
COL5A1 Promotes the Progression of Gastric Cancer by Acting as a ceRNA of miR-137-3p to Upregulate FSTL1 Expression

 ,
,
Abstract
:Simple Summary
Abstract

1. Introduction
2. Materials and Methods
2.1. Tumor Data Download and Differential Expression Analysis
2.2. Cox Proportional Hazards Regression Model Based on DEMs
2.3. Prediction of Target Genes of SRDEMs and Functional Enrichment Analysis of Differentially Expressed Target Genes (DETGs)
2.4. Construction and Analysis of PPI Networks with DETGs
2.5. Weighted Gene Co-Expression Network Analysis (WGCNA)
2.6. Cell Culture
2.7. Plasmid Construction
2.8. Transfection
2.9. RNA Extraction and Quantitative Real-Time PCR (qRT-PCR)
2.10. Western Blotting
2.11. Cell Counting Kit-8 (CCK-8) Assay
2.12. Ethynyldeoxyuridine (EdU) Proliferation Assay
2.13. Transwell Migration/Invasion Assay
2.14. Wound Healing Assay
2.15. Dual-Luciferase Reporter Assay
2.16. ESTIMATE and CIBERSORT
2.17. Statistical Analysis
3. Results
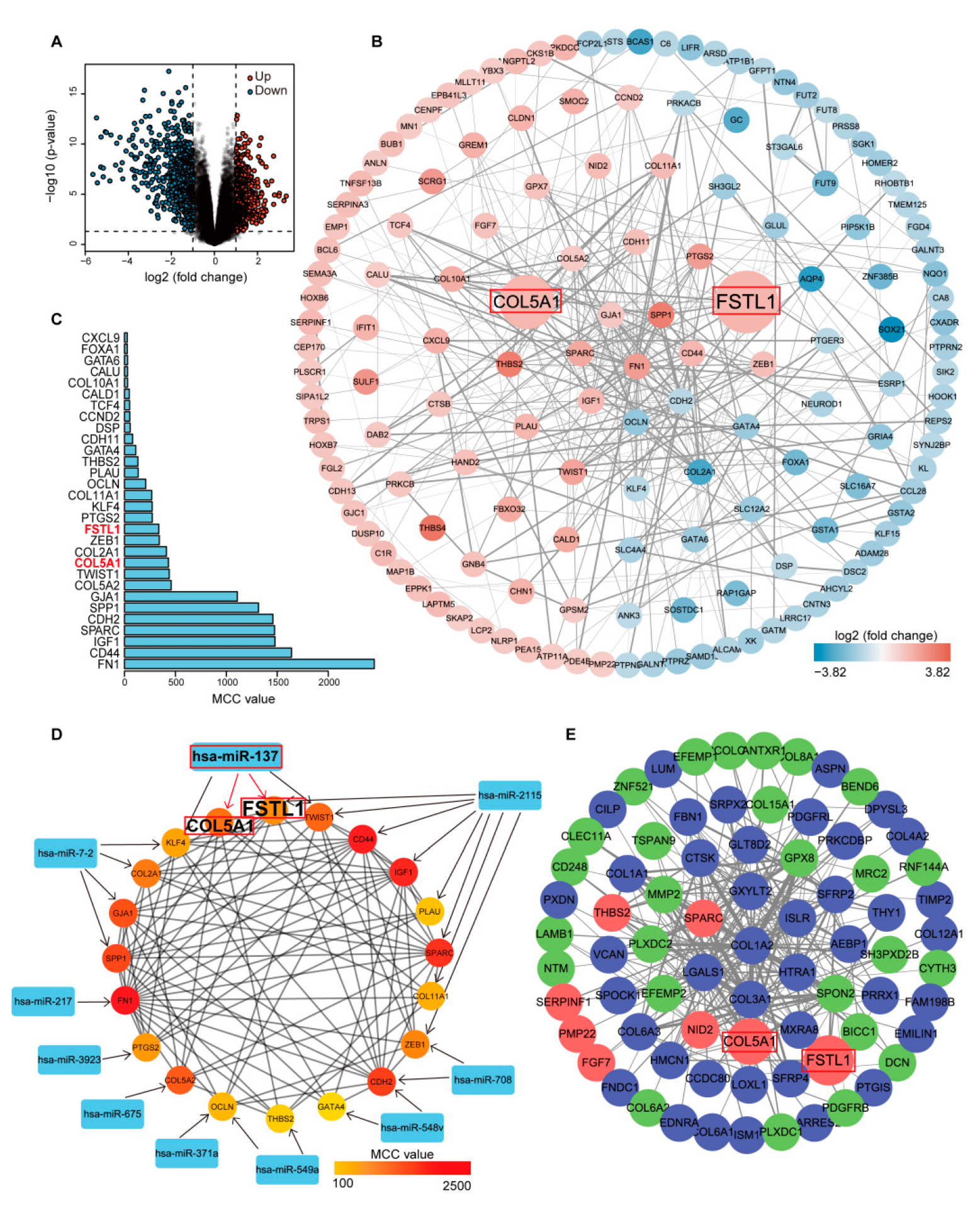
3.1. Cox Proportional Hazards Regression Model of DEMs
3.2. Prediction of Target Genes of SRDEMs and Functional Enrichment Analysis of DETGs
3.3. Identification of Hub Genes in DETGs
3.4. WGCNA Identification of the Hub Module
3.5. MiR-137-3p Suppressed the Proliferation, Invasion, and Migration of GC Cells
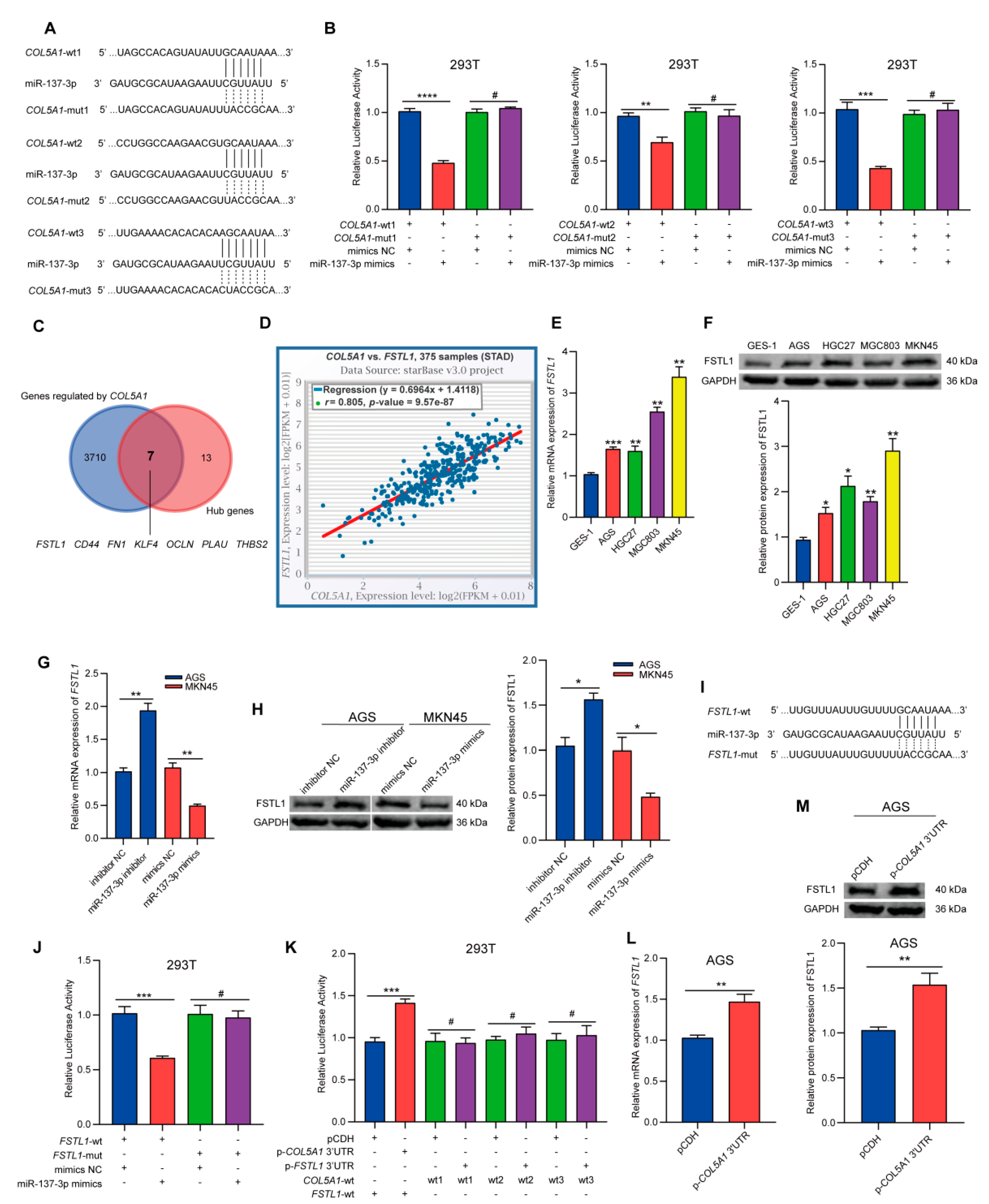
