Oleocanthal Attenuates Metastatic Castration-Resistant Prostate Cancer Progression and Recurrence by Targeting SMYD2

 , and
, and
Abstract
:Simple Summary
Abstract
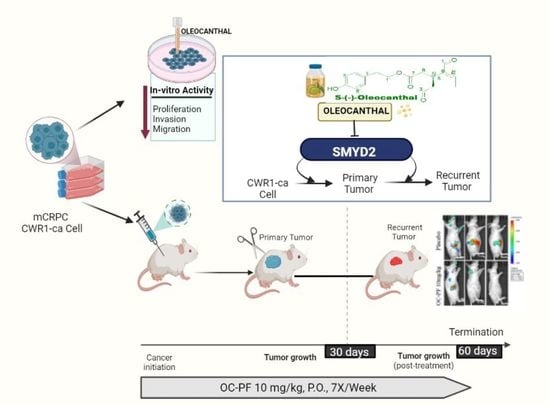
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Cell Lines and Culture Conditions
2.3. Experimental Treatments
2.4. Cell Viability Assay
2.5. CRISPR-Cas9 SMYD2 Knockout (KO)
2.6. Wound-Healing Assay
2.7. Colony Formation Assay
2.8. Transwell Migration Assay
2.9. Invasion Assay
2.10. Western Blot Analysis
2.11. Lentivirus Transduction-Aided Luciferase Labeling of CWR-R1ca Cells
2.12. Animal Models and Treatment
2.12.1. OC Suppressed mCRPC CWR-R1ca-Luc Cell Progression In Vivo in Nude Mouse Model
2.12.2. Oral Treatments with OC-PF Effectively Suppressed mCRPC CWR-R1ca-Luc Cells’ Recurrence in Nude Mouse Model after Primary Tumor Surgical Excision
2.13. Quantification of PSA Serum Levels Using ELISA Kit
2.14. Immunohistochemistry (IHC) Study
2.15. Hematoxylin and Eosin Y (H&E) Staining
2.16. Analysis of Clinical SMYD2 Gene Expression Data
2.17. Statistics
3. Results
3.1. Prognosis Analysis of SMYD Family mRNA Expression in Prostate Cancer Patients
3.2. Prognostic Analysis of SMYD2 mRNA Expression in PC Patients
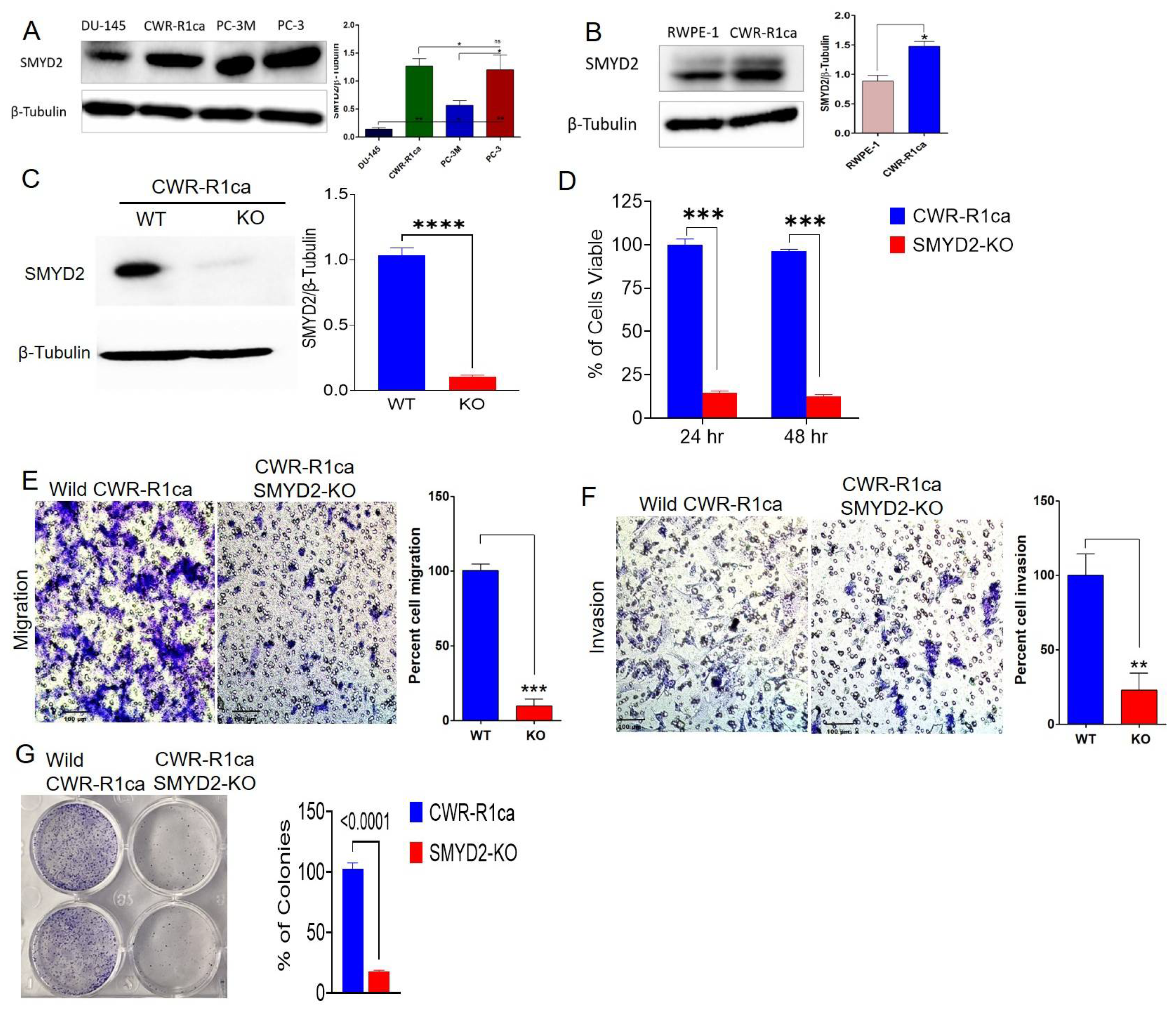
3.3. Exploration of SMYD2 Expression Pattern in PC and Non-Tumorigenic RWPE-1 Prostatic Epithelial Cells
3.4. SMYD2 Is Essential for mCRPC Cell Survival and Motility
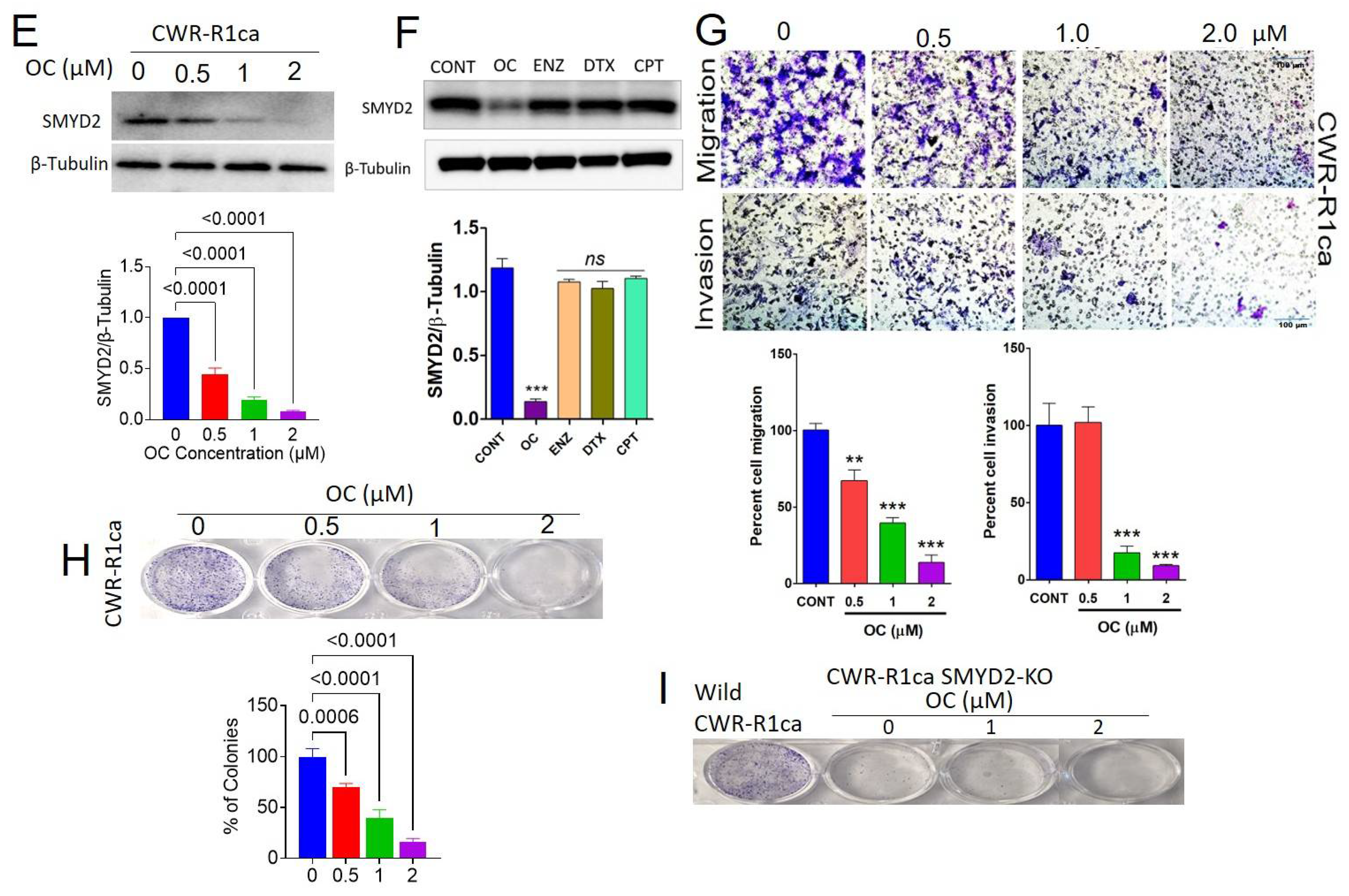
3.5. OC Inhibited SMYD2 Monomethylation in Biochemical Assays
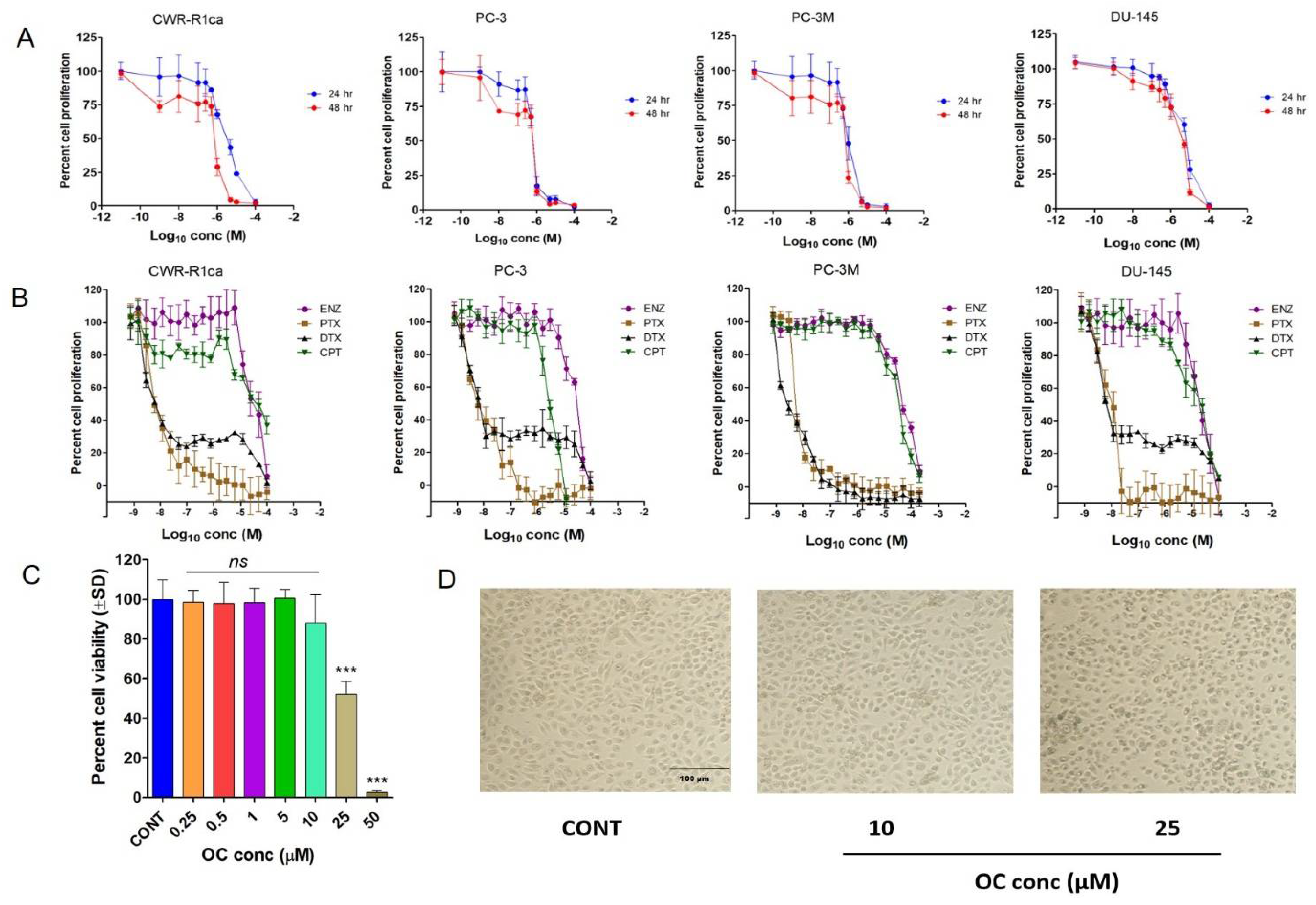
3.6. OC Treatments Selectively Suppressed the Viability of PC Cells by Reducing SMYD2 Expression but Did Not Adversely Affect the Non-Tumorigenic RWPE-1 Prostatic Epithelial Cells’ Viability at Therapeutic Doses
