
The Role of Metabolic Plasticity of Tumor-Associated Macrophages in Shaping the Tumor Microenvironment Immunity



Abstract
:Simple Summary
Abstract
1. Introduction
2. Function of Tumor-Associated Macrophages (TAM) in TME
2.1. TAM in Promoting Tumor Progression
2.2. TAMs Interaction with T Cells
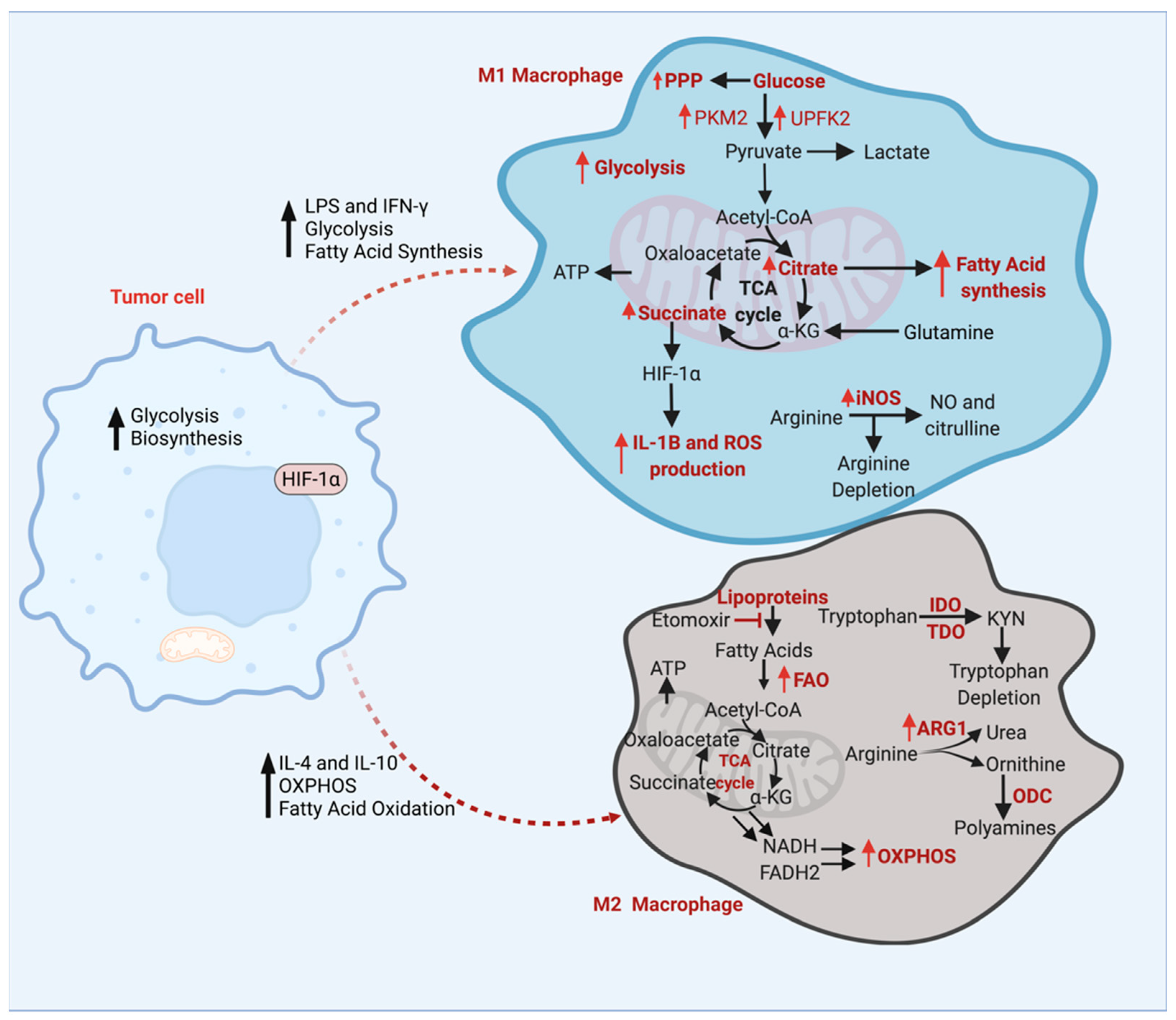
3. Energy Metabolism of TAMs
3.1. Glucose Metabolism of TAMs
3.2. Amino Acid Metabolism
3.3. Fatty Acid Metabolism
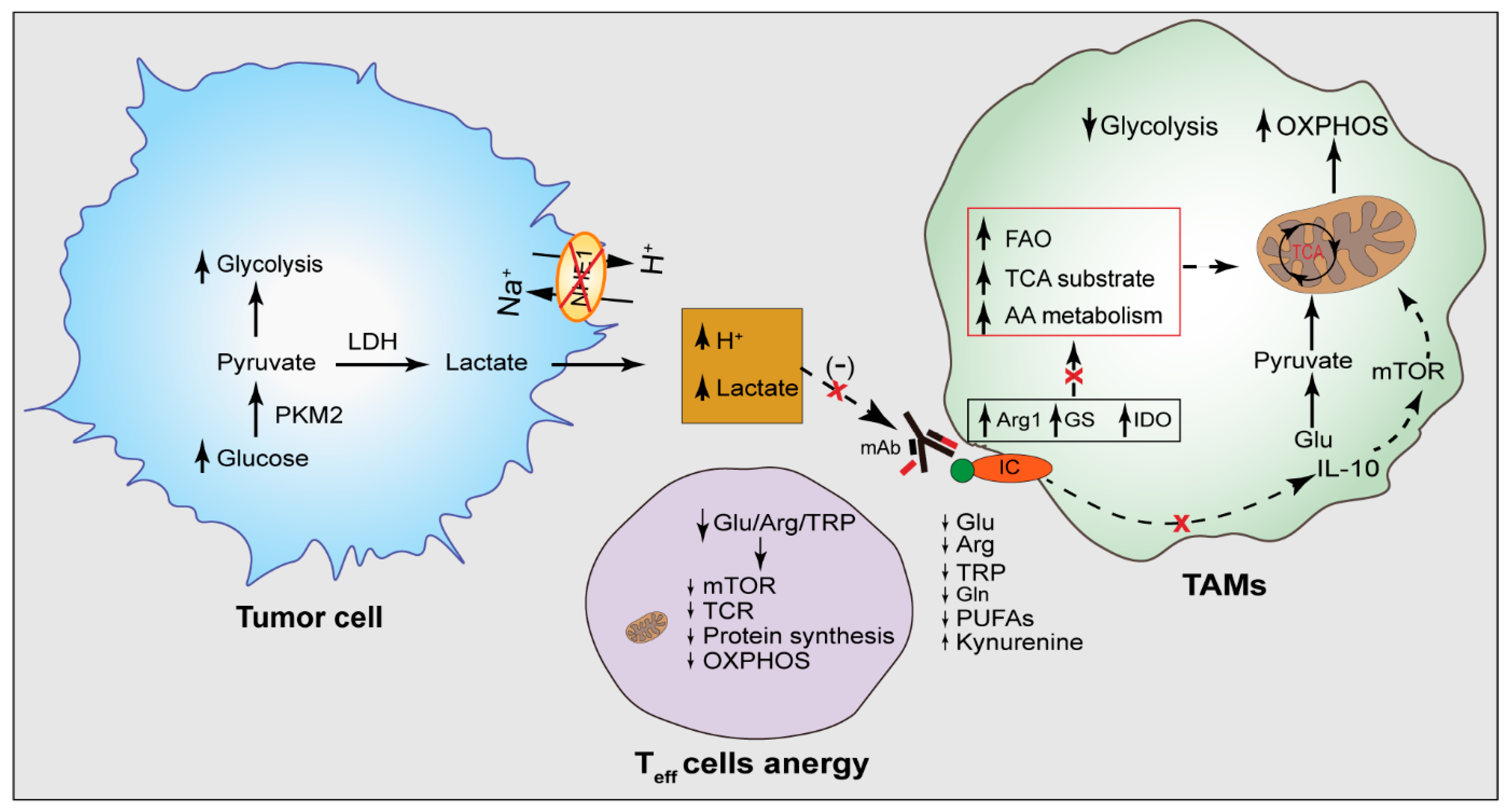
3.4. Tumor Acidity in Dysregulated TAM Metabolism
3.5. The Effect of TAMs Metabolism on T Lymphocytes
4. TAM Metabolism as a Target for Immunotherapy
4.1. Targeting TAM Metabolism for Improving Cancer Therapy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Target | Metabolic Effect | Disease | Effect on TAMs and TME |
|---|---|---|---|---|
| Metformin | AMPK (AMP-activated protein kinase) | mTOR inhibition, increased lipid breakdown, impaired glycolysis | Cancer | Decreased M2 polarization of macrophages [133,134] |
| Compound 1 | Lactate DHG | Decreased lactate production, interfering TME acidity | Cancer | Increased M1/M2 ratio in TME [120] |
| Methionine Sulfoximine | Glutamine Synthetase | Inhibition of glutamine metabolism, impaired glutamine usage by TAMs | Lewis Lung Carcinoma | Repolarization of M2 macrophages to M1 macrophages [70] |
| Etomoxir | FAO | Impaired lipid oxidation | Cancer | Decreased M2 activity [89] |
| Ranolozin | FAO | Impaired lipid oxidation | Cancer | Decreased M2 activity [89] |
| CB-1158, L-Norvaline | Arginase | Inhibition of arginine synthesis, decreasing arginine levels | Solid tumors, Alzheimer’s disease | Increased anti-tumoral T cell response in TME, Repolarization of TAMs [89] |
| Doxorubicin, Paclitaxel, Methodextrate | ABC Transporter | Disruption of efflux mechanism of cells leading to lipid accumulation in cells | Multidrug Resistant Cancer | Repolarization of TAMs [135] |
| Pralnacasan, NCX-4016, YVAD, VAD | Caspase-1 | Decreased lipid levels in inflammatory cells | Cancer, autoimmune diseases, rheumatoid arthritis, osteoarthritis | TAMs repolarization [136] |
| Rapamycin, RAD001 | mTOR | Decreased synthesis of proteins and nucleic acids | Amyotrophic lateral sclerosis, Glioma, Non-small cell lung cancer | Repolarization of TAMs [137] |
| PHGDH inhibitors | Phosphoglycerate dehydrogenase | Disruption of serine synthesis | Breast cancer, lung cancer, malign melanoma | Increased anti-tumoral T cell response in TME [138,139,140] |
| 2-deoxyglucose (2-DG) | Hexokinase 2 | Aerobic glycolysis inhibition by effecting the first step of glycolysis | Cancer | Repolarization of TAMs [141] |
| CB-839 | Glutaminase-1 | Negative effect on glutamine metabolism | Breast Cancer | Decreased glutamate levels in TME [142,143] |
4.2. Na+/H+ Exchanger (NHE1) as a Modulator of TAM Metabolism and Immune Checkpoint Therapy
4.3. Targeting TAM Metabolism to Improve Immune Checkpoint Therapy
4.4. Effect of Tumor Acidity on the Activity of Therapeutic Antibodies
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunk, P.R.; Dougherty, S.C.; Lynch, K.; Whitehair, R.; Meneveau, M.; Obeid, J.M.; Winters, K.; Ju, J.Y.; Stelow, E.B.; Bauer, T.W.; et al. Myeloid Cell Infiltration Correlates With Prognosis in Cholangiocarcinoma and Varies Based on Tumor Location. J. Immunother. 2021, 44, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Oike, N.; Kawashima, H.; Ogose, A.; Hotta, T.; Hatano, H.; Ariizumi, T.; Sasaki, T.; Yamagishi, T.; Umezu, H.; Endo, N. Prognostic impact of the tumor immune microenvironment in synovial sarcoma. Cancer Sci. 2018, 109, 3043–3054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chanmee, T.; Ontong, P.; Konno, K.; Itano, N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers 2014, 6, 1670–1690. [Google Scholar] [CrossRef] [Green Version]
- Nagarsheth, N.; Wicha, M.S.; Zou, W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol. 2017, 17, 559–572. [Google Scholar] [CrossRef] [Green Version]
- Zou, W.; Chen, L. Inhibitory B7-family molecules in the tumour microenvironment. Nat. Rev. Immunol. 2008, 8, 467–477. [Google Scholar] [CrossRef]
- Yan, D.; Kowal, J.; Akkari, L.; Schuhmacher, A.J.; Huse, J.T.; West, B.L.; Joyce, J.A. Inhibition of colony stimulating factor-1 receptor abrogates microenvironment-mediated therapeutic resistance in gliomas. Oncogene 2017, 36, 6049–6058. [Google Scholar] [CrossRef]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef] [Green Version]
- Sieow, J.L.; Gun, S.Y.; Wong, S.C. The Sweet Surrender: How Myeloid Cell Metabolic Plasticity Shapes the Tumor Microenvironment. Front. Cell Dev. Biol. 2018, 6, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talty, R.; Olino, K. Metabolism of Innate Immune Cells in Cancer. Cancers 2021, 13, 904. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Caslin, H.L.; Abebayehu, D.; Pinette, J.A.; Ryan, J.J. Lactate Is a Metabolic Mediator That Shapes Immune Cell Fate and Function. Front. Physiol. 2021, 12, 688485. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Roy, A.; Dwarakanath, B.S. Metabolic Cooperation and Competition in the Tumor Microenvironment: Implications for Therapy. Front. Oncol. 2017, 7, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitale, I.; Manic, G.; Coussens, L.M.; Kroemer, G.; Galluzzi, L. Macrophages and Metabolism in the Tumor Microenvironment. Cell Metab. 2019, 30, 36–50. [Google Scholar] [CrossRef] [PubMed]
