1. Introduction
Estimates indicate that around 77% of primary brain tumors are gliomas and that glioblastoma (GB) is the most malignant and frequent type of glioma [
1], with more than 10,000 cases annually diagnosed and a 95% 5-year mortality [
2]. To this poor prognosis accounts GB pathophysiology heterogeneity, treatment refractoriness, and a high rate of recurrence due to the extensive invasiveness of the surrounding cerebral parenchyma, which renders it difficult to complete surgical resections [
3].
Within a broad pool of redundant pathways and potential targets [
4], amplification and mutation of the epidermal growth factor receptor (
EGFR or
HER1) gene arise in about 60% of primary GB, thus becoming an attractive therapeutic target [
5]. EGFR overexpression has been associated with decontrolled cell proliferation and inhibition of apoptosis, as well as with the establishment of malignancy by inducing the progression of low-grade to high-grade glioma [
6]. Thus, a search for new EGFR tyrosine kinase inhibitors (TKIs) has been pursued [
7], but monotherapy with these agents has not increased disease-free survival [
8]. Several studies have pointed out that activation of compensatory pathways that generate multiple inputs to downstream phosphatidylinositol-3-kinase (PI3K) signaling may confer insensitivity to EGFR-targeted therapies, unraveling new opportunities for multitargeting in malignant and resistant brain tumors [
9]. Actually, the PI3K/AKT/mTOR pathway is overexpressed or constitutively hyperactivated in about 78% of GB cases, strongly contributing to tumor malignancy and invasiveness [
10]. The least studied PI3Kp110β subunit was recently described as the principal activator of downstream AKT signaling with a strong association with tumors’ recurrence rate and prognosis [
11]. However, there are no published successful results indicating the relevance of PI3Kp110β inhibition in glioma cells or proposing EGFR/PI3K dual inhibitors.
Most drugs showing pre-clinical potential against GB cannot achieve therapeutic concentrations at the tumor site due to low blood–brain barrier (BBB) penetration [
12]. The anatomic basis of the BBB is formed by a tightly sealed monolayer of brain microvascular endothelial cells. These cells have unique features such as the expression of complex intercellular junctions that are responsible for high transendothelial electrical resistance (TEER) and low paracellular flux, which reflect a restricted permeability [
13]. So, approximately 100% of large molecules and 98% of small molecules do not cross the BBB [
13], dampening the treatment of most brain pathologies.
Only low-molecular-weight (<700 Da but typically <300 Da) lipophilic molecules may diffuse through BBB’s endothelium and enter the brain passively [
14]. However, drugs with high lipid-solubility bind to plasma proteins such as albumin, with a low off-rate, which restricts their delivery to the brain [
15]. Moreover, increased lipophilicity raises the likelihood of the drug becoming a substrate for BBB efflux transporters, such as the highly expressed P-glycoprotein (P-gp), restricting the drug’s accumulation in the brain [
16]. These facts have attracted attention to study the BBB as an obstacle to overcome during the drug discovery phase, which could represent the key for the successful treatment of neuro-pathologies such as GB [
17].
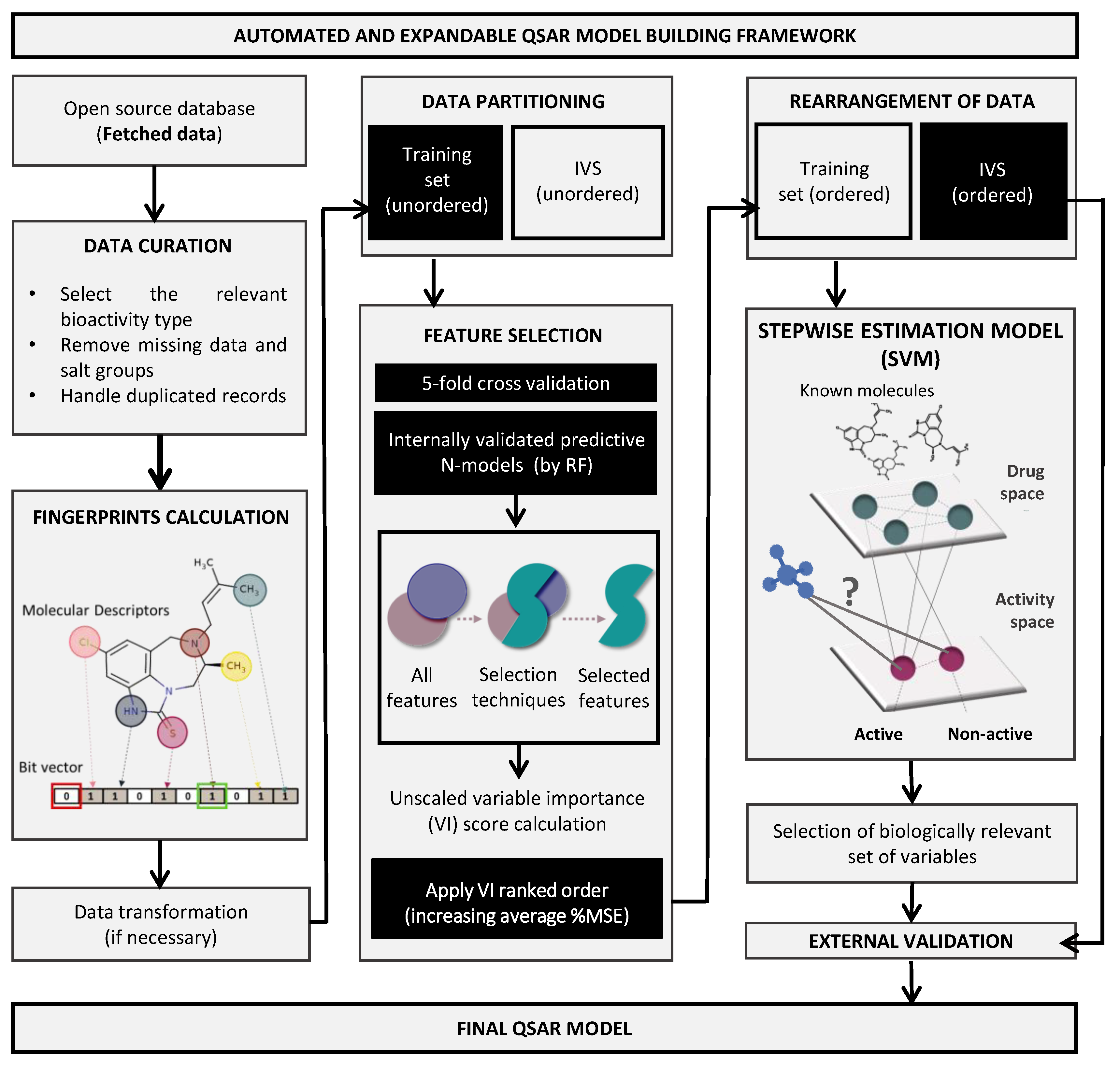
