
High-Throughput CRISPR Screening in Hematological Neoplasms

 , , , ,
, , , ,  , and
, and
Abstract
:Simple Summary
Abstract

1. Introduction
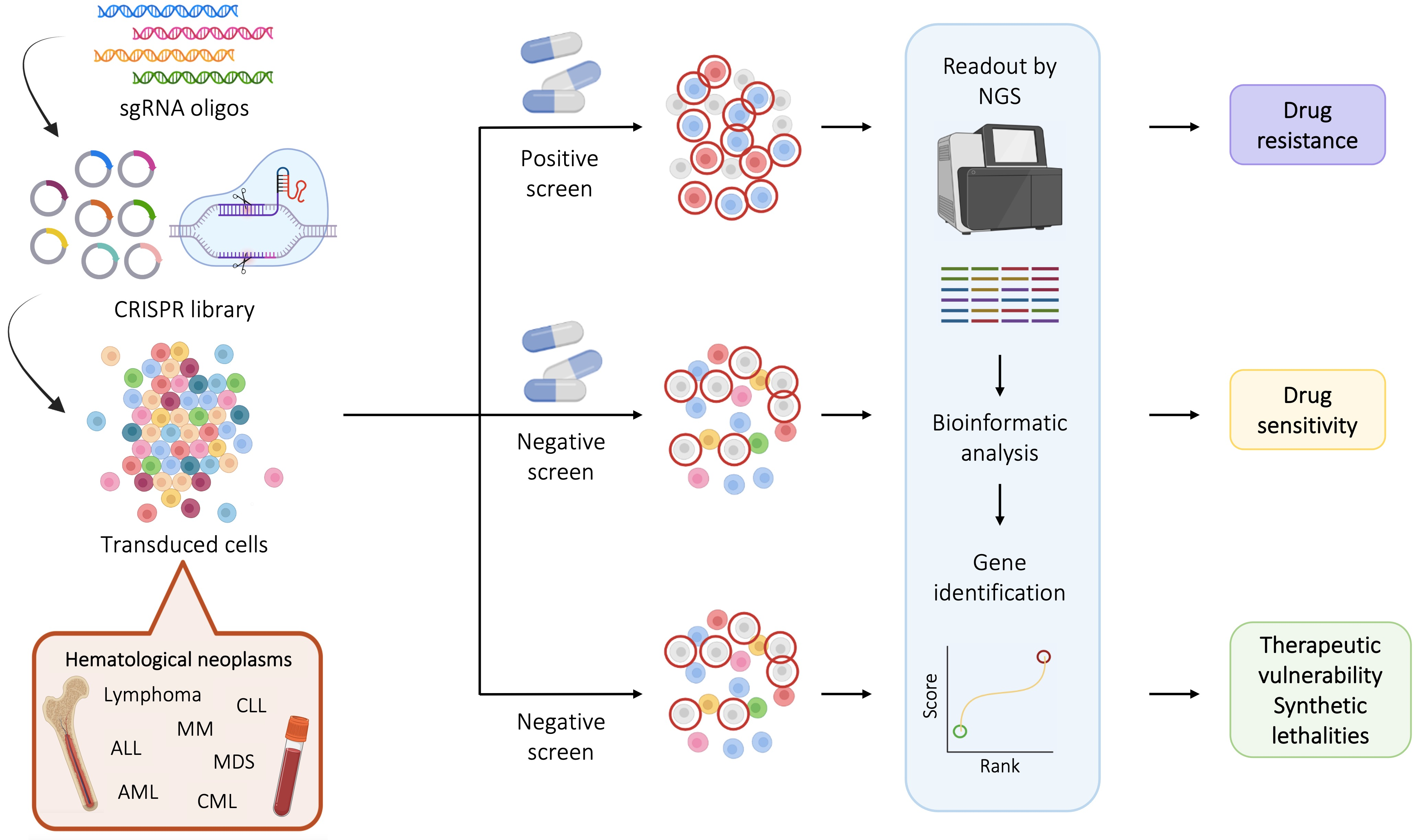
2. The CRISPR Screening Libraries Used for the Study of Hematological Malignancies
2.1. Genome-Wide Libraries According to the Aim of the Study
2.1.1. CRISPR Knockout
2.1.2. CRISPR Activation

{kind=link}
{kind=link}
{kind=link}
| Library-Name | Library Type | Library Size | Total sgRNAs (Targeted Genes) | gRNAs Per Gene | Addgene Reference | Used in Hematology | Aim of the Study |
|---|---|---|---|---|---|---|---|
| Sanjana et al. [23]—GeCKO | Knockout | Genome-wide | 123,411 (19,050 genes and 1864 miRNAs) | 6 | #1000000048 | ALL [41,42,43], AML [44,45,46,47,48,49], CML [50], HL [51], NHL [52,53,54], MDS [55], MM [56,57,58,59,60] | Drug resistance and sensitivity, therapeutic vulnerability, synthetic lethality |
| Doench et al. [28]—Brunello | Knockout | Genome-wide | 76,441 (19,114) | 4 | #73179 | AML [61,62], CLL [63], CML [64], NHL [65,66,67,68], MM [69] | Drug resistance and sensitivity, therapeutic vulnerability, synthetic lethality |
| Tzelepis et al. [32]—Human improved genome-wide library | Knockout | Genome-wide | 90,709 (18,010) | ~5 | #67989 | AML [32,70,71,72,73,74,75], NHL [76] | Drug resistance and sensitivity, therapeutic vulnerabilities, synthetic lethality |