- Patsoukis, N.; Bardhan, K.; Chatterjee, P.; Sari, D.; Liu, B.; Bell, L.N.; Karoly, E.D.; Freeman, G.J.; Petkova, V.; Seth, P.; et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 2015, 6, 6692. [Google Scholar] [CrossRef] [Green Version]
- Gubin, M.M.; Esaulova, E.; Ward, J.P.; Malkova, O.N.; Runci, D.; Wong, P.; Noguchi, T.; Arthur, C.D.; Meng, W.; Alspach, E.; et al. High-Dimensional Analysis Delineates Myeloid and Lymphoid Compartment Remodeling during Successful Immune-Checkpoint Cancer Therapy. Cell 2018, 175, 1014–1030.e1019. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Green, M.D.; Lang, X.; Lazarus, J.; Parsels, J.D.; Wei, S.; Parsels, L.A.; Shi, J.; Ramnath, N.; Wahl, D.R.; et al. Inhibition of ATM Increases Interferon Signaling and Sensitizes Pancreatic Cancer to Immune Checkpoint Blockade Therapy. Cancer Res. 2019, 79, 3940–3951. [Google Scholar] [CrossRef]
- de Goede, K.E.; Driessen, A.J.M.; Van den Bossche, J. Metabolic Cancer-Macrophage Crosstalk in the Tumor Microenvironment. Biology 2020, 9, 380. [Google Scholar] [CrossRef]
- Yang, Q.; Zhang, H.; Wei, T.; Lin, A.; Sun, Y.; Luo, P.; Zhang, J. Single-Cell RNA Sequencing Reveals the Heterogeneity of Tumor-Associated Macrophage in Non-Small Cell Lung Cancer and Differences Between Sexes. Front. Immunol. 2021, 12, 756722. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Sheng, L.; Pan, D.; Jiang, S.; Ding, L.; Ma, X.; Liu, Y.; Jia, D. Single-Cell Transcriptomic Analysis Revealed a Critical Role of SPP1/CD44-Mediated Crosstalk Between Macrophages and Cancer Cells in Glioma. Front. Cell Dev. Biol. 2021, 9, 779319. [Google Scholar] [CrossRef] [PubMed]
- De Kleer, I.; Willems, F.; Lambrecht, B.; Goriely, S. Ontogeny of myeloid cells. Front. Immunol. 2014, 5, 423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lankadasari, M.B.; Mukhopadhyay, P.; Mohammed, S.; Harikumar, K.B. TAMing pancreatic cancer: Combat with a double edged sword. Mol. Cancer 2019, 18, 48. [Google Scholar] [CrossRef] [Green Version]
- Herz, J.; Filiano, A.J.; Smith, A.; Yogev, N.; Kipnis, J. Myeloid Cells in the Central Nervous System. Immunity 2017, 46, 943–956. [Google Scholar] [CrossRef] [Green Version]
- Haas, L.; Obenauf, A.C. Allies or Enemies-The Multifaceted Role of Myeloid Cells in the Tumor Microenvironment. Front. Immunol. 2019, 10, 2746. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Schioppa, T.; Porta, C.; Allavena, P.; Sica, A. Role of tumor-associated macrophages in tumor progression and invasion. Cancer Metastasis Rev. 2006, 25, 315–322. [Google Scholar] [CrossRef]
- Zhou, J.; Tang, Z.; Gao, S.; Li, C.; Feng, Y.; Zhou, X. Tumor-Associated Macrophages: Recent Insights and Therapies. Front. Oncol. 2020, 10, 188. [Google Scholar] [CrossRef]
- Franklin, R.A.; Liao, W.; Sarkar, A.; Kim, M.V.; Bivona, M.R.; Liu, K.; Pamer, E.G.; Li, M.O. The cellular and molecular origin of tumor-associated macrophages. Science 2014, 344, 921–925. [Google Scholar] [CrossRef] [Green Version]
- Ginhoux, F.; Lim, S.; Hoeffel, G.; Low, D.; Huber, T. Origin and differentiation of microglia. Front. Cell Neurosci. 2013, 7, 45. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Barres, B.A. Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol. 2018, 18, 225–242. [Google Scholar] [CrossRef] [PubMed]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, T.; Wang, Y.; Nan, Z.; Wu, J.; Li, A.; Zhang, T.; Qu, X.; Li, C. Interaction Between Macrophage Extracellular Traps and Colon Cancer Cells Promotes Colon Cancer Invasion and Correlates With Unfavorable Prognosis. Front. Immunol. 2021, 12, 779325. [Google Scholar] [CrossRef]
- Canioni, D.; Salles, G.; Mounier, N.; Brousse, N.; Keuppens, M.; Morchhauser, F.; Lamy, T.; Sonet, A.; Rousselet, M.C.; Foussard, C.; et al. High numbers of tumor-associated macrophages have an adverse prognostic value that can be circumvented by rituximab in patients with follicular lymphoma enrolled onto the GELA-GOELAMS FL-2000 trial. J. Clin. Oncol. 2008, 26, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Cendrowicz, E.; Sas, Z.; Bremer, E.; Rygiel, T.P. The Role of Macrophages in Cancer Development and Therapy. Cancers 2021, 13, 1946. [Google Scholar] [CrossRef] [PubMed]
- Tiainen, S.; Tumelius, R.; Rilla, K.; Hamalainen, K.; Tammi, M.; Tammi, R.; Kosma, V.M.; Oikari, S.; Auvinen, P. High numbers of macrophages, especially M2-like (CD163-positive), correlate with hyaluronan accumulation and poor outcome in breast cancer. Histopathology 2015, 66, 873–883. [Google Scholar] [CrossRef]
- Tong, N.; He, Z.; Ma, Y.; Wang, Z.; Huang, Z.; Cao, H.; Xu, L.; Zou, Y.; Wang, W.; Yi, C.; et al. Tumor Associated Macrophages, as the Dominant Immune Cells, Are an Indispensable Target for Immunologically Cold Tumor-Glioma Therapy? Front. Cell Dev. Biol. 2021, 9, 706286. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Yu, Y.; Wang, X.; Zhang, T. Tumor-Associated Macrophages in Tumor Immunity. Front. Immunol. 2020, 11, 583084. [Google Scholar] [CrossRef]
- Gunderson, A.J.; Yamazaki, T.; McCarty, K.; Fox, N.; Phillips, M.; Alice, A.; Blair, T.; Whiteford, M.; O’Brien, D.; Ahmad, R.; et al. TGFbeta suppresses CD8(+) T cell expression of CXCR3 and tumor trafficking. Nat. Commun. 2020, 11, 1749. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Xu, J.; Lan, H. Tumor-associated macrophages in tumor metastasis: Biological roles and clinical therapeutic applications. J. Hematol. Oncol. 2019, 12, 76. [Google Scholar] [CrossRef]
