
Potential Therapeutics Targeting Upstream Regulators and Interactors of EHMT1/2

 , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Structure and Function of EHMTs
3. EHMT1/GLP and EHMT2/G9a Dysregulation in Cancer
4. Pharmacological Inhibitors of EHMTs and Their Limitations
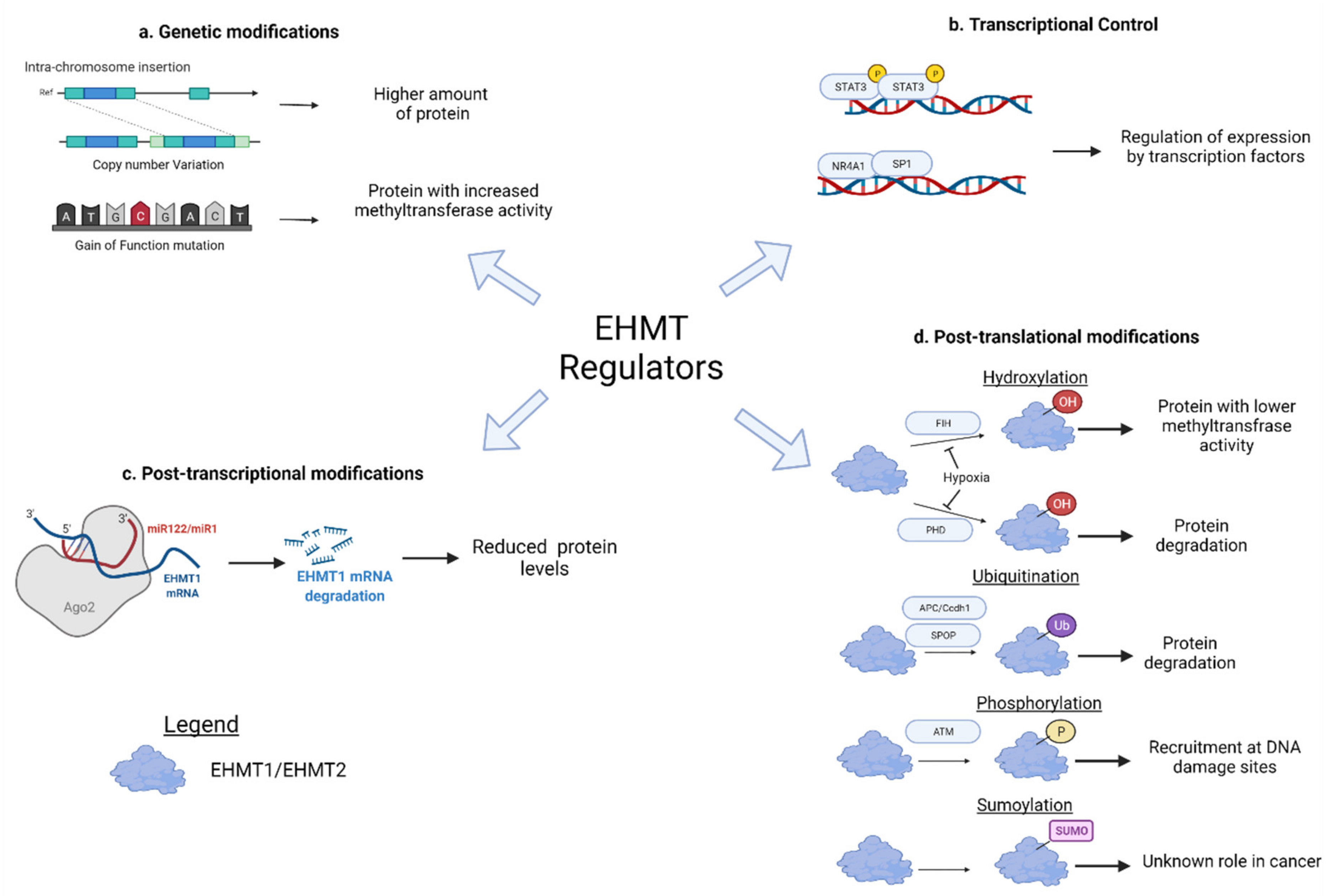
5. Upstream Regulators of EHMT1/2
5.1. Mutation and Copy Number Alterations
5.2. Transcriptional Regulation
5.3. Post-Transcriptional Regulation
5.4. Post-Translational Regulation
6. Interacting Partners of EHMT1/2

{kind=link}
{kind=link}
| Interactors | G9a/GLP | Function | Cancer Type | Phenotype | Potential Therapeutics |
|---|---|---|---|---|---|
| Transcription factors | |||||
| MDM2 | GLP | Cancer | Osteosarcoma [86] | Avoid p53-induced cell death | Nutlin analogs [87] MI-219 [88] |
| P53 | GLP and G9a | Cancer | CRC [89] HCC [17] | Cell cycle progression Escaping apoptosis | Nutlin analogs [87] MI-219 [88] |
| Lung cancer (activator) [90] | Enhance apoptosis and reduce colony formation | Nutlin analogs [87] MI-219 [88] KJ-pyr-9 [91] Omomyc [92] | |||
| MYC | G9a | Cancer | Breast cancer [93,94] | Cell proliferation | |
| STAT3 | G9a | Cancer | GC [95] Breast cancer [96] | Evading autophagy EMT and CSC maintenance | SH003 [97] STA-21 [98] Stattic [99] IS3295 [100] Cisplatin [100] |
| FOXO1 | G9a | Cancer | CRC [101] | Cell proliferation | Troglitazone [102] Gallic acid [103] Skp2E3LIs [104] NSC689857 [105] Linichlorin A [106] |
| RUNX3 | G9a | Cancer | GC [107] | Cell proliferation suppresses apoptosis and immune response | - |
| RUNX2 | G9a | Cancer | Breast cancer [108] Prostate cancer [108] | Metastasis | - |
| TBX2 | G9a | Cancer | Breast cancer [109] | Cell proliferation | - |
| NKX3.1 | G9a | Cancer | Prostate cancer [110] | Inhibit cell differentiation | - |
| Zinc finger proteins | |||||
| WIZ | G9a and GLP | Maintenance of pluripotency | - | - | - |
| Snail | G9a | Cancer | Breast cancer [111] | EMT Cell proliferation Metabolic reprogramming CSC maintenance | SD-093 [112] LY2157299 [113] AP12009 [114] ISTH0036 |
| Slug | G9a | Cancer | HCC [115] Lung cancer [115] | EMT | SD-093 [112] LY2157299 [113] AP12009 [114] ISTH0036 [116] |
| ZNF644 | G9a | Neurodevelopment, maintenance of pluripotency | - | - | - |
| ZNF518B | G9a | Cancer | CRC [117] | Cell proliferation | - |
| Non-transcription factor proteins | |||||
| Cyclin D | G9a | Cancer | Breast cancer [118] | Cell proliferation | - |
| RPA | G9a | Cancer | CRC [119] | Radio and chemoresistance | - |
| MT1h | GLP | Cancer | HCC [120] Prostate cancer [120] | Reduce cell cycle Reduce Migration and invasion Reduce colony formation | - |
| Epigenetic regulators | |||||
| EZH2 | G9a | Cancer | Breast cancer [121] | Cell proliferation | GSK343 [121] GSK2816126 [122] |
| GLP | Repressive complex | - | - | - | |
| HDACs | G9a | Cancer | HCC [115] | EMT Migration and invasion | TSA [123] |
| DNMTs | G9a | Cancer | Hematological malignancies [124] | Cell proliferation Inhibit apoptosis | CM-272 [124] Azacytidine [71] Decitabine [72] |
| CDYL | G9a and GLP | Cancer | Osteosarcoma [125] HCC [126] | Cell proliferation | D03 [127] |
| CDYL2 | G9a and GLP | Cancer | Breast cancer [128] | Migration Sphere formation | - |
| Long non-coding RNAs | |||||
| TERNA1 | G9a | Cancer | HCC [129] Osteosarcoma [129] | EMT Migration and invasion | - |
| NEAT1 | G9a | Cancer | HCC [130] | EMT Migration and invasion | - |
| HOTAIRM1 | G9a | Cancer | Osteosarcoma [131] GBM [131] | Cell proliferation Migration and invasion Reduce apoptosis | - |
6.1. Transcription Factors
6.1.1. P53
6.1.2. MYC
6.1.3. STAT3
6.1.4. Other Transcription Factors
6.2. Zinc Finger Proteins
6.3. Non-Transcription Factor Protein
6.4. Epigenetic Regulators
CDYL
6.5. lncRNA
7. Targeting Regulators and EHMT Interactors in Cancer
7.1. Upstream Regulators
7.1.1. Copy Number Gains and Gain-of-Function Mutation
7.1.2. EGFR Signaling
7.1.3. NR4A1
7.1.4. miRNAs
7.1.5. Post-Translational Modifiers
7.2. Targeting EHMT Interactors in Cancer
7.3. Targeting Transcription Factors
7.3.1. Inhibition of MDM2/Activation of p53
7.3.2. Inhibition of Myc
7.3.3. Inhibition of STAT3
7.3.4. Activation of FOXO1
7.4. Targeting Zinc Finger Transcription Factors
7.5. Targeting Non-Transcription Factors
Inhibition of Cyclin D
7.6. Targeting Epigenetic Regulators
Inhibition of CDYL
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Maleszewska, M.; Wojtas, B.; Kaminska, B. Deregulation of Epigenetic Mechanisms in Cancer. Postępy Biochem. 2018, 64, 148–156. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Zhang, R. Epigenetics in Autoimmune Diseases: Pathogenesis and Prospects for Therapy. Autoimmun. Rev. 2015, 14, 854–863. [Google Scholar] [CrossRef]
- Mirabella, A.C.; Foster, B.M.; Bartke, T. Chromatin Deregulation in Disease. Chromosoma 2016, 125, 75–93. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Shilatifard, A. Epigenetic Modifications of Histones in Cancer. Genome Biol. 2019, 20, 245. [Google Scholar] [CrossRef]
- Murray, K. The Occurrence of Iε-N-Methyl Lysine in Histones. Biochemistry 1964, 3, 10–15. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. Regulation of Chromatin by Histone Modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Husmann, D.; Gozani, O. Histone Lysine Methyltransferases in Biology and Disease. Nat. Struct. Mol. Biol. 2019, 26, 880–889. [Google Scholar] [CrossRef]
- Tachibana, M.; Sugimoto, K.; Fukushima, T.; Shinkai, Y. SET Domain-Containing Protein, G9a, Is a Novel Lysine-Preferring Mammalian Histone Methyltransferase with Hyperactivity and Specific Selectivity to Lysines 9 and 27 of Histone H3. J. Biol. Chem. 2001, 276, 25309–25317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rice, J.C.; Briggs, S.D.; Ueberheide, B.; Barber, C.M.; Shabanowitz, J.; Hunt, D.F.; Shinkai, Y.; Allis, C.D. Histone Methyltransferases Direct Different Degrees of Methylation to Define Distinct Chromatin Domains. Mol. Cell 2003, 12, 1591–1598. [Google Scholar] [CrossRef]
- Bittencourt, D.; Wu, D.-Y.; Jeong, K.W.; Gerke, D.S.; Herviou, L.; Ianculescu, I.; Chodankar, R.; Siegmund, K.D.; Stallcup, M.R. G9a Functions as a Molecular Scaffold for Assembly of Transcriptional Coactivators on a Subset of Glucocorticoid Receptor Target Genes. Proc. Natl. Acad. Sci. USA 2012, 109, 19673–19678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michalak, E.M.; Visvader, J.E. Dysregulation of Histone Methyltransferases in Breast Cancer—Opportunities for New Targeted Therapies? Mol. Oncol. 2016, 10, 1497–1515. [Google Scholar] [CrossRef] [Green Version]
- Pangeni, R.P.; Yang, L.; Zhang, K.; Wang, J.; Li, W.; Guo, C.; Yun, X.; Sun, T.; Wang, J.; Raz, D.J. G9a Regulates Tumorigenicity and Stemness through Genome-Wide DNA Methylation Reprogramming in Non-Small Cell Lung Cancer. Clin. Epigenetics 2020, 12, 88. [Google Scholar] [CrossRef]
- Souza, B.K.; Freire, N.H.; Jaeger, M.; de Farias, C.B.; Brunetto, A.L.; Brunetto, A.T.; Roesler, R. EHMT2/G9a as an Epigenetic Target in Pediatric and Adult Brain Tumors. Int. J. Mol. Sci. 2021, 22, 11292. [Google Scholar] [CrossRef]
- Hua, K.-T.; Wang, M.-Y.; Chen, M.-W.; Wei, L.-H.; Chen, C.-K.; Ko, C.-H.; Jeng, Y.-M.; Sung, P.-L.; Jan, Y.-H.; Hsiao, M.; et al. The H3K9 Methyltransferase G9a Is a Marker of Aggressive Ovarian Cancer That Promotes Peritoneal Metastasis. Mol. Cancer 2014, 13, 189. [Google Scholar] [CrossRef] [Green Version]
