
At the Crossroads of Life and Death: The Proteins That Influence Cell Fate Decisions


{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction: The Cellular Responses to Damage
2. p53 as a Master Regulator of Damage Pathways
3. The Apoptotic Pathways
4. Non-Apoptotic Cell Death
5. Cellular Senescence
6. Changing Senescent Cell Fates with Therapeutic Purposes
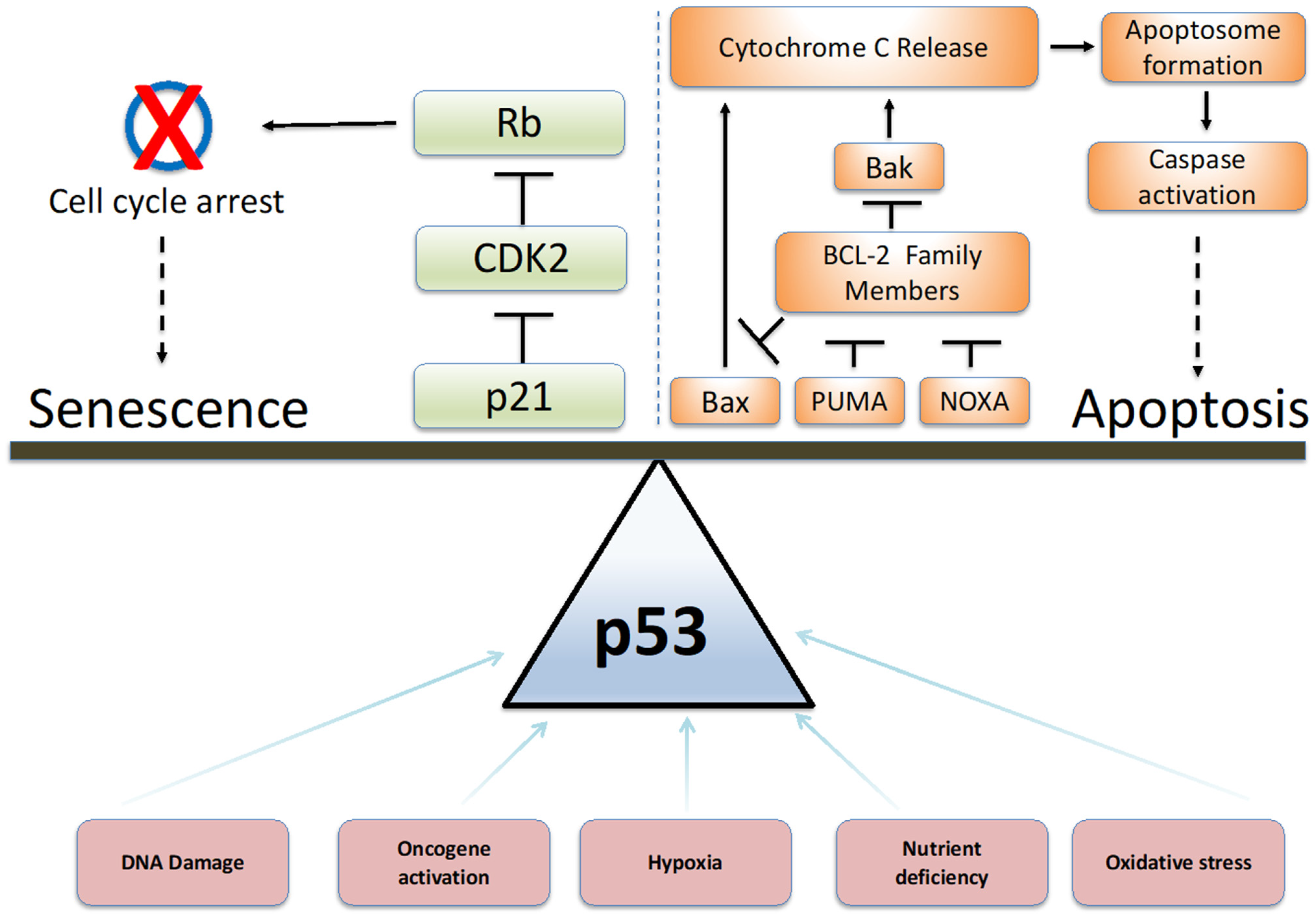
7. Role of p53 in Cell Fate Decisions
8. ROS Involvement in Cell Fate Decisions
9. BCL-2 Family Proteins Role in Cell Fate Decisions
10. Conclusions: The Complex World of Cell Fate Decisions
Author Contributions
Funding
Conflicts of Interest
References
- Lukin, D.J.; Carvajal, L.A.; Liu, W.J.; Resnick-Silverman, L.; Manfredi, J.J. p53 Promotes cell survival due to the reversibility of its cell-cycle checkpoints. Mol. Cancer Res. 2015, 13, 16–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Staschke, K.A.; Wek, R.C. Adapting to cell stress from inside and out. Nat. Cell Biol. 2019, 21, 799–800. [Google Scholar] [CrossRef] [PubMed]
- Okorokov, A.L.; Sherman, M.B.; Plisson, C.; Grinkevich, V.; Sigmundsson, K.; Selivanova, G.; Milner, J.; Orlova, E.V. The structure of p53 tumour suppressor protein reveals the basis for its functional plasticity. EMBO J. 2006, 25, 5191–5200. [Google Scholar] [CrossRef] [Green Version]
- Kannappan, R.; Mattapally, S.; Wagle, P.A.; Zhang, J. Transactivation domain of p53 regulates DNA repair and integrity in human iPS cells. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H512–H521. [Google Scholar] [CrossRef]
- Walker, K.K.; Levine, A.J. Identification of a novel p53 functional domain that is necessary for efficient growth suppression. Proc. Natl. Acad. Sci. USA 1996, 93, 15335–15340. [Google Scholar] [CrossRef] [Green Version]
- Hafner, A.; Bulyk, M.L.; Jambhekar, A.; Lahav, G. The multiple mechanisms that regulate p53 activity and cell fate. Nat. Rev. Mol. Cell Biol. 2019, 20, 199–210. [Google Scholar] [CrossRef]
- Shieh, S.Y.; Ikeda, M.; Taya, Y.; Prives, C. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 1997, 91, 325–334. [Google Scholar] [CrossRef] [Green Version]
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef] [Green Version]
- Maréchal, A.; Zou, L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef]
- Pichierri, P.; Rosselli, F. The DNA crosslink-induced S-phase checkpoint depends on ATR–CHK1 and ATR–NBS1–FANCD2 pathways. EMBO J. 2004, 23, 1178–1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, R.T. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001, 15, 2177–2196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beishline, K.; Azizkhan-Clifford, J. Interplay between the cell cycle and double-strand break response in mammalian cells. Cell Cycle Control. 2014, 1170, 41–59. [Google Scholar]
- Qian, Y.; Chen, X. Tumor suppression by p53: Making cells senescent. Histol. Histopathol. 2010, 25, 515. [Google Scholar]
- Herbig, U.; Jobling, W.A.; Chen, B.P.; Chen, D.J.; Sedivy, J.M. Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21CIP1, but not p16INK4a. Mol. Cell 2004, 14, 501–513. [Google Scholar] [CrossRef]