3.7. OC Treatments Suppressed the Migration and Invasion of the CRPC CWR-R1ca Cells
3.8. OC Treatments Inhibited the Colony Formation of the Wild but Not the SMYD2-KO CWR-R1ca Cells
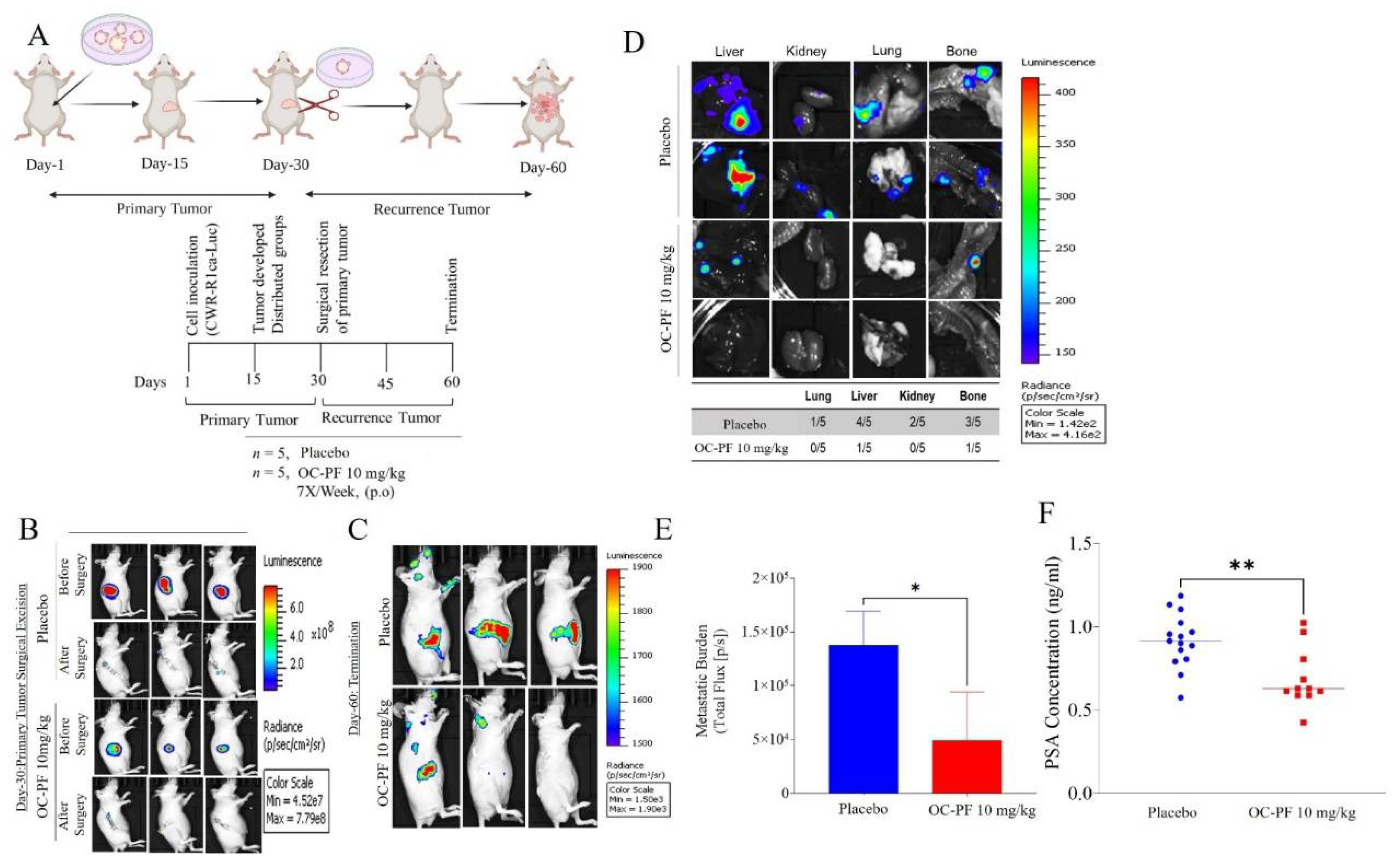
3.9. Oral OC-PF Treatments Suppressed the Progression of CWR-R1ca-Luc Cells Engrafted into Nude Mice
3.10. Immunofluorsence Expression Comparison of ki67 and CD31 in OC Versus Placebo Control Treatments in Collected mCRPC Primary Tumors
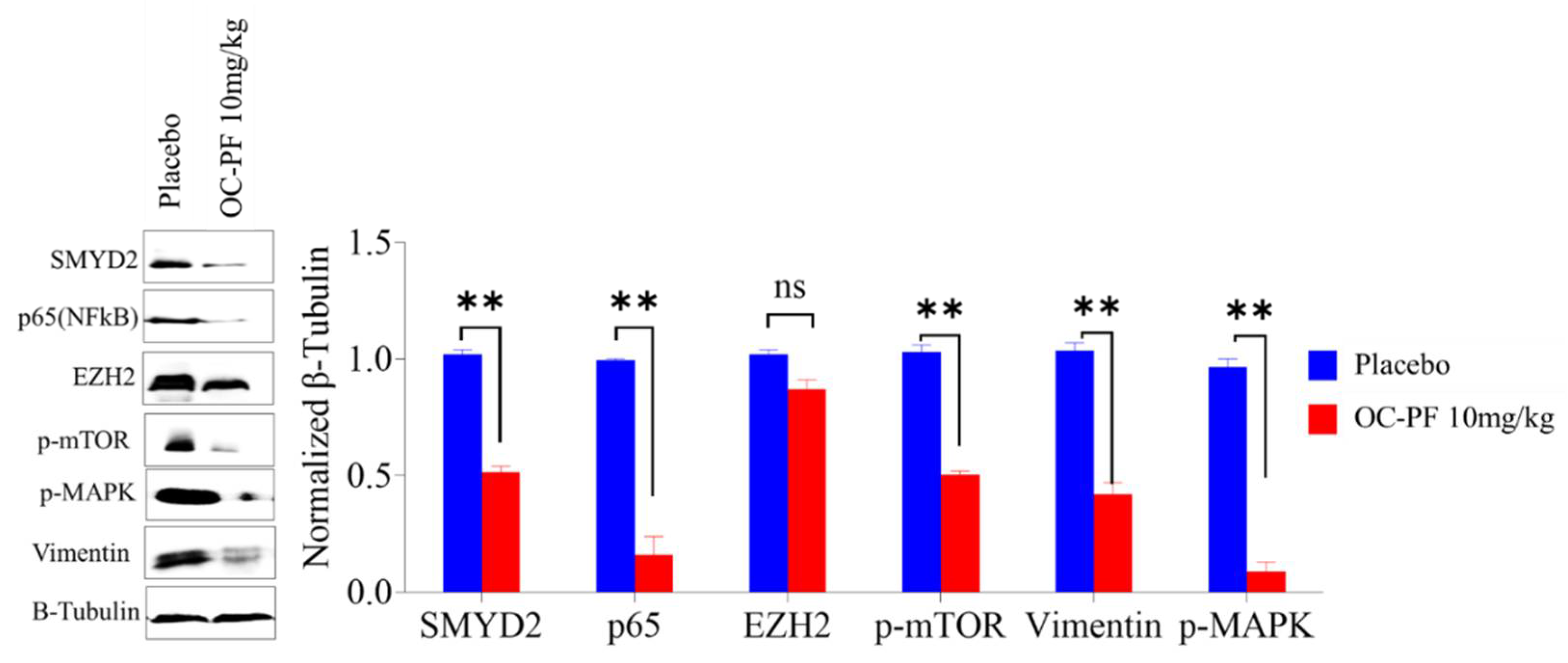
3.11. OC Treatments Reduced the Expression of SMYD2 and Downstream Signals in Collected mCRPC Primary Tumors
3.12. Oleocanthal PF Treatment Inhibited Locoregional Recurrence of the mCRPC CWR-R1ca Cells in Nude Mice after Primary Tumor Surgical Excision
3.13. In Vivo Safety of OC-PF Treatments
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Giri, V.N.; Morgan, T.M.; Morris, D.S.; Berchuck, J.E.; Hyatt, C.; Taplin, M.E. Genetic testing in prostate cancer management: Considerations informing primary care. CA Cancer J. Clin. 2022, 72, 360–371. [Google Scholar] [CrossRef] [PubMed]
- Gandaglia, G.; Abdollah, F.; Schiffmann, J.; Trudeau, V.; Shariat, S.F.; Kim, S.P.; Perrotte, P.; Montorsi, F.; Briganti, A.; Trinh, Q.D.; et al. Distribution of metastatic sites in patients with prostate cancer: A population-based analysis. Prostate 2014, 74, 210–216. [Google Scholar] [CrossRef]
- Giri, V.N.; Knudsen, K.E.; Kelly, W.K.; Cheng, H.H.; Cooney, K.A.; Cookson, M.S.; Dahut, W.; Weissman, S.; Soule, H.R.; Petrylak, D.P.; et al. Implementation of germline testing for prostate cancer: Philadelphia Prostate Cancer Consensus Conference 2019. J. Clin. Oncol. 2020, 38, 2798–2811. [Google Scholar] [CrossRef]