3.6. COL5A1 Can Reversely Sponge miR-137-3p and Upregulate the Expression of FSTL1 through a ceRNA Mechanism
3.7. Knockdown of COL5A1 Inhibited the Proliferation, Migration, and Invasion of GC Cells, Which Can Be Rescued by miR-137-3p Inhibitor or Overexpression of FSTL1
3.8. FSTL1 Was Related to Immune Infiltration in the TME of GC Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Smyth, E.C.; Nilsson, M.; Grabsch, H.I.; van Grieken, N.C.; Lordick, F. Gastric cancer. Lancet 2020, 396, 635–648. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target Recognition and Regulatory Functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arvey, A.; Larsson, E.; Sander, C.; Leslie, C.S.; Marks, D.S. Target mRNA abundance dilutes microRNA and siRNA activity. Mol. Syst. Biol. 2010, 6, 363. [Google Scholar] [CrossRef] [PubMed]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA Hypothesis: The Rosetta Stone of a Hidden RNA Language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.-Z.; Cheng, T.-T.; He, Q.-J.; Lei, Z.-Y.; Chi, J.; Tang, Z.; Liao, Q.-X.; Zhang, H.; Zeng, L.-S.; Cui, S.-Z. LINC01133 as ceRNA inhibits gastric cancer progression by sponging miR-106a-3p to regulate APC expression and the Wnt/β-catenin pathway. Mol. Cancer 2018, 17, 126. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, S.; Wang, H.; Cao, J.; Huang, X.; Chen, Z.; Xu, P.; Sun, G.; Xu, J.; Lv, J.; et al. Circular RNA circNRIP1 acts as a microRNA-149-5p sponge to promote gastric cancer progression via the AKT1/mTOR pathway. Mol. Cancer 2019, 18, 20. [Google Scholar] [CrossRef] [Green Version]