- Llambi, F.; Moldoveanu, T.; Tait, S.W.; Bouchier-Hayes, L.; Temirov, J.; McCormick, L.L.; Dillon, C.P.; Green, D.R. A unified model of mammalian BCL-2 protein family interactions at the mitochondria. Mol. Cell 2011, 44, 517–531. [Google Scholar] [CrossRef] [Green Version]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [Green Version]
- D’Arcy, M.S. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef]
- Boatright, K.M.; Renatus, M.; Scott, F.L.; Sperandio, S.; Shin, H.; Pedersen, I.M.; Ricci, J.E.; Edris, W.A.; Sutherlin, D.P.; Green, D.R.; et al. A unified model for apical caspase activation. Mol. Cell 2003, 11, 529–541. [Google Scholar] [CrossRef]
- Danial, N.N.; Korsmeyer, S.J. Cell death: Critical control points. Cell 2004, 116, 205–219. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef]
- Brouwer, J.M.; Westphal, D.; Dewson, G.; Robin, A.Y.; Uren, R.T.; Bartolo, R.; Thompson, G.V.; Colman, P.M.; Kluck, R.M.; Czabotar, P.E. Bak core and latch domains separate during activation, and freed core domains form symmetric homodimers. Mol. Cell 2014, 55, 938–946. [Google Scholar] [CrossRef] [Green Version]
- Westphal, D.; Dewson, G.; Czabotar, P.E.; Kluck, R.M. Molecular biology of Bax and Bak activation and action. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 521–531. [Google Scholar] [CrossRef] [Green Version]
- Pietsch, E.C.; Perchiniak, E.; Canutescu, A.A.; Wang, G.; Dunbrack, R.L.; Murphy, M.E. Oligomerization of BAK by p53 utilizes conserved residues of the p53 DNA binding domain. J. Biol. Chem. 2008, 283, 21294–21304. [Google Scholar] [CrossRef] [Green Version]
- Uren, R.T.; O’Hely, M.; Iyer, S.; Bartolo, R.; Shi, M.X.; Brouwer, J.M.; Alsop, A.E.; Dewson, G.; Kluck, R.M. Disordered clusters of Bak dimers rupture mitochondria during apoptosis. Elife 2017, 6, e19944. [Google Scholar] [CrossRef]
- Kim, M.P.; Lozano, G. Mutant p53 partners in crime. Cell Death Differ. 2018, 25, 161–168. [Google Scholar] [CrossRef]
- Locksley, R.M.; Killeen, N.; Lenardo, M.J. The TNF and TNF receptor superfamilies: Integrating mammalian biology. Cell 2001, 104, 487–501. [Google Scholar] [CrossRef] [Green Version]
- Ashkenazi, A.; Dixit, V.M. Death receptors: Signaling and modulation. Science 1998, 281, 1305–1308. [Google Scholar] [CrossRef] [Green Version]
- Wajant, H. The Fas signaling pathway: More than a paradigm. Science 2002, 296, 1635–1636. [Google Scholar] [CrossRef]
- Kischkel, F.C.; Hellbardt, S.; Behrmann, I.; Germer, M.; Pawlita, M.; Krammer, P.H.; Peter, M.E. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J. 1995, 14, 5579–5588. [Google Scholar] [CrossRef]
- Dickens, L.S.; Boyd, R.S.; Jukes-Jones, R.; Hughes, M.A.; Robinson, G.L.; Fairall, L.; Schwabe, J.W.; Cain, K.; Macfarlane, M. A death effector domain chain DISC model reveals a crucial role for caspase-8 chain assembly in mediating apoptotic cell death. Mol. Cell 2012, 47, 291–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schleich, K.; Warnken, U.; Fricker, N.; Ozturk, S.; Richter, P.; Kammerer, K.; Schnolzer, M.; Krammer, P.H.; Lavrik, I.N. Stoichiometry of the CD95 death-inducing signaling complex: Experimental and modeling evidence for a death effector domain chain model. Mol. Cell 2012, 47, 306–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, J.L.; Hughes, M.A.; Meng, X.; Sarnowska, N.A.; Powley, I.R.; Jukes-Jones, R.; Dinsdale, D.; Ragan, T.J.; Fairall, L.; Schwabe, J.W.R.; et al. Cryo-EM structural analysis of FADD:Caspase-8 complexes defines the catalytic dimer architecture for co-ordinated control of cell fate. Nat. Commun. 2021, 12, 819. [Google Scholar] [CrossRef] [PubMed]