Aware that drug discovery is a challenging, time consuming, and expensive process, computational tools may act as a “virtual shortcut”, speeding up and potentially reducing the research cost. Through the use of specialized machine learning algorithms and access to large libraries of compounds, in silico testing allows the screening of a high number of molecules in search of those that fit optimal queries and cause the desired biologic response [
18]. Computational modeling is able to support multitargeting approaches by predicting BBB permeation, target-specificity, and ADMET (absorption, distribution, metabolism, excretion, and toxicity) properties of molecules, opening doors to a new dimensionality of targeted and combined therapies, with higher effectiveness, for many brain pathologies, namely GB [
19].
Considering the continuous amount of molecular data being added to compound activity databases such as ChEMBL or PubChem, there are emerging opportunities to build well-trained and properly validated empirical models [
20]. A common molecular representation used in these models relies on the use of chemical fingerprints that generally identify molecular fragments such as molecular descriptors encoded as bit vectors, where each bit position is associated with a presence or absence of a specific substructure pattern [
21]. Several supervised machine learning tools are able to use this type of representation for pattern recognition algorithms, allowing to build automated and robust Quantitative Structure–Activity Relationship (QSAR) models by correlation of molecular structural information with biological activities [
22]. Such models have limitations from the limited data from which they were fitted. So, the concept of an applicability domain (AD) defines the structural boundaries to which a model can be reliably applied [
23]. Moreover, there is a trend to improve the quality of in silico predictions by allying the empirical results of QSAR models and similarity searches to 3D-docking simulations, providing a direct target–ligand interaction study to unravel the energetically most favorable binding mode [
24,
25]. The combination of computational approaches with experimental practices and clinical trials has revealed strategic primacies to overcome medicinal chemistry challenges and assess the efficacy and safety of new medical interventions, minimizing the ethical concerns, and boosting pharmaceutical research productivity and quality [
20,
26].
The aim of this work is to identify drug candidates that predictably overcome the BBB and inhibit the EGFR/PI3Kp110β pathway as a multi-targeting approach against GB. We used an in silico strategy, relying on automated QSAR models and docking simulations, to screen libraries of millions of molecules, as well as using relevant in vitro models and biological assays to validate the obtained computational results. We succeeded in identifying promising drug candidates, in this way opening new avenues for GB treatment.
3. Results
3.1. Internal and External Validation of Final QSAR Models Supported Their High Predictive Performance
The performance of the presented QSAR modeling workflow was assessed by an unbiased protocol to support optimized predictive models for all chemical problems (
Figure 2). The internal lowest RMSE in predicting BBB permeation (0.513), EGFR inhibition (0.193), and PI3Kp110β inhibition (0.175) was achieved by using AtomPair 1024-bit vectors, considering the 83 best-ranked features in BBB’s model and 150 features in the last two models (
Figure 2A).