- Casciello, F.; Windloch, K.; Gannon, F.; Lee, J.S. Functional Role of G9a Histone Methyltransferase in Cancer. Front. Immunol. 2015, 6, 487. [Google Scholar] [CrossRef] [Green Version]
- Kondo, Y.; Shen, L.; Ahmed, S.; Boumber, Y.; Sekido, Y.; Haddad, B.R.; Issa, J.-P.J. Downregulation of Histone H3 Lysine 9 Methyltransferase G9a Induces Centrosome Disruption and Chromosome Instability in Cancer Cells. PLoS ONE 2008, 3, e2037. [Google Scholar] [CrossRef] [Green Version]
- Rada, M.; Vasileva, E.; Lezina, L.; Marouco, D.; Antonov, A.V.; Macip, S.; Melino, G.; Barlev, N.A. Human EHMT2/G9a Activates P53 through Methylation-Independent Mechanism. Oncogene 2017, 36, 922–932. [Google Scholar] [CrossRef] [Green Version]
- Cao, H.; Li, L.; Yang, D.; Zeng, L.; Yewei, X.; Yu, B.; Liao, G.; Chen, J. Recent Progress in Histone Methyltransferase (G9a) Inhibitors as Anticancer Agents. Eur. J. Med. Chem. 2019, 179, 537–546. [Google Scholar] [CrossRef]
- Jan, S.; Dar, M.I.; Wani, R.; Sandey, J.; Mushtaq, I.; Lateef, S.; Syed, S.H. Targeting EHMT2/G9a for Cancer Therapy: Progress and Perspective. Eur. J. Pharmacol. 2021, 893, 173827. [Google Scholar] [CrossRef]
- Tachibana, M.; Ueda, J.; Fukuda, M.; Takeda, N.; Ohta, T.; Iwanari, H.; Sakihama, T.; Kodama, T.; Hamakubo, T.; Shinkai, Y. Histone Methyltransferases G9a and GLP Form Heteromeric Complexes and Are Both Crucial for Methylation of Euchromatin at H3-K9. Genes Dev. 2005, 19, 815–826. [Google Scholar] [CrossRef] [Green Version]
- Poulard, C.; Noureddine, L.M.; Pruvost, L.; Le Romancer, M. Structure, Activity, and Function of the Protein Lysine Methyltransferase G9a. Life 2021, 11, 1082. [Google Scholar] [CrossRef] [PubMed]
- Trievel, R.C.; Beach, B.M.; Dirk, L.M.A.; Houtz, R.L.; Hurley, J.H. Structure and Catalytic Mechanism of a SET Domain Protein Methyltransferase. Cell 2002, 111, 91–103. [Google Scholar] [CrossRef] [Green Version]
- Collins, R.E.; Northrop, J.P.; Horton, J.R.; Lee, D.Y.; Zhang, X.; Stallcup, M.R.; Cheng, X. The Ankyrin Repeats of G9a and GLP Histone Methyltransferases Are Mono- and Dimethyllysine Binding Modules. Nat. Struct. Mol. Biol. 2008, 15, 245–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estève, P.-O.; Patnaik, D.; Chin, H.G.; Benner, J.; Teitell, M.A.; Pradhan, S. Functional Analysis of the N- and C-Terminus of Mammalian G9a Histone H3 Methyltransferase. Nucleic Acids Res. 2005, 33, 3211–3223. [Google Scholar] [CrossRef] [Green Version]
- Shinkai, Y.; Tachibana, M. H3K9 Methyltransferase G9a and the Related Molecule GLP. Genes Dev. 2011, 25, 781–788. [Google Scholar] [CrossRef] [Green Version]
- Ling, B.M.T.; Bharathy, N.; Chung, T.-K.; Kok, W.K.; Li, S.; Tan, Y.H.; Rao, V.K.; Gopinadhan, S.; Sartorelli, V.; Walsh, M.J.; et al. Lysine Methyltransferase G9a Methylates the Transcription Factor MyoD and Regulates Skeletal Muscle Differentiation. Proc. Natl. Acad. Sci. USA 2012, 109, 841–846. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.; Chiu, D.K.-C.; Tsang, F.H.-C.; Law, C.-T.; Cheng, C.L.-H.; Au, S.L.-K.; Lee, J.M.-F.; Wong, C.C.-L.; Ng, I.O.-L.; Wong, C.-M. Histone Methyltransferase G9a Promotes Liver Cancer Development by Epigenetic Silencing of Tumor Suppressor Gene RARRES3. J. Hepatol. 2017, 67, 758–769. [Google Scholar] [CrossRef]
- Dang, N.-N.; Jiao, J.; Meng, X.; An, Y.; Han, C.; Huang, S. Abnormal Overexpression of G9a in Melanoma Cells Promotes Cancer Progression via Upregulation of the Notch1 Signaling Pathway. Aging 2020, 12, 2393–2407. [Google Scholar] [CrossRef]
- Hu, L.; Zang, M.; Wang, H.; Zhang, B.; Wang, Z.; Fan, Z.; Wu, H.; Li, J.; Su, L.; Yan, M.; et al. G9A Promotes Gastric Cancer Metastasis by Upregulating ITGB3 in a SET Domain-Independent Manner. Cell Death Dis. 2018, 9, 278. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Park, S.; Park, S.-Y.; Lee, C.-Y.; Eum, D.-Y.; Shim, J.-W.; Choi, S.-H.; Choi, Y.-J.; Park, S.-J.; Heo, K. G9a Knockdown Suppresses Cancer Aggressiveness by Facilitating Smad Protein Phosphorylation through Increasing BMP5 Expression in Luminal A Type Breast Cancer. Int. J. Mol. Sci. 2022, 23, 589. [Google Scholar] [CrossRef]
- Liu, S.; Ye, D.; Guo, W.; Yu, W.; He, Y.; Hu, J.; Wang, Y.; Zhang, L.; Liao, Y.; Song, H.; et al. G9a Is Essential for EMT-Mediated Metastasis and Maintenance of Cancer Stem Cell-like Characters in Head and Neck Squamous Cell Carcinoma. Oncotarget 2015, 6, 6887–6901. [Google Scholar] [CrossRef] [Green Version]
- Hsiao, S.-M.; Chen, M.-W.; Chen, C.-A.; Chien, M.-H.; Hua, K.-T.; Hsiao, M.; Kuo, M.-L.; Wei, L.-H. The H3K9 Methyltransferase G9a Represses E-Cadherin and Is Associated with Myometrial Invasion in Endometrial Cancer. Ann. Surg. Oncol. 2015, 22 (Suppl. S3), S1556–S1565. [Google Scholar] [CrossRef]
- Sun, T.; Zhang, K.; Pangeni, R.P.; Wu, J.; Li, W.; Du, Y.; Guo, Y.; Chaurasiya, S.; Arvanitis, L.; Raz, D.J. G9a Promotes Invasion and Metastasis of Non-Small Cell Lung Cancer through Enhancing Focal Adhesion Kinase Activation via NF-ΚB Signaling Pathway. Mol. Cancer Res. MCR 2021, 19, 429–440. [Google Scholar] [CrossRef]
- Rowbotham, S.P.; Li, F.; Dost, A.F.M.; Louie, S.M.; Marsh, B.P.; Pessina, P.; Anbarasu, C.R.; Brainson, C.F.; Tuminello, S.J.; Lieberman, A.; et al. H3K9 Methyltransferases and Demethylases Control Lung Tumor-Propagating Cells and Lung Cancer Progression. Nat. Commun. 2018, 9, 4559. [Google Scholar] [CrossRef]
- Guan, X.; Zhong, X.; Men, W.; Gong, S.; Zhang, L.; Han, Y. Analysis of EHMT1 Expression and Its Correlations with Clinical Significance in Esophageal Squamous Cell Cancer. Mol. Clin. Oncol. 2014, 2, 76–80. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.-C.; Chang, J.; Huang, S.C.-C.; Lin, H.-C.; Ho, A.-S.; Lim, K.-H.; Chang, C.-C.; Huang, L.; Chang, Y.-C.; Chang, Y.-F.; et al. YM155 as an Inhibitor of Cancer Stemness Simultaneously Inhibits Autophosphorylation of Epidermal Growth Factor Receptor and G9a-Mediated Stemness in Lung Cancer Cells. PLoS ONE 2017, 12, e0182149. [Google Scholar] [CrossRef]
- Yang, Y.; Shen, J.; Yan, D.; Yuan, B.; Zhang, S.; Wei, J.; Du, T. Euchromatic Histone Lysine Methyltransferase 1 Regulates Cancer Development in Human Gastric Cancer by Regulating E-Cadherin. Oncol. Lett. 2018, 15, 9480–9486. [Google Scholar] [CrossRef] [Green Version]
- Nachiyappan, A.; Soon, J.L.J.; Lim, H.J.; Lee, V.K.; Taneja, R. EHMT1 Promotes Tumor Progression and Maintains Stemness by Regulating ALDH1A1 Expression in Alveolar Rhabdomyosarcoma. J. Pathol. 2022, 256, 349–362. [Google Scholar] [CrossRef]
- Nachiyappan, A.; Gupta, N.; Taneja, R. EHMT1/EHMT2 in EMT, Cancer Stemness and Drug Resistance: Emerging Evidence and Mechanisms. FEBS J. 2022, 289, 1329–1351. [Google Scholar] [CrossRef]
- Wen, B.; Wu, H.; Shinkai, Y.; Irizarry, R.A.; Feinberg, A.P. Large Histone H3 Lysine 9 Dimethylated Chromatin Blocks Distinguish Differentiated from Embryonic Stem Cells. Nat. Genet. 2009, 41, 246–250. [Google Scholar] [CrossRef] [Green Version]
- Berman, B.P.; Weisenberger, D.J.; Aman, J.F.; Hinoue, T.; Ramjan, Z.; Liu, Y.; Noushmehr, H.; Lange, C.P.E.; Van Dijk, C.M.; Tollenaar, R.A.E.M.; et al. Regions of Focal DNA Hypermethylation and Long-Range Hypomethylation in Colorectal Cancer Coincide with Nuclear Lamina–Associated Domains. Nat. Genet. 2012, 44, 40–46. [Google Scholar] [CrossRef] [Green Version]