- Fu, T.M.; Li, Y.; Lu, A.; Li, Z.; Vajjhala, P.R.; Cruz, A.C.; Srivastava, D.B.; DiMaio, F.; Penczek, P.A.; Siegel, R.M.; et al. Cryo-EM Structure of Caspase-8 Tandem DED Filament Reveals Assembly and Regulation Mechanisms of the Death-Inducing Signaling Complex. Mol. Cell 2016, 64, 236–250. [Google Scholar] [CrossRef] [Green Version]
- Fridman, J.S.; Lowe, S.W. Control of apoptosis by p53. Oncogene 2003, 22, 9030–9040. [Google Scholar] [CrossRef] [Green Version]
- Mihara, M.; Erster, S.; Zaika, A.; Petrenko, O.; Chittenden, T.; Pancoska, P.; Moll, U.M. p53 has a direct apoptogenic role at the mitochondria. Mol. Cell 2003, 11, 577–590. [Google Scholar] [CrossRef]
- Zaitceva, V.; Kopeina, G.S.; Zhivotovsky, B. Anastasis: Return Journey from Cell Death. Cancers 2021, 13, 3671. [Google Scholar] [CrossRef]
- Sun, G.; Guzman, E.; Balasanyan, V.; Conner, C.M.; Wong, K.; Zhou, H.R.; Kosik, K.S.; Montell, D.J. A molecular signature for anastasis, recovery from the brink of apoptotic cell death. J. Cell Biol. 2017, 216, 3355–3368. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.M.; Talbot, C.C., Jr.; Fung, M.C.; Tang, H.L. Molecular signature of anastasis for reversal of apoptosis. F1000 Res. 2017, 6, 43. [Google Scholar] [CrossRef]
- Geske, F.J.; Lieberman, R.; Strange, R.; Gerschenson, L.E. Early stages of p53-induced apoptosis are reversible. Cell Death Differ. 2001, 8, 182–191. [Google Scholar] [CrossRef] [Green Version]
- Ichim, G.; Lopez, J.; Ahmed, S.U.; Muthalagu, N.; Giampazolias, E.; Delgado, M.E.; Haller, M.; Riley, J.S.; Mason, S.M.; Athineos, D.; et al. Limited mitochondrial permeabilization causes DNA damage and genomic instability in the absence of cell death. Mol. Cell 2015, 57, 860–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fresquet, V.; Rieger, M.; Carolis, C.; Garcia-Barchino, M.J.; Martinez-Climent, J.A. Acquired mutations in BCL2 family proteins conferring resistance to the BH3 mimetic ABT-199 in lymphoma. Blood 2014, 123, 4111–4119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godefroy, N.; Lemaire, C.; Renaud, F.; Rincheval, V.; Perez, S.; Parvu-Ferecatu, I.; Mignotte, B.; Vayssiere, J.L. p53 can promote mitochondria- and caspase-independent apoptosis. Cell Death Differ. 2004, 11, 785–787. [Google Scholar] [CrossRef] [PubMed]
- Montero, J.; Dutta, C.; van Bodegom, D.; Weinstock, D.; Letai, A. p53 regulates a non-apoptotic death induced by ROS. Cell Death Differ. 2013, 20, 1465–1474. [Google Scholar] [CrossRef] [Green Version]