After selecting the best set of features and defining QSAR patterns, SF-models’ external validation was performed using an independent validation set (
Figure 2B). The BBB permeation model was trained with a dataset of fewer than 1000 molecules, whereas EGFR and PI3K models were trained with 1870 and 7134 molecules (
Supplementary Table S1), respectively.
The comparison between the performance of externally validated full-models (without feature selection) and final SF-models (
Figure 2C) confirmed the effectiveness of feature selection, thus corroborating previous results [
20]. The number of selected features in SF-models ranged between 8.1 and 14.6% of the total processed features considered in full models. The PVE score of full-models ranged between 0.43 and 0.59 while in final SF-models were in the 0.55–0.71 range. Moreover, error analysis of predictive models showed an averaged RMSE of full-models of 0.40, while the final SF-models had an averaged RMSE of 0.24.
A retroactive validation of the SF-model was accomplished using the IVS, showing an r
2 coefficient of 0.577 in the BBB permeation model, 0.8387 in the EGFR model, and 0.8864 in the PI3Kp110β model (
Figure 2D).
3.2. ZINC15 Screening by Developed QSAR Models Pointed out Unswerving Candidates
The ZINC15 screening allowed the selection of: 31 molecules for EGFR with LogBB ≥ 0.2 and -pIC
50 ≥ 7; 14 candidates for PI3Kp110β with LogBB ≥ 0 and -pIC
50 ≥ 6.5; and 7 candidates for dual targeting that maximized the scaled values of IC
50 of both targets (spIC
50 (EGFR) x spIC
50 (PI3Kp110β) ≥ 0.1) and assumed LogBB ≥ −0.2 (
Figure 3). Most molecules were predicted as weakly BBB permeant.
Of the 52 identified molecules, 14 were found present in the ChEMBL database, meaning that about 73.1% correspond to new molecules without any information about their activity in potential targets.
3.3. Self- and Cross-Docking Analysis Supported High Correlation between Activity Values and Unbiased Docking Score Using Optimal 3D-Models
Self-docking results showed that MOE AlphaHB and GOLD ChemPLP scoring functions reproduced the co-crystallized ligands more accurately across both EGFR and PI3Kp110β structures (RMSD < 2.5 Å) (
Figure 4A1). For those protocols, the most representative 3D-models were based on PDBID:3w2s (EGFR) and PDBID:4bfr (PI3Kp110β) structures, assuming RMSD values between docked and X-ray poses of 1.251 and 0.342 Å, respectively. These findings were supported by cross-docking results since 3w2s and 4bfr were the structures able to accommodate the higher number of ligands with a pose overlapping the co-crystallized one (
Figure 4A2). MOE AlphaHB and GOLD ChemPLP functions also showed the best results in cross-docking simulations both on scoring and ranking power, by reproducing the crystallographic pose (0.24–2.45 Å and 0.22–2.20 Å RMSD ranges, respectively) and displaying the highest correlation between score and IC
50 values. Considering the protein–ligand interactions, these docking protocols successfully reproduced the consensus H-bonding residues involved in EGFR- and PI3-kinase domain-binding interface, namely, the residues Lys745, Asp855, Leu718, Met793, Phe856, and Gly857 for EGFR (
Figure 4B1), and Val848, Ile930, Met773, and Met92 for PI3Kp110β (
Figure 4B2).
Validated docking protocols consistently correlated standard molecules’ -pIC
50 values, within a representative range, with GOLD ChemPLP score for EGFR 3w2s structure and MOE AlphaHB score for PI3Kp110β 4bfr structure, assuming a linear correlation coefficient r
2 of 0.9036 (
Figure 4C1) and 0.881 (
Figure 4C2), respectively.
3.4. Unbiased in Silico Testing Supported Experimental Testing of 27 Molecules
The 52 screened molecules from ZINC15 were reduced to 27 molecules of interest, including 18 EGFR inhibitors (molecules 1–18), 6 PI3Kp110β inhibitors (molecules 19–24), and 3 dual inhibitors (molecules 25–27). The most relevant information about those molecules is summarized in
Table 1, namely, regarding the QSAR model’s predictions, and their docking profiling.
Overall selected molecules have good values of predicted BBB transport and molecular activity in respective targets. EGFR candidates assumed higher values of logBB ranging from 0.203 to 0.571 when compared to PI3Kp110β (0.0097–0.162) or dual target (−0.0718 to −0.156) candidates. However, when comparing the values of -pIC50 (>7) with the docking binding energies, we observed lower score values for EGFR candidates than would be expected. Even though, all EGFR candidates with low binding energies (<60) assumed a high ligand efficiency (>3). Overwhelming, PI3Kp110β candidates performed much better than the other candidates in docking simulations fitting the pocket of the target with the lowest energy resources probably due to their flexible chains.
