
Extracellular Prostaglandins E1 and E2 and Inflammatory Cytokines Are Regulated by the Senescence Program in Potentially Premalignant Oral Keratinocytes

 , , , ,
, , , ,
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cell Lines Used in the Study
2.3. Collection of the Conditioned Medium and Cell Pellets for Analysis of the Extracellular Metabolites and Proteins
2.4. Enzyme-Linked Immunosorbent Assay (ELISA)
2.5. Knockdown of p53, COX-1 and COX-2 in OKF6
2.6. Western Blotting
2.7. Metabolomic Analysis, Normalisation and Data Presentation as Scaled Intensity
2.8. Drug Treatment of the Keratinocyte Cultures
2.9. Statistical Analysis
3. Results
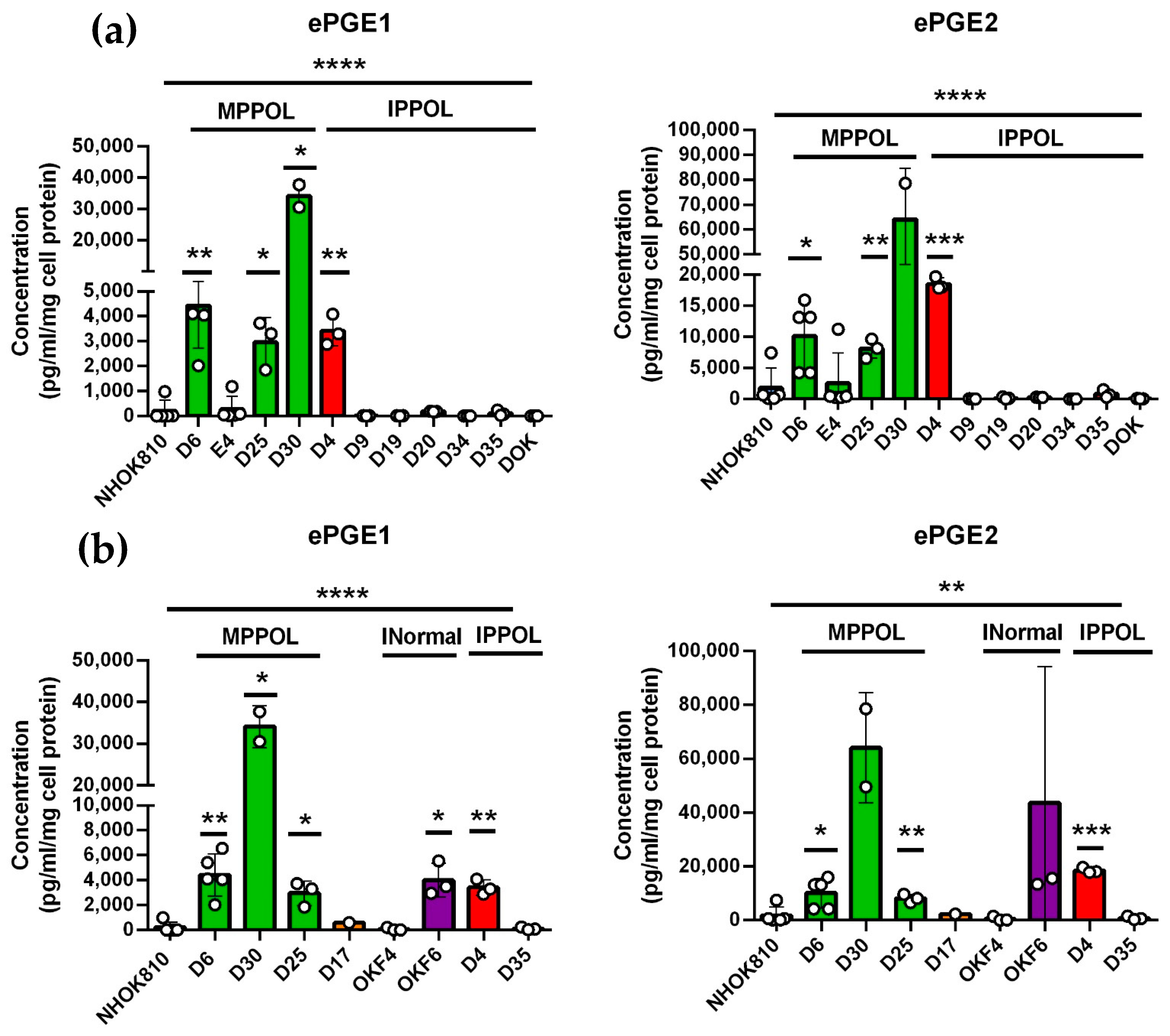
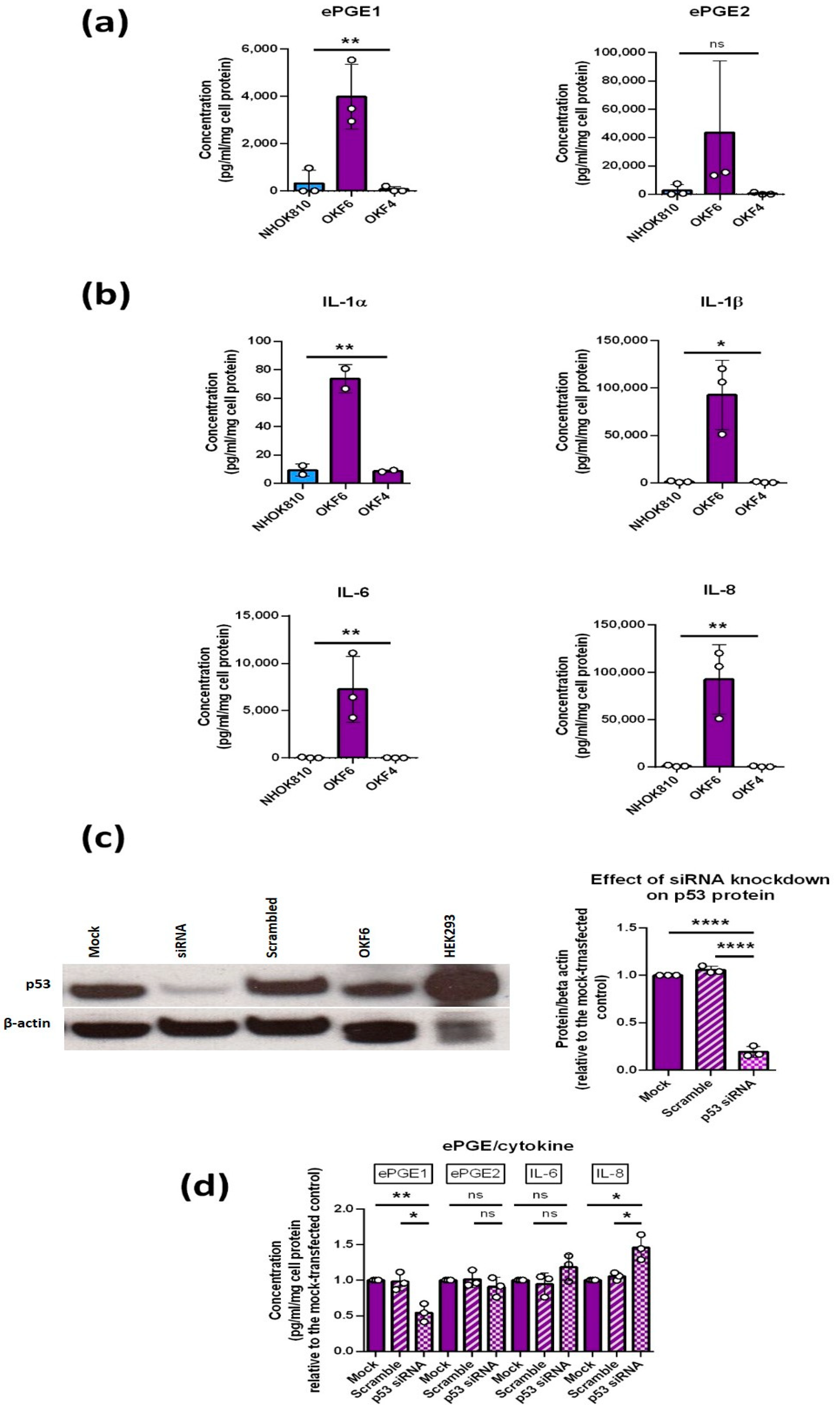
3.1. Extracellular PGEs 1 and 2 (ePGEs 1 and 2) Are Upregulated in MPPOLs
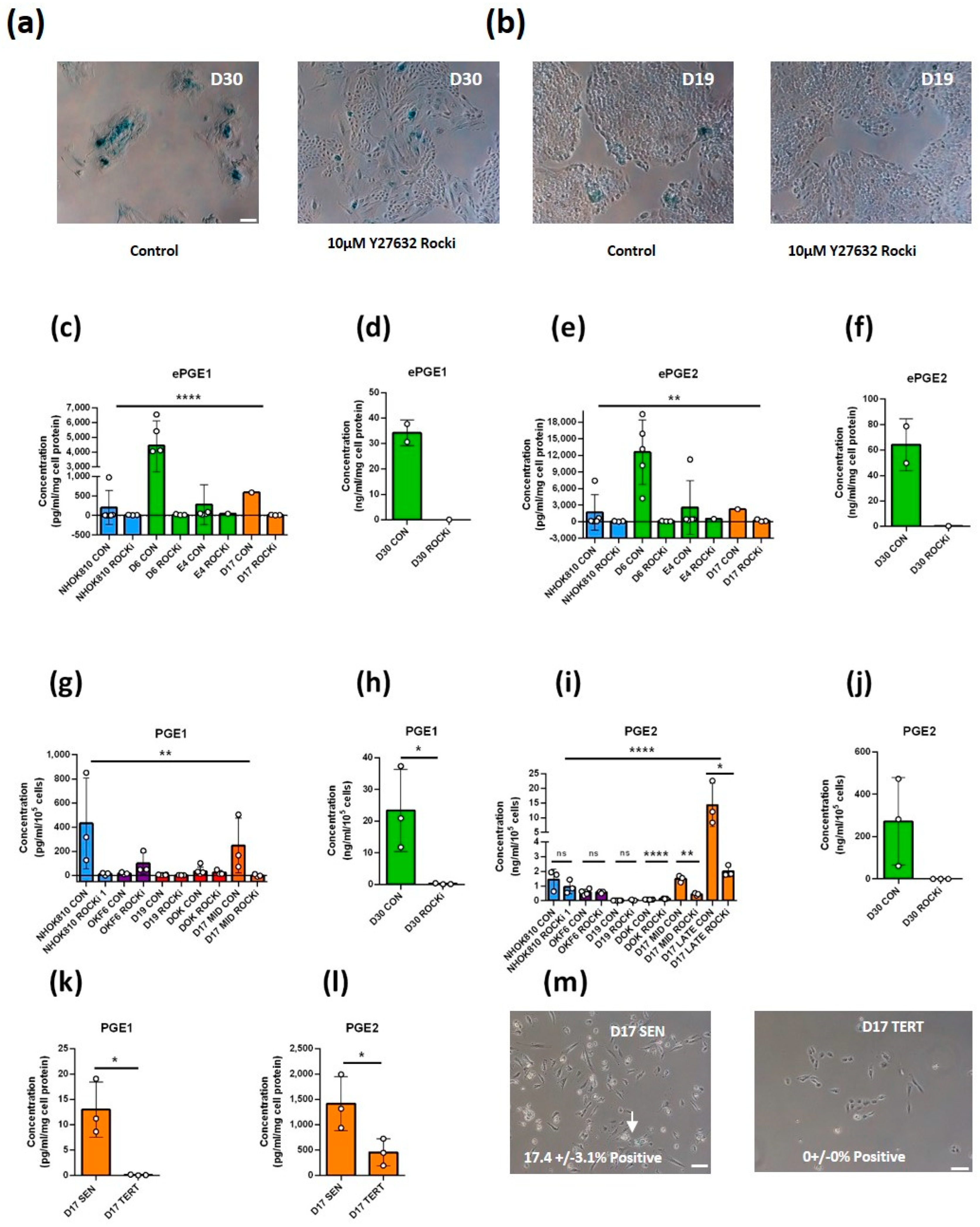
3.2. ePGEs 1 and 2 Are Regulated by Senescence and Its Breakdown
3.2.1. Ablation of Senescence with the Rho-Activated Kinase Inhibitor Y 27632 (ROCKi) Abolishes ePGE Expression in MPPOL Keratinocytes
3.2.2. Ablation of Replicative Senescence with ROCKi Abolishes ePGE Expression in p16INK4A-deficient Line D17
3.2.3. Ectopic TERT Expression in the Absence of p16INK4A Bypasses Replicative Senescence and Suppresses the Production of ePGEs
3.2.4. Ectopic TERT Expression in the Absence of p16INK4A Is Insufficient to Completely Suppress the Production of ePGEs
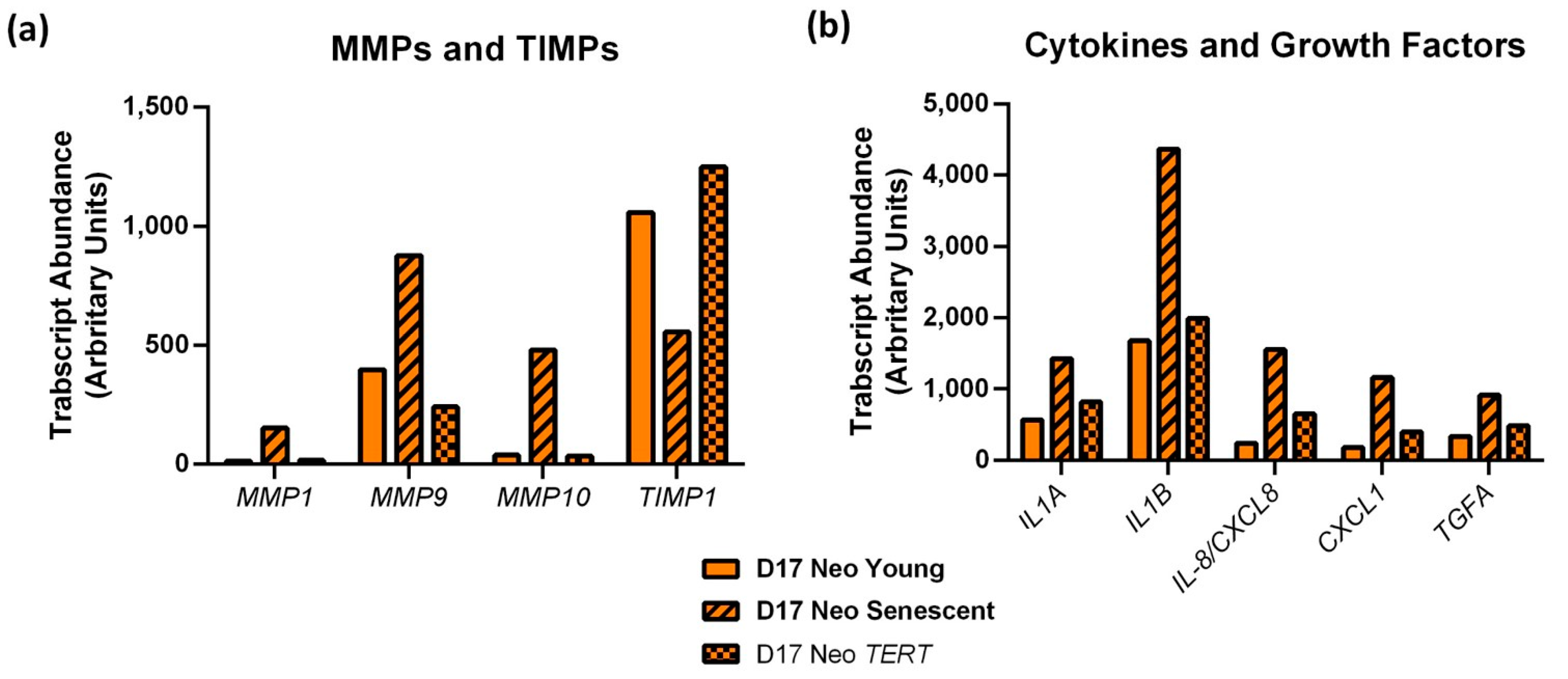
3.3. MPPOLs Do Not Display Increased Levels of All Senescence Markers but Do Show Reduced Levels of the Licensing Factor MCM2/7 and Increased Levels of Some but Not All SASP Factors
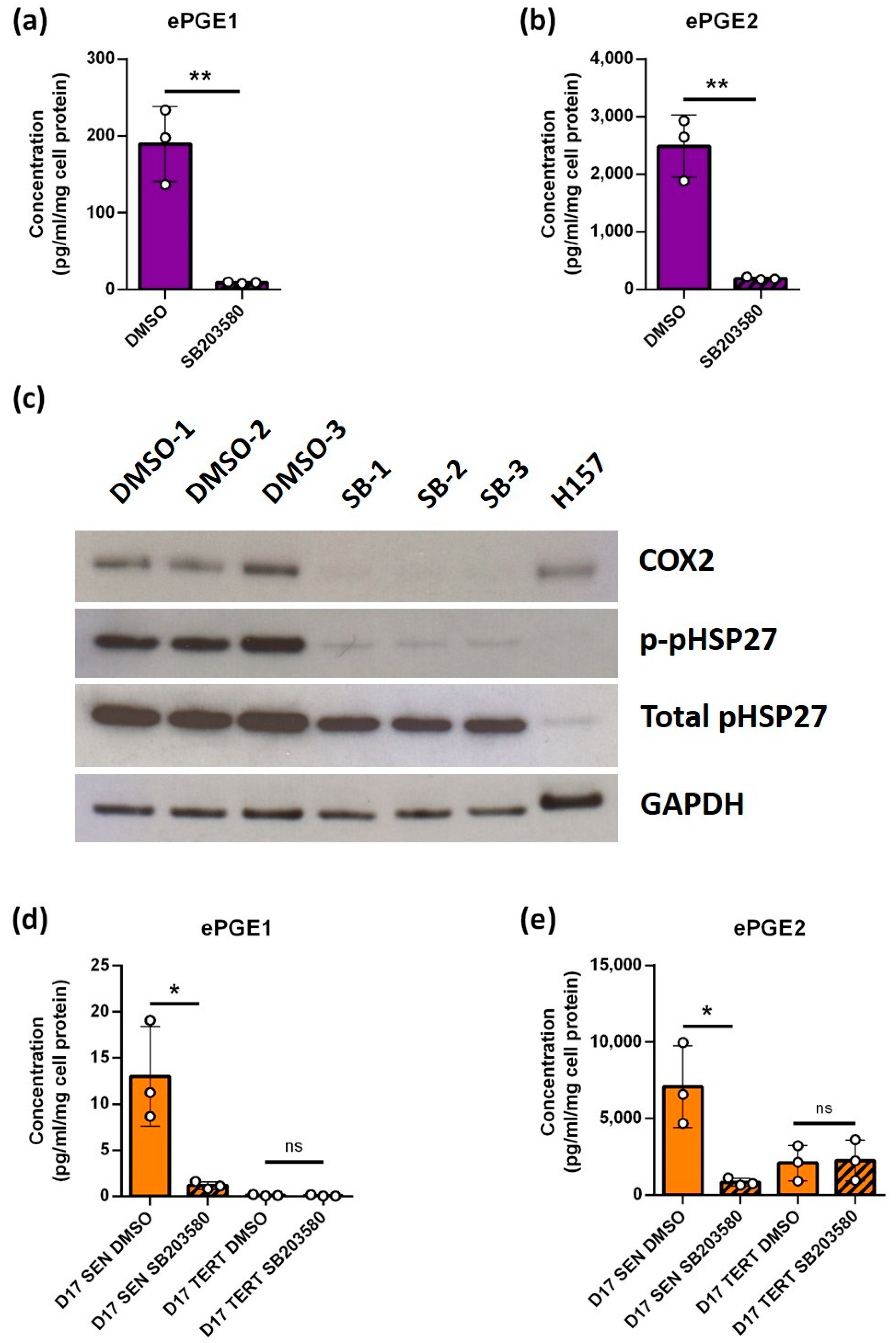
3.4. The Upregulation of the ePGEs Is Dependent on Cyclo-Oxygenase 2 (COX2) but Not Cyclo-Oxygenase 1 (COX-1)
3.5. The Upregulation of the ePGEs Is Dependent on p38 Mitogen-Activated Kinase (p38MAPK) a Known Regulator of COX2
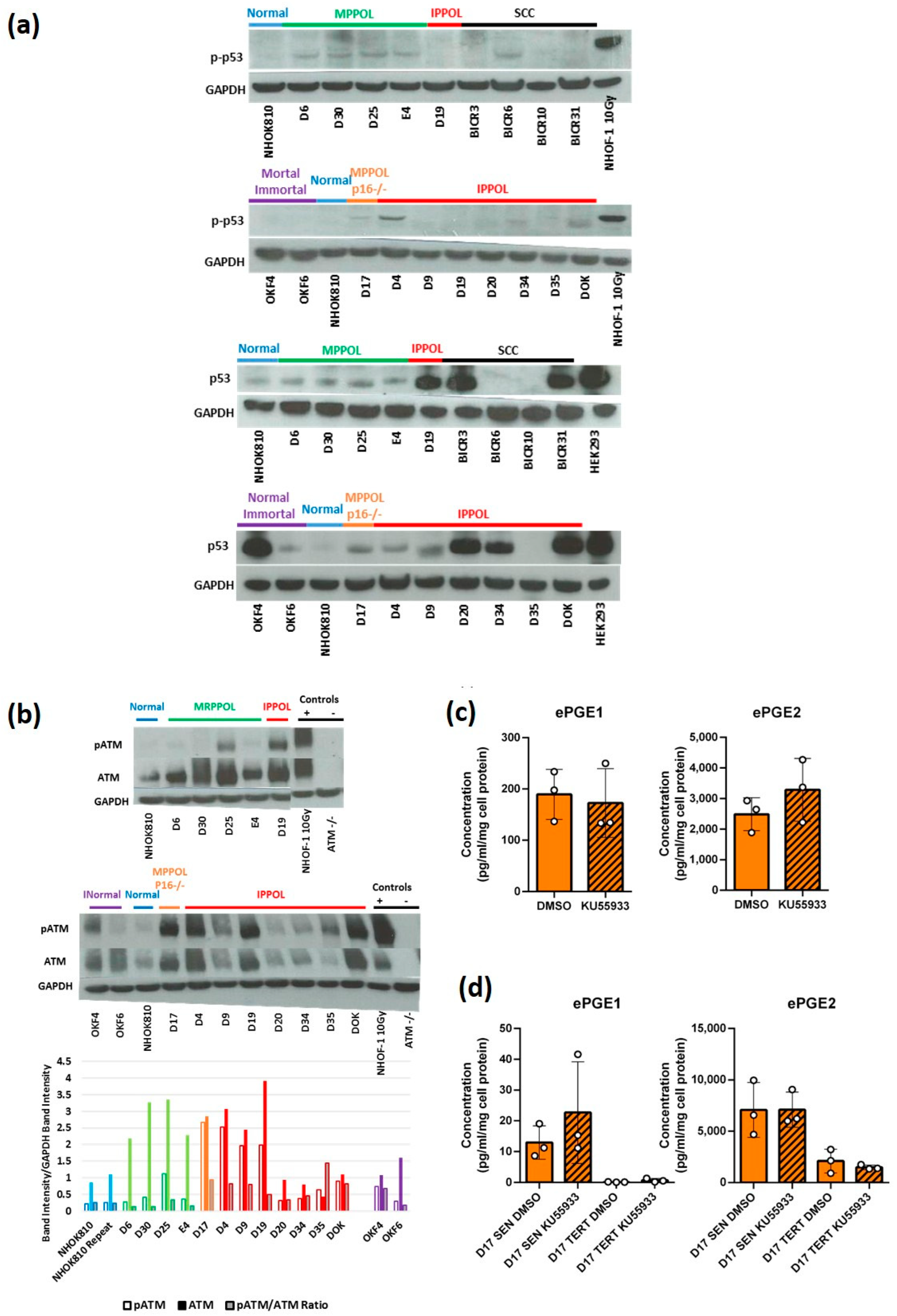
3.6. The DNA Damage Response (DDR) Is Not Specifically Elevated in MPPOL Relative to IPPOL Keratinocytes
3.7. Regulation of the PGEs by p53
p53 Is Necessary for the Upregulation of ePGE1 but Not ePGE2 in OKF6 Cells
3.8. PPOL Keratinocyte Senescence in the Absence of p16INK4A Induces ePGE and a Distinct Set of SASP Factor Transcripts That Are Reversed by Telomerase
3.9. ePGEs Are Not Inversely Related to the Levels of PGE Receptors
3.10. The Effect of Hydrocortisone (HC) on the Regulation of Extracellular PGEs
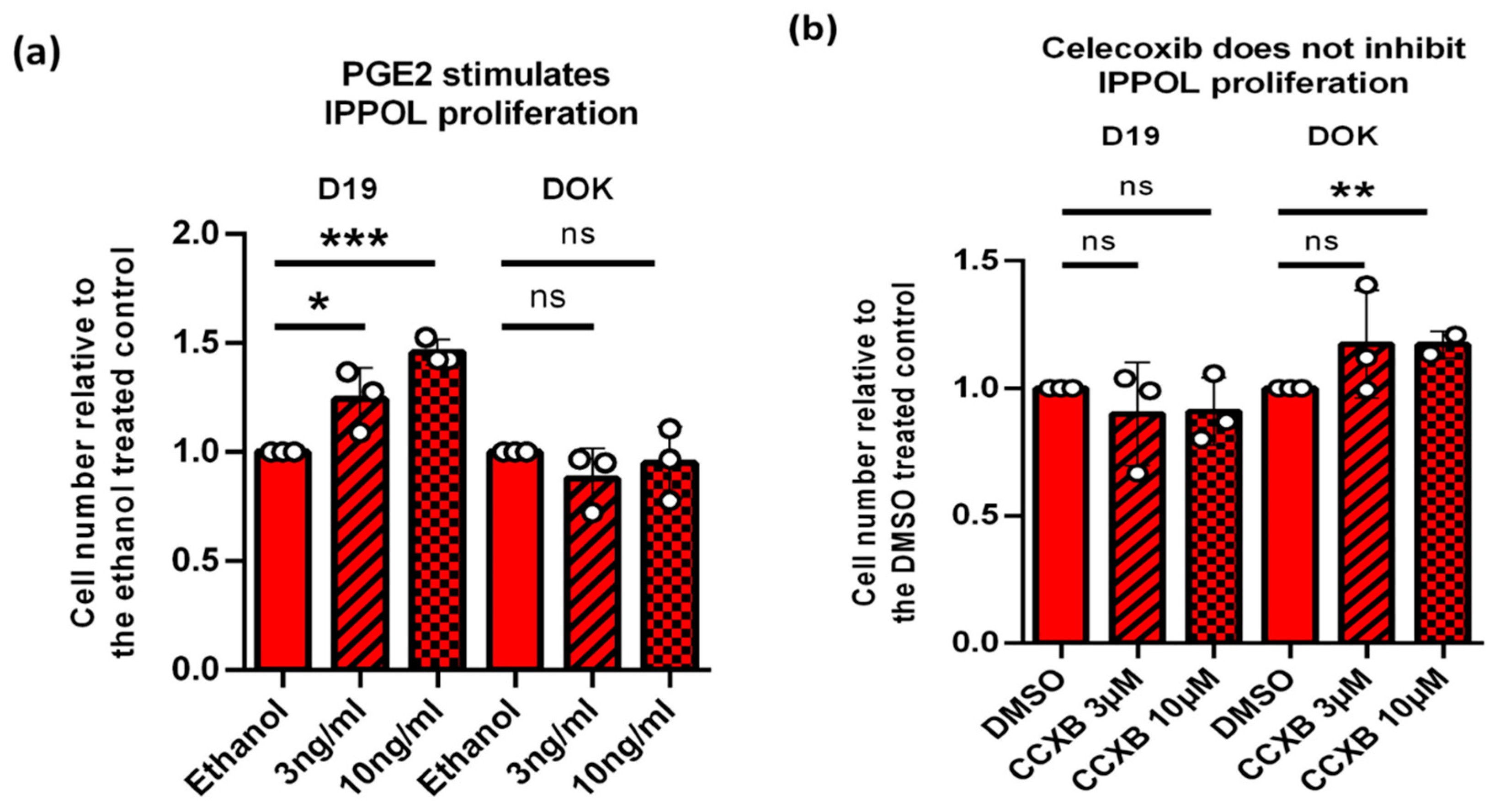
3.11. PGE2 but Not PGE1 Stimulates Proliferation in Some IPPOL Lines Lacking Autocrine Secretion of the PGEs