- Abida, W.; Patnaik, A.; Campbell, D.; Shapiro, J.; Bryce, A.H.; McDermott, R.; Sautois, B.; Vogelzang, N.J.; Bambury, R.M.; Voog, E.; et al. Triton2 investigators. Rucaparib in men with metastatic castration-resistant prostate cancer harboring a BRCA1 or BRCA2 gene alteration. J. Clin. Oncol. 2020, 38, 3763–3772. [Google Scholar] [CrossRef]
- de Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for metastatic castration-resistant prostate cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef]
- Yi, X.; Jiang, X.J.; Fang, Z.M. Histone methyltransferase SMYD2: Ubiquitous regulator of disease. Clin. Epigenetics 2019, 11, 112. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Li, L.; Zhang, H.; Luo, X.; Dai, J.; Zhou, S.; Gu, J.; Zhu, J.; Atadja, P.; Lu, C.; et al. Structure of human SMYD2 protein reveals the basis of p53 tumor suppressor methylation. J. Biol. Chem. 2011, 286, 38725–38737. [Google Scholar] [CrossRef] [Green Version]
- Zipin-Roitman, A.; Aqaqe, N.; Yassin, M.; Biechonski, S.; Amar, M.; van Delft, M.F.; Gan, O.I.; McDermott, S.P.; Buzina, A.; Ketela, T.; et al. SMYD2 lysine methyltransferase regulates leukemia cell growth and regeneration after genotoxic stress. Oncotarget 2017, 8, 16712–16727. [Google Scholar] [CrossRef] [Green Version]
- Yan, L.; Ding, B.; Liu, H.; Zhang, Y.; Zeng, J.; Hu, J.; Yao, W.; Yu, G.; An, R.; Chen, Z.; et al. Inhibition of SMYD2 suppresses tumor progression by down-regulating microRNA-125b and attenuates multi-drug resistance in renal cell carcinoma. Theranostics 2019, 9, 8377–8391. [Google Scholar] [CrossRef]
- Skawran, B.; Steinemann, D.; Weigmann, A.; Flemming, P.; Becker, T.; Flik, J.; Kreipe, H.; Schlegelberger, B.; Wilkens, L. Gene expression profiling in hepatocellular carcinoma: Upregulation of genes in amplified chromosome regions. Mod. Pathol. 2008, 21, 505–516. [Google Scholar] [CrossRef] [Green Version]