| Doench et al. [28]—Avana | Knockout | Genome-wide | 73,782 (18,547) | 4 | NA | ALL [77], AML [78] | Drug sensitivity, therapeutic vulnerability |
| Hart et al. [17]—Toronto KnockOut (TKO) | Knockout | Genome-wide | 176,500 (17,661) | 6 | #1000000069 | CLL [79] | Synthetic lethality |
| Jaiswal et al. [80] | Knockout | Custom | 268 (36 RBP genes) | ~4 | NA | ALL [80] | Therapeutic vulnerability |
| Gabra M et al. [81]—miRKO library | Knockout | Custom | 6835 (1795 miRNAs) | 3 to 4 | NA | AML [81] | Therapeutic vulnerability |
| Lin S. et al. [82] | Knockout | Custom | 1320 (~200) | 6 | NA | AML [82] | Therapeutic vulnerability |
| Liss et al. [83] | Knockout | Custom | NA | NA | NA | AML [83] | Therapeutic vulnerability |
| Lin C.H. et al. [84] | Knockout | Custom | NA | NA | NA | AML [84] | Therapeutic vulnerability |
| Lin K.H. et al. [85] | Knockout | Custom | 11,610 (2322) | 5 | NA | AML [85] | Therapeutic vulnerability |
| Ott et al. [86] | Knockout | Custom | ~3500 (147 TFs) | ~7 | NA | CLL [86] | Therapeutic vulnerability |
| Kazimierska et al. [64]—MYC-CRISPR library | Knockout | Custom | 46,354 (24,981) | ~2 | #173195 | CML [64] | Therapeutic vulnerability |
| Han et al. [87]–Double-sgRNA library | Knockout | Custom | ~490,000 double-sgRNAs (21,321) | up to 9 | NA | CML [87] | Synthetic lethality |
| Wei et al. [88]—Ubiquitin regulator-focused library | Knockout | Custom | ~1300 (800) | 10 | NA | HL [88,89] | Drug sensitivity, therapeutic vulnerability |