3.12. Exogenous PGE2 Does Not Induce Oral Fibroblast Senescence
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gupta, S.; Kong, W.; Peng, Y.; Miao, Q.; Mackillop, W.J. Temporal trends in the incidence and survival of cancers of the upper aerodigestive tract in Ontario and the United States. Int. J. Cancer 2009, 125, 2159–2165. [Google Scholar] [CrossRef] [PubMed]
- Braakhuis, B.J.; Tabor, M.P.; Kummer, J.A.; Leemans, C.R.; Brakenhoff, R.H. A genetic explanation of Slaughter’s concept of field cancerization: Evidence and clinical implications. Cancer Res. 2003, 63, 1727–1730. [Google Scholar] [PubMed]
- Weber, R.G.; Scheer, M.; Born, I.A.; Joos, S.; Cobbers, J.M.; Hofele, C.; Reifenberger, G.; Zoller, J.E.; Lichter, P. Recurrent chromosomal imbalances detected in biopsy material from oral premalignant and malignant lesions by combined tissue microdissection, universal DNA amplification, and comparative genomic hybridization. Am. J. Pathol. 1998, 153, 295–303. [Google Scholar] [CrossRef] [Green Version]
- de Bruin, E.C.; McGranahan, N.; Mitter, R.; Salm, M.; Wedge, D.C.; Yates, L.; Jamal-Hanjani, M.; Shafi, S.; Murugaesu, N.; Rowan, A.J.; et al. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science 2014, 346, 251–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, H.M.; Daly, C.; Chalkley, R.; Senguven, B.; Ross, L.; Egan, P.; Chengot, P.; Graham, J.; Sethi, N.; Ong, T.K.; et al. The genomic road to invasion-examining the similarities and differences in the genomes of associated oral pre-cancer and cancer samples. Genome Med. 2017, 9, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burd, C.E.; Sorrentino, J.A.; Clark, K.S.; Darr, D.B.; Krishnamurthy, J.; Deal, A.M.; Bardeesy, N.; Castrillon, D.H.; Beach, D.H.; Sharpless, N.E. Monitoring tumorigenesis and senescence in vivo with a p16(INK4a)-luciferase model. Cell 2013, 152, 340–351. [Google Scholar] [CrossRef] [Green Version]
- McGregor, F.; Muntoni, A.; Fleming, J.; Brown, J.; Felix, D.H.; MacDonald, D.G.; Parkinson, E.K.; Harrison, P.R. Molecular changes associated with oral dysplasia progression and acquisition of immortality: Potential for its reversal by 5-azacytidine. Cancer Res. 2002, 62, 4757–4766. [Google Scholar]
- Edington, K.G.; Loughran, O.P.; Berry, I.J.; Parkinson, E.K. Cellular immortality: A late event in the progression of human squamous cell carcinoma of the head and neck associated with p53 alteration and a high frequency of allele loss. Mol. Carcinog. 1995, 13, 254–265. [Google Scholar] [CrossRef]