3.5. ADMET Properties Assessment Supported Molecules’ Drug-Likeness
For the 27 selected molecules, most descriptors in silico estimated by QikProp were within the range of values for 95% of known drugs (
Table 1). Above 78% of the molecules naturally exhibit great pharmacological properties according to the Lipinski rule and are more likely to be membrane permeable and easily absorbed via passive diffusion in the human intestine. The molecular weight (MW) of considered molecules was below 600 Da, where about 74% presented optimal values under 400 Da.
All molecules were predicted to have central nervous system (CNS) activity only assuming passive BBB diffusion, except for one molecule (CNS activity: −2). Regarding octanol/water partition coefficient (QPlogPo/w) calculations, almost all candidates were at the higher range of the recommended values (>4), which translates to optimal lipidic solubility and high passive diffusion ability. All molecules showed low propension for binding human serum albumin (QplogKhsa) and forming non-BBB permeable complexes (QplogS). The apparent gut–blood barrier diffusional ability was also predicted (QPPCaco), supporting great absorption properties (>500 nm/s). All molecules presented a low number of reactive functional groups (#rtvFG), which can lead to false positives in screening assays and to decomposition, reactivity, or toxicity problems in vivo, supporting the effectiveness of the PAINS filter applied.
According with the ADMETsar2.0 analysis, most molecules were pointed as free of toxic/carcinogenic secondary effects and, as such, easily absorbed orally.
For each candidate are represented the following parameters: SMILES, QSAR model’s predictions of LogBB and –pIC50 for each target; probabilistic surface of molecular activity (PMSA) in respective targets; resultant score of the validated functions of docking simulation (CHEMPLP scoring for EGFR model and Alpha HB scoring for PI3Kp110β model); and respective ligand efficiency (score/number of heavy atoms).
3.6. Screening Analysis of Molecules’ Cytotoxicity in GB Cells Validated In Silico Predictions and Supported Low Range EC50 for 6 Promising Molecules
To establish the cytotoxic profile of the selected 27 molecules, cell viability was assessed (MTT test) in both U87MG and U87MG-wtEGFR cell lines (
Supplementary Figure S1). About 92% of the molecules caused a significant decrease in cell viability in at least one concentration and/or cell line. Most molecules targeting EGFR (1–18) performed better in U87MG-wtEGFR, supporting their specificity for the considered target and the higher sensitivity of this cell-line for TKI action. The cytotoxic effect observed for some molecules (e.g., 6–8), reached a plateau at very low concentrations, and it was especially evident in the U87MG-wtEGFR cell-line, suggesting some type of saturation.
When the threshold was set at 50% of cell viability, molecules 8, 17, 19, 20, 25, and 27 showed remarkable effects on both cell lines. For five of those molecules, the percentage of viable cells decreased in a dose-dependent manner, although the degree of cell viability impairment varied. Molecule 25 showed a remarkable cytotoxicity at 100 µM, with nearly 0% of viable cells. For the stated six molecules, additional assays were performed to establish the 24 h EC
50 for each GB cell-line (
Figure 5). Most molecules assumed lower EC
50 values in U87MG-wtEGFR when compared with the parental cell-line U87MG. Noteworthy, dual targeting candidates assumed the lowest values of EC
50 in both cell-lines (11–23 µM), inducing more than 80% of cell viability impairment.
3.7. Selected Molecules Treatment Led to Decreased EGFR and/or AKT Phosphorylation and Increased U87MG Cells Apoptosis
Activation of EGFR signaling is associated with its phosphorylation, as well as of downstream players. Thus, the expression of phosphorylated EGFR and AKT (downstream substrate of PI3K) in U87MG was analyzed by immunofluorescence after incubation with each selected molecule. Specifically, the status of T1068 residue of the EGFR-kinase domain and the Ser473 residue of AKT were analyzed.
For EGFR targeting molecules, 8 and 17, a relevant decrease in phospho-EGFR expression was observed after 1 h of exposure (
Figure 6A), reaching about 30% of that observed in control at 9 h (
p < 0.001,
Figure 6B). Moreover, an increase in nuclei with morphological changes typical of apoptosis, such as condensation of chromatin and nuclear fragmentation, was observed along time (
Figure 6A). By counting the percentage of apoptotic cells (
Figure 6C), both molecules induced almost 40% of cell death at 9 h (
p < 0.001). Even though, molecule 17 seems to have a faster anti-tumor effect by causing more apoptosis at early stages (
p < 0.001, 1 h) with statistical differences between treatments observed until 6 h (
p < 0.01, 1 h;
p < 0.001, 3 h and 6 h).
Analysis of phosphorylated AKT revealed that molecules 19 and 20 lead to a decreased expression as compared with the control (
Figure 7A), indicating that these molecules effectively inhibited AKT signaling pathway. Semi-quantitative analysis of the labeling intensity (
Figure 7B) showed a prevailing effect of molecule 19 (
p < 0.05, 1 h;
p < 0.01, 3 h;
p < 0.001, 6 h)
, indicating a faster action on the target than molecule 20. This translated into a higher apoptotic cell death (
Figure 7C), achieving about 35% at 9 h (
p < 0.001), as compared with about 15% observed for molecule 20 (
p < 0.001). Moreover, molecule 19 not only caused cell shrinkage and apoptotic bodies, but also caused nuclear blebbing and membrane lysis at later stages, consistent with an inflammatory process possibly delivered by necrosis (
Figure 7A, 6–9 h).