- Ge, Z.; Ding, S. The Crosstalk Between Tumor-Associated Macrophages (TAMs) and Tumor Cells and the Corresponding Targeted Therapy. Front. Oncol. 2020, 10, 590941. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.P.; Tang, B.; Wang, Y.; Duran, C.L.; Karagiannis, G.S.; Xue, E.A.; Entenberg, D.; Borriello, L.; Coste, A.; Eddy, R.J.; et al. Live tumor imaging shows macrophage induction and TMEM-mediated enrichment of cancer stem cells during metastatic dissemination. Nat. Commun. 2021, 12, 7300. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Clauser, K.R.; Tam, W.L.; Frose, J.; Ye, X.; Eaton, E.N.; Reinhardt, F.; Donnenberg, V.S.; Bhargava, R.; Carr, S.A.; et al. A breast cancer stem cell niche supported by juxtacrine signalling from monocytes and macrophages. Nat. Cell Biol. 2014, 16, 1105–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, X.; Szulzewsky, F.; Yerevanian, A.; Chen, Z.; Heinzmann, D.; Rasmussen, R.D.; Alvarez-Garcia, V.; Kim, Y.; Wang, B.; Tamagno, I.; et al. Loss of CX3CR1 increases accumulation of inflammatory monocytes and promotes gliomagenesis. Oncotarget 2015, 6, 15077–15094. [Google Scholar] [CrossRef] [Green Version]
- Aramini, B.; Masciale, V.; Grisendi, G.; Banchelli, F.; D’Amico, R.; Maiorana, A.; Morandi, U.; Dominici, M.; Haider, K.H. Cancer stem cells and macrophages: Molecular connections and future perspectives against cancer. Oncotarget 2021, 12, 230–250. [Google Scholar] [CrossRef]
- Raggi, C.; Mousa, H.S.; Correnti, M.; Sica, A.; Invernizzi, P. Cancer stem cells and tumor-associated macrophages: A roadmap for multitargeting strategies. Oncogene 2016, 35, 671–682. [Google Scholar] [CrossRef]
- Gaggero, S.; Witt, K.; Carlsten, M.; Mitra, S. Cytokines Orchestrating the Natural Killer-Myeloid Cell Crosstalk in the Tumor Microenvironment: Implications for Natural Killer Cell-Based Cancer Immunotherapy. Front. Immunol. 2020, 11, 621225. [Google Scholar] [CrossRef]
- Nunez, S.Y.; Ziblat, A.; Secchiari, F.; Torres, N.I.; Sierra, J.M.; Raffo Iraolagoitia, X.L.; Araya, R.E.; Domaica, C.I.; Fuertes, M.B.; Zwirner, N.W. Human M2 Macrophages Limit NK Cell Effector Functions through Secretion of TGF-beta and Engagement of CD85j. J. Immunol. 2018, 200, 1008–1015. [Google Scholar] [CrossRef] [Green Version]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Proto, J.D.; Doran, A.C.; Gusarova, G.; Yurdagul, A., Jr.; Sozen, E.; Subramanian, M.; Islam, M.N.; Rymond, C.C.; Du, J.; Hook, J.; et al. Regulatory T Cells Promote Macrophage Efferocytosis during Inflammation Resolution. Immunity 2018, 49, 666–677.e666. [Google Scholar] [CrossRef] [Green Version]
- Konkel, J.E.; Zhang, D.; Zanvit, P.; Chia, C.; Zangarle-Murray, T.; Jin, W.; Wang, S.; Chen, W. Transforming Growth Factor-beta Signaling in Regulatory T Cells Controls T Helper-17 Cells and Tissue-Specific Immune Responses. Immunity 2017, 46, 660–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batlle, E.; Massague, J. Transforming Growth Factor-beta Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, D.; Cang, H.; Guo, B. Crosstalk between cancer and immune cells: Role of tumor-associated macrophages in the tumor microenvironment. Cancer Med. 2019, 8, 4709–4721. [Google Scholar] [CrossRef]
- Tu, S.; Lin, X.; Qiu, J.; Zhou, J.; Wang, H.; Hu, S.; Yao, Y.; Wang, Y.; Deng, Y.; Zhou, Y.; et al. Crosstalk Between Tumor-Associated Microglia/Macrophages and CD8-Positive T Cells Plays a Key Role in Glioblastoma. Front. Immunol. 2021, 12, 650105. [Google Scholar] [CrossRef]
- Thomas, D.A.; Massague, J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 2005, 8, 369–380. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, S.; Alexander, M.; Misharin, A.V.; Budinger, G.R.S. The role of macrophages in the resolution of inflammation. J. Clin. Investig. 2019, 129, 2619–2628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, M.; Keshav, S.; Harris, N.; Gordon, S. Interleukin 4 potently enhances murine macrophage mannose receptor activity: A marker of alternative immunologic macrophage activation. J. Exp. Med. 1992, 176, 287–292. [Google Scholar] [CrossRef]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [Green Version]
- Oshi, M.; Tokumaru, Y.; Asaoka, M.; Yan, L.; Satyananda, V.; Matsuyama, R.; Matsuhashi, N.; Futamura, M.; Ishikawa, T.; Yoshida, K.; et al. M1 Macrophage and M1/M2 ratio defined by transcriptomic signatures resemble only part of their conventional clinical characteristics in breast cancer. Sci. Rep. 2020, 10, 16554. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, M.H.; Sun, I.H.; Zhao, L.; Leone, R.D.; Sun, I.M.; Xu, W.; Collins, S.L.; Tam, A.J.; Blosser, R.L.; Patel, C.H.; et al. Targeting glutamine metabolism enhances tumor-specific immunity by modulating suppressive myeloid cells. J. Clin. Investig. 2020, 130, 3865–3884. [Google Scholar] [CrossRef]
- Lu, J.; Xu, Z.; Duan, H.; Ji, H.; Zhen, Z.; Li, B.; Wang, H.; Tang, H.; Zhou, J.; Guo, T.; et al. Tumor-associated macrophage interleukin-beta promotes glycerol-3-phosphate dehydrogenase activation, glycolysis and tumorigenesis in glioma cells. Cancer Sci. 2020, 111, 1979–1990. [Google Scholar] [CrossRef] [PubMed]
- Bando, H.; Atsumi, T.; Nishio, T.; Niwa, H.; Mishima, S.; Shimizu, C.; Yoshioka, N.; Bucala, R.; Koike, T. Phosphorylation of the 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatase/PFKFB3 family of glycolytic regulators in human cancer. Clin. Cancer Res. 2005, 11, 5784–5792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Zhang, X.; Zheng, L.; Zhao, H.; Yan, G.; Zhang, Q.; Zhou, Y.; Lei, J.; Zhang, J.; Wang, J.; et al. RIPK3 Orchestrates Fatty Acid Metabolism in Tumor-Associated Macrophages and Hepatocarcinogenesis. Cancer Immunol. Res. 2020, 8, 710–721. [Google Scholar] [CrossRef] [Green Version]