- Rheinwald, J.G.; Beckett, M.A. Tumorigenic keratinocyte lines requiring anchorage and fibroblast support cultured from human squamous cell carcinomas. Cancer Res. 1981, 41, 1657–1663. [Google Scholar]
- Hunter, K.D.; Thurlow, J.K.; Fleming, J.; Drake, P.J.; Vass, J.K.; Kalna, G.; Higham, D.J.; Herzyk, P.; Macdonald, D.G.; Parkinson, E.K.; et al. Divergent routes to oral cancer. Cancer Res. 2006, 66, 7405–7413. [Google Scholar] [CrossRef] [Green Version]
- Karen-Ng, L.P.; James, E.L.; Stephen, A.; Bennett, M.H.; Mycielska, M.E.; Parkinson, E.K. The Extracellular Metabolome Stratifies Low and High Risk Potentially Premalignant Oral Keratinocytes and Identifies Citrate as a Potential Non-Invasive Marker of Tumour Progression. Cancers 2021, 13, 4212. [Google Scholar] [CrossRef] [PubMed]
- Veeramachaneni, R.; Walker, T.; Revil, T.; Weck, A.; Badescu, D.; O’Sullivan, J.; Higgins, C.; Elliott, L.; Liloglou, T.; Risk, J.M.; et al. Analysis of head and neck carcinoma progression reveals novel and relevant stage-specific changes associated with immortalisation and malignancy. Sci. Rep. 2019, 9, 11992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Boer, D.V.; Brink, A.; Buijze, M.; Stigter-van Walsum, M.; Hunter, K.D.; Ylstra, B.; Bloemena, E.; Leemans, C.R.; Brakenhoff, R.H. Establishment and Genetic Landscape of Precancer Cell Model Systems from the Head and Neck Mucosal Lining. Mol. Cancer Res. 2019, 17, 120–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickson, M.A.; Hahn, W.C.; Ino, Y.; Ronfard, V.; Wu, J.Y.; Weinberg, R.A.; Louis, D.N.; Li, F.P.; Rheinwald, J.G. Human keratinocytes that express hTERT and also bypass a p16(INK4a)-enforced mechanism that limits life span become immortal yet retain normal growth and differentiation characteristics. Mol. Cell. Biol. 2000, 20, 1436–1447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muntoni, A.; Fleming, J.; Gordon, K.E.; Hunter, K.; McGregor, F.; Parkinson, E.K.; Harrison, P.R. Senescing oral dysplasias are not immortalized by ectopic expression of hTERT alone without other molecular changes, such as loss of INK4A and/or retinoic acid receptor-beta: But p53 mutations are not necessarily required. Oncogene 2003, 22, 7804–7808. [Google Scholar] [CrossRef] [Green Version]
- Hassona, Y.; Cirillo, N.; Lim, K.P.; Herman, A.; Mellone, M.; Thomas, G.J.; Pitiyage, G.N.; Parkinson, E.K.; Prime, S.S. Progression of genotype-specific oral cancer leads to senescence of cancer-associated fibroblasts and is mediated by oxidative stress and TGF-beta. Carcinogenesis 2013, 34, 1286–1295. [Google Scholar] [CrossRef] [Green Version]