The effect of the dual targeting molecules, 25 and 27, in both phospho-EGFR and -AKT, was also assessed by double-labeling immunofluorescence analysis (
Figure 8). Immunofluorescence analysis indicated cell membrane alterations, noted by the rounded cell shape, detachment from the substrate, and fragmentation predominantly after 6 h incubation (
Figure 8A). Likewise, analysis of phospho-EGFR and phospho-AKT immunostaining intensity along time (
Figure 8B,C, respectively) suggests that both molecules inhibit the phosphorylated target expression mainly after 6 h exposure (>50%,
p < 0.001). Moreover, molecule 25 appears more specific for EGFR phosphorylation inhibition than molecule 27 (
p < 0.001, 3 h and 9 h), reducing the mean intensity of its phosphorylated form in 92.6% vs. 66.5% at 9 h (
p < 0.001).
Regarding apoptotic cell death (
Figure 8D), a 12.8% increase at 6 h (
p < 0.001) and a further rise to 31.9% at 9 h (
p < 0.001) were observed for molecule 27. Although, the anti-tumor effect of molecule 25 was groundbreaking when compared with molecule 27-induced cell death (
p < 0.001, 3–9 h), reaching almost 85% of apoptosis after 9 h exposure (
p < 0.001), which supports the previous screening results. Seemingly to molecule 19, molecule 25 induced several necrosis-like phenotypical markers. The effects of molecule 25-treatment occur in a short time window since apoptotic bodies and cell lysis were predominantly observed at 3 h and 6 h, whereas at 9 h almost no cells were found and the few that remained were clearly apoptotic (
Figure 8A).
These results validate the in silico predictions by showing that molecules with predicted ability to inhibit EGFR indeed compromise its activation as indicated by the decreased expression of the phosphorylated form of the receptor, and that those identified as inhibitors of PI3K reduce the activation of its downstream signaling molecule, AKT, as shown by the decreased expression of the phosphorylated form. Moreover, they showed that the dual inhibitors block the activation of both EGFR and PI3K signaling. Collectively, these results demonstrate the reliability of the QSAR model to reveal novel drug candidates for inhibition of EGFR and PI3K activation for a multitargeting therapeutics for GB.
3.8. Analysis of HBMEC Junctional Tightness, Morphology, and Efflux Activity Supported three Candidates as Tailored for Brain Delivery
MTT cell viability assay (
Figure 9A) revealed a lack of cytotoxicity of molecules 8, 19, and 20 in HBMEC at a concentration higher than the EC
50 (100 µM). Contrarywise, molecules 17, 25, and 27 showed cytotoxicity, impairing cell viability in about 65%, 100%, and 58%, respectively (
p < 0.001).
The junctional protein β-catenin expression was also analyzed to understand the effects of the molecules at EC
50 on endothelium integrity (
Figure 9B). In control cells, β-catenin exhibited a continuous line at the cell–cell contacts; however, after molecule 17 or 27 treatment, this junctional protein assumed an irregular and discontinuous appearance (
Figure 9B1). Molecules 17 and 27 disrupted barrier integrity (78% and 82% of control,
p < 0.001, respectively,
Figure 9B2), increasing the number and size of monolayer gaps (
Figure 9B3). Moreover, the distribution of β-catenin evaluated via densitometric analysis (
Figure 9B4), showed that after molecules 17 and 27 treatments HBMEC exhibited more cytoplasmatic (
p < 0.05 and
p < 0.001, respectively) and less membrane β-catenin (
p < 0.05) compared to controls, supporting a higher internalization of β-catenin and consequent compromise of barrier properties. Simultaneously, the results supported a high cytotoxic effect of molecule 25, inducing 100% of HBMEC detachment (
Figure 9B1,B2), and the absence of changes in β-catenin expression and location by the treatment with molecules 8, 19, and 20.
After a morphological study (
Figure 9B5), no significant changes in HBMEC shape were detected by molecules 8, 19, and 20 treatments. Contrarywise, molecules 17 and 27 increased cell roundness (
p < 0.001 and
p < 0.01, respectively). The morphological changes in HBMEC after the molecules treatment were also considered by F-actin cytoskeleton protein expression analysis (
Figure 9C1). Consistently with previous results, molecules 17 and 27 highly increased the number of stress fibers/cell, especially at the ventral cell surface, but molecule 20 also caused a slight intensification relative to the control baseline (
Figure 9C2). Molecule 25 again proved its high cytotoxicity by inducing all-cells detachment (
Figure 9C1).