- Tu, H.C.; Ren, D.; Wang, G.X.; Chen, D.Y.; Westergard, T.D.; Kim, H.; Sasagawa, S.; Hsieh, J.J.; Cheng, E.H. The p53-cathepsin axis cooperates with ROS to activate programmed necrotic death upon DNA damage. Proc. Natl. Acad. Sci. USA 2009, 106, 1093–1098. [Google Scholar] [CrossRef] [Green Version]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Shay, J.W.; Wright, W.E. Hayflick, his limit, and cellular ageing. Nat. Rev. Mol. Cell Biol. 2000, 1, 72–76. [Google Scholar] [CrossRef]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G. Cellular senescence: Defining a path forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- Campisi, J.; Di Fagagna, F.D.A. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef]
- Narita, M.; Nuñez, S.; Heard, E.; Narita, M.; Lin, A.W.; Hearn, S.A.; Spector, D.L.; Hannon, G.J.; Lowe, S.W. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 2003, 113, 703–716. [Google Scholar] [CrossRef] [Green Version]
- Bracken, A.P.; Kleine-Kohlbrecher, D.; Dietrich, N.; Pasini, D.; Gargiulo, G.; Beekman, C.; Theilgaard-Mönch, K.; Minucci, S.; Porse, B.T.; Marine, J.-C. The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev. 2007, 21, 525–530. [Google Scholar] [CrossRef] [Green Version]
- Munro, J.; Barr, N.I.; Ireland, H.; Morrison, V.; Parkinson, E.K. Histone deacetylase inhibitors induce a senescence-like state in human cells by a p16-dependent mechanism that is independent of a mitotic clock. Exp. Cell Res. 2004, 295, 525–538. [Google Scholar] [CrossRef] [PubMed]
- Di Fagagna, F.d.A. Living on a break: Cellular senescence as a DNA-damage response. Nat. Rev. Cancer 2008, 8, 512–522. [Google Scholar] [CrossRef] [PubMed]
- Shay, J.W.; Roninson, I.B. Hallmarks of senescence in carcinogenesis and cancer therapy. Oncogene 2004, 23, 2919–2933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, K.; Mori, I.; Nakayama, Y.; Miyakoda, M.; Kodama, S.; Watanabe, M. Radiation-induced senescence-like growth arrest requires TP53 function but not telomere shortening. Radiat. Res. 2001, 155, 248–253. [Google Scholar] [CrossRef]
- Larsson, O.; Scheele, C.; Liang, Z.; Moll, J.; Karlsson, C.; Wahlestedt, C. Kinetics of senescence-associated changes of gene expression in an epithelial, temperature-sensitive SV40 large T antigen model. Cancer Res. 2004, 64, 482–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calcinotto, A.; Kohli, J.; Zagato, E.; Pellegrini, L.; Demaria, M.; Alimonti, A. Cellular senescence: Aging, cancer, and injury. Physiol. Rev. 2019, 99, 1047–1078. [Google Scholar] [CrossRef]
- Bos, J.L. Ras oncogenes in human cancer: A review. Cancer Res. 1989, 49, 4682–4689. [Google Scholar]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef] [Green Version]
- Michaloglou, C.; Vredeveld, L.C.; Soengas, M.S.; Denoyelle, C.; Kuilman, T.; Van Der Horst, C.M.; Majoor, D.M.; Shay, J.W.; Mooi, W.J.; Peeper, D.S. BRAF E600-associated senescence-like cell cycle arrest of human naevi. Nature 2005, 436, 720–724. [Google Scholar] [CrossRef]
- Ruan, S.; Okcu, M.F.; Ren, J.P.; Chiao, P.; Andreeff, M.; Levin, V.; Zhang, W. Overexpressed WAF1/Cip1 renders glioblastoma cells resistant to chemotherapy agents 1, 3-bis (2-chloroethyl)-1-nitrosourea and cisplatin. Cancer Res. 1998, 58, 1538–1543. [Google Scholar]
- Hsu, C.-H.; Altschuler, S.J.; Wu, L.F. Patterns of early p21 dynamics determine proliferation-senescence cell fate after chemotherapy. Cell 2019, 178, 361–373. [Google Scholar] [CrossRef] [PubMed]