- Reynoird, N.; Mazur, P.K.; Stellfeld, T.; Flores, N.M.; Lofgren, S.M.; Carlson, S.M.; Brambilla, E.; Hainaut, P.; Kaznowska, E.B.; Arrowsmith, C.H.; et al. Coordination of stress signals by the lysine methyltransferase SMYD2 promotes pancreatic cancer. Genes Dev. 2016, 30, 772–785. [Google Scholar] [CrossRef]
- Meng, F.; Liu, X.; Lin, C.; Xu, L.; Liu, J.; Zhang, P.; Zhang, X.; Song, J.; Yan, Y.; Ren, Z.; et al. SMYD2 suppresses APC2 expression to activate the Wnt/beta-catenin pathway and promotes epithelial-mesenchymal transition in colorectal cancer. Am. J. Cancer Res. 2020, 10, 997–1011. [Google Scholar]
- Li, L.X.; Zhou, J.X.; Calvet, J.P.; Godwin, A.K.; Jensen, R.A.; Li, X. Lysine methyltransferase SMYD2 promotes triple negative breast cancer progression. Cell Death Dis. 2018, 9, 326. [Google Scholar] [CrossRef]
- Kukita, A.; Sone, K.; Oda, K.; Hamamoto, R.; Kaneko, S.; Komatsu, M.; Wada, M.; Honjoh, H.; Kawata, Y.; Kojima, M.; et al. Histone methyltransferase SMYD2 selective inhibitor LLY-507 in combination with poly ADP ribose polymerase inhibitor has therapeutic potential against high-grade serous ovarian carcinomas. Biochem. Biophys. Res. Commun. 2019, 513, 340–346. [Google Scholar] [CrossRef]
- Komatsu, S.; Imoto, I.; Tsuda, H.; Kozaki, K.I.; Muramatsu, T.; Shimada, Y.; Aiko, S.; Yoshizumi, Y.; Ichikawa, D.; Otsuji, E.; et al. Overexpression of SMYD2 relates to tumor cell proliferation and malignant outcome of esophageal squamous cell carcinoma. Carcinogenesis 2009, 30, 1139–1146. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.S.; Hayami, S.; Toyokawa, G.; Maejima, K.; Yamane, Y.; Suzuki, T.; Dohmae, N.; Kogure, M.; Kang, D.; Neal, D.E.; et al. RB1 methylation by SMYD2 enhances cell cycle progression through an increase of RB1 phosphorylation. Neoplasia 2012, 14, 476–786. [Google Scholar] [CrossRef] [Green Version]