- Prime, S.S.; Cirillo, N.; Hassona, Y.; Lambert, D.W.; Paterson, I.C.; Mellone, M.; Thomas, G.J.; James, E.N.; Parkinson, E.K. Fibroblast activation and senescence in oral cancer. J. Oral Pathol. Med. 2017, 46, 82–88. [Google Scholar] [CrossRef]
- Rheinwald, J.G.; Hahn, W.C.; Ramsey, M.R.; Wu, J.Y.; Guo, Z.; Tsao, H.; De Luca, M.; Catricala, C.; O’Toole, K.M. A two-stage, p16(INK4A)- and p53-dependent keratinocyte senescence mechanism that limits replicative potential independent of telomere status. Mol. Cell. Biol. 2002, 22, 5157–5172. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef] [Green Version]
- Loughran, O.; Clark, L.J.; Bond, J.; Baker, A.; Berry, I.J.; Edington, K.G.; Ly, I.S.; Simmons, R.; Haw, R.; Black, D.M.; et al. Evidence for the inactivation of multiple replicative lifespan genes in immortal human squamous cell carcinoma keratinocytes. Oncogene 1997, 14, 1955–1964. [Google Scholar] [CrossRef] [Green Version]
- Chapman, S.; Liu, X.; Meyers, C.; Schlegel, R.; McBride, A.A. Human keratinocytes are efficiently immortalized by a Rho kinase inhibitor. J. Clin. Investig. 2010, 120, 2619–2626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, A.L.; Morton, J.P.; Manoharan, I.; Nelson, D.M.; Jamieson, N.B.; Pawlikowski, J.S.; McBryan, T.; Doyle, B.; McKay, C.; Oien, K.A.; et al. Activation of the PIK3CA/AKT pathway suppresses senescence induced by an activated RAS oncogene to promote tumorigenesis. Mol. Cell 2011, 42, 36–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuilman, T.; Michaloglou, C.; Vredeveld, L.C.; Douma, S.; van Doorn, R.; Desmet, C.J.; Aarden, L.A.; Mooi, W.J.; Peeper, D.S. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 2008, 133, 1019–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef]
- Kang, T.W.; Yevsa, T.; Woller, N.; Hoenicke, L.; Wuestefeld, T.; Dauch, D.; Hohmeyer, A.; Gereke, M.; Rudalska, R.; Potapova, A.; et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 2011, 479, 547–551. [Google Scholar] [CrossRef]
- Qian, M.; Qian, D.; Jing, H.; Li, Y.; Ma, C.; Zhou, Y. Combined cetuximab and celecoxib treatment exhibits a synergistic anticancer effect on human oral squamous cell carcinoma in vitro and in vivo. Oncol. Rep. 2014, 32, 1681–1688. [Google Scholar] [CrossRef] [Green Version]
- Saba, N.F.; Hurwitz, S.J.; Kono, S.A.; Yang, C.S.; Zhao, Y.; Chen, Z.; Sica, G.; Muller, S.; Moreno-Williams, R.; Lewis, M.; et al. Chemoprevention of head and neck cancer with celecoxib and erlotinib: Results of a phase ib and pharmacokinetic study. Cancer Prev. Res. 2014, 7, 283–291. [Google Scholar] [CrossRef] [Green Version]