- Hansen, K.D.; Timp, W.; Bravo, H.C.; Sabunciyan, S.; Langmead, B.; McDonald, O.G.; Wen, B.; Wu, H.; Liu, Y.; Diep, D.; et al. Increased Methylation Variation in Epigenetic Domains across Cancer Types. Nat. Genet. 2011, 43, 768–775. [Google Scholar] [CrossRef] [Green Version]
- McDonald, O.G.; Wu, H.; Timp, W.; Doi, A.; Feinberg, A.P. Genome-Scale Epigenetic Reprogramming during Epithelial-to-Mesenchymal Transition. Nat. Struct. Mol. Biol. 2011, 18, 867–874. [Google Scholar] [CrossRef]
- Rugo, H.S.; Jacobs, I.; Sharma, S.; Scappaticci, F.; Paul, T.A.; Jensen-Pergakes, K.; Malouf, G.G. The Promise for Histone Methyltransferase Inhibitors for Epigenetic Therapy in Clinical Oncology: A Narrative Review. Adv. Ther. 2020, 37, 3059–3082. [Google Scholar] [CrossRef]
- Yuan, Y.; Wang, Q.; Paulk, J.; Kubicek, S.; Kemp, M.M.; Adams, D.J.; Shamji, A.F.; Wagner, B.K.; Schreiber, S.L. A Small-Molecule Probe of the Histone Methyltransferase G9a Induces Cellular Senescence in Pancreatic Adenocarcinoma. ACS Chem. Biol. 2012, 7, 1152–1157. [Google Scholar] [CrossRef]
- Halby, L.; Champion, C.; Sénamaud-Beaufort, C.; Ajjan, S.; Drujon, T.; Rajavelu, A.; Ceccaldi, A.; Jurkowska, R.; Lequin, O.; Nelson, W.G.; et al. Rapid Synthesis of New DNMT Inhibitors Derivatives of Procainamide. Chembiochem Eur. J. Chem. Biol. 2012, 13, 157–165. [Google Scholar] [CrossRef]
- Kubicek, S.; O’Sullivan, R.J.; August, E.M.; Hickey, E.R.; Zhang, Q.; Teodoro, M.L.; Rea, S.; Mechtler, K.; Kowalski, J.A.; Homon, C.A.; et al. Reversal of H3K9me2 by a Small-Molecule Inhibitor for the G9a Histone Methyltransferase. Mol. Cell 2007, 25, 473–481. [Google Scholar] [CrossRef]
- Liu, F.; Chen, X.; Allali-Hassani, A.; Quinn, A.M.; Wasney, G.A.; Dong, A.; Barsyte, D.; Kozieradzki, I.; Senisterra, G.; Chau, I.; et al. Discovery of a 2,4-Diamino-7-Aminoalkoxyquinazoline as a Potent and Selective Inhibitor of Histone Lysine Methyltransferase G9a. J. Med. Chem. 2009, 52, 7950–7953. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Chen, X.; Allali-Hassani, A.; Quinn, A.M.; Wigle, T.J.; Wasney, G.A.; Dong, A.; Senisterra, G.; Chau, I.; Siarheyeva, A.; et al. Protein Lysine Methyltransferase G9a Inhibitors: Design, Synthesis, and Structure Activity Relationships of 2,4-Diamino-7-Aminoalkoxy-Quinazolines. J. Med. Chem. 2010, 53, 5844–5857. [Google Scholar] [CrossRef] [Green Version]
- Vedadi, M.; Barsyte-Lovejoy, D.; Liu, F.; Rival-Gervier, S.; Allali-Hassani, A.; Labrie, V.; Wigle, T.J.; Dimaggio, P.A.; Wasney, G.A.; Siarheyeva, A.; et al. A Chemical Probe Selectively Inhibits G9a and GLP Methyltransferase Activity in Cells. Nat. Chem. Biol. 2011, 7, 566–574. [Google Scholar] [CrossRef]
- Deimling, S.J.; Olsen, J.B.; Tropepe, V. The Expanding Role of the Ehmt2/G9a Complex in Neurodevelopment. Neurogenesis 2017, 4, e1316888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, S.; Weng, Q.Y.; Insco, M.L.; Chen, K.Y.; Muralidhar, S.; Pozniak, J.; Diaz, J.M.S.; Drier, Y.; Nguyen, N.; Lo, J.A.; et al. Gain-of-Function Genetic Alterations of G9a Drive Oncogenesis. Cancer Discov. 2020, 10, 980–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaj, T.; Sirk, S.J.; Shui, S.; Liu, J. Genome-Editing Technologies: Principles and Applications. Cold Spring Harb. Perspect. Biol. 2016, 8, a023754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; Kim, J.-S. A Guide to Genome Engineering with Programmable Nucleases. Nat. Rev. Genet. 2014, 15, 321–334. [Google Scholar] [CrossRef]
- Li, H.; Yang, Y.; Hong, W.; Huang, M.; Wu, M.; Zhao, X. Applications of Genome Editing Technology in the Targeted Therapy of Human Diseases: Mechanisms, Advances and Prospects. Signal Transduct. Target. Ther. 2020, 5, 1–23. [Google Scholar] [CrossRef]
- Cyranoski, D. CRISPR Gene-Editing Tested in a Person for the First Time. Nature 2016, 539, 479. [Google Scholar] [CrossRef]
- Bergin, C.J.; Benoit, Y.D. G9a Is SETting the Stage for Colorectal Oncogenesis. Genes 2020, 11, 616. [Google Scholar] [CrossRef]
- Chang, Y.-F.; Lim, K.-H.; Chiang, Y.-W.; Sie, Z.-L.; Chang, J.; Ho, A.-S.; Cheng, C.-C. STAT3 Induces G9a to Exacerbate HER3 Expression for the Survival of Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitors in Lung Cancers. BMC Cancer 2019, 19, 959. [Google Scholar] [CrossRef]
- Ayati, A.; Moghimi, S.; Salarinejad, S.; Safavi, M.; Pouramiri, B.; Foroumadi, A. A Review on Progression of Epidermal Growth Factor Receptor (EGFR) Inhibitors as an Efficient Approach in Cancer Targeted Therapy. Bioorganic Chem. 2020, 99, 103811. [Google Scholar] [CrossRef]
- Abourehab, M.A.S.; Alqahtani, A.M.; Youssif, B.G.M.; Gouda, A.M. Globally Approved EGFR Inhibitors: Insights into Their Syntheses, Target Kinases, Biological Activities, Receptor Interactions, and Metabolism. Molecules 2021, 26, 6677. [Google Scholar] [CrossRef]
- Shrestha, R.; Mohankumar, K.; Jin, U.-H.; Martin, G.; Safe, S. The Histone Methyltransferase Gene G9A Is Regulated by Nuclear Receptor 4A1 in Alveolar Rhabdomyosarcoma Cells. Mol. Cancer Ther. 2021, 20, 612–622. [Google Scholar] [CrossRef]
- Lee, S.-O.; Li, X.; Hedrick, E.; Jin, U.-H.; Tjalkens, R.B.; Backos, D.S.; Li, L.; Zhang, Y.; Wu, Q.; Safe, S. Diindolylmethane Analogs Bind NR4A1 and Are NR4A1 Antagonists in Colon Cancer Cells. Mol. Endocrinol. 2014, 28, 1729–1739. [Google Scholar] [CrossRef] [Green Version]
- Köhler, J.; Erlenkamp, G.; Eberlin, A.; Rumpf, T.; Slynko, I.; Metzger, E.; Schüle, R.; Sippl, W.; Jung, M. Lestaurtinib Inhibits Histone Phosphorylation and Androgen-Dependent Gene Expression in Prostate Cancer Cells. PLoS ONE 2012, 7, e34973. [Google Scholar] [CrossRef]
- Yuan, L.-T.; Lee, W.-J.; Yang, Y.-C.; Chen, B.-R.; Yang, C.-Y.; Chen, M.-W.; Chen, J.-Q.; Hsiao, M.; Chien, M.-H.; Hua, K.-T. Histone Methyltransferase G9a-Promoted Progression of Hepatocellular Carcinoma Is Targeted by Liver-Specific Hsa-MiR-122. Cancers 2021, 13, 2376. [Google Scholar] [CrossRef]
- Kargbo, R.B. RIBOTACs: Small Molecules Selectively Destroy Cancer-Associated RNA. ACS Med. Chem. Lett. 2021, 12, 1872–1873. [Google Scholar] [CrossRef]
- Kang, J.; Shin, S.-H.; Yoon, H.; Huh, J.; Shin, H.-W.; Chun, Y.-S.; Park, J.-W. FIH Is an Oxygen Sensor in Ovarian Cancer for G9a/GLP-Driven Epigenetic Regulation of Metastasis-Related Genes. Cancer Res. 2018, 78, 1184–1199. [Google Scholar] [CrossRef] [Green Version]
- Fuchs-Tarlovsky, V. Role of Antioxidants in Cancer Therapy. Nutrition 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Hielscher, A.; Gerecht, S. Hypoxia and Free Radicals: Role in Tumor Progression and the Use of Engineering-Based Platforms to Address These Relationships. Free Radic. Biol. Med. 2015, 79, 281–291. [Google Scholar] [CrossRef] [Green Version]
- Casciello, F.; Al-Ejeh, F.; Kelly, G.; Brennan, D.J.; Ngiow, S.F.; Young, A.; Stoll, T.; Windloch, K.; Hill, M.M.; Smyth, M.J.; et al. G9a Drives Hypoxia-Mediated Gene Repression for Breast Cancer Cell Survival and Tumorigenesis. Proc. Natl. Acad. Sci. USA 2017, 114, 7077–7082. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Gao, K.; Xie, H.; Wang, D.; Zhang, P.; Wei, T.; Yan, Y.; Pan, Y.; Ye, W.; Chen, H.; et al. SPOP Mutation Induces DNA Methylation via Stabilizing GLP/G9a. Nat. Commun. 2021, 12, 5716. [Google Scholar] [CrossRef]
- Coulouarn, C.; Factor, V.M.; Andersen, J.B.; Durkin, M.E.; Thorgeirsson, S.S. Loss of MiR-122 Expression in Liver Cancer Correlates with Suppression of the Hepatic Phenotype and Gain of Metastatic Properties. Oncogene 2009, 28, 3526–3536. [Google Scholar] [CrossRef] [Green Version]