- Shin, V.Y.; Chu, K.M. MiRNA as potential biomarkers and therapeutic targets for gastric cancer. World J. Gastroenterol. 2014, 20, 10432–10439. [Google Scholar] [CrossRef]
- So, J.B.Y.; Kapoor, R.; Zhu, F.; Koh, C.; Zhou, L.; Zou, R.; Tang, Y.C.; Goo, P.C.K.; Rha, S.Y.; Chung, H.C.; et al. Development and validation of a serum microRNA biomarker panel for detecting gastric cancer in a high-risk population. Gut 2021, 70, 829–837. [Google Scholar] [CrossRef]
- Luscombe, N.M.; Greenbaum, D.; Gerstein, M. What is Bioinformatics? A Proposed Definition and Overview of the Field. Methods Inf. Med. 2001, 40, 346–358. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing:: Vienna, Austria, 2019; Available online: https://www.R-project.org/ (accessed on 26 August 2020).
- Li, L.; Zhu, Z.; Zhao, Y.; Zhang, Q.; Wu, X.; Miao, B.; Cao, J.; Fei, S. FN1, SPARC, and SERPINE1 are highly expressed and significantly related to a poor prognosis of gastric adenocarcinoma revealed by microarray and bioinformatics. Sci. Rep. 2019, 9, 7827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Therneau T (2022). _A Package for Survival Analysis in R_. R Package Version 3.3-1. Available online: https://CRAN.R-project.org/package=survival (accessed on 27 August 2020).