- Niklander, S.E.; Crane, H.L.; Darda, L.; Lambert, D.W.; Hunter, K.D. The role of icIL-1RA in keratinocyte senescence and development of the senescence-associated secretory phenotype. J. Cell Sci. 2021, 134, jcs252080. [Google Scholar] [CrossRef]
- Stacey, M.; Thacker, S.; Taylor, A.M. Cultured skin keratinocytes from both normal individuals and basal cell naevus syndrome patients are more resistant to gamma-rays and UV light compared with cultured skin fibroblasts. Int. J. Radiat. Biol. 1989, 56, 45–58. [Google Scholar] [CrossRef]
- Prime, S.S.; Nixon, S.V.; Crane, I.J.; Stone, A.; Matthews, J.B.; Maitland, N.J.; Remnant, L.; Powell, S.K.; Game, S.M.; Scully, C. The behaviour of human oral squamous cell carcinoma in cell culture. J. Pathol. 1990, 160, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Loughran, O.; Malliri, A.; Owens, D.; Gallimore, P.H.; Stanley, M.A.; Ozanne, B.; Frame, M.C.; Parkinson, E.K. Association of CDKN2A/p16INK4A with human head and neck keratinocyte replicative senescence: Relationship of dysfunction to immortality and neoplasia. Oncogene 1996, 13, 561–568. [Google Scholar] [PubMed]
- Wrede, D.; Tidy, J.A.; Crook, T.; Lane, D.; Vousden, K.H. Expression of RB and p53 proteins in HPV-positive and HPV-negative cervical carcinoma cell lines. Mol. Carcinog. 1991, 4, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Pitiyage, G.N.; Slijepcevic, P.; Gabrani, A.; Chianea, Y.G.; Lim, K.P.; Prime, S.S.; Tilakaratne, W.M.; Fortune, F.; Parkinson, E.K. Senescent mesenchymal cells accumulate in human fibrosis by a telomere-independent mechanism and ameliorate fibrosis through matrix metalloproteinases. J. Pathol. 2011, 223, 604–617. [Google Scholar] [CrossRef]
- James, E.L.; Lane, J.A.; Michalek, R.D.; Karoly, E.D.; Parkinson, E.K. Replicatively senescent human fibroblasts reveal a distinct intracellular metabolic profile with alterations in NAD+ and nicotinamide metabolism. Sci. Rep. 2016, 6, 38489. [Google Scholar] [CrossRef] [Green Version]
- James, E.L.; Michalek, R.D.; Pitiyage, G.N.; de Castro, A.M.; Vignola, K.S.; Jones, J.; Mohney, R.P.; Karoly, E.D.; Prime, S.S.; Parkinson, E.K. Senescent human fibroblasts show increased glycolysis and redox homeostasis with extracellular metabolomes that overlap with those of irreparable DNA damage, aging, and disease. J. Proteome Res. 2015, 14, 1854–1871. [Google Scholar] [CrossRef]
- Jackson, A.L.; Linsley, P.S. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat. Rev. Drug Discov. 2010, 9, 57–67. [Google Scholar] [CrossRef]