- Cheng, D.; Deng, J.; Zhang, B.; He, X.; Meng, Z.; Li, G.; Ye, H.; Zheng, S.; Wei, L.; Deng, X.; et al. LncRNA HOTAIR Epigenetically Suppresses MiR-122 Expression in Hepatocellular Carcinoma via DNA Methylation. EBioMedicine 2018, 36, 159–170. [Google Scholar] [CrossRef] [Green Version]
- Neklesa, T.K.; Winkler, J.D.; Crews, C.M. Targeted Protein Degradation by PROTACs. Pharmacol. Ther. 2017, 174, 138–144. [Google Scholar] [CrossRef]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [Green Version]
- Ginjala, V.; Rodriguez-Colon, L.; Ganguly, B.; Gangidi, P.; Gallina, P.; Al-Hraishawi, H.; Kulkarni, A.; Tang, J.; Gheeya, J.; Simhadri, S.; et al. Protein-Lysine Methyltransferases G9a and GLP1 Promote Responses to DNA Damage. Sci. Rep. 2017, 7, 16613. [Google Scholar] [CrossRef]
- Liu, L.; Kimball, S.; Liu, H.; Holowatyj, A.; Yang, Z.-Q. Genetic Alterations of Histone Lysine Methyltransferases and Their Significance in Breast Cancer. Oncotarget 2014, 6, 2466–2482. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Zhang, H.-Y.; Fei, L.-R.; Zhang, M.-Y.; Wang, C.-C.; Luo, Y.; Han, Y.-C. SATB2 Suppresses Non-Small Cell Lung Cancer Invasiveness by G9a. Clin. Exp. Med. 2018, 18, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Chin, H.G.; Estève, P.-O.; Pradhan, M.; Benner, J.; Patnaik, D.; Carey, M.F.; Pradhan, S. Automethylation of G9a and Its Implication in Wider Substrate Specificity and HP1 Binding. Nucleic Acids Res. 2007, 35, 7313–7323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poulard, C.; Baulu, E.; Lee, B.H.; Pufall, M.A.; Stallcup, M.R. Increasing G9a Automethylation Sensitizes B Acute Lymphoblastic Leukemia Cells to Glucocorticoid-Induced Death. Cell Death Dis. 2018, 9, 1038. [Google Scholar] [CrossRef] [PubMed]
- Wyld, L.; Bellantuono, I.; Tchkonia, T.; Morgan, J.; Turner, O.; Foss, F.; George, J.; Danson, S.; Kirkland, J.L. Senescence and Cancer: A Review of Clinical Implications of Senescence and Senotherapies. Cancers 2020, 12, 2134. [Google Scholar] [CrossRef] [PubMed]
- DNA Damage Signaling Triggers Degradation of Histone Methyltransferases through APC/C(Cdh1) in Senescent Cells—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/22178396/ (accessed on 27 April 2022).
- Srinivasan, S.; Shankar, S.R.; Wang, Y.; Taneja, R. SUMOylation of G9a Regulates Its Function as an Activator of Myoblast Proliferation. Cell Death Dis. 2019, 10, 250. [Google Scholar] [CrossRef]
- Scheer, S.; Zaph, C. The Lysine Methyltransferase G9a in Immune Cell Differentiation and Function. Front. Immunol. 2017, 8, 429. [Google Scholar] [CrossRef] [Green Version]
- Purcell, D.J.; Jeong, K.W.; Bittencourt, D.; Gerke, D.S.; Stallcup, M.R. A Distinct Mechanism for Coactivator versus Corepressor Function by Histone Methyltransferase G9a in Transcriptional Regulation. J. Biol. Chem. 2011, 286, 41963–41971. [Google Scholar] [CrossRef] [Green Version]
- Oh, S.-T.; Kim, K.-B.; Chae, Y.-C.; Kang, J.-Y.; Hahn, Y.; Seo, S.-B. H3K9 Histone Methyltransferase G9a-Mediated Transcriptional Activation of P21. FEBS Lett. 2014, 588, 685–691. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Li, Z.; Zwolinska, A.K.; Smith, M.A.; Cross, B.; Koomen, J.; Yuan, Z.-M.; Jenuwein, T.; Marine, J.-C.; Wright, K.L.; et al. MDM2 Recruitment of Lysine Methyltransferases Regulates P53 Transcriptional Output. EMBO J. 2010, 29, 2538–2552. [Google Scholar] [CrossRef] [Green Version]
- Beloglazkina, A.; Zyk, N.; Majouga, A.; Beloglazkina, E. Recent Small-Molecule Inhibitors of the P53–MDM2 Protein–Protein Interaction. Molecules 2020, 25, 1211. [Google Scholar] [CrossRef] [Green Version]
- Shangary, S.; Wang, S. Small-Molecule Inhibitors of the MDM2-P53 Protein-Protein Interaction to Reactivate P53 Function: A Novel Approach for Cancer Therapy. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 223–241. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Wang, Y.; Shen, Y.; He, P.; Ding, J.; Chen, Y. G9a Stimulates CRC Growth by Inducing P53 Lys373 Dimethylation-Dependent Activation of Plk1. Theranostics 2018, 8, 2884–2895. [Google Scholar] [CrossRef]
- Schlereth, K.; Beinoraviciute-Kellner, R.; Zeitlinger, M.K.; Bretz, A.C.; Sauer, M.; Charles, J.P.; Vogiatzi, F.; Leich, E.; Samans, B.; Eilers, M.; et al. DNA Binding Cooperativity of P53 Modulates the Decision between Cell-Cycle Arrest and Apoptosis. Mol. Cell 2010, 38, 356–368. [Google Scholar] [CrossRef]
- Jung, K.-Y.; Wang, H.; Teriete, P.; Yap, J.L.; Chen, L.; Lanning, M.E.; Hu, A.; Lambert, L.J.; Holien, T.; Sundan, A.; et al. Perturbation of the C-Myc–Max Protein–Protein Interaction via Synthetic α-Helix Mimetics. J. Med. Chem. 2015, 58, 3002–3024. [Google Scholar] [CrossRef] [Green Version]
- Madden, S.K.; De Araujo, A.D.; Gerhardt, M.; Fairlie, D.P.; Mason, J.M. Taking the Myc out of Cancer: Toward Therapeutic Strategies to Directly Inhibit c-Myc. Mol. Cancer 2021, 20, 3. [Google Scholar] [CrossRef]
- Tu, W.B.; Shiah, Y.-J.; Lourenco, C.; Mullen, P.J.; Dingar, D.; Redel, C.; Tamachi, A.; Ba-Alawi, W.; Aman, A.; Al-awar, R.; et al. MYC Interacts with the G9a Histone Methyltransferase to Drive Transcriptional Repression and Tumorigenesis. Cancer Cell 2018, 34, 579–595.e8. [Google Scholar] [CrossRef] [Green Version]
- Mabe, N.W.; Garcia, N.M.G.; Wolery, S.E.; Newcomb, R.; Meingasner, R.C.; Vilona, B.A.; Lupo, R.; Lin, C.-C.; Chi, J.-T.; Alvarez, J.V. G9a Promotes Breast Cancer Recurrence through Repression of a Pro-Inflammatory Program. Cell Rep. 2020, 33, 108341. [Google Scholar] [CrossRef]
- Kim, T.W.; Cheon, C.; Ko, S.-G. SH003 Activates Autophagic Cell Death by Activating ATF4 and Inhibiting G9a under Hypoxia in Gastric Cancer Cells. Cell Death Dis. 2020, 11, 717. [Google Scholar] [CrossRef]
- Chang, C.-C.; Wu, M.-J.; Yang, J.-Y.; Camarillo, I.G.; Chang, C.-J. Leptin–STAT3–G9a Signaling Promotes Obesity-Mediated Breast Cancer Progression. Cancer Res. 2015, 75, 2375–2386. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.K.; Cho, S.-G.; Woo, S.-M.; Yun, Y.J.; Park, S.; Shin, Y.C.; Ko, S.-G. Herbal Extract SH003 Suppresses Tumor Growth and Metastasis of MDA-MB-231 Breast Cancer Cells by Inhibiting STAT3-IL-6 Signaling. Mediat. Inflamm. 2014, 2014, 492173. [Google Scholar] [CrossRef] [Green Version]
- Song, H.; Wang, R.; Wang, S.; Lin, J. A Low-Molecular-Weight Compound Discovered through Virtual Database Screening Inhibits Stat3 Function in Breast Cancer Cells. Proc. Natl. Acad. Sci. USA 2005, 102, 4700–4705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, J.; Cheng, X.-D.; Zhang, W.-D.; Qin, J.-J. Recent Update on Development of Small-Molecule STAT3 Inhibitors for Cancer Therapy: From Phosphorylation Inhibition to Protein Degradation. J. Med. Chem. 2021, 64, 8884–8915. [Google Scholar] [CrossRef]
- Zhao, M.; Jiang, B.; Gao, F.-H. Small Molecule Inhibitors of STAT3 for Cancer Therapy. Curr. Med. Chem. 2011, 18, 4012–4018. [Google Scholar] [CrossRef]
- Chae, Y.-C.; Kim, J.-Y.; Park, J.W.; Kim, K.-B.; Oh, H.; Lee, K.-H.; Seo, S.-B. FOXO1 Degradation via G9a-Mediated Methylation Promotes Cell Proliferation in Colon Cancer. Nucleic Acids Res. 2019, 47, 1692–1705. [Google Scholar] [CrossRef] [Green Version]
