
Brain Gliomas and Ollier Disease: Molecular Findings as Predictive Risk Factors?

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Background
2. Methods
3. Results
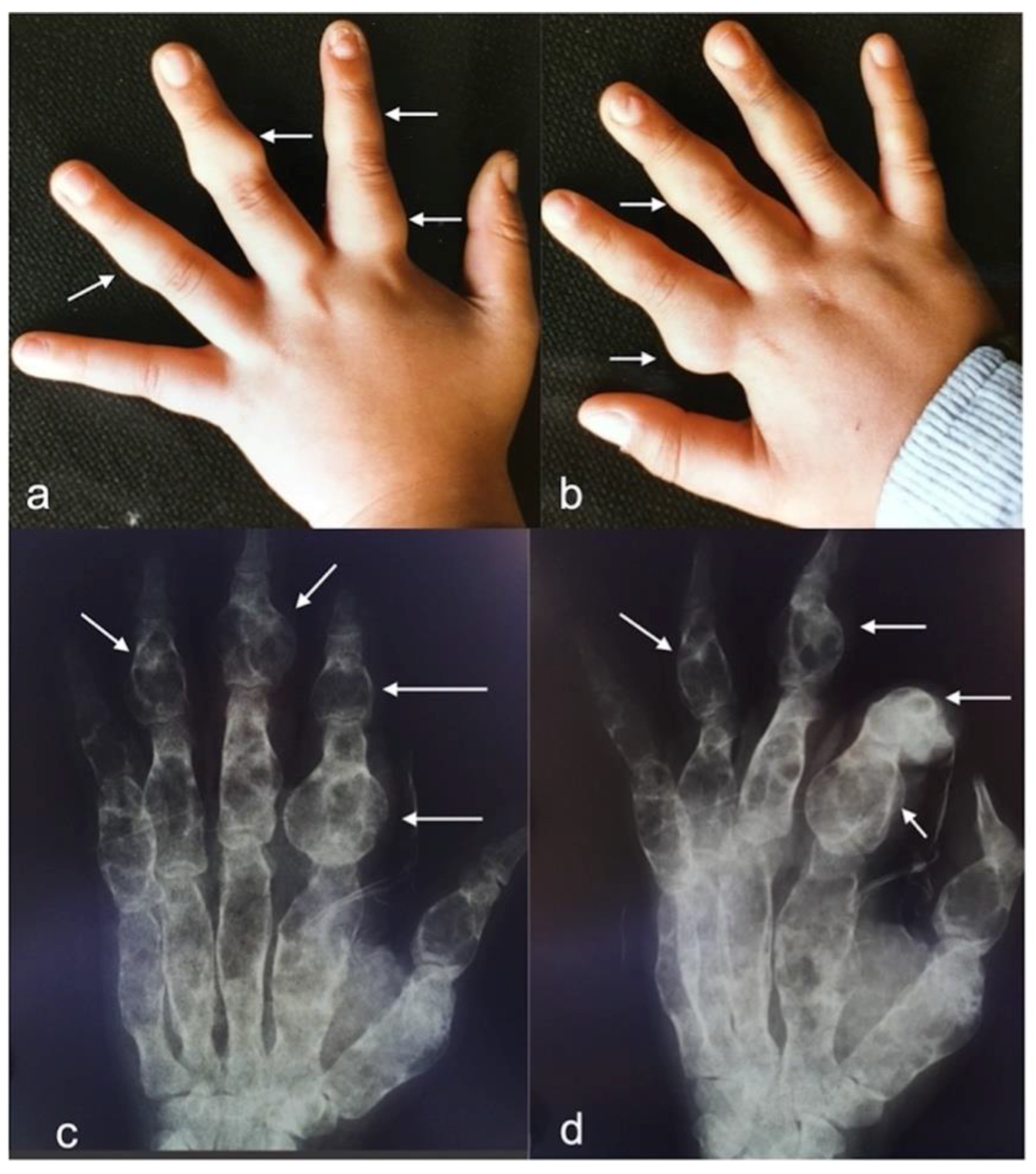
3.1. Case Description
3.2. Diagnosis
3.3. Literature Review
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Limitations of the Study
Advantages
Abbreviations
| OD | Ollier Disease |
| CT | Computed Tomography |
| MRI | Magnetic Resonance Imaging |
| WHO | World Health Organization |
| CNS | Central Nervous System |
| PFS | Progression Free Survival |
| OS | Overall Survival |
| MPFS | Malignant Progression-Free Survival |
References
- D’Angelo, L.; Massimi, L.; Narducci, A.; Di Rocco, C. Ollier disease. Childs Nerv. Syst. 2009, 25, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Silve, C.; Jüppner, H. Ollier disease. Orphanet. J. Rare Dis. 2006, 1, 37. [Google Scholar] [CrossRef] [PubMed]
- White, M.S.; Martin, P.L.; McLean, T.W. Acute myelogenous leukemia associated with Ollier disease. Pediatr. Blood Cancer 2008, 50, 645–646. [Google Scholar] [CrossRef] [PubMed]
- Tamimi, H.K.; Bolen, J.W. Enchondromatosis (Ollier’s disease) and ovarian juvenile granulosa cell tumor. Cancer 1984, 53, 1605–1608. [Google Scholar] [CrossRef]
- Chang, S.; Prados, M.D. Identical twins with Ollier’s disease and intracranial gliomas: Case report. Neurosurgery 1994, 34, 903–906, discussion 906. [Google Scholar] [CrossRef]
- Hofman, S.; Heeg, M.; Klein, J.P.; Krikke, A.P. Simultaneous occurrence of a supra- and an infratentorial glioma in a patient with Ollier’s disease: More evidence for non-mesodermal tumor predisposition in multiple enchondromatosis. Skelet. Radiol. 1998, 27, 688–691. [Google Scholar] [CrossRef]
- Mahafza, W.S. Multiple enchondromatosis Ollier’s disease with two primary brain tumors. Saudi Med. J. 2004, 25, 1261–1263. [Google Scholar]
- Nishio, S.; Morioka, T.; Suzuki, S.; Takeshita, I.; Fukui, M. Thalamic gliomas: A clinicopathologic analysis of 20 cases with reference to patient age. Acta Neurochir. 1997, 139, 336–342. [Google Scholar] [CrossRef]
- Rawlings, C.E.; Bullard, D.E.; Burger, P.C.; Friedman, A.H. A case of Ollier’s disease associated with two intracranial gliomas. Neurosurgery 1987, 21, 400–403. [Google Scholar] [CrossRef]
- Gajavelli, S.; Nakhla, J.; Nasser, R.; Yassari, R.; Weidenheim, K.M.; Graber, J. Ollier disease with anaplastic astrocytoma: A review of the literature and a unique case. Surg. Neurol. Int. 2016, 7, S607–S611. [Google Scholar] [CrossRef]