- Laberge, R.M.; Zhou, L.; Sarantos, M.R.; Rodier, F.; Freund, A.; de Keizer, P.L.; Liu, S.; Demaria, M.; Cong, Y.S.; Kapahi, P.; et al. Glucocorticoids suppress selected components of the senescence-associated secretory phenotype. Aging Cell 2012, 11, 569–578. [Google Scholar] [CrossRef] [Green Version]
- Abdul-Aziz, A.M.; Sun, Y.; Hellmich, C.; Marlein, C.R.; Mistry, J.; Forde, E.; Piddock, R.E.; Shafat, M.S.; Morfakis, A.; Mehta, T.; et al. Acute myeloid leukemia induces protumoral p16INK4a-driven senescence in the bone marrow microenvironment. Blood 2019, 133, 446–456. [Google Scholar] [CrossRef] [Green Version]
- Bachelor, M.A.; Silvers, A.L.; Bowden, G.T. The role of p38 in UVA-induced cyclooxygenase-2 expression in the human keratinocyte cell line, HaCaT. Oncogene 2002, 21, 7092–7099. [Google Scholar] [CrossRef] [Green Version]
- Matsuura, H.; Sakaue, M.; Subbaramaiah, K.; Kamitani, H.; Eling, T.E.; Dannenberg, A.J.; Tanabe, T.; Inoue, H.; Arata, J.; Jetten, A.M. Regulation of cyclooxygenase-2 by interferon gamma and transforming growth factor alpha in normal human epidermal keratinocytes and squamous carcinoma cells. Role of mitogen-activated protein kinases. J. Biol. Chem. 1999, 274, 29138–29148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tivey, H.S.; Brook, A.J.; Rokicki, M.J.; Kipling, D.; Davis, T. p38 (MAPK) stress signalling in replicative senescence in fibroblasts from progeroid and genomic instability syndromes. Biogerontology 2013, 14, 47–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodier, F.; Coppe, J.P.; Patil, C.K.; Hoeijmakers, W.A.; Munoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 2009, 11, 973–979. [Google Scholar] [CrossRef]
- Fumagalli, M.; Rossiello, F.; Clerici, M.; Barozzi, S.; Cittaro, D.; Kaplunov, J.M.; Bucci, G.; Dobreva, M.; Matti, V.; Beausejour, C.M.; et al. Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat. Cell Biol. 2012, 14, 355–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hewitt, G.; Jurk, D.; Marques, F.D.; Correia-Melo, C.; Hardy, T.; Gackowska, A.; Anderson, R.; Taschuk, M.; Mann, J.; Passos, J.F. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat. Commun. 2012, 3, 708. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Horejsi, Z.; Koed, K.; Kramer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Sethy, R.; Rakesh, R.; Patne, K.; Arya, V.; Sharma, T.; Haokip, D.T.; Kumari, R.; Muthuswami, R. Regulation of ATM and ATR by SMARCAL1 and BRG1. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech. 2018, 1861, 1076–1092. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.S.; Bohrson, C.; Pike, A.M.; Wheelan, S.J.; Greider, C.W. ATM Kinase Is Required for Telomere Elongation in Mouse and Human Cells. Cell Rep. 2015, 13, 1623–1632. [Google Scholar] [CrossRef] [Green Version]