- Friedman, J.; Hastie, T.; Tibshirani, R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J. Stat. Softw. 2010, 33, 1–22. [Google Scholar] [CrossRef] [Green Version]
- E Ahmed, F.; Vos, P.W.; Holbert, D. Modeling survival in colon cancer: A methodological review. Mol. Cancer 2007, 6, 15. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Gao, C.; Liu, Y.; Wen, Y.; Hong, X.; Huang, Z. Bioinformatics Analysis of Prognostic miRNA Signature and Potential Critical Genes in Colon Cancer. Front. Genet. 2020, 11, 478. [Google Scholar] [CrossRef]
- Heagerty, P.J.; Zheng, Y. Survival Model Predictive Accuracy and ROC Curves. Biometrics 2005, 61, 92–105. [Google Scholar] [CrossRef] [Green Version]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA sequences to function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef]
- Agarwal, V.; Bell, G.W.; Nam, J.-W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, e05005. [Google Scholar] [CrossRef]
- Wong, N.; Wang, X. miRDB: An online resource for microRNA target prediction and functional annotations. Nucleic Acids Res. 2015, 43, D146–D152. [Google Scholar] [CrossRef]
- Hanbo Chen (2022). VennDiagram: Generate High-Resolution Venn and Euler Plots. R package version 1.7.3. Available online: https://CRAN.R-project.org/package=VennDiagram (accessed on 28 August 2020).
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein–protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.-H.; Chen, S.-H.; Wu, H.-H.; Ho, C.-W.; Ko, M.-T.; Lin, C.-Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8 (Suppl. 4), S11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshihara, K.; Shahmoradgoli, M.; Martínez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Trevino, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, D.; Ye, Z.; Shen, R.; Yu, G.; Wu, J.; Xiong, Y.; Zhou, R.; Qiu, W.; Huang, N.; Sun, L.; et al. IOBR: Multi-Omics Immuno-Oncology Biological Research to Decode Tumor Microenvironment and Signatures. Front. Immunol. 2021, 12, 687975. [Google Scholar] [CrossRef]
- Bosson, A.D.; Zamudio, J.R.; Sharp, P.A. Endogenous miRNA and Target Concentrations Determine Susceptibility to Potential ceRNA Competition. Mol. Cell 2014, 56, 347–359. [Google Scholar] [CrossRef] [Green Version]
- Denzler, R.; McGeary, S.E.; Title, A.C.; Agarwal, V.; Bartel, D.P.; Stoffel, M. Impact of MicroRNA Levels, Target-Site Complementarity, and Cooperativity on Competing Endogenous RNA-Regulated Gene Expression. Mol. Cell 2016, 64, 565–579. [Google Scholar] [CrossRef] [Green Version]
- Li, J.-H.; Liu, S.; Zhou, H.; Qu, L.-H.; Yang, J.-H. starBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein–RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014, 42, D92–D97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Z.; Chen, L.; Meng, L.; Han, W.; Huang, L.; Xu, A. LncRNA HOTAIR promotes the growth and metastasis of gastric cancer by sponging miR-1277-5p and upregulating COL5A1. Gastric Cancer 2020, 23, 1018–1032. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Lin, J.; Luo, X.; Chen, Y.; Li, Z.; Ma, G.; Li, K. miR-137: A New Player in Schizophrenia. Int. J. Mol. Sci. 2014, 15, 3262–3271. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, R.; Zhou, X.; Guo, R.; Yin, J.; Li, Y.; Ma, G. miR-137: A Novel Therapeutic Target for Human Glioma. Mol. Ther. Nucleic Acids 2020, 21, 614–622. [Google Scholar] [CrossRef]