- Mellon, C.D.; Carter, J.E.; Owen, D.B. Ollier’s disease and Maffucci’s syndrome: Distinct entities or a continuum. Case report: Enchondromatosis complicated by an intracranial glioma. J. Neurol. 1988, 235, 376–378. [Google Scholar] [CrossRef] [PubMed]
- Becker, W.; Thron, A. Dyschondroplasia with glioma of the brain. Third histologically verified case (author’s transl). Arch. Orthop. Trauma. Surg. 1979, 93, 141–144. [Google Scholar] [CrossRef] [PubMed]
- Balcer, L.J.; Galetta, S.L.; Cornblath, W.T.; Liu, G.T. Neuro-ophthalmologic manifestations of Maffucci’s syndrome and Ollier’s disease. J. Neuroophthalmol. 1999, 19, 62–66. [Google Scholar] [CrossRef]
- Bathla, G.; Gupta, S.; Ong, C.K. Multifocal intracranial astrocytoma in a pediatric patient with Ollier disease. Indian J. Radiol. Imaging 2012, 22, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Bendel, C.J.; Gelmers, H.J. Multiple enchondromatosis (Ollier’s disease) complicated by malignant astrocytoma. Eur. J. Radiol. 1991, 12, 135–137. [Google Scholar] [CrossRef]
- Walid, M.S.; Troup, E.C. Cerebellar anaplastic astrocytoma in a teenager with Ollier Disease. J. Neurooncol. 2008, 89, 59–62. [Google Scholar] [CrossRef]
- Karabulut, A.K.; Türk, S.; Tamsel, İ.; Kim, J.; Argın, M. Diffuse midline glioma in Ollier disease: A case report and a brief review of the literature. Radiol. Case Rep. 2021, 16, 2299–2305. [Google Scholar] [CrossRef]
- Schwartz, H.S.; Zimmerman, N.B.; Simon, M.A.; Wroble, R.R.; Millar, E.A.; Bonfiglio, M. The malignant potential of enchondromatosis. J. Bone Jt. Surg. Am. 1987, 69, 269–274. [Google Scholar] [CrossRef]
- Patt, S.; Weigel, K.; Mayer, H.M. A case of dyschondroplasia associated with brain stem glioma: Diagnosis by stereotactic biopsy. Neurosurgery 1990, 27, 487–491. [Google Scholar] [CrossRef]
- Frappaz, D.; Ricci, A.C.; Kohler, R.; Bret, P.; Mottolese, C. Diffuse brain stem tumor in an adolescent with multiple enchondromatosis (Ollier’s disease). Childs Nerv. Syst. 1999, 15, 222–225. [Google Scholar] [CrossRef]
- Ranger, A.; Szymczak, A.; Hammond, R.R.; Zelcer, S. Pediatric thalamic glioblastoma associated with Ollier disease (multiple enchondromatosis): A rare case of concurrence. J. Neurosurg. Pediatr. 2009, 4, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Hori, K.; Matsumine, A.; Niimi, R.; Maeda, M.; Uchida, K.; Nakamura, T.; Sudo, A. Diffuse gliomas in an adolescent with multiple enchondromatosis (Ollier’s disease). Oncol. Lett. 2010, 1, 595–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Achiha, T.; Arita, H.; Kagawa, N.; Murase, T.; Ikeda, J.I.; Morii, E.; Kanemura, Y.; Fujimoto, Y.; Kishima, H. Enchondromatosis-associated oligodendroglioma: Case report and literature review. Brain Tumor Pathol. 2018, 35, 36–40. [Google Scholar] [CrossRef]