- Coppe, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Munoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef]
- Abrahao, A.C.; Castilho, R.M.; Squarize, C.H.; Molinolo, A.A.; dos Santos-Pinto, D., Jr.; Gutkind, J.S. A role for COX2-derived PGE2 and PGE2-receptor subtypes in head and neck squamous carcinoma cell proliferation. Oral Oncol. 2010, 46, 880–887. [Google Scholar] [CrossRef] [Green Version]
- Dagouassat, M.; Gagliolo, J.M.; Chrusciel, S.; Bourin, M.C.; Duprez, C.; Caramelle, P.; Boyer, L.; Hue, S.; Stern, J.B.; Validire, P.; et al. The cyclooxygenase-2-prostaglandin E2 pathway maintains senescence of chronic obstructive pulmonary disease fibroblasts. Am. J. Respir. Crit. Care Med. 2013, 187, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Rhen, T.; Cidlowski, J.A. Antiinflammatory action of glucocorticoids--new mechanisms for old drugs. N. Engl. J. Med. 2005, 353, 1711–1723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, J.; Watt, F.M.; Speight, P.M. Changes in the expression of alpha v integrins in oral squamous cell carcinomas. J. Oral Pathol. Med. 1997, 26, 63–68. [Google Scholar] [CrossRef]
- Goertz, O.; Ring, A.; Buschhaus, B.; Hirsch, T.; Daigeler, A.; Steinstraesser, L.; Steinau, H.U.; Langer, S. Influence of anti-inflammatory and vasoactive drugs on microcirculation and angiogenesis after burn in mice. Burns 2011, 37, 656–664. [Google Scholar] [CrossRef]
- Huang, Y.; Marui, A.; Sakaguchi, H.; Esaki, J.; Arai, Y.; Hirose, K.; Bir, S.C.; Horiuchi, H.; Maruyama, T.; Ikeda, T.; et al. Sustained release of prostaglandin E1 potentiates the impaired therapeutic angiogenesis by basic fibroblast growth factor in diabetic murine hindlimb ischemia. Circ. J. 2008, 72, 1693–1699. [Google Scholar] [CrossRef] [Green Version]
- Coppe, J.P.; Boysen, M.; Sun, C.H.; Wong, B.J.; Kang, M.K.; Park, N.H.; Desprez, P.Y.; Campisi, J.; Krtolica, A. A role for fibroblasts in mediating the effects of tobacco-induced epithelial cell growth and invasion. Mol. Cancer Res. 2008, 6, 1085–1098. [Google Scholar] [CrossRef] [Green Version]
- Jiao, J.; Ishikawa, T.O.; Dumlao, D.S.; Norris, P.C.; Magyar, C.E.; Mikulec, C.; Catapang, A.; Dennis, E.A.; Fischer, S.M.; Herschman, H.R. Targeted deletion and lipidomic analysis identify epithelial cell COX-2 as a major driver of chemically induced skin cancer. Mol. Cancer Res. 2014, 12, 1677–1688. [Google Scholar] [CrossRef] [Green Version]