- Ding, F.; Zhang, S.; Gao, S.; Shang, J.; Li, Y.; Cui, N.; Zhao, Q. MiR-137 functions as a tumor suppressor in pancreatic cancer by targeting MRGBP. J. Cell. Biochem. 2018, 119, 4799–4807. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Wu, L.; Zhang, K.; Wang, H.; Zhang, T.; Gutierrez, L.; O’Connell, D.; Zhang, P.; Li, Y.; Gao, T.; et al. miR-137 regulates ferroptosis by targeting glutamine transporter SLC1A5 in melanoma. Cell Death Differ. 2018, 25, 1457–1472. [Google Scholar] [CrossRef] [Green Version]
- Gu, Q.; Zhang, J.; Hu, H.; Tan, Y.-E.; Shi, S.; Nian, Y. Clinical Significance of MiR-137 Expression in Patients with Gastric Cancer After Radical Gastrectomy. PLoS ONE 2015, 10, e0142377. [Google Scholar] [CrossRef]
- Cheng, Y.; Li, Y.; Liu, D.; Zhang, R.; Zhang, J. miR-137 effects on gastric carcinogenesis are mediated by targeting Cox-2-activated PI3K/AKT signaling pathway. FEBS Lett. 2014, 588, 3274–3281. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Duan, J.; Shi, W.; Lei, L.; Lv, P. Long Non-Coding RNA NCK1-AS1 Serves an Oncogenic Role in Gastric Cancer by Regulating miR-137/NUP43 Axis. OncoTargets Ther. 2020, 13, 9929–9939. [Google Scholar] [CrossRef]
- Lu, Y.; Li, L.; Li, L.; Wu, G.; Liu, G. Circular RNA circHECTD1 prevents Diosbulbin-B-sensitivity via miR-137/PBX3 axis in gastric cancer. Cancer Cell Int. 2021, 21, 264. [Google Scholar] [CrossRef]
- Ricard-Blum, S. The Collagen Family. Cold Spring Harb. Perspect. Biol. 2011, 3, a004978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Xu, H.; Wang, W.; Li, S.; Li, H.; Li, T.; Zhang, W.; Yu, X.; Liu, L. The role of collagen in cancer: From bench to bedside. J. Transl. Med. 2019, 17, 309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavaco, A.C.M.; Dâmaso, S.; Casimiro, S.; Costa, L. Collagen biology making inroads into prognosis and treatment of cancer progression and metastasis. Cancer Metastasis Rev. 2020, 39, 603–623. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Zhong, S.; Tong, Y.; Liang, Y.; Feng, G.; Zhou, X.; Zhang, Z.; Huang, G. Alteration and prognostic values of collagen gene expression in patients with gastric cancer under different treatments. Pathol. Res. Pr. 2020, 216, 152831. [Google Scholar] [CrossRef]
- Kauppila, S.; Stenbäck, F.; Risteli, J.; Jukkola, A.; Risteli, L. Aberrant type I and type III collagen gene expression in human breast cancer in vivo. J. Pathol. 1998, 186, 262–268. [Google Scholar] [CrossRef]
- Kita, Y.; Mimori, K.; Tanaka, F.; Matsumoto, T.; Haraguchi, N.; Ishikawa, K.; Matsuzaki, S.; Fukuyoshi, Y.; Inoue, H.; Natsugoe, S.; et al. Clinical significance of LAMB3 and COL7A1 mRNA in esophageal squamous cell carcinoma. Eur. J. Surg. Oncol. (EJSO) 2009, 35, 52–58. [Google Scholar] [CrossRef]