- Ménégaut, L.; Thomas, C.; Lagrost, L.; Masson, D. Fatty acid metabolism in macrophages: A target in cardio-metabolic diseases. Curr. Opin. Lipidol. 2017, 28, 19–26. [Google Scholar] [CrossRef]
- Connors, J.; Dawe, N.; Van Limbergen, J. The Role of Succinate in the Regulation of Intestinal Inflammation. Nutrients 2018, 11, 25. [Google Scholar] [CrossRef] [Green Version]
- Fultang, L.; Booth, S.; Yogev, O.; Martins da Costa, B.; Tubb, V.; Panetti, S.; Stavrou, V.; Scarpa, U.; Jankevics, A.; Lloyd, G.; et al. Metabolic engineering against the arginine microenvironment enhances CAR-T cell proliferation and therapeutic activity. Blood 2020, 136, 1155–1160. [Google Scholar] [CrossRef]
- Palmieri, E.M.; Menga, A.; Martín-Pérez, R.; Quinto, A.; Riera-Domingo, C.; De Tullio, G.; Hooper, D.C.; Lamers, W.H.; Ghesquière, B.; McVicar, D.W.; et al. Pharmacologic or Genetic Targeting of Glutamine Synthetase Skews Macrophages toward an M1-like Phenotype and Inhibits Tumor Metastasis. Cell Rep. 2017, 20, 1654–1666. [Google Scholar] [CrossRef] [Green Version]
- Palsson-McDermott, E.M.; Dyck, L.; Zaslona, Z.; Menon, D.; McGettrick, A.F.; Mills, K.H.G.; O’Neill, L.A. Pyruvate Kinase M2 Is Required for the Expression of the Immune Checkpoint PD-L1 in Immune Cells and Tumors. Front. Immunol. 2017, 8, 1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.L.; Lin, C.Y.; Kung, H.J. Targeting Mitochondrial OXPHOS and Their Regulatory Signals in Prostate Cancers. Int. J. Mol. Sci. 2021, 22, 3435. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Liu, H.; Li, Z.; Ming, A.L.; Chen, H. Mechanism of PKM2 affecting cancer immunity and metabolism in Tumor Microenvironment. J. Cancer 2021, 12, 3566–3574. [Google Scholar] [CrossRef]
- Palsson-McDermott, E.M.; Curtis, A.M.; Goel, G.; Lauterbach, M.A.; Sheedy, F.J.; Gleeson, L.E.; van den Bosch, M.W.; Quinn, S.R.; Domingo-Fernandez, R.; Johnston, D.G.; et al. Pyruvate kinase M2 regulates Hif-1α activity and IL-1β induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab. 2015, 21, 65–80. [Google Scholar] [CrossRef] [Green Version]
- Xia, Q.; Jia, J.; Hu, C.; Lu, J.; Li, J.; Xu, H.; Fang, J.; Feng, D.; Wang, L.; Chen, Y. Tumor-associated macrophages promote PD-L1 expression in tumor cells by regulating PKM2 nuclear translocation in pancreatic ductal adenocarcinoma. Oncogene 2022, 41, 865–877. [Google Scholar] [CrossRef] [PubMed]
- Carrasco-Pozo, C.; Tan, K.N.; Avery, V.M. Hemin Prevents Increased Glycolysis in Macrophages upon Activation: Protection by Microbiota-Derived Metabolites of Polyphenols. Antioxidants 2020, 9, 1109. [Google Scholar] [CrossRef]
- Bailey, J.D.; Diotallevi, M.; Nicol, T.; McNeill, E.; Shaw, A.; Chuaiphichai, S.; Hale, A.; Starr, A.; Nandi, M.; Stylianou, E.; et al. Nitric Oxide Modulates Metabolic Remodeling in Inflammatory Macrophages through TCA Cycle Regulation and Itaconate Accumulation. Cell Rep. 2019, 28, 218–230.e217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.Y.; Huang, T.W.; Hsieh, Y.T.; Wang, Y.F.; Yen, C.C.; Lee, G.L.; Yeh, C.C.; Peng, Y.J.; Kuo, Y.Y.; Wen, H.T.; et al. Cancer-Derived Succinate Promotes Macrophage Polarization and Cancer Metastasis via Succinate Receptor. Mol. Cell 2020, 77, 213–227.e215. [Google Scholar] [CrossRef]
- Fletcher, M.; Ramirez, M.E.; Sierra, R.A.; Raber, P.; Thevenot, P.; Al-Khami, A.A.; Sanchez-Pino, D.; Hernandez, C.; Wyczechowska, D.D.; Ochoa, A.C.; et al. l-Arginine depletion blunts antitumor T-cell responses by inducing myeloid-derived suppressor cells. Cancer Res. 2015, 75, 275–283. [Google Scholar] [CrossRef] [Green Version]
- Geiger, R.; Rieckmann, J.C.; Wolf, T.; Basso, C.; Feng, Y.; Fuhrer, T.; Kogadeeva, M.; Picotti, P.; Meissner, F.; Mann, M.; et al. L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity. Cell 2016, 167, 829–842.e813. [Google Scholar] [CrossRef] [Green Version]
- Matos, A.; Carvalho, M.; Bicho, M.; Ribeiro, R. Arginine and Arginases Modulate Metabolism, Tumor Microenvironment and Prostate Cancer Progression. Nutrients 2021, 13, 4503. [Google Scholar] [CrossRef] [PubMed]
- Arlauckas, S.P.; Garren, S.B.; Garris, C.S.; Kohler, R.H.; Oh, J.; Pittet, M.J.; Weissleder, R. Arg1 expression defines immunosuppressive subsets of tumor-associated macrophages. Theranostics 2018, 8, 5842–5854. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Gan, S.; Zhu, Q.; Dai, D.; Li, N.; Wang, H.; Chen, X.; Hou, D.; Wang, Y.; Pan, Q.; et al. Modulation of M2 macrophage polarization by the crosstalk between Stat6 and Trim24. Nat. Commun. 2019, 10, 4353. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Sun, J.; Li, Y.; Zhou, X.; Zhai, W.; Wu, Y.; Chen, G.; Gou, S.; Sui, X.; Zhao, W.; et al. Tryptophan 2,3-dioxygenase 2 controls M2 macrophages polarization to promote esophageal squamous cell carcinoma progression via AKT/GSK3β/IL-8 signaling pathway. Acta Pharm. Sin. B 2021, 11, 2835–2849. [Google Scholar] [CrossRef] [PubMed]
- Mondanelli, G.; Iacono, A.; Allegrucci, M.; Puccetti, P.; Grohmann, U. Immunoregulatory Interplay Between Arginine and Tryptophan Metabolism in Health and Disease. Front. Immunol. 2019, 10, 1565. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, E.M.; Menga, A.; Lebrun, A.; Hooper, D.C.; Butterfield, D.A.; Mazzone, M.; Castegna, A. Blockade of Glutamine Synthetase Enhances Inflammatory Response in Microglial Cells. Antioxid. Redox Signal. 2017, 26, 351–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzone, M.; Menga, A.; Castegna, A. Metabolism and TAM functions-it takes two to tango. FEBS J. 2018, 285, 700–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Vos, K.E.; Eliasson, P.; Proikas-Cezanne, T.; Vervoort, S.J.; van Boxtel, R.; Putker, M.; van Zutphen, I.J.; Mauthe, M.; Zellmer, S.; Pals, C.; et al. Modulation of glutamine metabolism by the PI(3)K-PKB-FOXO network regulates autophagy. Nature Cell Biol. 2012, 14, 829–837. [Google Scholar] [CrossRef]
