuan, X.; Zeng, Z.; Tunici, P.; Ng, H.; Abdulkadir, I.R.; Lu, L.; Irvin, D.; Black, K.L.; Yu, J.S. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol. Cancer 2006, 5, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garros-Regulez, L.; Aldaz, P.; Arrizabalaga, O.; Moncho-Amor, V.; Carrasco-Garcia, E.; Manterola, L.; Moreno-Cugnon, L.; Barrena, C.; Villanua, J.; Ruiz, I.; et al. mTOR inhibition decreases SOX2-SOX9 mediated glioma stem cell activity and temozolomide resistance. Expert Opin. Ther. Targets 2016, 20, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zhou, J.; Wang, C.; Chi, Y.; Wei, Q.; Fu, Z.; Lian, C.; Huang, Q.; Liao, C.; Yang, Z.; et al. LncRNA SOX2OT promotes temozolomide resistance by elevating SOX2 expression via ALKBH5-mediated epigenetic regulation in glioblastoma. Cell Death Dis. 2020, 11, 384. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Xiong, X.; Sun, Y. Functional characterization of SOX2 as an anticancer target. Signal Transduct. Target. Ther. 2020, 5, 135. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Sequence | Annealing Temperature (°C) |
|---|---|---|
| MSH2 | 60 | |
| for. | TTTACCCGGAGGAGAGACTGC | |
| rev. | TGCTCTCCCTTTTTGCCTTTC | |
| MSH6 | 60 | |
| for. | AGAGCAATGCAACGTGCAGA | |
| rev. | TTTGGCGGCTACTTCGCCTA | |
| CD133 | 60 | |
| for. | CAGAAGGCATATGAATCC | |
| rev. | CACCACATTTGTTACAGC | |
| CD44 | 60 | |
| for. | AGCACAATCCAGGCAACTCC | |
| rev. | CTGGTATGAGCTGAGGCTGC | |
| SOX2 | 60 | |
| for. | ACCGGCGGCAACCAGAAGAACAG | |
| rev. | GCGCCGCGGCCGGTATTTAT | |
| GAPDH | 60 | |
| for. | TCACCACCATGGAGAAGGC | |
| rev. | GCTAAGCAGTTGGTGGTGCA | |
| RPS13 | 60 | |
| for. | CGAAAGCATCTTGAGAGGAACA | |
| rev. | TCGAGCCAAACGGTGAATC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, Y.; Chen, Z.; Kim, S.N.; Gan, C.; Ryl, T.; Lesjak, M.S.; Rodemerk, J.; Zhong, R.D.; Wrede, K.; Dammann, P.; et al. Characterization of Temozolomide Resistance Using a Novel Acquired Resistance Model in Glioblastoma Cell Lines. Cancers 2022, 14, 2211. https://doi.org/10.3390/cancers14092211
Zhu Y, Chen Z, Kim SN, Gan C, Ryl T, Lesjak MS, Rodemerk J, Zhong RD, Wrede K, Dammann P, et al. Characterization of Temozolomide Resistance Using a Novel Acquired Resistance Model in Glioblastoma Cell Lines. Cancers. 2022; 14(9):2211. https://doi.org/10.3390/cancers14092211
Chicago/Turabian StyleZhu, Yuan, Zhen Chen, Su Na Kim, Chao Gan, Tatsiana Ryl, Michaela Silvia Lesjak, Jan Rodemerk, Rong De Zhong, Karsten Wrede, Philipp Dammann, and et al. 2022. "Characterization of Temozolomide Resistance Using a Novel Acquired Resistance Model in Glioblastoma Cell Lines" Cancers 14, no. 9: 2211. https://doi.org/10.3390/cancers14092211