- Javelaud, D.; Besançon, F. Inactivation of p21WAF1Sensitizes Cells to Apoptosis via an Increase of Both p14ARF and p53 Levels and an Alteration of the Bax/Bcl-2 Ratio. J. Biol. Chem. 2002, 277, 37949–37954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masgras, I.; Carrera, S.; de Verdier, P.J.; Brennan, P.; Majid, A.; Makhtar, W.; Tulchinsky, E.; Jones, G.D.D.; Roninson, I.B.; Macip, S. Reactive oxygen species and mitochondrial sensitivity to oxidative stress determine induction of cancer cell death by p21. J. Biol. Chem. 2012, 287, 9845–9854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bond, J.; Wyllie, F.; Wynford-Thomas, D. Escape from senescence in human diploid fibroblasts induced directly by mutant p53. Oncogene 1994, 9, 1885–1889. [Google Scholar] [PubMed]
- Dirac, A.M.; Bernards, R. Reversal of Senescence in Mouse Fibroblasts through Lentiviral Suppression of p53* 210. J. Biol. Chem. 2003, 278, 11731–11734. [Google Scholar] [CrossRef] [Green Version]
- Reyes, J.; Chen, J.-Y.; Stewart-Ornstein, J.; Karhohs, K.W.; Mock, C.S.; Lahav, G. Fluctuations in p53 signaling allow escape from cell-cycle arrest. Mol. Cell 2018, 71, 581–591. [Google Scholar] [CrossRef] [Green Version]
- Elmore, L.W.; Di, X.; Dumur, C.; Holt, S.E.; Gewirtz, D.A. Evasion of a single-step, chemotherapy-induced senescence in breast cancer cells: Implications for treatment response. Clin. Cancer Res. 2005, 11, 2637–2643. [Google Scholar] [CrossRef] [Green Version]
- Roberson, R.S.; Kussick, S.J.; Vallieres, E.; Chen, S.-Y.J.; Wu, D.Y. Escape from therapy-induced accelerated cellular senescence in p53-null lung cancer cells and in human lung cancers. Cancer Res. 2005, 65, 2795–2803. [Google Scholar] [CrossRef] [Green Version]
- Saleh, T.; Tyutyunyk-Massey, L.; Murray, G.F.; Alotaibi, M.R.; Kawale, A.S.; Elsayed, Z.; Henderson, S.C.; Yakovlev, V.; Elmore, L.W.; Toor, A. Tumor cell escape from therapy-induced senescence. Biochem. Pharmacol. 2019, 162, 202–212. [Google Scholar] [CrossRef]
- Wang, C.; Vegna, S.; Jin, H.; Benedict, B.; Lieftink, C.; Ramirez, C.; de Oliveira, R.L.; Morris, B.; Gadiot, J.; Wang, W. Inducing and exploiting vulnerabilities for the treatment of liver cancer. Nature 2019, 574, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Qing, Y.; Li, H.; Zhao, Y.; Hu, P.; Wang, X.; Yu, X.; Zhu, M.; Wang, H.; Wang, Z.; Guo, Q. One-Two Punch Therapy for the Treatment of T-Cell Malignancies Involving p53-Dependent Cellular Senescence. Oxidative Med. Cell. Longev. 2021, 2021, 5529518. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, C.; Feng, Z. Tumor suppressor p53 and its gain-of-function mutants in cancer. Acta Biochim. Biophys. Sin. 2014, 46, 170–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milner, J.; Medcalf, E.A. Cotranslation of activated mutant p53 with wild type drives the wild-type p53 protein into the mutant conformation. Cell 1991, 65, 765–774. [Google Scholar] [CrossRef]