- Koga, H. Troglitazone Induces P27Kip1-Associated Cell-Cycle Arrest through down-Regulating Skp2 in Human Hepatoma Cells. Hepatology 2003, 37, 1086–1096. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.-D.; Kao, S.-H.; Ou, T.-T.; Chen, Y.-J.; Li, Y.-J.; Wang, C.-J. Gallic Acid Induces G2/M Phase Arrest of Breast Cancer Cell MCF-7 through Stabilization of P27 Kip1 Attributed to Disruption of P27 Kip1/Skp2 Complex. J. Agric. Food Chem. 2011, 59, 1996–2003. [Google Scholar] [CrossRef] [PubMed]
- Ungermannova, D.; Lee, J.; Zhang, G.; Dallmann, H.G.; McHenry, C.S.; Liu, X. High-Throughput Screening AlphaScreen Assay for Identification of Small-Molecule Inhibitors of Ubiquitin E3 Ligase SCFSkp2-Cks1. J. Biomol. Screen. 2013, 18, 910–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.; Sran, A.; Carroll, D.C.; Huang, J.; Tsvetkov, L.; Zhou, X.; Sheung, J.; McLaughlin, J.; Issakani, S.D.; Payan, D.G.; et al. Developing Structure–Activity Relationships from an HTS Hit for Inhibition of the Cks1–Skp2 Protein–Protein Interaction. Bioorg. Med. Chem. Lett. 2015, 25, 5199–5202. [Google Scholar] [CrossRef]
- Ooi, L.-C.; Watanabe, N.; Futamura, Y.; Sulaiman, S.F.; Darah, I.; Osada, H. Identification of Small Molecule Inhibitors of P27Kip1 Ubiquitination by High-Throughput Screening. Cancer Sci. 2013, 104, 1461–1467. [Google Scholar] [CrossRef]
- Lee, S.H.; Hyeon, D.Y.; Yoon, S.-H.; Jeong, J.-H.; Han, S.-M.; Jang, J.-W.; Nguyen, M.P.; Chi, X.-Z.; An, S.; Hyun, K.; et al. RUNX3 Methylation Drives Hypoxia-Induced Cell Proliferation and Antiapoptosis in Early Tumorigenesis. Cell Death Differ. 2021, 28, 1251–1269. [Google Scholar] [CrossRef]
- Purcell, D.J.; Khalid, O.; Ou, C.-Y.; Little, G.H.; Frenkel, B.; Baniwal, S.K.; Stallcup, M.R. Recruitment of Coregulator G9a by Runx2 for Selective Enhancement or Suppression of Transcription. J. Cell. Biochem. 2012, 113, 2406–2414. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Miao, Z.; Wang, Z.; Zhao, T.; Xu, Y.; Song, Y.; Huang, J.; Zhang, J.; Xu, H.; Wu, J.; et al. TBX2 Overexpression Promotes Proliferation and Invasion through Epithelial-mesenchymal Transition and ERK Signaling Pathway. Exp. Ther. Med. 2018, 17, 723–729. [Google Scholar] [CrossRef] [Green Version]
- Dutta, A.; Le Magnen, C.; Mitrofanova, A.; Ouyang, X.; Califano, A.; Abate-Shen, C. Identification of an NKX3.1-G9a-UTY Transcriptional Regulatory Network That Controls Prostate Differentiation. Science 2016, 352, 1576–1580. [Google Scholar] [CrossRef] [Green Version]
- Dong, C.; Wu, Y.; Yao, J.; Wang, Y.; Yu, Y.; Rychahou, P.G.; Evers, B.M.; Zhou, B.P. G9a Interacts with Snail and Is Critical for Snail-Mediated E-Cadherin Repression in Human Breast Cancer. J. Clin. Investig. 2012, 122, 1469–1486. [Google Scholar] [CrossRef] [Green Version]
- Yingling, J.M.; McMillen, W.T.; Yan, L.; Huang, H.; Sawyer, J.S.; Graff, J.; Clawson, D.K.; Britt, K.S.; Anderson, B.D.; Beight, D.W.; et al. Preclinical Assessment of Galunisertib (LY2157299 Monohydrate), a First-in-Class Transforming Growth Factor-β Receptor Type I Inhibitor. Oncotarget 2018, 9, 6659–6677. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.-G.; Malek, E.; Choi, S.H.; Ignatz-Hoover, J.J.; Driscoll, J.J. Novel Therapies Emerging in Oncology to Target the TGF-β Pathway. J. Hematol. Oncol. 2021, 14, 55. [Google Scholar] [CrossRef]
- Hau, P.; Jachimczak, P.; Schlingensiepen, R.; Schulmeyer, F.; Jauch, T.; Steinbrecher, A.; Brawanski, A.; Proescholdt, M.; Schlaier, J.; Buchroithner, J.; et al. Inhibition of TGF-Beta2 with AP 12009 in Recurrent Malignant Gliomas: From Preclinical to Phase I/II Studies. Oligonucleotides 2007, 17, 201–212. [Google Scholar] [CrossRef]
- Hu, Y.; Zheng, Y.; Dai, M.; Wang, X.; Wu, J.; Yu, B.; Zhang, H.; Cui, Y.; Kong, W.; Wu, H.; et al. G9a and Histone Deacetylases Are Crucial for Snail2-mediated E-cadherin Repression and Metastasis in Hepatocellular Carcinoma. Cancer Sci. 2019, 110, 3442–3452. [Google Scholar] [CrossRef] [Green Version]
- Pfeiffer, N.; Voykov, B.; Renieri, G.; Bell, K.; Richter, P.; Weigel, M.; Thieme, H.; Wilhelm, B.; Lorenz, K.; Feindor, M.; et al. First-in-Human Phase I Study of ISTH0036, an Antisense Oligonucleotide Selectively Targeting Transforming Growth Factor Beta 2 (TGF-Β2), in Subjects with Open-Angle Glaucoma Undergoing Glaucoma Filtration Surgery. PLoS ONE 2017, 12, e0188899. [Google Scholar] [CrossRef] [Green Version]
- Gimeno-Valiente, F.; Riffo-Campos, Á.L.; Torres, L.; Tarazona, N.; Gambardella, V.; Cervantes, A.; López-Rodas, G.; Franco, L.; Castillo, J. Epigenetic Mechanisms Are Involved in the Oncogenic Properties of ZNF518B in Colorectal Cancer. Cancers 2021, 13, 1433. [Google Scholar] [CrossRef]
- Li, Z.; Jiao, X.; Di Sante, G.; Ertel, A.; Casimiro, M.C.; Wang, M.; Katiyar, S.; Ju, X.; Klopfenstein, D.V.; Tozeren, A.; et al. Cyclin D1 Integrates G9a-Mediated Histone Methylation. Oncogene 2019, 38, 4232–4249. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Zhu, Q.; Lu, X.; Du, Y.; Cao, L.; Shen, C.; Hou, T.; Li, M.; Li, Z.; Liu, C.; et al. G9a Coordinates with the RPA Complex to Promote DNA Damage Repair and Cell Survival. Proc. Natl. Acad. Sci. USA 2017, 114, E6054–E6063. [Google Scholar] [CrossRef] [Green Version]
- Si, M.; Lang, J. The Roles of Metallothioneins in Carcinogenesis. J. Hematol. Oncol. 2018, 11, 107. [Google Scholar] [CrossRef]
- Curry, E.; Green, I.; Chapman-Rothe, N.; Shamsaei, E.; Kandil, S.; Cherblanc, F.L.; Payne, L.; Bell, E.; Ganesh, T.; Srimongkolpithak, N.; et al. Dual EZH2 and EHMT2 Histone Methyltransferase Inhibition Increases Biological Efficacy in Breast Cancer Cells. Clin. Epigenetics 2015, 7, 84. [Google Scholar] [CrossRef] [Green Version]
- Ishiguro, K.; Kitajima, H.; Niinuma, T.; Maruyama, R.; Nishiyama, N.; Ohtani, H.; Sudo, G.; Toyota, M.; Sasaki, H.; Yamamoto, E.; et al. Dual EZH2 and G9a Inhibition Suppresses Multiple Myeloma Cell Proliferation by Regulating the Interferon Signal and IRF4-MYC Axis. Cell Death Discov. 2021, 7, 7. [Google Scholar] [CrossRef]
- Alao, J.P.; Lam, E.W.-F.; Ali, S.; Buluwela, L.; Bordogna, W.; Lockey, P.; Varshochi, R.; Stavropoulou, A.V.; Coombes, R.C.; Vigushin, D.M. Histone Deacetylase Inhibitor Trichostatin a Represses Estrogen Receptor α-Dependent Transcription and Promotes Proteasomal Degradation of Cyclin D1 in Human Breast Carcinoma Cell Lines. Clin. Cancer Res. 2004, 10, 8094–8104. [Google Scholar] [CrossRef] [Green Version]
- San José-Enériz, E.; Agirre, X.; Rabal, O.; Vilas-Zornoza, A.; Sanchez-Arias, J.A.; Miranda, E.; Ugarte, A.; Roa, S.; Paiva, B.; Estella-Hermoso de Mendoza, A.; et al. Discovery of First-in-Class Reversible Dual Small Molecule Inhibitors against G9a and DNMTs in Hematological Malignancies. Nat. Commun. 2017, 8, 15424. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Liu, S.; Yuan, S.; Yu, H.; Zhang, Y.; Yang, X.; Xie, G.; Chen, Z.; Li, W.; Xu, B.; et al. Chromodomain Protein CDYL Is Required for Transmission/Restoration of Repressive Histone Marks. J. Mol. Cell Biol. 2017, 9, 178–194. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Zhang, H.; Wang, P.; Mao, Z.; Feng, L.; Wang, Y.; Liu, C.; Xia, Q.; Li, B.; Zhao, H.; et al. Short-Form CDYLb but Not Long-Form CDYLa Functions Cooperatively with Histone Methyltransferase G9a in Hepatocellular Carcinomas. Genes Chromosomes Cancer 2013, 52, 644–655. [Google Scholar] [CrossRef]
- Yang, L.; Liu, Y.; Fan, M.; Zhu, G.; Jin, H.; Liang, J.; Liu, Z.; Huang, Z.; Zhang, L. Identification and Characterization of Benzo[d]Oxazol-2(3H)-One Derivatives as the First Potent and Selective Small-Molecule Inhibitors of Chromodomain Protein CDYL. Eur. J. Med. Chem. 2019, 182, 111656. [Google Scholar] [CrossRef]