- Fabini, E.; Manoni, E.; Ferroni, C.; Rio, A.D.; Bartolini, M. Small-molecule inhibitors of lysine methyltransferases SMYD2 and SMYD3: Current trends. Future Med. Chem. 2019, 11, 901–921. [Google Scholar] [CrossRef]
- Rubio-Tomás, T. Novel insights into SMYD2 and SMYD3 inhibitors: From potential anti-tumoural therapy to a variety of new applications. Mol. Biol. Rep. 2021, 48, 7499–7508. [Google Scholar] [CrossRef]
- Kaniskan, H.U.; Martini, M.L.; Jin, J. Inhibitors of protein methyltransferases and demethylases. Chem. Rev. 2018, 118, 989–1068. [Google Scholar] [CrossRef] [PubMed]
- Vieira, F.Q.; Costa-Pinheiro, P.; Almeida-Rios, D.; Graça, I.; Monteiro-Reis, S.; Simões-Sousa, S.; Carneiro, I.; Sousa, E.J.; Godinho, M.; Baltazar, F.; et al. SMYD3 contributes to a more aggressive phenotype of prostate cancer and targets Cyclin D2 through H4K20Me3. Oncotarget 2015, 6, 13644–13657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, K.L.; Chin, K.Y. The biological activities of oleocanthal from a molecular perspective. Nutrients 2018, 10, 570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beauchamp, G.K.; Keast, R.S.; Morel, D.; Lin, J.; Pika, J.; Han, Q.; Lee, C.H.; Smith, A.B.; Breslin, P.A. Phytochemistry: Ibuprofen-like activity in extra-virgin olive oil. Nature 2005, 437, 45–46. [Google Scholar] [CrossRef] [PubMed]
- Siddique, A.B.; Ayoub, N.M.; Tajmim, A.; Meyer, S.A.; Hill, R.A.; El Sayed, K.A. (−)-Oleocanthal prevents breast cancer locoregional recurrence after primary tumor surgical excision and neoadjuvant targeted therapy in orthotopic nude mouse models. Cancers 2019, 11, 637. [Google Scholar] [CrossRef] [Green Version]
- Siddique, A.B.; Kilgore, P.; Tajmim, A.; Singh, S.S.; Meyer, S.A.; Jois, S.D.; Cvek, U.; Trutschl, M.; El Sayed, K.A. (−)-Oleocanthal as a dual c-MET-COX2 inhibitor for the control of lung cancer. Nutrients 2020, 12, 1749. [Google Scholar] [CrossRef]