- Hossain, F.; Al-Khami, A.A.; Wyczechowska, D.; Hernandez, C.; Zheng, L.; Reiss, K.; Valle, L.D.; Trillo-Tinoco, J.; Maj, T.; Zou, W.; et al. Inhibition of Fatty Acid Oxidation Modulates Immunosuppressive Functions of Myeloid-Derived Suppressor Cells and Enhances Cancer Therapies. Cancer Immunol. Res. 2015, 3, 1236–1247. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Han, Y.; Rodriguez Sillke, Y.; Deng, H.; Siddiqui, S.; Treese, C.; Schmidt, F.; Friedrich, M.; Keye, J.; Wan, J.; et al. Lipid droplet-dependent fatty acid metabolism controls the immune suppressive phenotype of tumor-associated macrophages. EMBO Mol. Med. 2019, 11, e10698. [Google Scholar] [CrossRef]
- Su, P.; Wang, Q.; Bi, E.; Ma, X.; Liu, L.; Yang, M.; Qian, J.; Yi, Q. Enhanced Lipid Accumulation and Metabolism Are Required for the Differentiation and Activation of Tumor-Associated Macrophages. Cancer Res. 2020, 80, 1438–1450. [Google Scholar] [CrossRef] [PubMed]
- Xiang, W.; Shi, R.; Kang, X.; Zhang, X.; Chen, P.; Zhang, L.; Hou, A.; Wang, R.; Zhao, Y.; Zhao, K.; et al. Monoacylglycerol lipase regulates cannabinoid receptor 2-dependent macrophage activation and cancer progression. Nat. Commun. 2018, 9, 2574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, S.; Wu, D.; Lu, Q.; Qian, L.; Ding, Y.; Liu, G.; Jia, X.; Zhang, Y.; Xiao, W.; Gong, W. Angiopoietin-like 4 deficiency upregulates macrophage function through the dysregulation of cell-intrinsic fatty acid metabolism. Am. J. Cancer Res. 2020, 10, 595–609. [Google Scholar] [PubMed]
- Divakaruni, A.S.; Hsieh, W.Y.; Minarrieta, L.; Duong, T.N.; Kim, K.K.O.; Desousa, B.R.; Andreyev, A.Y.; Bowman, C.E.; Caradonna, K.; Dranka, B.P.; et al. Etomoxir Inhibits Macrophage Polarization by Disrupting CoA Homeostasis. Cell Metab. 2018, 28, 490–503.e497. [Google Scholar] [CrossRef] [Green Version]
- Hasan, M.N.; Luo, L.; Ding, D.; Song, S.; Bhuiyan, M.I.H.; Liu, R.; Foley, L.M.; Guan, X.; Kohanbash, G.; Hitchens, T.K.; et al. Blocking NHE1 stimulates glioma tumor immunity by restoring OXPHOS function of myeloid cells. Theranostics 2021, 11, 1295–1309. [Google Scholar] [CrossRef] [PubMed]
- Husain, Z.; Huang, Y.; Seth, P.; Sukhatme, V.P. Tumor-derived lactate modifies antitumor immune response: Effect on myeloid-derived suppressor cells and NK cells. J. Immunol. 2013, 191, 1486–1495. [Google Scholar] [CrossRef] [PubMed]
- Huber, V.; Camisaschi, C.; Berzi, A.; Ferro, S.; Lugini, L.; Triulzi, T.; Tuccitto, A.; Tagliabue, E.; Castelli, C.; Rivoltini, L. Cancer acidity: An ultimate frontier of tumor immune escape and a novel target of immunomodulation. Semin. Cancer Biol. 2017, 43, 74–89. [Google Scholar] [CrossRef]
- Wculek, S.K.; Dunphy, G.; Heras-Murillo, I.; Mastrangelo, A.; Sancho, D. Metabolism of tissue macrophages in homeostasis and pathology. Cell. Mol. Immunol. 2021. [Google Scholar] [CrossRef]
- Noe, J.T.; Rendon, B.E.; Geller, A.E.; Conroy, L.R.; Morrissey, S.M.; Young, L.E.A.; Bruntz, R.C.; Kim, E.J.; Wise-Mitchell, A.; Rizzo, M.B.d.S.; et al. Lactate supports a metabolic-epigenetic link in macrophage polarization. Sci. Adv. 2021, 7, eabi8602. [Google Scholar] [CrossRef]
- Colegio, O.R.; Chu, N.-Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef]
- Labadie, B.W.; Bao, R.; Luke, J.J. Reimagining IDO Pathway Inhibition in Cancer Immunotherapy via Downstream Focus on the Tryptophan-Kynurenine-Aryl Hydrocarbon Axis. Clin. Cancer Res. 2019, 25, 1462–1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, G.K.; Park, H.J.; Macleod, M.; Chandler, P.; Munn, D.H.; Mellor, A.L. Tryptophan deprivation sensitizes activated T cells to apoptosis prior to cell division. Immunology 2002, 107, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Siska, P.J.; Jiao, J.; Matos, C.; Singer, K.; Berger, R.S.; Dettmer, K.; Oefner, P.J.; Cully, M.D.; Wang, Z.; Quinn, I.W.; et al. Kynurenine induces T cell fat catabolism and has limited suppressive effects in vivo. EBioMedicine 2021, 74, 103734. [Google Scholar] [CrossRef] [PubMed]
- Cibrian, D.; Sanchez-Madrid, F. CD69: From activation marker to metabolic gatekeeper. Eur. J. Immunol. 2017, 47, 946–953. [Google Scholar] [CrossRef]
- Zhao, Q.; Kuang, D.M.; Wu, Y.; Xiao, X.; Li, X.F.; Li, T.J.; Zheng, L. Activated CD69+ T cells foster immune privilege by regulating IDO expression in tumor-associated macrophages. J. Immunol. 2012, 188, 1117–1124. [Google Scholar] [CrossRef] [Green Version]
- Mellor, A.L.; Munn, D.H. IDO expression by dendritic cells: Tolerance and tryptophan catabolism. Nat. Rev. Immunol. 2004, 4, 762–774. [Google Scholar] [CrossRef]
- Sosnowska, A.; Chlebowska-Tuz, J.; Matryba, P.; Pilch, Z.; Greig, A.; Wolny, A.; Grzywa, T.M.; Rydzynska, Z.; Sokolowska, O.; Rygiel, T.P.; et al. Inhibition of arginase modulates T-cell response in the tumor microenvironment of lung carcinoma. Oncoimmunology 2021, 10, 1956143. [Google Scholar] [CrossRef]
- Porta, C.; Sica, A.; Riboldi, E. Tumor-associated myeloid cells: New understandings on their metabolic regulation and their influence in cancer immunotherapy. FEBS J. 2018, 285, 717–733. [Google Scholar] [CrossRef] [Green Version]