- Pearce, P.; Robertson, T.; Ortiz-Gomez, J.D.; Rajah, T.; Tollesson, G. Multifocal supratentorial diffuse glioma in a young patient with Ollier disease. J. Clin. Neurosci. 2012, 19, 477–478. [Google Scholar] [CrossRef] [PubMed]
- Koc, F. Ollier Disease Anaplastic Mixed Oligoastrocytoma A Rare Association With Brain Tumors. Neurosurg. Q. 2006, 16, 195–197. [Google Scholar] [CrossRef]
- Simsek, S. Ollier’s disease with intracranial glioma. Child’s Nerv. Syst. 2002, 12, 66–69. [Google Scholar]
- van Nielen, K.M.; de Jong, B.M. A case of Ollier’s disease associated with two intracerebral low-grade gliomas. Clin. Neurol. Neurosurg. 1999, 101, 106–110. [Google Scholar] [CrossRef]
- El Abiad, J.M.; Robbins, S.M.; Cohen, B.; Levin, A.S.; Valle, D.L.; Morris, C.D.; de Macena Sobreira, N.L. Natural history of Ollier disease and Maffucci syndrome: Patient survey and review of clinical literature. Am. J. Med. Genet. A 2020, 182, 1093–1103. [Google Scholar] [CrossRef]
- Verdegaal, S.H.; Bovée, J.V.; Pansuriya, T.C.; Grimer, R.J.; Ozger, H.; Jutte, P.C.; San Julian, M.; Biau, D.J.; van der Geest, I.C.; Leithner, A.; et al. Incidence, predictive factors, and prognosis of chondrosarcoma in patients with Ollier disease and Maffucci syndrome: An international multicenter study of 161 patients. Oncologist 2011, 16, 1771–1779. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, C.; Thomas, L.; Psimaras, D.; Bielle, F.; Vauléon, E.; Loiseau, H.; Cartalat-Carel, S.; Meyronet, D.; Dehais, C.; Honnorat, J.; et al. Characteristics of gliomas in patients with somatic IDH mosaicism. Acta Neuropathol. Commun. 2016, 4, 31. [Google Scholar] [CrossRef]
- Capper, D.; Jones, D.T.W.; Sill, M.; Hovestadt, V.; Schrimpf, D.; Sturm, D.; Koelsche, C.; Sahm, F.; Chavez, L.; Reuss, D.E.; et al. DNA methylation-based classification of central nervous system tumours. Nature 2018, 555, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Della Monica, R.; Cuomo, M.; Buonaiuto, M.; Costabile, D.; Franca, R.A.; Del Basso De Caro, M.; Catapano, G.; Chiariotti, L.; Visconti, R. MGMT and Whole-Genome DNA Methylation Impacts on Diagnosis, Prognosis and Therapy of Glioblastoma Multiforme. Int. J. Mol. Sci. 2022, 23, 7148. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Louis, D. WHO Classification of Tumours of the Central Nervous System, 4th ed.; IARC, Ed.; International Agency for Research on Cancer: Lyon, France, 2016. [Google Scholar]