- Khanfar, M.A.; Bardaweel, S.K.; Akl, M.R.; El Sayed, K.A. Olive oil-derived oleocanthal as potent inhibitor of mammalian target of rapamycin: Biological evaluation and molecular modeling studies. Phytother. Res. 2015, 29, 1776–1782. [Google Scholar] [CrossRef] [Green Version]
- Pei, T.; Meng, Q.; Han, J.; Sun, H.; Li, L.; Song, R.; Sun, B.; Pan, S.; Liang, D.; Liu, L. (−)-Oleocanthal inhibits growth and metastasis by blocking activation of STAT3 in human hepatocellular carcinoma. Oncotarget 2016, 7, 43475–43491. [Google Scholar] [CrossRef] [Green Version]
- Tajmim, A.; Siddique, A.B.; El Sayed, K. Optimization of Taste-Masked (−)-Oleocanthal effervescent formulation with potent breast cancer progression and recurrence suppressive activities. Pharmaceutics 2019, 11, 515. [Google Scholar] [CrossRef] [Green Version]
- Qusa, M.H.; Siddique, A.B.; Nazzal, S.; El Sayed, K.A. Novel olive oil phenolic (−)-oleocanthal (+)-xylitol-based solid dispersion formulations with potent oral anti-breast cancer activities. Int. J. Pharm. 2019, 569, 118596. [Google Scholar] [CrossRef]
- Westaby, D.; Viscuse, P.V.; Ravilla, R.; de la Maza, M.D.F.; Hahn, A.; Sharp, A.; de Bono, J.; Aparicio, A.; Fleming, M.T. Beyond the androgen receptor: The sequence, the mutants, and new avengers in the treatment of castrate-resistant metastatic prostate cancer. Am. Soc. Clin. Oncol. Educ. Book 2021, 41, e190–e202. [Google Scholar] [CrossRef]
- Siddique, A.B.; Ebrahim, H.; Mohyeldin, M.; Qusa, M.; Batarseh, Y.; Fayyad, A.; Tajmim, A.; Nazzal, S.; Kaddoumi, A.; El Sayed, K.A. Novel liquid-liquid extraction and self-emulsion methods for simplified isolation of extra-virgin olive oil phenolics with emphasis on (−)-oleocanthal and its oral anti-breast cancer activity. PLoS ONE 2019, 14, e0214798. [Google Scholar] [CrossRef] [Green Version]