- Wang, S.; Wu, G.; Han, Y.; Song, P.; Chen, J.; Wu, Y.; Yang, J.; Liang, P. MiR-124 Regulates STAT3-mediated Cell Proliferation, Migration and Apoptosis in Bladder Cancer. Oncol. Lett. 2018, 16, 5875–5881. [Google Scholar] [CrossRef] [Green Version]
- Song, W.; Gu, Y.; Lu, S.; Wu, H.; Cheng, Z.; Hu, J.; Qian, Y.; Zheng, Y.; Fan, H. LncRNA TRERNA1 Facilitates Hepatocellular Carcinoma Metastasis by Dimethylating H3K9 in the CDH1 Promoter Region via the Recruitment of the EHMT2/SNAI1 Complex. Cell Prolif. 2019, 52, e12621. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Cheng, C. Long Noncoding RNA NEAT1 Promotes the Metastasis of Osteosarcoma via Interaction with the G9a-DNMT1-Snail Complex. Am. J. Cancer Res. 2018, 8, 81. [Google Scholar]
- Li, Q.; Dong, C.; Cui, J.; Wang, Y.; Hong, X. Over-Expressed LncRNA HOTAIRM1 Promotes Tumor Growth and Invasion through up-Regulating HOXA1 and Sequestering G9a/EZH2/Dnmts Away from the HOXA1 Gene in Glioblastoma Multiforme. J. Exp. Clin. Cancer Res. CR 2018, 37, 265. [Google Scholar] [CrossRef] [Green Version]
- West, L.E.; Gozani, O. Regulation of P53 Function by Lysine Methylation. Epigenomics 2011, 3, 361–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Dorsey, J.; Chuikov, S.; Zhang, X.; Jenuwein, T.; Reinberg, D.; Berger, S.L. G9a and Glp Methylate Lysine 373 in the Tumor Suppressor P53. J. Biol. Chem. 2010, 285, 9636–9641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakatsuka, T.; Tateishi, K.; Kato, H.; Fujiwara, H.; Yamamoto, K.; Kudo, Y.; Nakagawa, H.; Tanaka, Y.; Ijichi, H.; Ikenoue, T.; et al. Inhibition of Histone Methyltransferase G9a Attenuates Liver Cancer Initiation by Sensitizing DNA-Damaged Hepatocytes to P53-Induced Apoptosis. Cell Death Dis. 2021, 12, 99. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V. C-Myc Target Genes Involved in Cell Growth, Apoptosis, and Metabolism. Mol. Cell. Biol. 1999, 19, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ke, X.X.; Zhang, R.; Zhong, X.; Zhang, L.; Cui, H. Deficiency of G9a Inhibits Cell Proliferation and Activates Autophagy via Transcriptionally Regulating C-Myc Expression in Glioblastoma. Front. Cell Dev. Biol. 2020, 8, 593964. [Google Scholar] [CrossRef]
- Siveen, K.S.; Sikka, S.; Surana, R.; Dai, X.; Zhang, J.; Kumar, A.P.; Tan, B.K.H.; Sethi, G.; Bishayee, A. Targeting the STAT3 Signaling Pathway in Cancer: Role of Synthetic and Natural Inhibitors. Biochim. Biophys. Acta Rev. Cancer 2014, 1845, 136–154. [Google Scholar] [CrossRef] [Green Version]
- Ivanov, V.N.; Bhoumik, A.; Krasilnikov, M.; Raz, R.; Owen-Schaub, L.B.; Levy, D.; Horvath, C.M.; Ronai, Z. Cooperation between STAT3 and C-Jun Suppresses Fas Transcription. Mol. Cell 2001, 7, 517–528. [Google Scholar] [CrossRef] [Green Version]
- Tzivion, G.; Dobson, M.; Ramakrishnan, G. FoxO Transcription Factors; Regulation by AKT and 14-3-3 Proteins. Biochim. Biophys. Acta BBA Mol. Cell Res. 2011, 1813, 1938–1945. [Google Scholar] [CrossRef] [Green Version]
- Van der Heide, L.P.; Hoekman, M.F.M.; Smidt, M.P. The Ins and Outs of FoxO Shuttling: Mechanisms of FoxO Translocation and Transcriptional Regulation. Biochem. J. 2004, 380, 297–309. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Liu, X.; Bai, J.; Pei, D.; Zheng, J. The Emerging Role of RUNX3 in Cancer Metastasis (Review). Oncol. Rep. 2016, 35, 1227–1236. [Google Scholar] [CrossRef] [Green Version]
- Crawford, N.T.; McIntyre, A.J.; McCormick, A.; D’Costa, Z.C.; Buckley, N.E.; Mullan, P.B. TBX2 Interacts with Heterochromatin Protein 1 to Recruit a Novel Repression Complex to EGR1-Targeted Promoters to Drive the Proliferation of Breast Cancer Cells. Oncogene 2019, 38, 5971–5986. [Google Scholar] [CrossRef] [Green Version]
- Zhu, B.; Zhang, M.; Williams, E.M.; Keller, C.; Mansoor, A.; Davie, J.K. TBX2 Represses PTEN in Rhabdomyosarcoma and Skeletal Muscle. Oncogene 2016, 35, 4212–4224. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.-F.; Hu, Y.; Yang, C.-C.; Xu, X.-H.; Ning, T.-Y.; Wang, Z.-L.; Ye, J.-H.; Liu, L.-K. Snail Overexpression Induces an Epithelial to Mesenchymal Transition and Cancer Stem Cell-like Properties in SCC9 Cells. Lab. Investig. 2012, 92, 744–752. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Lv, R.; Qi, W.; Wu, D.; Xu, Y.; Liu, W.; Mou, Y.; Wang, L. Snail Contributes to the Maintenance of Stem Cell-Like Phenotype Cells in Human Pancreatic Cancer. PLoS ONE 2014, 9, e87409. [Google Scholar] [CrossRef]
- Wang, Y.; Shi, J.; Chai, K.; Ying, X.; Zhou, B.P. The Role of Snail in EMT and Tumorigenesis. Curr. Cancer Drug Targets 2013, 13, 963–972. [Google Scholar] [CrossRef]
- Dong, C.; Yuan, T.; Wu, Y.; Wang, Y.; Fan, T.W.M.; Miriyala, S.; Lin, Y.; Yao, J.; Shi, J.; Kang, T.; et al. Loss of FBP1 by Snail-Mediated Repression Provides Metabolic Advantages in Basal-like Breast Cancer. Cancer Cell 2013, 23, 316–331. [Google Scholar] [CrossRef] [Green Version]
- Walsh, H.R.; Cruickshank, B.M.; Brown, J.M.; Marcato, P. The Flick of a Switch: Conferring Survival Advantage to Breast Cancer Stem Cells Through Metabolic Plasticity. Front. Oncol. 2019, 9, 753. [Google Scholar] [CrossRef]
- Medici, D.; Hay, E.D.; Olsen, B.R. Snail and Slug Promote Epithelial-Mesenchymal Transition through -Catenin–T-Cell Factor-4-Dependent Expression of Transforming Growth Factor-3. Mol. Biol. Cell 2008, 19, 13. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Zheng, Y.; Dai, M.; Wu, J.; Yu, B.; Zhang, H.; Kong, W.; Wu, H.; Yu, X. Snail2 Induced E-Cadherin Suppression and Metastasis in Lung Carcinoma Facilitated by G9a and HDACs. Cell Adhes. Migr. 2019, 13, 284–291. [Google Scholar] [CrossRef] [Green Version]
- Bian, C.; Chen, Q.; Yu, X. The Zinc Finger Proteins ZNF644 and WIZ Regulate the G9a/GLP Complex for Gene Repression. eLife 2015, 4, e05606. [Google Scholar] [CrossRef] [Green Version]
- Mayer, D.; Stadler, M.B.; Rittirsch, M.; Hess, D.; Lukonin, I.; Winzi, M.; Smith, A.; Buchholz, F.; Betschinger, J. Zfp281 Orchestrates Interconversion of Pluripotent States by Engaging Ehmt1 and Zic2. EMBO J. 2020, 39, e102591. [Google Scholar] [CrossRef]
- Topacio, B.R.; Zatulovskiy, E.; Cristea, S.; Xie, S.; Tambo, C.S.; Rubin, S.M.; Sage, J.; Kõivomägi, M.; Skotheim, J.M. Cyclin D-Cdk4,6 Drives Cell-Cycle Progression via the Retinoblastoma Protein’s C-Terminal Helix. Mol. Cell 2019, 74, 758–770.e4. [Google Scholar] [CrossRef]
- Massagué, J. TGFβ in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [Green Version]
- Van Steensel, B.; Belmont, A.S. Lamina-Associated Domains: Links with Chromosome Architecture, Heterochromatin and Gene Repression. Cell 2017, 169, 780–791. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Guan, Y.; Chen, X.; Yang, J.; Cheng, Y. DNA Repair Pathways in Cancer Therapy and Resistance. Front. Pharmacol. 2021, 11, 629266. [Google Scholar] [CrossRef]
- Zou, Y.; Liu, Y.; Wu, X.; Shell, S.M. Functions of Human Replication Protein A (RPA): From DNA Replication to DNA Damage and Stress Responses. J. Cell. Physiol. 2006, 208, 267–273. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.-C.; Zheng, Z.-L.; Zuo, Z.-H.; Yu, Y.P.; Chen, R.; Tseng, G.C.; Nelson, J.B.; Luo, J.-H. Metallothionein 1 h Tumour Suppressor Activity in Prostate Cancer Is Mediated by Euchromatin Methyltransferase 1: MT1h Suppresses Prostate Cancer through Activation of EHMT1. J. Pathol. 2013, 230, 184–193. [Google Scholar] [CrossRef]