- Weller, M.; van den Bent, M.; Preusser, M.; Le Rhun, E.; Tonn, J.C.; Minniti, G.; Bendszus, M.; Balana, C.; Chinot, O.; Dirven, L.; et al. EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nat. Rev. Clin. Oncol. 2021, 18, 170–186. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef]
- Amary, M.F.; Bacsi, K.; Maggiani, F.; Damato, S.; Halai, D.; Berisha, F.; Pollock, R.; O’Donnell, P.; Grigoriadis, A.; Diss, T.; et al. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J. Pathol. 2011, 224, 334–343. [Google Scholar] [CrossRef]
- Amary, M.F.; Damato, S.; Halai, D.; Eskandarpour, M.; Berisha, F.; Bonar, F.; McCarthy, S.; Fantin, V.R.; Straley, K.S.; Lobo, S.; et al. Ollier disease and Maffucci syndrome are caused by somatic mosaic mutations of IDH1 and IDH2. Nat. Genet. 2011, 43, 1262–1265. [Google Scholar] [CrossRef]
- Pallud, J.; Fontaine, D.; Duffau, H.; Mandonnet, E.; Sanai, N.; Taillandier, L.; Peruzzi, P.; Guillevin, R.; Bauchet, L.; Bernier, V.; et al. Natural history of incidental World Health Organization grade II gliomas. Ann. Neurol. 2010, 68, 727–733. [Google Scholar] [CrossRef]
- Rice, T.; Lachance, D.H.; Molinaro, A.M.; Eckel-Passow, J.E.; Walsh, K.M.; Barnholtz-Sloan, J.; Ostrom, Q.T.; Francis, S.S.; Wiemels, J.; Jenkins, R.B.; et al. Understanding inherited genetic risk of adult glioma—a review. Neurooncol. Pract. 2016, 3, 10–16. [Google Scholar] [CrossRef] [Green Version]
- Brat, D.J.; Verhaak, R.G.; Aldape, K.D.; Yung, W.K.; Salama, S.R.; Cooper, L.A.; Rheinbay, E.; Miller, C.R.; Vitucci, M.; Morozova, O.; et al. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N. Engl. J. Med. 2015, 372, 2481–2498. [Google Scholar] [CrossRef] [Green Version]
- Pansuriya, T.C.; van Eijk, R.; d’Adamo, P.; van Ruler, M.A.; Kuijjer, M.L.; Oosting, J.; Cleton-Jansen, A.M.; van Oosterwijk, J.G.; Verbeke, S.L.; Meijer, D.; et al. Somatic mosaic IDH1 and IDH2 mutations are associated with enchondroma and spindle cell hemangioma in Ollier disease and Maffucci syndrome. Nat. Genet. 2011, 43, 1256–1261. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Mei, Q.; Li, H.; Ke, C.; Yu, J.; Chen, J. A survival analysis of surgically treated incidental low-grade glioma patients. Sci. Rep. 2021, 11, 8522. [Google Scholar] [CrossRef]