- Wu, Y.-H.; Chang, T.-H.; Huang, Y.-F.; Huang, H.-D.; Chou, C.-Y. COL11A1 promotes tumor progression and predicts poor clinical outcome in ovarian cancer. Oncogene 2014, 33, 3432–3440. [Google Scholar] [CrossRef]
- Mattiotti, A.; Prakash, S.; Barnett, P.; van den Hoff, M.J.B. Follistatin-like 1 in development and human diseases. Exp. Cell. Mol. Life Sci. 2018, 75, 2339–2354. [Google Scholar] [CrossRef]
- Chan, Q.K.; Ngan, H.Y.; Ip, P.P.; Liu, V.W.; Xue, W.C.; Cheung, A.N. Tumor suppressor effect of follistatin-like 1 in ovarian and endometrial carcinogenesis—a differential expression and functional analysis. Carcinogenesis 2009, 30, 114–121. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Han, X.; Yu, Y.; Ding, Y.; Ni, C.; Liu, W.; Hou, X.; Li, Z.; Hou, J.; Shen, D.; et al. A genetic polymorphism affects the risk and prognosis of renal cell carcinoma: Association with follistatin-like protein 1 expression. Sci. Rep. 2016, 6, 26689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, M.C.-C.; Ng, K.Y.; Wong, T.L.; Tong, M.; Lee, T.K.; Ming, X.-Y.; Law, S.; Lee, N.P.; Cheung, A.L.; Qin, Y.-R.; et al. FSTL1 Promotes Metastasis and Chemoresistance in Esophageal Squamous Cell Carcinoma through NFκB–BMP Signaling Cross-talk. Cancer Res. 2017, 77, 5886–5899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, X.; Wang, P.; Li, S.; Jiang, Y.; Wu, C. Follistatin-like protein 1 knockdown elicits human gastric cancer cell apoptosis via a STAT6-dependent pathway. Oncol. Rep. 2019, 42, 2806–2813. [Google Scholar] [CrossRef] [PubMed]
- Gu, C.; Wang, X.; Long, T.; Wang, X.; Zhong, Y.; Ma, Y.; Hu, Z.; Li, Z. FSTL1 interacts with VIM and promotes colorectal cancer metastasis via activating the focal adhesion signalling pathway. Cell Death Dis. 2018, 9, 654. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Huang, Y.; Lou, C.; He, Y.; Zhang, Y.; Zhang, Q. FSTL1 enhances chemoresistance and maintains stemness in breast cancer cells via integrin β3/Wnt signaling under miR-137 regulation. Cancer Biol. Ther. 2019, 20, 328–337. [Google Scholar] [CrossRef] [Green Version]
- Kudo-Saito, C.; Fuwa, T.; Murakami, K.; Kawakami, Y. Targeting FSTL1 Prevents Tumor Bone Metastasis and Consequent Immune Dysfunction. Cancer Res. 2013, 73, 6185–6193. [Google Scholar] [CrossRef] [Green Version]
- Kudo-Saito, C.; Ishida, A.; Shouya, Y.; Teramoto, K.; Igarashi, T.; Kon, R.; Saito, K.; Awada, C.; Ogiwara, Y.; Toyoura, M. Blocking the FSTL1-DIP2A Axis Improves Anti-tumor Immunity. Cell Rep. 2018, 24, 1790–1801. [Google Scholar] [CrossRef] [Green Version]
- Aran, D.; Sirota, M.; Butte, A.J. Systematic pan-cancer analysis of tumour purity. Nat. Commun. 2015, 6, 8971. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Cheng, W.; Ren, X.; Wang, Z.; Liu, X.; Li, G.; Han, S.; Jiang, T.; Wu, A. Tumor Purity as an Underlying Key Factor in Glioma. Clin. Cancer Res. 2017, 23, 6279–6291. [Google Scholar] [CrossRef] [Green Version]
- Murray, P.J. Macrophage Polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef]