- Zheng, Y.; Jiang, L.; Hu, Y.; Xiao, C.; Xu, N.; Zhou, J.; Zhou, X. Metallothionein 1H (MT1H) Functions as a Tumor Suppressor in Hepatocellular Carcinoma through Regulating Wnt/β-Catenin Signaling Pathway. BMC Cancer 2017, 17, 161. [Google Scholar] [CrossRef] [Green Version]
- Mozzetta, C.; Pontis, J.; Fritsch, L.; Robin, P.; Portoso, M.; Proux, C.; Margueron, R.; Ait-Si-Ali, S. The Histone H3 Lysine 9 Methyltransferases G9a and GLP Regulate Polycomb Repressive Complex 2-Mediated Gene Silencing. Mol. Cell 2014, 53, 277–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritsch, L.; Robin, P.; Mathieu, J.R.R.; Souidi, M.; Hinaux, H.; Rougeulle, C.; Harel-Bellan, A.; Ameyar-Zazoua, M.; Ait-Si-Ali, S. A Subset of the Histone H3 Lysine 9 Methyltransferases Suv39h1, G9a, GLP, and SETDB1 Participate in a Multimeric Complex. Mol. Cell 2010, 37, 46–56. [Google Scholar] [CrossRef] [Green Version]
- Estève, P.-O.; Chin, H.G.; Smallwood, A.; Feehery, G.R.; Gangisetty, O.; Karpf, A.R.; Carey, M.F.; Pradhan, S. Direct Interaction between DNMT1 and G9a Coordinates DNA and Histone Methylation during Replication. Genes Dev. 2006, 20, 3089–3103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boulias, K. Functional Role of G9a-Induced Histone Methylation in Small Heterodimer Partner-Mediated Transcriptional Repression. Nucleic Acids Res. 2004, 32, 6096–6103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulligan, P.; Westbrook, T.F.; Ottinger, M.; Pavlova, N.; Chang, B.; Macia, E.; Shi, Y.-J.; Barretina, J.; Liu, J.; Howley, P.M.; et al. CDYL Bridges REST and Histone Methyltransferases for Gene Repression and Suppression of Cellular Transformation. Mol. Cell 2008, 32, 718–726. [Google Scholar] [CrossRef]
- Caron, C.; Pivot-Pajot, C.; Van Grunsven, L.A.; Col, E.; Lestrat, C.; Rousseaux, S.; Khochbin, S. Cdyl: A New Transcriptional Co-repressor. EMBO Rep. 2003, 4, 877–882. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Z.; Zhu, W.; Meng, H.; Tong, L.; Li, X.; Luo, P.; Yi, L.; Zhang, X.; Guo, L.; Wei, T.; et al. CDYL Promotes the Chemoresistance of Small Cell Lung Cancer by Regulating H3K27 Trimethylation at the CDKN1C Promoter. Theranostics 2019, 9, 4717–4729. [Google Scholar] [CrossRef]
- Siouda, M.; Dujardin, A.D.; Barbollat-Boutrand, L.; Mendoza-Parra, M.A.; Gibert, B.; Ouzounova, M.; Bouaoud, J.; Tonon, L.; Robert, M.; Foy, J.-P.; et al. CDYL2 Epigenetically Regulates MIR124 to Control NF-ΚB/STAT3-Dependent Breast Cancer Cell Plasticity. iScience 2020, 23, 101141. [Google Scholar] [CrossRef]
- Jeong, D.; Kim, J.; Nam, J.; Sun, H.; Lee, Y.-H.; Lee, T.-J.; Aguiar, R.C.T.; Kim, S.-W. MicroRNA-124 Links P53 to the NF-ΚB Pathway in B-Cell Lymphomas. Leukemia 2015, 29, 1868–1874. [Google Scholar] [CrossRef]
- Ribeiro, D.M.; Zanzoni, A.; Cipriano, A.; Delli Ponti, R.; Spinelli, L.; Ballarino, M.; Bozzoni, I.; Tartaglia, G.G.; Brun, C. Protein Complex Scaffolding Predicted as a Prevalent Function of Long Non-Coding RNAs. Nucleic Acids Res. 2018, 46, 917–928. [Google Scholar] [CrossRef]
- Morriss, G.R.; Cooper, T.A. Protein Sequestration as a Normal Function of Long Noncoding RNAs and a Pathogenic Mechanism of RNAs Containing Nucleotide Repeat Expansions. Hum. Genet. 2017, 136, 1247–1263. [Google Scholar] [CrossRef]
- O’Driscoll, M.; Jeggo, P.A. The Role of Double-Strand Break Repair—Insights from Human Genetics. Nat. Rev. Genet. 2006, 7, 45–54. [Google Scholar] [CrossRef]
- Uddin, F.; Rudin, C.M.; Sen, T. CRISPR Gene Therapy: Applications, Limitations, and Implications for the Future. Front. Oncol. 2020, 10, 1387. [Google Scholar] [CrossRef]
- Yang, Y.; Xu, J.; Ge, S.; Lai, L. CRISPR/Cas: Advances, Limitations, and Applications for Precision Cancer Research. Front. Med. 2021, 8, 649896. [Google Scholar] [CrossRef]
- Saraon, P.; Pathmanathan, S.; Snider, J.; Lyakisheva, A.; Wong, V.; Stagljar, I. Receptor Tyrosine Kinases and Cancer: Oncogenic Mechanisms and Therapeutic Approaches. Oncogene 2021, 40, 4079–4093. [Google Scholar] [CrossRef]
- Normanno, N.; De Luca, A.; Bianco, C.; Strizzi, L.; Mancino, M.; Maiello, M.R.; Carotenuto, A.; De Feo, G.; Caponigro, F.; Salomon, D.S. Epidermal Growth Factor Receptor (EGFR) Signaling in Cancer. Gene 2006, 366, 2–16. [Google Scholar] [CrossRef]
- Parag-Sharma, K.; Tasoulas, J.; Musicant, A.M.; Nascimento-Filho, C.H.V.D.; Zhu, Z.; Twomey, C.; Liu, P.; Castilho, R.M.; Amelio, A.L. Synergistic Efficacy of Combined EGFR and HDAC Inhibitors Overcomes Tolerance to EGFR Monotherapy in Salivary Mucoepidermoid Carcinoma. Oral Oncol. 2021, 115, 105166. [Google Scholar] [CrossRef]
- Deutsch, A.J.A.; Angerer, H.; Fuchs, T.E.; Neumeister, P. The Nuclear Orphan Receptors NR4A as Therapeutic Target in Cancer Therapy. Anticancer Agents Med. Chem. 2012, 12, 1001–1014. [Google Scholar] [CrossRef]
- Song, J.; Diao, F.; Ma, X.; Xu, S.; Cui, Y.; Jiang, S.; Liu, J. Androgen Upregulates NR4A1 via the TFAP2A and ETS Signaling Networks. Int. J. Biochem. Cell Biol. 2019, 113, 1–7. [Google Scholar] [CrossRef]
- Yang, X.; Sun, L.; Wang, L.; Yao, B.; Mo, H.; Yang, W. LncRNA SNHG7 Accelerates the Proliferation, Migration and Invasion of Hepatocellular Carcinoma Cells via Regulating MiR-122-5p and RPL4. Biomed. Pharmacother. 2019, 118, 109386. [Google Scholar] [CrossRef]
- Dey, S.K.; Jaffrey, S.R. RIBOTACs: Small Molecules Target RNA for Degradation. Cell Chem. Biol. 2019, 26, 1047–1049. [Google Scholar] [CrossRef]
- Hirota, K. HIF-α Prolyl Hydroxylase Inhibitors and Their Implications for Biomedicine: A Comprehensive Review. Biomedicines 2021, 9, 468. [Google Scholar] [CrossRef]
- Lu, H.; Zhou, Q.; He, J.; Jiang, Z.; Peng, C.; Tong, R.; Shi, J. Recent Advances in the Development of Protein–Protein Interactions Modulators: Mechanisms and Clinical Trials. Signal Transduct. Target. Ther. 2020, 5, 213. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Kumar, S.; Zhang, X.; Qin, D.; Kang, S.; King, K.; Qiu, S.; Liu, M.; Nikolovska-Coleska, Z.; McEachern, D.; et al. Preclinical Characterization of MI-219, A Novel, Potent and Orally Active Small Molecule Inhibitor of the MDM2-P53 Interaction. Cancer Res. 2007, 67, LB-365. [Google Scholar]
- Whitfield, J.R.; Beaulieu, M.-E.; Soucek, L. Strategies to Inhibit Myc and Their Clinical Applicability. Front. Cell Dev. Biol. 2017, 5, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, H.; Jain, A.D.; Truica, M.I.; Izquierdo-Ferrer, J.; Anker, J.F.; Lysy, B.; Sagar, V.; Luan, Y.; Chalmers, Z.R.; Unno, K.; et al. Small-Molecule MYC Inhibitors Suppress Tumor Growth and Enhance Immunotherapy. Cancer Cell 2019, 36, 483–497.e15. [Google Scholar] [CrossRef]
- Hart, J.R.; Garner, A.L.; Yu, J.; Ito, Y.; Sun, M.; Ueno, L.; Rhee, J.-K.; Baksh, M.M.; Stefan, E.; Hartl, M.; et al. Inhibitor of MYC Identified in a Kröhnke Pyridine Library. Proc. Natl. Acad. Sci. USA 2014, 111, 12556–12561. [Google Scholar] [CrossRef] [Green Version]
- Demma, M.J.; Mapelli, C.; Sun, A.; Bodea, S.; Ruprecht, B.; Javaid, S.; Wiswell, D.; Muise, E.; Chen, S.; Zelina, J.; et al. Omomyc Reveals New Mechanisms to Inhibit the MYC Oncogene. Mol. Cell. Biol. 2019, 39, e00248-19. [Google Scholar] [CrossRef] [Green Version]
- Ali, A.; Shafarin, J.; Unnikannan, H.; Al-Jabi, N.; Jabal, R.A.; Bajbouj, K.; Muhammad, J.S.; Hamad, M. Co-Targeting BET Bromodomain BRD4 and RAC1 Suppresses Growth, Stemness and Tumorigenesis by Disrupting the c-MYC-G9a-FTH1axis and Downregulation of HDAC1 in Molecular Subtypes of Breast Cancer. Int. J. Biol. Sci. 2021, 17, 4474–4492. [Google Scholar] [CrossRef]