- Potts, M.B.; Smith, J.S.; Molinaro, A.M.; Berger, M.S. Natural history and surgical management of incidentally discovered low-grade gliomas. J. Neurosurg. 2012, 116, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Pallud, J.; Capelle, L.; Taillandier, L.; Badoual, M.; Duffau, H.; Mandonnet, E. The silent phase of diffuse low-grade gliomas. Is it when we missed the action? Acta Neurochir. 2013, 155, 2237–2242. [Google Scholar] [CrossRef] [PubMed]
- Satar, Z.; Hotton, G.; Samandouras, G. Systematic review-Time to malignant transformation in low-grade gliomas: Predicting a catastrophic event with clinical, neuroimaging, and molecular markers. Neurooncol. Adv. 2021, 3, vdab101. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| N. of Cases | Authors/Year | Sex/Age | CNS Tumor | Molecular Analysis | Surgery at Initial Diagnosis | Adjuvant Treatment | Progression/Recurrence | Outcome | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Histology | Location | Glioma | Enchon-Droma | |||||||
| 1 | Becker et al. [7]. 1979 | n.s./26 | Oligoastrocytoma (grade II) | r. frontal | n.a. | n.a. | n.s. | n.s. | n.s. | n.s. |
| 2 | Rawlings et al. [34]. 1987 | M/29 | anaplastic astrocytoma (grade III) | multicentric (r. cerebellum, r. frontal) | n.a. | n.a. | biopsy | RT | n.s. | n.s. |
| 3 | Mellon et al. [25]. 1988 | M/34 | astrocytoma (grade II) | r. frontal | n.a. | n.a. | craniotomy | n.s. | n.s. | n.s. |
| 4 | Schwartz et al. [36]. 1987 | M/38 | malignant astrocytoma (grade III) | temporo- parietal | n.a. | n.a. | n.s. | n.s. | n.s. | dead |
| 5 | Patt et al. [30]. 1990 | M/24 | Astrocytoma (grade II) | brainstem | n.a. | n.a. | biopsy | n.s. | n.s. | n.s. |
| 6 | Bendel et al. [8]. 1991 | F/29 | high grade astrocytoma (grade III) | l. frontal | n.a. | n.a. | n.s. | n.s. | n.s. | n.s. |
| 7 | Chang et al. [12]. 1994 | M/23 | anaplastic astrocytoma (grade III) | Multicentric (both hemispheres) | n.a. | n.a. | biopsy | WBRT | no at 3 y | Alive at 3 y |
| 8 | Chang et al. [12]. 1994 | M/25 | oligodendroglioma | r. frontal | n.a. | n.a. | Craniotomy (STR) | RT | no at 8 mo. | Alive at 8 mo |
| 9 | Chang et al. [12]. 1994 | M/46 | oligoastrocytoma | Multicentric (both frontal lobes) | n.a. | n.a. | biopsy | RT | no at 4 mo. | Alive at 4 mo. |
| 10 | Hofman et al. [18]. 1998 | M/28 | Astrocytoma (grade II) | multicentric (l. temporal, brainstem) | n.a. | n.a. | biopsy | RT | no at 1 y | Alive at 1 y |
| 11 | Balcer et al. [5]. 1999 | F/23 | astrocytoma | pons | n.a. | n.a. | none | RT | n.s. | n.s. |
| 12 | Frappaz et al. [16]. 1999 | M/16 | astrocytoma | brainstem | n.a. | n.a. | none | RT | no at 7 mo. | Alive at 7 mo. |
| 13 | Van Nielen et al. [37]. 1999 | M/28 | Astrocytoma (grade II) | multicentric (l. temporal, brainstem) | n.a. | n.a. | biopsy | RT | n.s. | n.s. |