- Piccolo, V.; Curina, A.; Genua, M.; Ghisletti, S.; Simonatto, M.; Sabò, A.; Amati, B.; Ostuni, R.; Natoli, G. Opposing macrophage polarization programs show extensive epigenomic and transcriptional cross-talk. Nat. Immunol. 2017, 18, 530–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Zhang, Y. Tumor-associated macrophages: From basic research to clinical application. J. Hematol. Oncol. 2017, 10, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarosz-Biej, M.; Kamińska, N.; Matuszczak, S.; Cichoń, T.; Pamuła-Piłat, J.; Czapla, J.; Smolarczyk, R.; Skwarzyńska, D.; Kulik, K.; Szala, S. M1-like macrophages change tumor blood vessels and microenvironment in murine melanoma. PLoS ONE 2018, 13, e0191012. [Google Scholar] [CrossRef] [PubMed]
- Caux, C.; Ramos, R.N.; Prendergast, G.C.; Bendriss-Vermare, N.; Ménétrier-Caux, C. A Milestone Review on How Macrophages Affect Tumor Growth. Cancer Res. 2016, 76, 6439–6442. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Zhang, S.; Wang, Q.; Zhang, X. Tumor-recruited M2 macrophages promote gastric and breast cancer metastasis via M2 macrophage-secreted CHI3L1 protein. J. Hematol. Oncol. 2017, 10, 36. [Google Scholar] [CrossRef] [Green Version]
- Sousa, S.; Brion, R.; Lintunen, M.; Kronqvist, P.; Sandholm, J.; Mönkkönen, J.; Kellokumpu-Lehtinen, P.-L.; Lauttia, S.; Tynninen, O.; Joensuu, H.; et al. Human breast cancer cells educate macrophages toward the M2 activation status. Breast Cancer Res. 2015, 17, 101. [Google Scholar] [CrossRef] [Green Version]
- De Palma, M.; Lewis, C.E. Cancer: Macrophages limit chemotherapy. Nature 2011, 472, 303–304. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRNA | HR | p-Value |
|---|---|---|
| hsa-miR-7-2 | 0.84 (0.75–0.93) | <0.001 |
| hsa-miR-328 | 1.26 (1.09–1.47) | 0.002 |
| hsa-miR-3923 | 1.24 (1.08–1.43) | 0.002 |
| hsa-miR-675 | 1.10 (1.03–1.16) | 0.003 |
| hsa-miR-7-3 | 0.86 (0.78–0.96) | 0.006 |
| hsa-miR-549a | 0.84 (0.74–0.96) | 0.009 |
| hsa-miR-125a | 1.27 (1.06–1.52) | 0.009 |
| hsa-miR-708 | 1.16 (1.03–1.31) | 0.017 |
| hsa-miR-217 | 1.12 (1.02–1.24) | 0.018 |
| hsa-miR-137 | 1.14 (1.02–1.27) | 0.018 |
| hsa-miR-100 | 1.12 (1.02–1.23) | 0.019 |
| hsa-miR-2115 | 0.82 (0.69–0.98) | 0.029 |
| hsa-miR-548v | 1.14 (1.01–1.28) | 0.029 |
| hsa-miR-6511b-1 | 1.20 (1.01–1.42) | 0.034 |
| hsa-miR-187 | 1.07 (1.00–1.13) | 0.035 |
| hsa-miR-145 | 1.10 (1.00–1.20) | 0.043 |
| hsa-miR-371a | 1.11 (1.00–1.24) | 0.045 |
| hsa-miR-216a | 1.11 (1.00–1.24) | 0.049 |
| SRDEM | DETGs |
|---|---|
| hsa-miR-328 | ESRP1, IFIT1, MICALL1, MLLT11, MN1, PRDM16 |
| hsa-miR-549a | ATP11A, C1R, CDH11, EPPK1, FUT9, GLUL, GPD1L, LAPTM5, NIPAL1, OCLN, PPP2R3A, PRELID2, RAI14, SGK1, SH3BGRL2, SOX21, ST3GAL6, SYNJ2BP, TFCP2L1, THBS2 |
| hsa-miR-708 | AADAC, ABHD2, AHCYL2, ANK3, AQP4, BCL6, CAP2, CCL28, CCND2, CDH2, CMTM4, COL10A1, EMP1, EPB41L3, ETNK1, FGD4, FUT2, GABRP, GPR155, IFIT1, KIAA1958, MAGI3, MAP1B, NLRP1, PAFAH2, PLSCR1, PTGER3, REPS2, RGN, RHOBTB1, SCRG1, SIK2, STS, TMEM161B, UGT8, XK, ZEB1, ZNF462 |
| hsa-miR-217 | ABCC9,ANK3, ANLN, ATP1B1, CALD1, CORO1C, ESRRG, FN1, HOMER2, SEMA3A, SLC4A4, TMEM246, UBL3 |
| hsa-miR-371a | ABHD2,CALU, ELOVL6, HAND2, MSRB3, NEUROD1, OCLN, SGPP2, SH3BGRL2, SLC16A7, SYNJ2BP, TMEM185B, TOB1 |