- Shourideh, M.; DePriest, A.; Mohler, J.L.; Wilson, E.M.; Koochekpour, S. Characterization of fibroblast-free CWR-R1ca castration-recurrent prostate cancer cell line. Prostate 2016, 76, 1067–1077. [Google Scholar] [CrossRef]
- Siddique, A.B.; Ebrahim, H.Y.; Akl, M.R.; Ayoub, N.M.; Goda, A.A.; Mohyeldin, M.M.; Nagumalli, S.K.; Hananeh, W.M.; Liu, Y.Y.; Meyer, S.A.; et al. (−)-Oleocanthal combined with lapatinib treatment synergized against HER-2 positive breast cancer in vitro and in vivo. Nutrients 2019, 11, 412. [Google Scholar] [CrossRef] [Green Version]
- Tajmim, A.; Cuevas-Ocampo, A.K.; Siddique, A.B.; Qusa, M.H.; King, J.A.; Abdelwahed, K.S.; Sonju, J.J.; El Sayed, K.A. (−)-Oleocanthal nutraceuticals for Alzheimer’s disease amyloid pathology: Novel oral formulations, therapeutic, and molecular insights in 5xFAD transgenic mice model. Nutrients 2021, 13, 1702. [Google Scholar] [CrossRef]
- MedixGraph Website. Available online: http://medixgraph.com/cfuscope (accessed on 28 February 2022).
- Clark, A.J.; Fakurnejad, S.; Ma, Q.; Hashizume, R. Bioluminescence imaging of an immunocompetent animal model for glioblastoma. J. Vis. Exp. 2016, 107, e53287. [Google Scholar] [CrossRef] [Green Version]
- University of Alabama-Birmingham Website. Available online: http://ualcan.path.uab.edu (accessed on 28 February 2022).
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.; Varambally, S. UALCAN: A portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [Green Version]
- Gepia-Cancer Website. Available online: http://gepia.cancer-pku.cn/ (accessed on 28 February 2022).
- cBioportal Website. Available online: http://cbioportal.org (accessed on 28 February 2022).