- Zhang, H.-F.; Lai, R. STAT3 in Cancer—Friend or Foe? Cancers 2014, 6, 1408–1440. [Google Scholar] [CrossRef] [Green Version]
- Seo, H.-S.; Ku, J.M.; Lee, H.-J.; Woo, J.-K.; Cheon, C.; Kim, M.; Jang, B.-H.; Shin, Y.C.; Ko, S.-G. SH003 Reverses Drug Resistance by Blocking Signal Transducer and Activator of Transcription 3 (STAT3) Signaling in Breast Cancer Cells. Biosci. Rep. 2017, 37, BSR20170125. [Google Scholar] [CrossRef] [Green Version]
- Cai, Z.; Moten, A.; Peng, D.; Hsu, C.-C.; Pan, B.-S.; Manne, R.; Li, H.; Lin, H.-K. The Skp2 Pathway: A Critical Target for Cancer Therapy. Semin. Cancer Biol. 2020, 67, 16–33. [Google Scholar] [CrossRef]
- Chen, Q.; Xie, W.; Kuhn, D.J.; Voorhees, P.M.; Lopez-Girona, A.; Mendy, D.; Corral, L.G.; Krenitsky, V.P.; Xu, W.; Moutouh-de Parseval, L.; et al. Targeting the P27 E3 Ligase SCFSkp2 Results in P27- and Skp2-Mediated Cell-Cycle Arrest and Activation of Autophagy. Blood 2008, 111, 4690–4699. [Google Scholar] [CrossRef] [Green Version]
- Meng, J.; Ding, Y.; Shen, A.; Yan, M.; He, F.; Ji, H.; Zou, L.; Liu, Y.; Wang, Y.; Lu, X.; et al. Overexpression of PPARγ Can Down-Regulate Skp2 Expression in MDA-MB-231 Breast Tumor Cells. Mol. Cell. Biochem. 2010, 345, 171–180. [Google Scholar] [CrossRef]
- Chan, C.-H.; Morrow, J.K.; Li, C.-F.; Gao, Y.; Jin, G.; Moten, A.; Stagg, L.J.; Ladbury, J.E.; Cai, Z.; Xu, D.; et al. Pharmacological Inactivation of Skp2 SCF Ubiquitin Ligase Restricts Cancer Stem Cell Traits and Cancer Progression. Cell 2013, 154, 556–568. [Google Scholar] [CrossRef] [Green Version]
- Pavlides, S.C.; Huang, K.-T.; Reid, D.A.; Wu, L.; Blank, S.V.; Mittal, K.; Guo, L.; Rothenberg, E.; Rueda, B.; Cardozo, T.; et al. Inhibitors of SCF-Skp2/Cks1 E3 Ligase Block Estrogen-Induced Growth Stimulation and Degradation of Nuclear P27kip1: Therapeutic Potential for Endometrial Cancer. Endocrinology 2013, 154, 4030–4045. [Google Scholar] [CrossRef]
- Harney, A.S.; Lee, J.; Manus, L.M.; Wang, P.; Ballweg, D.M.; LaBonne, C.; Meade, T.J. Targeted Inhibition of Snail Family Zinc Finger Transcription Factors by Oligonucleotide-Co(III) Schiff Base Conjugate. Proc. Natl. Acad. Sci. USA 2009, 106, 13667–13672. [Google Scholar] [CrossRef] [Green Version]
- Nagaraj, N.S.; Datta, P.K. Targeting the Transforming Growth Factor-β Signaling Pathway in Human Cancer. Expert Opin. Investig. Drugs 2010, 19, 77–91. [Google Scholar] [CrossRef] [Green Version]
- Brandl, M.; Seidler, B.; Haller, F.; Adamski, J.; Schmid, R.M.; Saur, D.; Schneider, G. IKKα Controls Canonical TGFβ–SMAD Signaling to Regulate Genes Expressing SNAIL and SLUG during EMT in Panc1 Cells. J. Cell Sci. 2013, 126, 2747. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.-Y.; Chung, C.-L.; Hu, T.-H.; Chen, J.-J.; Liu, P.-F.; Chen, C.-L. Recent Progress in TGF-β Inhibitors for Cancer Therapy. Biomed. Pharmacother. 2021, 134, 111046. [Google Scholar] [CrossRef]
- Morris, J.C.; Tan, A.R.; Olencki, T.E.; Shapiro, G.I.; Dezube, B.J.; Reiss, M.; Hsu, F.J.; Berzofsky, J.A.; Lawrence, D.P. Phase I Study of GC1008 (Fresolimumab): A Human Anti-Transforming Growth Factor-Beta (TGFβ) Monoclonal Antibody in Patients with Advanced Malignant Melanoma or Renal Cell Carcinoma. PLoS ONE 2014, 9, e90353. [Google Scholar] [CrossRef]
- Langenfeld, J.; Kiyokawa, H.; Sekula, D.; Boyle, J.; Dmitrovsky, E. Posttranslational Regulation of Cyclin D1 by Retinoic Acid: A Chemoprevention Mechanism. Proc. Natl. Acad. Sci. USA 1997, 94, 12070–12074. [Google Scholar] [CrossRef] [Green Version]
- Mao, X.; Cao, B.; Wood, T.E.; Hurren, R.; Tong, J.; Wang, X.; Wang, W.; Li, J.; Jin, Y.; Sun, W.; et al. A Small-Molecule Inhibitor of D-Cyclin Transactivation Displays Preclinical Efficacy in Myeloma and Leukemia via Phosphoinositide 3-Kinase Pathway. Blood 2011, 117, 1986–1997. [Google Scholar] [CrossRef]
- Mao, X.; Stewart, A.K.; Hurren, R.; Datti, A.; Zhu, X.; Zhu, Y.; Shi, C.; Lee, K.; Tiedemann, R.; Eberhard, Y.; et al. A Chemical Biology Screen Identifies Glucocorticoids That Regulate C-Maf Expression by Increasing Its Proteasomal Degradation through up-Regulation of Ubiquitin. Blood 2007, 110, 4047–4054. [Google Scholar] [CrossRef] [Green Version]
- Hurt, E.M.; Wiestner, A.; Rosenwald, A.; Shaffer, A.L.; Campo, E.; Grogan, T.; Bergsagel, P.L.; Kuehl, W.M.; Staudt, L.M. Overexpression of C-Maf Is a Frequent Oncogenic Event in Multiple Myeloma That Promotes Proliferation and Pathological Interactions with Bone Marrow Stroma. Cancer Cell 2004, 5, 191–199. [Google Scholar] [CrossRef] [Green Version]
- Tiedemann, R.E.; Mao, X.; Shi, C.-X.; Zhu, Y.X.; Palmer, S.E.; Sebag, M.; Marler, R.; Chesi, M.; Fonseca, R.; Bergsagel, P.L.; et al. Identification of Kinetin Riboside as a Repressor of CCND1 and CCND2 with Preclinical Antimyeloma Activity. J. Clin. Investig. 2008, 118, 1750–1764. [Google Scholar] [CrossRef] [Green Version]

| Upstream Regulators | Molecule | Disease | Phenotype | Potential Therapeutics |
|---|---|---|---|---|
| Genetic dysregulation | ||||
| Copy number gains | G9a (6p21) | Melanoma [52] | Proliferation | Gene therapy (yet to be explored) [53,54] |
| HCC [27] | Proliferation and migration | |||
| Gain of function | G9a (Glycine 1069) | Melanoma [52] | Proliferation | Mutant-specific inhibition (yet to be explored) [55,56] |
| CRC [57] | Migration and invasion | |||
| Transcriptional Dysregulation | ||||
| EGFR | G9a | Breast cancer [58] | Proliferation and survival | Lapatinib [59] Neratinib [60] |
| STAT3 | G9a | Breast cancer [58] | Proliferation and survival | BB1608 [58] |
| NR4A1 | G9a | ARMS [61] Breast cancer [61] Lung cancer [61] | Proliferation, Tumorigenesis | CDIM8 [62] Diindolylmethane analogues [62] Lestaurtinib [63] |
| miR-122 | G9a | HCC [27,64] | Reduces invasion and survival | RIBOTACS [65] |
| miR-1 | ||||
| Post-translational dysregulation | ||||
| FIH | G9a/GLP | Ovarian cancer [66] | Reduce migration and dissemination | Carotenoids [67] Ascorbic acid [68] |
| PHD1 | G9a | Breast cancer [69] | Reduce proliferation and metastasis | Tocopherol [67] |
| SPOP | GLP | Prostate cancer [70] | Reduce proliferation and survival | Potential activation by DNMT inhibitors [71,72] PROTAC [73] |
| APC/Ccdh1 | G9a/GLP | Cancers [74] | senescence | PROTAC [73] |
| ATM | G9a | Cancers [75] | DNA repair | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ang, G.C.K.; Gupta, A.; Yap, S.X.L.; Surana, U.; Taneja, R. Potential Therapeutics Targeting Upstream Regulators and Interactors of EHMT1/2. Cancers 2022, 14, 2855. https://doi.org/10.3390/cancers14122855
Ang GCK, Gupta A, Yap SXL, Surana U, Taneja R. Potential Therapeutics Targeting Upstream Regulators and Interactors of EHMT1/2. Cancers. 2022; 14(12):2855. https://doi.org/10.3390/cancers14122855
Chicago/Turabian StyleAng, Gareth Chin Khye, Amogh Gupta, Shirlyn Xue Ling Yap, Uttam Surana, and Reshma Taneja. 2022. "Potential Therapeutics Targeting Upstream Regulators and Interactors of EHMT1/2" Cancers 14, no. 12: 2855. https://doi.org/10.3390/cancers14122855
APA StyleAng, G. C. K., Gupta, A., Yap, S. X. L., Surana, U., & Taneja, R. (2022). Potential Therapeutics Targeting Upstream Regulators and Interactors of EHMT1/2. Cancers, 14(12), 2855. https://doi.org/10.3390/cancers14122855