| 14 | Simsek et al. [38]. 2002 | F/7 | Astrocytoma (grade II) | r. frontal | n.a. | n.a. | n.s. | n.s. | n.s. | n.s. |
| 15 | Mahafza et al. [24]. 2004 | F/21 | Astrocytoma (grade II) | r. frontal, brainstem | n.a. | n.a. | biopsy | n.s. | n.s. | n.s. |
| 16 | Koc et al. [21]. 2006 | F/28 | anaplastic oligoastrocytoma | r. frontal | n.a. | n.a. | craniotomy | RT at recurrence | recurrence at 6 y, reoperation | alive at 10 y |
| (grade III) | ||||||||||
| 17 | Ranger et al. [33]. 2009 | F/6 | glioblastoma | l. thalamus | n.a. | n.a. | biopsy | RT + CHT | progression | dead at 11 mo |
| 18 | Walid et al. [39]. 2008 | M/14 | anaplastic astrocytoma (grade III) | cerebellum | n.a. | n.a. | n.s. | n.s. | n.s. | n.s. |
| 19 | Hori et al. [19]. 2010 | M/19 | anaplastic astrocytoma (grade III) | Multicentric (brain, brainstem) | n.a. | n.a. | craniotomy | RT + CHT (TMZ) | stable | alive |
| 20 | Bathla et al. [6]. 2012 | M/16 | Astrocytoma (grade II) | multicentric (both frontal parietal lobes) | IDH1 R132H | n.a. | biopsy | CT and craniotomy at progression | progression to anaplastic astrocytoma (grade III) at 3 y, reoperation | Dead at 8 y |
| 21 | Pearce et al. [31]. 2012 | M/19 | Oligoastrocytoma (grade II) | multicentric (both hemispheres) | no mutation | n.a. | biopsy | no | progression at 9 mo. after biopsy, craniotomy | n.s. |
| 22 | Gajavelli et al. [17]. 2016 | F/55 | anaplastic astrocytoma (grade III) | multicentric (l. frontal, temporal, parietal) | IDH1 R132H | n.a. | craniotomy(STR) | no | stable at 3 y | alive at 3 y |
| 23 | Bonnet et al. [9]. 2016 | F/28 | Oligoastrocytoma (grade II) | multicentric | IDH1 R132H | n.a | biopsy | n.s. | n.s. | alive at 2.5 y |
| (temporal, frontal) | ||||||||||
| 24 | Bonnet et al. [9]. 2016 | M/26 | n.s. | frontal | n.s. | n.a | biopsy | n.s. | n.s. | alive at 1 y |
| 25 | Bonnet et al. [9]. 2016 | F/30 | Oligodendroglioma (grade II) | multicentric (frontal, temporal | IDH1 R132H | n.a | biopsy | n.s. | n.s. | Alive at 4 y |
| 26 | Bonnet et al. [9]. 2016 | M/31 | Glioblastoma (grade IV) | multicentric (frontal, parietal | IDH1 R132H | n.a | biopsy | n.s. | n.s. | alive at 9 mo |
| 27 | Bonnet et al. [9]. 2016 | F/31 | Oligoastrocytoma (grade III) | frontal | IDH1 R132H | IDH1 R132H | biopsy | n.s. | n.s. | alive at 1.5 y |
| 28 | Achiha et al. [1]. 2017 | M/32 | oligodendroglioma | l. frontal | IDH1 R132H | IDH1 R132H | n.s. | n.s. | n.s. | n.s. |