- Ko, A.; Han, S.Y.; Choi, C.H.; Cho, H.; Lee, M.S.; Kim, S.Y.; Song, J.S.; Hong, K.M.; Lee, H.W.; Hewitt, S.M.; et al. Oncogene-induced senescence mediated by c-Myc requires USP10 dependent deubiquitination and stabilization of p14ARF. Cell Death Differ. 2018, 25, 1050–1062. [Google Scholar] [CrossRef] [Green Version]
- Weber, J.D.; Taylor, L.J.; Roussel, M.F.; Sherr, C.J.; Bar-Sagi, D. Nucleolar Arf sequesters Mdm2 and activates p53. Nat. Cell Biol. 1999, 1, 20–26. [Google Scholar] [CrossRef]
- Polyak, K.; Xia, Y.; Zweier, J.L.; Kinzler, K.W.; Vogelstein, B. A model for p53-induced apoptosis. Nature 1997, 389, 300–305. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, K.; Shi, Y.; Shao, C. The tango of ROS and p53 in tissue stem cells. Cell Death Differ. 2018, 25, 639–641. [Google Scholar] [CrossRef] [Green Version]
- Macip, S.; Igarashi, M.; Fang, L.; Chen, A.; Pan, Z.Q.; Lee, S.W.; Aaronson, S.A. Inhibition of p21-mediated ROS accumulation can rescue p21-induced senescence. EMBO J. 2002, 21, 2180–2188. [Google Scholar] [CrossRef]
- Zuckerman, V.; Wolyniec, K.; Sionov, R.V.; Haupt, S.; Haupt, Y. Tumour suppression by p53: The importance of apoptosis and cellular senescence. J. Pathol. 2009, 219, 3–15. [Google Scholar] [CrossRef]
- Passos, J.F.; Nelson, G.; Wang, C.; Richter, T.; Simillion, C.; Proctor, C.J.; Miwa, S.; Olijslagers, S.; Hallinan, J.; Wipat, A. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol. Syst. Biol. 2010, 6, 347. [Google Scholar] [CrossRef] [PubMed]
- Castrogiovanni, C.; Waterschoot, B.; De Backer, O.; Dumont, P. Serine 392 phosphorylation modulates p53 mitochondrial translocation and transcription-independent apoptosis. Cell Death Differ. 2018, 25, 190–203. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Guo, W.; Zhou, H.; Luo, N.; Nie, C.; Zhao, X.; Yuan, Z.; Liu, X.; Wei, Y. Mitochondrial p53 phosphorylation induces Bak-mediated and caspase-independent cell death. Oncotarget 2015, 6, 17192–17205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Su, Z.; Zou, Z.; Tan, H.; Cai, D.; Su, L.; Gu, Z. Ser46 phosphorylation of p53 is an essential event in prolyl-isomerase Pin1-mediated p53-independent apoptosis in response to heat stress. Cell Death Dis. 2019, 10, 96. [Google Scholar] [CrossRef] [Green Version]
- Cesnekova, J.; Spacilova, J.; Hansikova, H.; Houstek, J.; Zeman, J.; Stiburek, L. LACE1 interacts with p53 and mediates its mitochondrial translocation and apoptosis. Oncotarget 2016, 7, 47687–47698. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, H.; Woods, N.T.; Piluso, L.G.; Lee, H.H.; Chen, J.; Bhalla, K.N.; Monteiro, A.; Liu, X.; Hung, M.C.; Wang, H.G. p53 acetylation is crucial for its transcription-independent proapoptotic functions. J. Biol. Chem. 2009, 284, 11171–11183. [Google Scholar] [CrossRef] [Green Version]
- Sot, B.; Freund, S.M.; Fersht, A.R. Comparative biophysical characterization of p53 with the pro-apoptotic BAK and the anti-apoptotic BCL-xL. J. Biol. Chem. 2007, 282, 29193–29200. [Google Scholar] [CrossRef] [Green Version]
- Wei, H.; Qu, L.; Dai, S.; Li, Y.; Wang, H.; Feng, Y.; Chen, X.; Jiang, L.; Guo, M.; Li, J.; et al. Structural insight into the molecular mechanism of p53-mediated mitochondrial apoptosis. Nat. Commun. 2021, 12, 2280. [Google Scholar] [CrossRef]