| hsa-miR-7-2 | BUB1, CALU, COL2A1, CTSB, DSP, EHF, EPB41L3, ESRRG, GALNT3, GATA6, GATM, GJA1, GJC1, GREM1, HOMER2, IL20RA, KLF4, MAP1B, NTN4, PDE4B, PDZRN4, PRKCB, PTGER3, SGK1, SH3BGRL2, SKAP2, SLC16A7, SLC4A4, SPP1, SYNJ2BP, TCF4 |
| hsa-miR-675 | ABCC9, ANKRD22, ATP11A, C6, CDH11, CDH13, COBLL1, COL5A2, COLCA1, CORO1C, DMRTA1, GC, ISPD, MYBL1, PIGR, PIP5K1B, SERPINF1, SLC12A2, SMOC2, STS, TFEC, UGT8, USP53 |
| hsa-miR-137 | ABCC9,ABHD2, AHCYL2, AJUBA, ANGPTL2, ATP1B1, CNTN3, COL5A1, CXADR, DSP, DUSP10, DUSP4, EHBP1L1, ELOVL6, ESRRG, FAT3, FBXO32, FGD4, FGL2, FRMD6, FSTL1, FUT9, GFPT1, GJC1, GPX7, GULP1, KIAA1958, KLF15, KLF4, LBH, LCP2, MPC1, MSRB3, NEUROD1, NIPAL1, NT5DC2, PDLIM3, PKDCC, PLEKHO2, RCN3, RFTN1, RHOBTB1, RRAGD, SERPINA3, SIK2, SIPA1L2, SLC12A2, TBC1D1, TCF4, THBS4, TMEM125, TMEM56, TRPS1, TWIST1, YBX3, ZNF385B |
| hsa-miR-548v | ABHD2, APOBEC1, ATP1B1, CCND2, CDH2, CHN1, CLDN1, CMTM4, FUT9, GALNT3, GATA4, GPSM2, HOXB7, MAP4K4, MRAP2, NAT1, NNT, PRKACB, PRSS8, PTPRZ1, RAB27B, SLC16A7, SLC4A4, SYNC, TMEM92, TNFSF13B |
| hsa-miR-2115 | ABCC5, ADAM28, ALCAM, ALDH6A1, ARSD, ATP11A, BCAS1, BEX5, C6orf58, CA8, CCL28, CD44, CDS1, CENPF, CEP170, CKS1B, CNTN3, COL11A1, CORO1C, CRISPLD1, CSGALNACT2, CXADR, CXCL9, DAB2, DSP, DUSP10, EHF, EMILIN2, EMP1, FAM83F, FAT3, FGF7, FOXA1, FSTL1, FUT8, FUT9, GALNT6, GNB4, GPD1L, GPR155, GPSM2, GSTA1, GSTA2, HOOK1, HOXB6, IGF1, KL, LRRC17, NEURL1B, NID2, NQO1, NSUN7, PDE4B, PEA15, PLAU, PMP22, PRELID2, PRKCB, PTPN3, PTPRN2, RAB11FIP2, RAB27B, RAP1GAP, RHOBTB1, SEMA3A, SGK1, SH3GL2, SIK2, SIPA1L2, SLC16A7, SMOC2, SPARC, SULF1, SYNJ2BP, TFCP2L1, TMEM161B, TMEM229A, TRPS1, TSPAN12, TWIST1, UGT8, ZEB1 |
| hsa-miR-3923 | ATP1B1, DSC2, GKN1, GRIA4, KL, LIFR, MSR1, NEUROD1, ODAM, PIP5K1B, PTGS2, SAMD13, SOSTDC1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, M.; Lu, Z.; Yu, B.; Zhao, J.; Li, L.; Zhu, K.; Ma, M.; Long, F.; Wu, R.; Hu, G.; et al. COL5A1 Promotes the Progression of Gastric Cancer by Acting as a ceRNA of miR-137-3p to Upregulate FSTL1 Expression. Cancers 2022, 14, 3244. https://doi.org/10.3390/cancers14133244
Yang M, Lu Z, Yu B, Zhao J, Li L, Zhu K, Ma M, Long F, Wu R, Hu G, et al. COL5A1 Promotes the Progression of Gastric Cancer by Acting as a ceRNA of miR-137-3p to Upregulate FSTL1 Expression. Cancers. 2022; 14(13):3244. https://doi.org/10.3390/cancers14133244
Chicago/Turabian StyleYang, Ming, Zhixing Lu, Bowen Yu, Jiajia Zhao, Liang Li, Kaiyu Zhu, Min Ma, Fei Long, Runliu Wu, Gui Hu, and et al. 2022. "COL5A1 Promotes the Progression of Gastric Cancer by Acting as a ceRNA of miR-137-3p to Upregulate FSTL1 Expression" Cancers 14, no. 13: 3244. https://doi.org/10.3390/cancers14133244
APA StyleYang, M., Lu, Z., Yu, B., Zhao, J., Li, L., Zhu, K., Ma, M., Long, F., Wu, R., Hu, G., Huang, L., Chou, J., Gong, N., Yang, K., Li, X., Zhang, Y., & Lin, C. (2022). COL5A1 Promotes the Progression of Gastric Cancer by Acting as a ceRNA of miR-137-3p to Upregulate FSTL1 Expression. Cancers, 14(13), 3244. https://doi.org/10.3390/cancers14133244