Knowing the influence of efflux transporters in brain drug delivery, the expression of P-gp in HBMEC was also analyzed (
Supplementary Figure S2). According to prior in silico ADMET predictions, molecules 17 and 27 were pointed out as being more likely substrates of P-gp, which was validated by a significant increase in P-gp mean intensity after molecules treatment (
p < 0.05 and
p < 0.001, respectively). No significant changes in P-gp expression were detected after molecule 8, 19, and 20 treatments.
Overall, molecules 17, 25, and 27 were cytotoxic for HBMEC, while molecules 8, 19, and 20 were safe for direct drug delivery, thus proceeding to further assays of BBB transport.
3.9. Selected Molecules Proved to Efficiently Cross the BBB Reinforcing In Silico Predictions
The Transwell-based in vitro BBB model was used (
Figure 10A1) to assess the integrity of the HBMEC monolayer during assays with molecules 8, 19, and 20 (
Figure 10A2). None of the molecules markedly changed the TEER at 30 min of exposure, but unexpectedly molecule 20 caused a TEER decrease at 120 min (
p < 0.05;
Figure 10B1). This TEER decrease did not cause barrier leakiness as demonstrated by immunostaining for ZO-1 and vimentin, junctional, and cytoskeleton proteins, respectively (
Figure 10B2). Similarly, analysis of HBMEC permeability to sodium fluorescein (
Figure 10B3) indicated no statistical differences between treated cells and control, presenting an index of paracellular diffusion (Pe) in line with previous findings [
37].
Taking advantage of the validated in vitro model of the human BBB, we confirmed by UPLC tandem mass spectrometry that molecules 8, 19, and 20 could be transported across the BBB endothelium. Results show the different retention times and the well-resolved peaks, where respective integration of peak areas (PA) enabled to estimate the concentration of each molecule in both chambers either after 30 or 120 min incubation (
Figure 10C1,C2). These data suggest similar levels of molecules 8 and 19 in upper and lower chambers at 30 min, supporting a higher BBB permeation when compared to molecule 20. Moreover, we observed that at 120 min, molecules 8 and 19 were mostly found in lower compartments, whereas such a difference was less evident for molecule 20. Interestingly, a trend of an overall decrease in time of total PA in both compartments was noted, suggesting that HBMEC can metabolize a fraction of the molecules (data not shown).
Analysis of endothelial transport efficiency showed that molecules 8 and 19 are transported across the HBMEC monolayer in a time-dependent manner (
p < 0.001, molecule 8;
p < 0.05, molecule 19, 30 vs. 120 min), while molecule 20 transport did not increase significantly from 30 to 120 min of incubation (
Figure 10C3). All molecules proved a higher-off range transport relatively to well-known BBB permeable compounds.
LogBB values were also calculated as a measure of brain penetration extent. Mean values of LogBB experimentally obtained at 30 and 120 min were impressively correlated with in silico predictions (
Table 1), translated by high linear correlation coefficients (
Figure 10D).
3.10. Selected Molecules Crossed the BBB Endothelium and Induced Cytotoxicity in GB Cells in a Representative Co-Culture Model
Molecules’ transport and cytotoxicity were assessed in an archetypical BBB-GB co-culture model, implemented in our laboratory (
Figure 11A1), and through a basolateral conditioned experiment (
Figure 11A2). Since molecule 8 is specific for EGFR inhibition, its effect was analyzed both in HBMEC:U87MG and HBMEC:U87MG-wtEGFR co-culture systems, while, molecules 19 and 20 only were evaluated in HBMEC:U87MG co-culture. The drugs were added to the “blood side” and their effect in GB cells in the “brain side” was assessed following 2 h of incubation, a timepoint at which drugs were able to cross the BBB endothelium (
Figure 10).
Compared to HBMEC mono-cultured, the presence of U87MG or U87MG-wtEGFR induced a slight decrease in barrier tightness, indicated by the decreased staining for ZO-1 (
Figure 11B). Semi-quantitative analysis of ZO-1 mean intensity (
Figure 11C) revealed a decrease in the co-culture system free of drug treatment (
p < 0.001), especially in the presence of U87MG-wtEGFR (
p < 0.05). As expected, TEER readings were not decreased by molecules 8, 19, and 20, supporting their non-cytotoxic profile for HBMEC (
Figure 11D), and corroborated by the unaltered ZO-1 immunostaining (
Figure 11B). In line with ZO-1 expression, a slight TEER decrease in HBMEC co-incubated with GB cells vs. mono-cultured was observed, although not significant.
The three molecules impaired U87MG B cell viability in the co-culture system (
p < 0.001, molecule 8;
p < 0.01, molecule 19;
p < 0.05, molecule 20;
Figure 11E), and Mol8 also impaired U87MG-wtEGFR in co-cultures (
p < 0.001), confirming their BBB permeation. However, none of the molecules induced a statistically significant difference in cell viability of U87MG incubated with basolateral conditioned medium of HBMEC in the BBB model, with a significant difference only observed for U87MG-wtEGFR incubated with molecule 8 (
p < 0.05). The marked decrease in GB cell viability in the co-culture system suggests that GB cells release soluble factors that increase BBB’s permeability, which may turn into an advantage for drug delivery for GB treatment. Overall, these studies indicate that the studied drug candidates may permeate across the BBB endothelium and reach therapeutic concentrations at the target GB cells in the “brain side” of the experimental system.