- Yao, H.; Mi, S.; Gong, W.; Lin, J.; Xu, N.; Perrett, S.; Xia, B.; Wang, J.; Feng, Y. Anti-apoptosis proteins Mcl-1 and Bcl-xL have different p53-binding profiles. Biochemistry 2013, 52, 6324–6334. [Google Scholar] [CrossRef]
- Hensley, K.; Floyd, R.A. Reactive oxygen species and protein oxidation in aging: A look back, a look ahead. Arch Biochem. Biophys. 2002, 397, 377–383. [Google Scholar] [CrossRef]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling. Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Fukai, T.; Ushio-Fukai, M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mailloux, R.J.; McBride, S.L.; Harper, M.-E. Unearthing the secrets of mitochondrial ROS and glutathione in bioenergetics. Trends Biochem. Sci. 2013, 38, 592–602. [Google Scholar] [CrossRef]
- Jones, D.P. [11] Redox potential of GSH/GSSG couple: Assay and biological significance. Methods Enzymol. 2002, 348, 93–112. [Google Scholar]
- Srinivas, U.S.; Tan, B.W.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA damage response in cancer. Redox Biol. 2019, 25, 101084. [Google Scholar] [CrossRef]
- Trachootham, D.; Lu, W.; Ogasawara, M.A.; Valle, N.R.-D.; Huang, P. Redox regulation of cell survival. Antioxid. Redox Signal. 2008, 10, 1343–1374. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Ames, B.N. Senescence-like growth arrest induced by hydrogen peroxide in human diploid fibroblast F65 cells. Proc. Natl. Acad. Sci. USA 1994, 91, 4130–4134. [Google Scholar] [CrossRef] [Green Version]
- Hagen, T.M.; Yowe, D.L.; Bartholomew, J.C.; Wehr, C.M.; Do, K.L.; Park, J.-Y.; Ames, B.N. Mitochondrial decay in hepatocytes from old rats: Membrane potential declines, heterogeneity and oxidants increase. Proc. Natl. Acad. Sci. USA 1997, 94, 3064–3069. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.M.; Bartholomew, J.C.; Campisi, J.; Acosta, M.; Reagan, J.D.; Ames, B.N. Molecular analysis of H2O2-induced senescent-like growth arrest in normal human fibroblasts: p53 and Rb control G1 arrest but not cell replication. Biochem. J. 1998, 332, 43–50. [Google Scholar] [CrossRef] [Green Version]
- Macip, S.; Igarashi, M.; Berggren, P.; Yu, J.; Lee, S.W.; Aaronson, S.A. Influence of induced reactive oxygen species in p53-mediated cell fate decisions. Mol. Cell Biol. 2003, 23, 8576–8585. [Google Scholar] [CrossRef] [Green Version]
- Bladier, C.; Wolvetang, E.J.; Hutchinson, P.; de Haan, J.B.; Kola, I. Response of a primary human fibroblast cell line to H2O2: Senescence-like growth arrest or apoptosis? Cell Growth Differ. 1997, 8, 589–598. [Google Scholar] [PubMed]
- Crescenzi, E.; Palumbo, G.; Brady, H.J. Bcl-2 activates a programme of premature senescence in human carcinoma cells. Biochem. J. 2003, 375, 263–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lind, E.F.; Wayne, J.; Wang, Q.Z.; Staeva, T.; Stolzer, A.; Petrie, H.T. Bcl-2-induced changes in E2F regulatory complexes reveal the potential for integrated cell cycle and cell death functions. J. Immunol. 1999, 162, 5374–5379. [Google Scholar] [PubMed]
- Vairo, G.; Soos, T.J.; Upton, T.M.; Zalvide, J.; DeCaprio, J.A.; Ewen, M.E.; Koff, A.; Adams, J.M. Bcl-2 retards cell cycle entry through p27(Kip1), pRB relative p130, and altered E2F regulation. Mol. Cell Biol. 2000, 20, 4745–4753. [Google Scholar] [CrossRef] [Green Version]