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, 11. [Google Scholar] [CrossRef] [Green Version]
- Friedl, P.; Wolf, K. Tumour-cell invasion and migration: Diversity and escape mechanisms. Nat. Rev. Cancer 2003, 3, 362–374. [Google Scholar] [CrossRef]
- Franken, N.A.; Rodermond, H.M.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef]
- Nguyen, H.; Allali-Hassani, A.; Antonysamy, S.; Chang, S.; Chen, L.H.; Curtis, C.; Emtage, S.; Fan, L.; Gheyi, T.; Li, F.; et al. LLY-507, a cell-active, potent, and selective inhibitor of protein-lysine methyltransferase SMYD2. J. Biol. Chem. 2015, 290, 13641–13653. [Google Scholar] [CrossRef] [Green Version]
- Elnagar, A.Y.; Sylvester, P.W.; El Sayed, K.A. (−)-Oleocanthal as a c-Met inhibitor for the control of metastatic breast and prostate cancers. Planta Med. 2011, 77, 1013–1019. [Google Scholar] [CrossRef]
- Chandrasekar, T.; Yang, J.C.; Gao, A.C.; Evans, C.P. Mechanisms of resistance in castration-resistant prostate cancer (CRPC). Transl. Androl. Urol. 2015, 4, 365–380. [Google Scholar]
- Zeng, Y.; Qiu, R.; Yang, Y.; Gao, T.; Zheng, Y.; Huang, W.; Gao, J.; Zhang, K.; Liu, R.; Wang, S.; et al. Regulation of EZH2 by SMYD2-mediated lysine methylation is implicated in tumorigenesis. Cell Rep. 2019, 29, 1482–1498.e4. [Google Scholar] [CrossRef]
- Komatsu, S.; Ichikawa, D.; Hirajima, S.; Nagata, H.; Nishimura, Y.; Kawaguchi, T.; Miyamae, M.; Okajima, W.; Ohashi, T.; Konishi, H.; et al. Overexpression of SMYD2 contributes to malignant outcome in gastric cancer. Br. J. Cancer 2015, 112, 357–364. [Google Scholar] [CrossRef] [Green Version]
- Wagner, T.; Jung, M. New lysine methyltransferase drug targets in cancer. Nat. Biotechnol. 2012, 30, 622–623. [Google Scholar] [CrossRef]
- Gottlieb, P.D.; Pierce, S.A.; Sims, R.J.; Yamagishi, H.; Weihe, E.K.; Harriss, J.V.; Maika, S.D.; Kuziel, W.A.; King, H.L.; Olson, E.N.; et al. Bop encodes a muscle-restricted protein containing MYND and SET domains and is essential for cardiac differentiation and morphogenesis. Nat. Genet. 2002, 31, 25–32. [Google Scholar] [CrossRef]
- Hamamoto, R.; Furukawa, Y.; Morita, M.; Iimura, Y.; Silva, F.P.; Li, M.; Yagyu, R.; Nakamura, Y. SMYD3 encodes a histone methyltransferase involved in the proliferation of cancer cells. Nat. Cell Biol. 2004, 6, 731–740. [Google Scholar] [CrossRef]
- Hu, L.; Zhu, Y.T.; Qi, C.; Zhu, Y.J. Identification of Smyd4 as a potential tumor suppressor gene involved in breast cancer development. Cancer Res. 2009, 69, 4067–4072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stender, J.D.; Pascual, G.; Liu, W.; Kaikkonen, M.U.; Do, K.; Spann, N.J.; Boutros, M.; Perrimon, N.; Rosenfeld, M.G.; Glass, C.K. Control of proinflammatory gene programs by regulated trimethylation and demethylation of histone H4K20. Mol. Cell 2012, 48, 28–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akl, M.R.; Busnena1, B.A.; Mohyeldin, M.M.; Foudah, A.I.; Ayoub, N.M.; Liu, Y.Y.; El Sayed, K.A. Olive phenolics as c-Met inhibitors: (−)-Oleocanthal attenuates cell proliferation, invasiveness, and tumor growth in breast cancer models. PLoS ONE 2014, 9, e97622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comen, E.; Norton, L.; Massagu, E.J. Clinical implications of cancer self-seeding. Nat. Rev. Clin. Oncol. 2011, 8, 369–377. [Google Scholar] [CrossRef]
- Pienta, K.J.; Robertson, B.A.; Coffey, D.S.; Taichman, R.S. The cancer diaspora: Metastasis beyond the seed and soil hypothesis. Clin. Cancer Res. 2013, 19, 5849–5855. [Google Scholar] [CrossRef] [Green Version]
- Siddique, A.B.; King, J.A.; Meyer, S.A.; Abdelwahed, K.S.; Busnena, B.; El Sayed, K.A. Safety evaluations of single dose of the olive secoiridoid S-(−)-oleocanthal in Swiss albino mice. Nutrients 2020, 12, 314. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Oleocanthal | Paclitaxel | Docetaxel | Enzalutamide | Cisplatin | |
|---|---|---|---|---|---|---|
| IC50 µM | IC50 nM | IC50 nM | IC50 µM | IC50 µM | ||
| (24 h) | (48 h) | (48 h) | (48 h) | (48 h) | (48 h) | |
| CWR-R1ca | 3.59 | 1.08 | 4.9 | 2.2 | 84.2 | 24.3 |
| PC-3 | 0.64 | 0.58 | 6.6 | 2.0 | 70.1 | 6.1 |
| PC-3M | 1.03 | 0.80 | 3.7 | 2.8 | 90.1 | 50.8 |
| DU-145 | 5.35 | 3.32 | 5.7 | 2.6 | 47.0 | 15.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siddique, A.B.; Ebrahim, H.Y.; Tajmim, A.; King, J.A.; Abdelwahed, K.S.; Abd Elmageed, Z.Y.; El Sayed, K.A. Oleocanthal Attenuates Metastatic Castration-Resistant Prostate Cancer Progression and Recurrence by Targeting SMYD2. Cancers 2022, 14, 3542. https://doi.org/10.3390/cancers14143542
Siddique AB, Ebrahim HY, Tajmim A, King JA, Abdelwahed KS, Abd Elmageed ZY, El Sayed KA. Oleocanthal Attenuates Metastatic Castration-Resistant Prostate Cancer Progression and Recurrence by Targeting SMYD2. Cancers. 2022; 14(14):3542. https://doi.org/10.3390/cancers14143542
Chicago/Turabian StyleSiddique, Abu Bakar, Hassan Y. Ebrahim, Afsana Tajmim, Judy Ann King, Khaldoun S. Abdelwahed, Zakaria Y. Abd Elmageed, and Khalid A. El Sayed. 2022. "Oleocanthal Attenuates Metastatic Castration-Resistant Prostate Cancer Progression and Recurrence by Targeting SMYD2" Cancers 14, no. 14: 3542. https://doi.org/10.3390/cancers14143542