4. Discussion
An automated and extendable platform based on our previous work [
20] was used as a QSAR modeling pipeline to obtain optimized predictive models for three different chemical problems, namely overcoming the BBB and targeting EGFR and PI3Kp110β signaling.
The obtained results of the developed BBB permeation QSAR model largely exceed the quality of the most recent published empirical models, most of which lack biological validation [
39,
40]. Still, the developed BBB’s permeation model is especially better in predicting compounds that can cross the BBB than the non-permeable ones, which makes it more tailored to be incorporated in drug discovery programs for brain delivery than non-CNS contexts. Concerning EGFR and PI3Kp110β inhibition models, they represent pioneering efforts in the data mining field since no targeted drug for brain tumors elected by regression QSAR models met the conditions to achieve clinical trials. Importantly, no QSAR model predicting the specific inhibition of PI3K subunit p110β has ever been built, because most efforts have focused on subunit p110α, which also supports the relevance of our models for further drug discovery programs. It is important to mention that although the identified molecules are predicted to specifically inhibit the β isoform, inhibition of the α isoform may not be disregarded. Anyway, considering that both isoforms lead to AKT activation, any contribution of the α isoform would be reflected in the phospho-AKT expression.
The produced models were used to screen the ZINC15 database. The latest version of ZINC15 accounts for more than 200 million “drug-like” molecules readily available for commercial purchase, some of which are associated with specific targets or diseases, opening new-fangled possibilities for their repurposing for novel clinical applications. From ZINC15 screening, five Food and Drug Administration (FDA)-approved compounds were found (canertinib, naquotinib, tesevatinib, poziotinib, and PKI-166), with potential to be repurposed in the GB clinical context. Moreover, the other candidates represent new options never explored before.
Allying the straightforward results from ZINC15 screening with docking studies represents a powerful strategy that has helped unravel the structural and energetic bases of the interaction between targets and candidate molecules. Overall, both EGFR and PI3Kp110β 3D-models allowed a great reproducibility of ligands binding mode and exhibit great performance in the establishment of a relationship between score and inhibitory capacity, clearly distinguishing between active and non-active compounds. By visual inspection of the molecules in the respective pocket and analysis of established bounding, we observed that most interactions were assured. These results, together with the predicted ADMET profile and drug-like phenotype of selected small molecules, strongly support the studied molecules as robust and qualified candidates for experimental testing.
The cytotoxic potential of the selected 27 molecules (18 EGFR inhibitors, 6 PI3Kp110β inhibitors, and 3 dual inhibitors) in two well-recognized GB cell lines, namely U87MG (parental) and U87MG overexpressing EGFR (U87MG-wtEGFR), was explored by cell viability analysis. Most tested molecules promoted tumor cell death, from which two EGFR inhibitors (molecules 8 and 17), two PI3Kp110β inhibitors (molecules 19 and 20), and two dual inhibitors (molecules 25 and 27) caused more than 50% of the cell death. Their respective dose–response curves fetched EC50 values in the 16–130 µM and 0.09–70 µM range for U87MG and U87MG-wtEGFR, respectively, which largely outperform drugs currently under investigation.
The effectiveness of the EGFR TKI gefitinib was tested in recurrent GB, but the downstream effectors remained constitutively active, leading to unsatisfactory effectiveness [
41]. More recently, efforts were focused on the use of dacomitinib, an oral, irreversible, second-generation, pan-HER TKI [
8], but poor global results in recurrent GB were obtained in a phase II clinical trial [
42]. Moreover, most of those molecules showed poor tumor-suppressive effects in pre-clinical studies under concentrations of 100 µM [
43,
44], which render our EGFR candidates powerful interviewees in the current market.
Regarding PI3K inhibitors, the new generation of pan-PI3K inhibitors, such as BKM120 and PX-866, exhibit improved drug properties such as high stability and low side effects [
45]. However, their use has been halted in clinical trials due to toxicity, poor pharmacodynamics, and selectivity [
46]. Despite recent advances in the study of selective PI3K inhibitors, such as alpelisib and idelalisib, no clinical study is currently ongoing focusing on the inhibition of the p110β subunit. The key factor that may emerge is that although several dual PI3K/mTOR inhibitors are being investigated (XL765, PKI-587), no dual EGFR/PI3K has been known until now. Consequently, our results point to a new field of therapeutic opportunities for GB treatment. Besides all efforts to develop new medicines, the alkylating agent temozolomide (TMZ) remains the standard of care for GB patients [
4]. Surprisingly, several cell-based studies have proved that TMZ dosage is far beyond the therapeutic recommended concentrations, where more than 50% cytotoxicity was only seen in up to 4000 µM doses [
47,
48]. Moreover, a recent systematic review of the sensitivity to TMZ of several glioma cell lines revealed the IC
50 values in U87 cells within the 124–230 µM range, depending on the incubation time, and that the median IC
50 at 72 h for patient-derived cell lines was 800 μM [
49]. This strongly wires the potential of our drug candidates, which revealed much lower EC
50 values. Moreover, they raise the hypothesis that a combined therapy with TMZ and one of the best drug candidates may represent a possible multitargeting therapeutic strategy for GB.