- Viola, A.; Munari, F.; Sanchez-Rodriguez, R.; Scolaro, T.; Castegna, A. The Metabolic Signature of Macrophage Responses. Front. Immunol. 2019, 10, 1462. [Google Scholar] [CrossRef] [Green Version]
- Erbel, C.; Rupp, G.; Domschke, G.; Linden, F.; Akhavanpoor, M.; Doesch, A.O.; Katus, H.A.; Gleissner, C.A. Differential regulation of aldose reductase expression during macrophage polarization depends on hyperglycemia. Innate Immun. 2016, 22, 230–237. [Google Scholar] [CrossRef] [Green Version]
- Penny, H.L.; Sieow, J.L.; Gun, S.Y.; Lau, M.C.; Lee, B.; Tan, J.; Phua, C.; Toh, F.; Nga, Y.; Yeap, W.H.; et al. Targeting Glycolysis in Macrophages Confers Protection Against Pancreatic Ductal Adenocarcinoma. Int. J. Mol. Sci. 2021, 22, 6350. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Zhang, S.; Gong, Z.; Li, X.; Cao, K.; Deng, H.; He, Y.; et al. The role of microenvironment in tumor angiogenesis. J. Exp. Clin. Cancer Res. 2020, 39, 204. [Google Scholar] [CrossRef] [PubMed]
- Zanotelli, M.R.; Reinhart-King, C.A. Mechanical Forces in Tumor Angiogenesis. Adv. Exp. Med. Biol. 2018, 1092, 91–112. [Google Scholar] [CrossRef]
- Henze, A.T.; Mazzone, M. The impact of hypoxia on tumor-associated macrophages. J. Clin. Investig. 2016, 126, 3672–3679. [Google Scholar] [CrossRef]
- Leblond, M.M.; Gerault, A.N.; Corroyer-Dulmont, A.; MacKenzie, E.T.; Petit, E.; Bernaudin, M.; Valable, S. Hypoxia induces macrophage polarization and re-education toward an M2 phenotype in U87 and U251 glioblastoma models. Oncoimmunology 2016, 5, e1056442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tariq, M.; Zhang, J.Q.; Liang, G.K.; He, Q.J.; Ding, L.; Yang, B. Gefitinib inhibits M2-like polarization of tumor-associated macrophages in Lewis lung cancer by targeting the STAT6 signaling pathway. Acta Pharmacol. Sin. 2017, 38, 1501–1511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Ma, T.; Shen, X.N.; Xia, X.F.; Xu, G.D.; Bai, X.L.; Liang, T.B. Macrophage-induced tumor angiogenesis is regulated by the TSC2-mTOR pathway. Cancer Res. 2012, 72, 1363–1372. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Gao, A.; Tu, B.; Wang, Y.; Yu, X.; Wang, Y.; Xiu, Y.; Wang, B.; Wan, Y.; Huang, Y. Metabolic modulation via mTOR pathway and anti-angiogenesis remodels tumor microenvironment using PD-L1-targeting codelivery. Biomaterials 2020, 255, 120187. [Google Scholar] [CrossRef]
- Ding, L.; Liang, G.; Yao, Z.; Zhang, J.; Liu, R.; Chen, H.; Zhou, Y.; Wu, H.; Yang, B.; He, Q. Metformin prevents cancer metastasis by inhibiting M2-like polarization of tumor associated macrophages. Oncotarget 2015, 6, 36441–36455. [Google Scholar] [CrossRef] [Green Version]
- Seth, P.; Csizmadia, E.; Hedblom, A.; Vuerich, M.; Xie, H.; Li, M.; Longhi, M.S.; Wegiel, B. Deletion of Lactate Dehydrogenase-A in Myeloid Cells Triggers Antitumor Immunity. Cancer Res. 2017, 77, 3632–3643. [Google Scholar] [CrossRef] [Green Version]
- Frank, A.C.; Raue, R.; Fuhrmann, D.C.; Sirait-Fischer, E.; Reuse, C.; Weigert, A.; Lutjohann, D.; Hiller, K.; Syed, S.N.; Brune, B. Lactate dehydrogenase B regulates macrophage metabolism in the tumor microenvironment. Theranostics 2021, 11, 7570–7588. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.J.; Kim, A.; Kim, G.T.; Yu, H.Y.; Lee, E.S.; Park, M.J.; Kim, Y.J.; Shim, S.M.; Park, T.S. Inhibition of lactate dehydrogenase A suppresses inflammatory response in RAW 264.7 macrophages. Mol. Med. Rep. 2019, 19, 629–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selvanesan, B.C.; Chandra, D.; Quispe-Tintaya, W.; Jahangir, A.; Patel, A.; Meena, K.; Alves Da Silva, R.A.; Friedman, M.; Gabor, L.; Khouri, O.; et al. Listeria delivers tetanus toxoid protein to pancreatic tumors and induces cancer cell death in mice. Sci. Transl. Med. 2022, 14, eabc1600. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, R.; Gu, H.; Zhang, E.; Qu, J.; Cao, W.; Huang, X.; Yan, H.; He, J.; Cai, Z. Metabolic reprogramming in macrophage responses. Biomarker Res. 2021, 9, 1. [Google Scholar] [CrossRef] [PubMed]
- Platten, M.; Nollen, E.A.A.; Rohrig, U.F.; Fallarino, F.; Opitz, C.A. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat. Rev. Drug Discov. 2019, 18, 379–401. [Google Scholar] [CrossRef]
- Zhai, L.; Bell, A.; Ladomersky, E.; Lauing, K.L.; Bollu, L.; Sosman, J.A.; Zhang, B.; Wu, J.D.; Miller, S.D.; Meeks, J.J.; et al. Immunosuppressive IDO in Cancer: Mechanisms of Action, Animal Models, and Targeting Strategies. Front. Immunol. 2020, 11, 1185. [Google Scholar] [CrossRef]
- Prendergast, G.C.; Malachowski, W.J.; Mondal, A.; Scherle, P.; Muller, A.J. Indoleamine 2,3-Dioxygenase and Its Therapeutic Inhibition in Cancer. Int. Rev. Cell Mol. Biol. 2018, 336, 175–203. [Google Scholar] [CrossRef]
- Yue, E.W.; Sparks, R.; Polam, P.; Modi, D.; Douty, B.; Wayland, B.; Glass, B.; Takvorian, A.; Glenn, J.; Zhu, W.; et al. INCB24360 (Epacadostat), a Highly Potent and Selective Indoleamine-2,3-dioxygenase 1 (IDO1) Inhibitor for Immuno-oncology. ACS Med. Chem. Lett. 2017, 8, 486–491. [Google Scholar] [CrossRef]
- Su, Q.; Wang, C.; Song, H.; Zhang, C.; Liu, J.; Huang, P.; Zhang, Y.; Zhang, J.; Wang, W. Co-delivery of anionic epitope/CpG vaccine and IDO inhibitor by self-assembled cationic liposomes for combination melanoma immunotherapy. J. Mater. Chem. B 2021, 9, 3892–3899. [Google Scholar] [CrossRef]
- Zou, S.; Wang, X.; Liu, P.; Ke, C.; Xu, S. Arginine metabolism and deprivation in cancer therapy. Biomed. Pharmacother. 2019, 118, 109210. [Google Scholar] [CrossRef]