- Alimirah, F.; Pulido, T.; Valdovinos, A.; Alptekin, S.; Chang, E.; Jones, E.; Diaz, D.A.; Flores, J.; Velarde, M.C.; Demaria, M.; et al. Cellular Senescence Promotes Skin Carcinogenesis through p38MAPK and p44/42MAPK Signaling. Cancer Res. 2020, 80, 3606–3619. [Google Scholar] [CrossRef]
- Shibata, M.; Kodani, I.; Osaki, M.; Araki, K.; Adachi, H.; Ryoke, K.; Ito, H. Cyclo-oxygenase-1 and -2 expression in human oral mucosa, dysplasias and squamous cell carcinomas and their pathological significance. Oral Oncol. 2005, 41, 304–312. [Google Scholar] [CrossRef] [Green Version]
- Natarajan, E.; Saeb, M.; Crum, C.P.; Woo, S.B.; McKee, P.H.; Rheinwald, J.G. Co-expression of p16(INK4A) and laminin 5 gamma2 by microinvasive and superficial squamous cell carcinomas in vivo and by migrating wound and senescent keratinocytes in culture. Am. J. Pathol. 2003, 163, 477–491. [Google Scholar] [CrossRef]
- Azazmeh, N.; Assouline, B.; Winter, E.; Ruppo, S.; Nevo, Y.; Maly, A.; Meir, K.; Witkiewicz, A.K.; Cohen, J.; Rizou, S.V.; et al. Chronic expression of p16(INK4a) in the epidermis induces Wnt-mediated hyperplasia and promotes tumor initiation. Nat. Commun. 2020, 11, 2711. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Tchkonia, T.; Jiang, J.; Kirkland, J.L.; Sun, Y. Targeting Senescent Cells for a Healthier Aging: Challenges and Opportunities. Adv. Sci. 2020, 7, 2002611. [Google Scholar] [CrossRef]
- Puram, S.V.; Tirosh, I.; Parikh, A.S.; Patel, A.P.; Yizhak, K.; Gillespie, S.; Rodman, C.; Luo, C.L.; Mroz, E.A.; Emerick, K.S.; et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 2017, 171, 1611–1624.e24. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karen-Ng, L.P.; Ahmad, U.S.; Gomes, L.; Hunter, K.D.; Wan, H.; Hagi-Pavli, E.; Parkinson, E.K. Extracellular Prostaglandins E1 and E2 and Inflammatory Cytokines Are Regulated by the Senescence Program in Potentially Premalignant Oral Keratinocytes. Cancers 2022, 14, 2636. https://doi.org/10.3390/cancers14112636
Karen-Ng LP, Ahmad US, Gomes L, Hunter KD, Wan H, Hagi-Pavli E, Parkinson EK. Extracellular Prostaglandins E1 and E2 and Inflammatory Cytokines Are Regulated by the Senescence Program in Potentially Premalignant Oral Keratinocytes. Cancers. 2022; 14(11):2636. https://doi.org/10.3390/cancers14112636
Chicago/Turabian StyleKaren-Ng, Lee Peng, Usama Sharif Ahmad, Luis Gomes, Keith David Hunter, Hong Wan, Eleni Hagi-Pavli, and Eric Kenneth Parkinson. 2022. "Extracellular Prostaglandins E1 and E2 and Inflammatory Cytokines Are Regulated by the Senescence Program in Potentially Premalignant Oral Keratinocytes" Cancers 14, no. 11: 2636. https://doi.org/10.3390/cancers14112636