The immunofluorescence analysis supported the direct impact of the identified drug candidates in the proliferation and viability of tumor cells, translated by a reduction in the cell number and an increase in apoptotic markers along time. The key point of these results may rely on the fact that from all six selected candidates, molecule 25 (canertinib) is approved for esophageal squamous cell carcinoma, which may emerge as an actual opportunity for repurposing.
As previously mentioned, many TKIs displayed excellent in vitro efficacy but failed in pre-clinical and clinical trials because of their great toxicity and high concentrations, which results in heavy side effects and toxicities [
50]. This attests the need of considering off-target effects in the drug discovery and development process. This issue was considered in the present work by including ADMET predictions in the computational studies. It was further addressed through analysis of the safety of the most promising drug candidates to resident cells. In particular, we analysed the drugs’ effects in HBMEC, considering that the focus of this work is on brain pathology and BBB permeation. Unfortunately, molecules 17, 25, and 27 showed significant cytotoxic effects against HBMEC, but they can be pursued in further studies involving GB-targeted nanomedicines, to achieve a therapeutic concentration of the drug at the intended target site, with a lower exposure in non-target organs. Contrastingly, molecules 8, 19, and 20 under the <100 µM range did not elicit toxicity, in line with the ADMET predictions. Indeed, they did not induce changes in the integrity and physiological properties of the BBB, as indicated by unaltered cell viability and morphology of endothelial cells. The results were validated by the integrity of the endothelial paracellular permeability, as shown by the absence of changes in β-catenin expression, TEER, and Pe to sodium fluorescein.
It is known that most brain targeting drugs are likely activators of BBB efflux transporters, namely, the highly expressed P-gp, which can lead to a multidrug resistance phenotype and dump any expected therapeutic effect. This is a major challenge in GB treatment since most targeted drugs developed so far are actively effluxed out of the brain, namely, some alkylating agents (e.g., TMZ), and TKI (e.g., erlotinib, dasatinib, and imatinib) [
51]. In this challenging context, our results arose in accordance with the in silico predictions, where molecules 17 and 27 appeared as potential P-gp substrates by increasing its expression, while molecules 8, 19, and 20 were poorly associated with P-gp alterations, which points to them as promising choices for brain accessibility.
To evaluate BBB’s permeation of the selected molecules based on their safety to the BBB (8, 19, and 20), a well-validated Transwell-based in vitro BBB model was used [
37]. The transport of these molecules across the BBB was unequivocally demonstrated within the tested concentration range, through a selective and sensitive UPLC-MS/MS method. When looking to transport efficiency results, we observed an impressive higher rate of brain penetration compared to other known BBB permeable compounds, especially for molecules 8 and 19. Curiously, molecule 20 transport revealed itself as non-time-dependent, which may reflect the involvement of other transport mechanisms subjected to transporter saturation. The chemical ability of some metabolites to establish hydrogen bonds with functional groups of biomolecules could suggest a propensity for an active transport mechanism in BBB. This was especially indicated by molecule 20 in silico descriptors, and experimental transport assay results. The chief factor may rely on the remarkable correlation between QSAR predicted values and experimental validated values of LogBB, which supports the accuracy and feasibility of our model in predicting BBB’s permeability.
In line with preceding findings, co-culture experiments proved that after exposing the luminal (“blood”) side of HBMEC to molecules 8, 19, and 20, increased delivery and cytotoxicity in GB cells growing on the “brain side” were observed compared to GB cells only incubated with the basolateral conditioned medium. These results suggest that GB cells are releasing soluble factors that may decrease BBB’s tightness and increase its permeability. Several studies have reported that vessels surrounding GB are leaky, in particular those with endothelial hyperplasia, commonly showing abnormal structural features, such as loss and/or abnormal morphology of TJs [
52]. As such, it is not surprising that GB cells may have tried to subvert HBMEC cells to switch to a defective phenotype as supported by a decreased expression of ZO-1 and lower values of TEER obtained in HBMEC co-cultured with GB cells.
The overall results point to the discovered molecules as a step forward in GB treatment regarding delivery across the BBB, and strong anti-tumor activity against GB cells and non-identified collateral toxicities. Therefore, the in vivo efficacy of those molecules should be investigated in pre-clinical settings.


 ,
, 











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}