- Yang, J.S.; Wang, C.C.; Qiu, J.D.; Ren, B.; You, L. Arginine metabolism: A potential target in pancreatic cancer therapy. Chin. Med. J. 2020, 134, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Steggerda, S.M.; Bennett, M.K.; Chen, J.; Emberley, E.; Huang, T.; Janes, J.R.; Li, W.; MacKinnon, A.L.; Makkouk, A.; Marguier, G.; et al. Inhibition of arginase by CB-1158 blocks myeloid cell-mediated immune suppression in the tumor microenvironment. J. Immunother. Cancer 2017, 5, 101. [Google Scholar] [CrossRef] [PubMed]
- Incio, J.; Suboj, P.; Chin, S.M.; Vardam-Kaur, T.; Liu, H.; Hato, T.; Babykutty, S.; Chen, I.; Deshpande, V.; Jain, R.K.; et al. Metformin Reduces Desmoplasia in Pancreatic Cancer by Reprogramming Stellate Cells and Tumor-Associated Macrophages. PLoS ONE 2015, 10, e0141392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uehara, T.; Eikawa, S.; Nishida, M.; Kunisada, Y.; Yoshida, A.; Fujiwara, T.; Kunisada, T.; Ozaki, T.; Udono, H. Metformin induces CD11b+-cell-mediated growth inhibition of an osteosarcoma: Implications for metabolic reprogramming of myeloid cells and anti-tumor effects. Int. Immunol. 2019, 31, 187–198. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.H.; Yu, A.M. ABC transporters in multidrug resistance and pharmacokinetics, and strategies for drug development. Curr. Pharm. Des. 2014, 20, 793–807. [Google Scholar] [CrossRef]
- Niu, Z.; Shi, Q.; Zhang, W.; Shu, Y.; Yang, N.; Chen, B.; Wang, Q.; Zhao, X.; Chen, J.; Cheng, N.; et al. Caspase-1 cleaves PPARgamma for potentiating the pro-tumor action of TAMs. Nat. Commun. 2017, 8, 766. [Google Scholar] [CrossRef] [Green Version]
- Ko, J.H.; Yoon, S.O.; Lee, H.J.; Oh, J.Y. Rapamycin regulates macrophage activation by inhibiting NLRP3 inflammasome-p38 MAPK-NFkappaB pathways in autophagy- and p62-dependent manners. Oncotarget 2017, 8, 40817–40831. [Google Scholar] [CrossRef] [Green Version]
- Locasale, J.W.; Grassian, A.R.; Melman, T.; Lyssiotis, C.A.; Mattaini, K.R.; Bass, A.J.; Heffron, G.; Metallo, C.M.; Muranen, T.; Sharfi, H.; et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat. Genet. 2011, 43, 869–874. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Zheng, A.; Hydbring, P.; Ambroise, G.; Ouchida, A.T.; Goiny, M.; Vakifahmetoglu-Norberg, H.; Norberg, E. PHGDH Defines a Metabolic Subtype in Lung Adenocarcinomas with Poor Prognosis. Cell Rep. 2017, 19, 2289–2303. [Google Scholar] [CrossRef] [Green Version]
- Possemato, R.; Marks, K.M.; Shaul, Y.D.; Pacold, M.E.; Kim, D.; Birsoy, K.; Sethumadhavan, S.; Woo, H.K.; Jang, H.G.; Jha, A.K.; et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 2011, 476, 346–350. [Google Scholar] [CrossRef] [Green Version]
- Penny, H.L.; Sieow, J.L.; Adriani, G.; Yeap, W.H.; See Chi Ee, P.; San Luis, B.; Lee, B.; Lee, T.; Mak, S.Y.; Ho, Y.S.; et al. Warburg metabolism in tumor-conditioned macrophages promotes metastasis in human pancreatic ductal adenocarcinoma. Oncoimmunology 2016, 5, e1191731. [Google Scholar] [CrossRef] [PubMed]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuneva, M.; Zamboni, N.; Oefner, P.; Sachidanandam, R.; Lazebnik, Y. Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. J. Cell Biol. 2007, 178, 93–105. [Google Scholar] [CrossRef] [Green Version]
- Fliegel, L. Role of Genetic Mutations of the Na(+)/H(+) Exchanger Isoform 1, in Human Disease and Protein Targeting and Activity. Mol. Cell Biochem. 2021, 476, 1221–1232. [Google Scholar] [CrossRef] [PubMed]
- Cong, D.; Zhu, W.; Shi, Y.; Pointer, K.B.; Clark, P.A.; Shen, H.; Kuo, J.S.; Hu, S.; Sun, D. Upregulation of NHE1 protein expression enables glioblastoma cells to escape TMZ-mediated toxicity via increased H(+) extrusion, cell migration and survival. Carcinogenesis 2014, 35, 2014–2024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huc, L.; Tekpli, X.; Holme, J.A.; Rissel, M.; Solhaug, A.; Gardyn, C.; Le Moigne, G.; Gorria, M.; Dimanche-Boitrel, M.T.; Lagadic-Gossmann, D. c-Jun NH2-terminal kinase-related Na+/H+ exchanger isoform 1 activation controls hexokinase II expression in benzo(a)pyrene-induced apoptosis. Cancer Res. 2007, 67, 1696–1705. [Google Scholar] [CrossRef] [Green Version]
- Singh, Y.; Zhou, Y.; Shi, X.; Zhang, S.; Umbach, A.T.; Salker, M.S.; Lang, K.S.; Lang, F. Alkaline Cytosolic pH and High Sodium Hydrogen Exchanger 1 (NHE1) Activity in Th9 Cells. J. Biol. Chem. 2016, 291, 23662–23671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, X.; Luo, L.; Begum, G.; Kohanbash, G.; Song, Q.; Rao, A.; Amankulor, N.; Sun, B.; Sun, D.; Jia, W. Elevated Na/H exchanger 1 (SLC9A1) emerges as a marker for tumorigenesis and prognosis in gliomas. J. Exp. Clin. Cancer Res. 2018, 37, 255. [Google Scholar] [CrossRef] [Green Version]
- Amith, S.R.; Fliegel, L. Na(+)/H(+) exchanger-mediated hydrogen ion extrusion as a carcinogenic signal in triple-negative breast cancer etiopathogenesis and prospects for its inhibition in therapeutics. Semin. Cancer Biol. 2017, 43, 35–41. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Long, X.; Wang, D.; Lou, M.; Zou, D.; Chen, R.; Nian, W.; Zhou, Q. Increased expression of Na(+)/H(+) exchanger isoform 1 predicts tumor aggressiveness and unfavorable prognosis in epithelial ovarian cancer. Oncol. Lett. 2018, 16, 6713–6720. [Google Scholar] [CrossRef] [Green Version]