| 29 | Al Rumeh et al. [2]. 2020 | F/23 | diffuse astrocytoma (grade II) | multicentric (l. frontal) | IDH1 R132C | n.a. | craniotomy | n.s. | n.s. | n.s. |
| 30 | Karabulut et al. [20]. 2021 | F/23 | diffuse midline glioma | pons | n.a. | n.a. | none | RT + CHT | stable | alive |
| 31 | Present case | M/33 | diffuse astrocytoma (grade II) | multicentric (both hemispheres) | IDH1 R132H | IDH1 R132H | biopsy | RT + CHT (TMZ) at initial diagnosis | progression at 48 mo. after biopsy, reoperation + RT | alive at 5 y |
| Covariates | Overall Series |
|---|---|
| (31 Patients) | |
| Age | 6–55 y |
| (median 26 y) | |
| Sex | 30 * |
| F | 12 (40%) |
| M | 18 (60%) |
| Location | 31 * |
| Single lesion | 16 (51%) |
| supratentorial | 11 |
| brainstem | 4 |
| cerebellar | 1 |
| Multicentric | 15 (49%) |
| only supratentorial | 10 |
| supra- and infratentorial | 5 |
| Pathology | |
| WHO grade | 24 * |
| II | 13 |
| III | 9 |
| IV | 2 |
| Histological type | 29 * |
| astrocytic | 18 |
| oligodendroglial | 3 |
| oligo-astrocytic | 6 |
| glioblastoma | 2 |
| Molecular biology | 10 * gliomas |
| Detection of IDH1 R132H mutation: | 3 * enchondromas |
| glioma | 8/10 |
| enchondroma | 3/3 |
| Covariates | Overall Series |
|---|---|
| (31 Patients) | |
| Surgery | 25 * |
| biopsy | 16 |
| craniotomy | 6 |
| none | 3 |
| Radiotherapy | 16 * |
| at initial diagnosis | 12 |
| at progression | 2 |
| none | 2 |
| Chemotherapy | 7 * |
| at initial diagnosis | 3 |
| at progression | 2 |
| none | 2 |
| Glioma evolution | 13 * |
| Stable | 9 (from 3 to 36 mo.) |
| Progression | 4 (from 9 to 72 mo.) |
| Outcome | 18 * |
| Alive | 15 (from 4 to 187 mo.) |
| Dead | 3 (from 11 to 96 mo.) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Corvino, S.; Mariniello, G.; Corazzelli, G.; Franca, R.A.; Del Basso De Caro, M.; Della Monica, R.; Chiariotti, L.; Maiuri, F. Brain Gliomas and Ollier Disease: Molecular Findings as Predictive Risk Factors? Cancers 2022, 14, 3464. https://doi.org/10.3390/cancers14143464
Corvino S, Mariniello G, Corazzelli G, Franca RA, Del Basso De Caro M, Della Monica R, Chiariotti L, Maiuri F. Brain Gliomas and Ollier Disease: Molecular Findings as Predictive Risk Factors? Cancers. 2022; 14(14):3464. https://doi.org/10.3390/cancers14143464
Chicago/Turabian StyleCorvino, Sergio, Giuseppe Mariniello, Giuseppe Corazzelli, Raduan Ahmed Franca, Marialaura Del Basso De Caro, Rosa Della Monica, Lorenzo Chiariotti, and Francesco Maiuri. 2022. "Brain Gliomas and Ollier Disease: Molecular Findings as Predictive Risk Factors?" Cancers 14, no. 14: 3464. https://doi.org/10.3390/cancers14143464