- Rincheval, V.; Renaud, F.; Lemaire, C.; Godefroy, N.; Trotot, P.; Boulo, V.; Mignotte, B.; Vayssiere, J.L. Bcl-2 can promote p53-dependent senescence versus apoptosis without affecting the G1/S transition. Biochem. Biophys. Res. Commun. 2002, 298, 282–288. [Google Scholar] [CrossRef]
- Wang, W.; Wang, D.; Li, H. Initiation of premature senescence by Bcl-2 in hypoxic condition. Int. J. Clin. Exp. Pathol. 2014, 7, 2446–2453. [Google Scholar]
- Bluwstein, A.; Kumar, N.; Leger, K.; Traenkle, J.; Oostrum, J.; Rehrauer, H.; Baudis, M.; Hottiger, M.O. PKC signaling prevents irradiation-induced apoptosis of primary human fibroblasts. Cell Death Dis. 2013, 4, e498. [Google Scholar] [CrossRef] [Green Version]
- Hayward, R.L.; Macpherson, J.S.; Cummings, J.; Monia, B.P.; Smyth, J.F.; Jodrell, D.I. Antisense Bcl-xl down-regulation switches the response to topoisomerase I inhibition from senescence to apoptosis in colorectal cancer cells, enhancing global cytotoxicity. Clin. Cancer Res. 2003, 9, 2856–2865. [Google Scholar]
- Bolesta, E.; Pfannenstiel, L.W.; Demelash, A.; Lesniewski, M.L.; Tobin, M.; Schlanger, S.E.; Nallar, S.C.; Papadimitriou, J.C.; Kalvakolanu, D.V.; Gastman, B.R. Inhibition of Mcl-1 promotes senescence in cancer cells: Implications for preventing tumor growth and chemotherapy resistance. Mol. Cell Biol. 2012, 32, 1879–1892. [Google Scholar] [CrossRef] [Green Version]
- Demelash, A.; Pfannenstiel, L.W.; Tannenbaum, C.S.; Li, X.; Kalady, M.F.; DeVecchio, J.; Gastman, B.R. Structure-Function Analysis of the Mcl-1 Protein Identifies a Novel Senescence-regulating Domain. J. Biol. Chem. 2015, 290, 21962–21975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vousden, K.H. p53: Death star. Cell 2000, 103, 691–694. [Google Scholar] [CrossRef] [Green Version]
- Murray-Zmijewski, F.; Slee, E.A.; Lu, X. A complex barcode underlies the heterogeneous response of p53 to stress. Nat. Rev. Mol. Cell Biol. 2008, 9, 702–712. [Google Scholar] [CrossRef] [PubMed]
- Tabasso, A.F.S.; Jones, D.J.L.; Jones, G.D.D.; Macip, S. Radiotherapy-Induced Senescence and its Effects on Responses to Treatment. Clin. Oncol. 2019, 31, 283–289. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dhokia, V.; Moss, J.A.Y.; Macip, S.; Fox, J.L. At the Crossroads of Life and Death: The Proteins That Influence Cell Fate Decisions. Cancers 2022, 14, 2745. https://doi.org/10.3390/cancers14112745
Dhokia V, Moss JAY, Macip S, Fox JL. At the Crossroads of Life and Death: The Proteins That Influence Cell Fate Decisions. Cancers. 2022; 14(11):2745. https://doi.org/10.3390/cancers14112745
Chicago/Turabian StyleDhokia, Vinesh, John A. Y. Moss, Salvador Macip, and Joanna L. Fox. 2022. "At the Crossroads of Life and Death: The Proteins That Influence Cell Fate Decisions" Cancers 14, no. 11: 2745. https://doi.org/10.3390/cancers14112745
APA StyleDhokia, V., Moss, J. A. Y., Macip, S., & Fox, J. L. (2022). At the Crossroads of Life and Death: The Proteins That Influence Cell Fate Decisions. Cancers, 14(11), 2745. https://doi.org/10.3390/cancers14112745