- Guan, X.; Hasan, M.N.; Begum, G.; Kohanbash, G.; Carney, K.E.; Pigott, V.M.; Persson, A.I.; Castro, M.G.; Jia, W.; Sun, D. Blockade of Na/H exchanger stimulates glioma tumor immunogenicity and enhances combinatorial TMZ and anti-PD-1 therapy. Cell Death Dis. 2018, 9, 1010. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Tu, W.; Chen, Y.; Zhu, M.; Jin, H.; Huang, T.; Zou, Z.; Xia, Q. The combination of PKM2 overexpression and M2 macrophages infiltration confers a poor prognosis for PDAC patients. J. Cancer 2020, 11, 2022–2031. [Google Scholar] [CrossRef] [PubMed]
- Mo, L.; Xu, L.; Jia, M.; Su, B.; Hu, Y.; Hu, Z.; Li, H.; Zhao, C.; Zhao, Z.; Li, J. Shikonin suppresses the epithelial-to-mesenchymal transition by downregulating NHE1 in bladder cancer cells. J. Cancer 2021, 12, 6814–6824. [Google Scholar] [CrossRef] [PubMed]
- Geeraerts, X.; Fernandez-Garcia, J.; Hartmann, F.J.; de Goede, K.E.; Martens, L.; Elkrim, Y.; Debraekeleer, A.; Stijlemans, B.; Vandekeere, A.; Rinaldi, G.; et al. Macrophages are metabolically heterogeneous within the tumor microenvironment. Cell Rep. 2021, 37, 110171. [Google Scholar] [CrossRef] [PubMed]
- Cassetta, L.; Pollard, J.W. Targeting macrophages: Therapeutic approaches in cancer. Nat. Rev. Drug Discov. 2018, 17, 887–904. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, J.; Yadav, D.K.; Bai, X.; Liang, T. Glucose Metabolism: The Metabolic Signature of Tumor Associated Macrophage. Front. Immunol. 2021, 12, 702580. [Google Scholar] [CrossRef]
- Chen, H.M.; van der Touw, W.; Wang, Y.S.; Kang, K.; Mai, S.; Zhang, J.; Alsina-Beauchamp, D.; Duty, J.A.; Mungamuri, S.K.; Zhang, B.; et al. Blocking immunoinhibitory receptor LILRB2 reprograms tumor-associated myeloid cells and promotes antitumor immunity. J. Clin. Investig. 2018, 128, 5647–5662. [Google Scholar] [CrossRef]
- Sun, Y.; Zhao, J.; Sun, X.; Ma, G. Identification of TNFAIP8 as an Immune-Related Biomarker Associated with Tumorigenesis and Prognosis in Cutaneous Melanoma Patients. Front. Genet. 2021, 12, 783672. [Google Scholar] [CrossRef]
- Li, T.; Wang, W.; Gong, S.; Sun, H.; Zhang, H.; Yang, A.G.; Chen, Y.H.; Li, X. Genome-wide analysis reveals TNFAIP8L2 as an immune checkpoint regulator of inflammation and metabolism. Mol. Immunol. 2018, 99, 154–162. [Google Scholar] [CrossRef]
- Strauss, L.; Mahmoud, M.A.A.; Weaver, J.D.; Tijaro-Ovalle, N.M.; Christofides, A.; Wang, Q.; Pal, R.; Yuan, M.; Asara, J.; Patsoukis, N.; et al. Targeted deletion of PD-1 in myeloid cells induces antitumor immunity. Sci. Immunol. 2020, 5. [Google Scholar] [CrossRef]
- Zhang, J.; Hou, C.; Dou, S.; Li, G.; Wang, Z.; Liu, Y.; Zhang, Y.; Wang, R.; Shen, B.; Han, G. T cell immunoglobulin and mucin domain protein 3 inhibits glycolysis in RAW 264.7 macrophages through Hexokinase 2. Scand. J. Immunol. 2021, 93, e12981. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Du, W.; Yi, L.; Wu, S.; He, C.; Zhai, W.; Yue, C.; Sun, R.; Menk, A.V.; Delgoffe, G.M.; et al. Checkpoint molecules coordinately restrain hyperactivated effector T cells in the tumor microenvironment. Oncoimmunology 2020, 9, 1708064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ando, H.; Emam, S.E.; Kawaguchi, Y.; Shimizu, T.; Ishima, Y.; Eshima, K.; Ishida, T. Increasing Tumor Extracellular pH by an Oral Alkalinizing Agent Improves Antitumor Responses of Anti-PD-1 Antibody: Implication of Relationships between Serum Bicarbonate Concentrations, Urinary pH, and Therapeutic Outcomes. Biol. Pharm. Bull. 2021, 44, 844–852. [Google Scholar] [CrossRef] [PubMed]
- Leone, R.D.; Powell, J.D. Metabolism of immune cells in cancer. Nat. Rev. Cancer 2020, 20, 516–531. [Google Scholar] [CrossRef]
- Chan, J.C.Y.; Diakos, C.I.; Engel, A.; Chan, D.L.H.; Pavlakis, N.; Gill, A.; Clarke, S.J. Serum bicarbonate is a marker of peri-operative mortality but is not associated with long term survival in colorectal cancer. PLoS ONE 2020, 15, e0228466. [Google Scholar] [CrossRef] [Green Version]
- Pilon-Thomas, S.; Kodumudi, K.N.; El-Kenawi, A.E.; Russell, S.; Weber, A.M.; Luddy, K.; Damaghi, M.; Wojtkowiak, J.W.; Mule, J.J.; Ibrahim-Hashim, A.; et al. Neutralization of Tumor Acidity Improves Antitumor Responses to Immunotherapy. Cancer Res. 2016, 76, 1381–1390. [Google Scholar] [CrossRef] [Green Version]
- Kaul, M.; Loos, M. Dissection of C1q capability of interacting with IgG. Time-dependent formation of a tight and only partly reversible association. J. Biol. Chem. 1997, 272, 33234–33244. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.W.; Frey, G.; Liu, H.; Xing, C.; Steinman, L.; Boyle, W.J.; Short, J.M. Generating tumor-selective conditionally active biologic anti-CTLA4 antibodies via protein-associated chemical switches. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hasan, M.N.; Capuk, O.; Patel, S.M.; Sun, D. The Role of Metabolic Plasticity of Tumor-Associated Macrophages in Shaping the Tumor Microenvironment Immunity. Cancers 2022, 14, 3331. https://doi.org/10.3390/cancers14143331
Hasan MN, Capuk O, Patel SM, Sun D. The Role of Metabolic Plasticity of Tumor-Associated Macrophages in Shaping the Tumor Microenvironment Immunity. Cancers. 2022; 14(14):3331. https://doi.org/10.3390/cancers14143331
Chicago/Turabian StyleHasan, Md Nabiul, Okan Capuk, Shivani M. Patel, and Dandan Sun. 2022. "The Role of Metabolic Plasticity of Tumor-Associated Macrophages in Shaping the Tumor Microenvironment Immunity" Cancers 14, no. 14: 3331. https://doi.org/10.3390/cancers14143331
APA StyleHasan, M. N., Capuk, O., Patel, S. M., & Sun, D. (2022). The Role of Metabolic Plasticity of Tumor-Associated Macrophages in Shaping the Tumor Microenvironment Immunity. Cancers, 14(14), 3331. https://doi.org/10.3390/cancers14143331