| Mo et al. [90] | Knockout | Custom | 19,011 | 4 to 8 | NA | NHL [90] | Drug resistance |
| Bohl et al. [91] | Knockout | Custom | 745 (177) | ~4 | NA | MM [91] | Drug resistance, drug sensitivity |
| Shen et al. [92] | Knockout | Custom | 30 (3) | 10 | NA | MM [92] | Therapeutic vulnerability |
| Wang et al. [26]—Kinase gRNA library | Knockout | Custom | 73,151 (7114) | 10 | #51044 | ALL [27] | Drug sensitivity |
| Gilbert et al. [35]—CRISPRi | Interference | Genome-wide | 206,421 (15,977) | 10 | #62217 | NHL [38] | Drug resistance |
| Gilbert et al. [35]—CRISPRa | Activation | Genome-wide | 198,810 (15,977) | 10 | #60956 | AML [40], NHL [38] | Drug resistance |
| Konermann et al. [34]—SAM | Activation | Genome-wide | 70,290 (23,430) | 3 | #1000000078 | AML [39] | Drug resistance |
| Bester et al. [40]— CaLR | Activation | Custom | 88,444 (14,701 lncRNA genes) | ~4 | NA | AML [40] | Drug resistance |
2.1.3. CRISPR Interference
2.2. Custom CRISPR Libraries
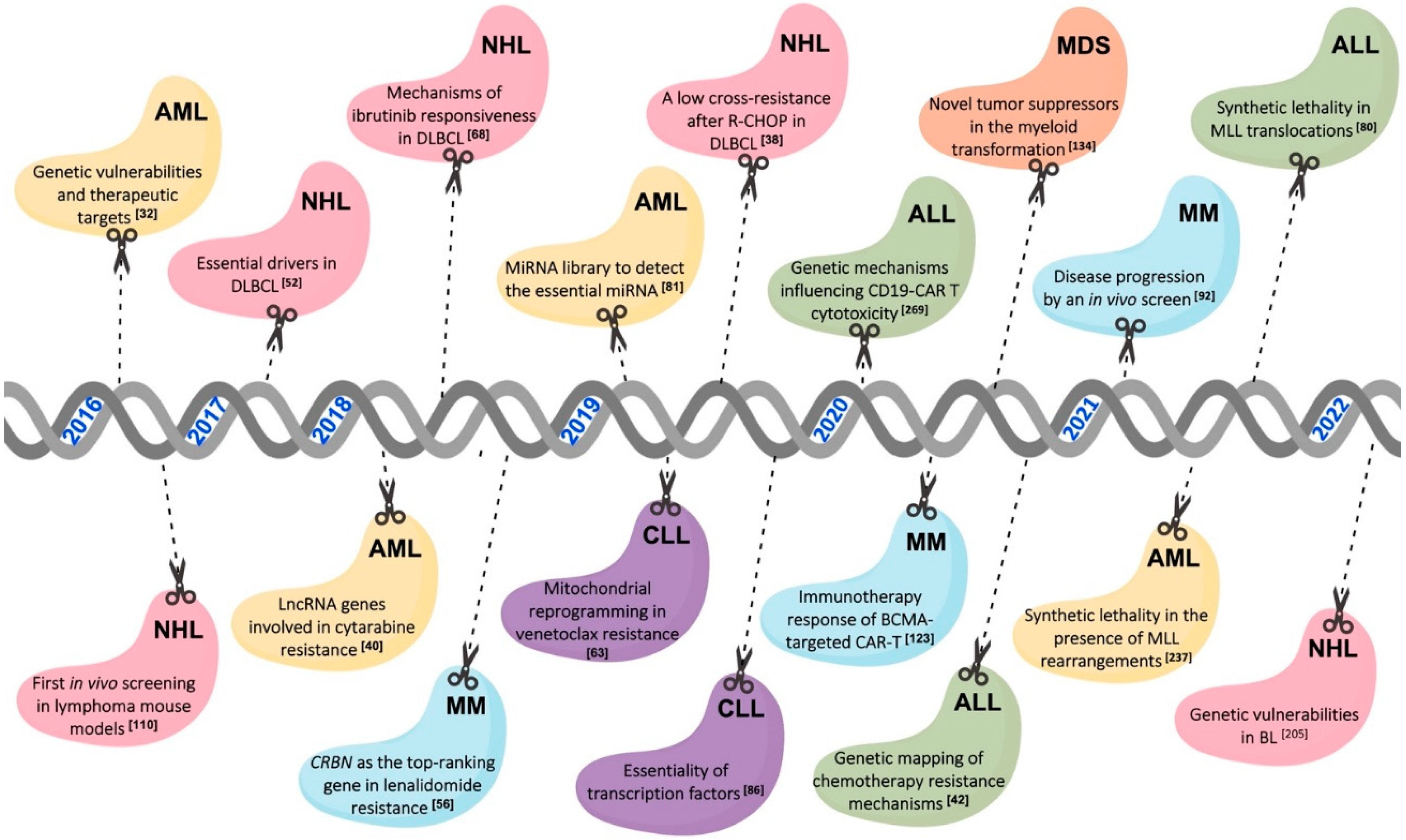
2.3. In Vivo CRISPR Screenings
3. Bioinformatic Tools in CRISPR Screening of Hematological Disorders
3.1. Algorithms
3.2. CRISPR Screening Databases
4. Mechanisms of Drug Resistance Uncovered by CRISPR High-Throughput Screening in Hematological Malignancies
4.1. Lymphoid Neoplasms
4.1.1. Lymphoma
4.1.2. Acute Lymphoblastic Leukemia
4.1.3. Chronic Lymphocytic Leukemia
4.1.4. Multiple Myeloma
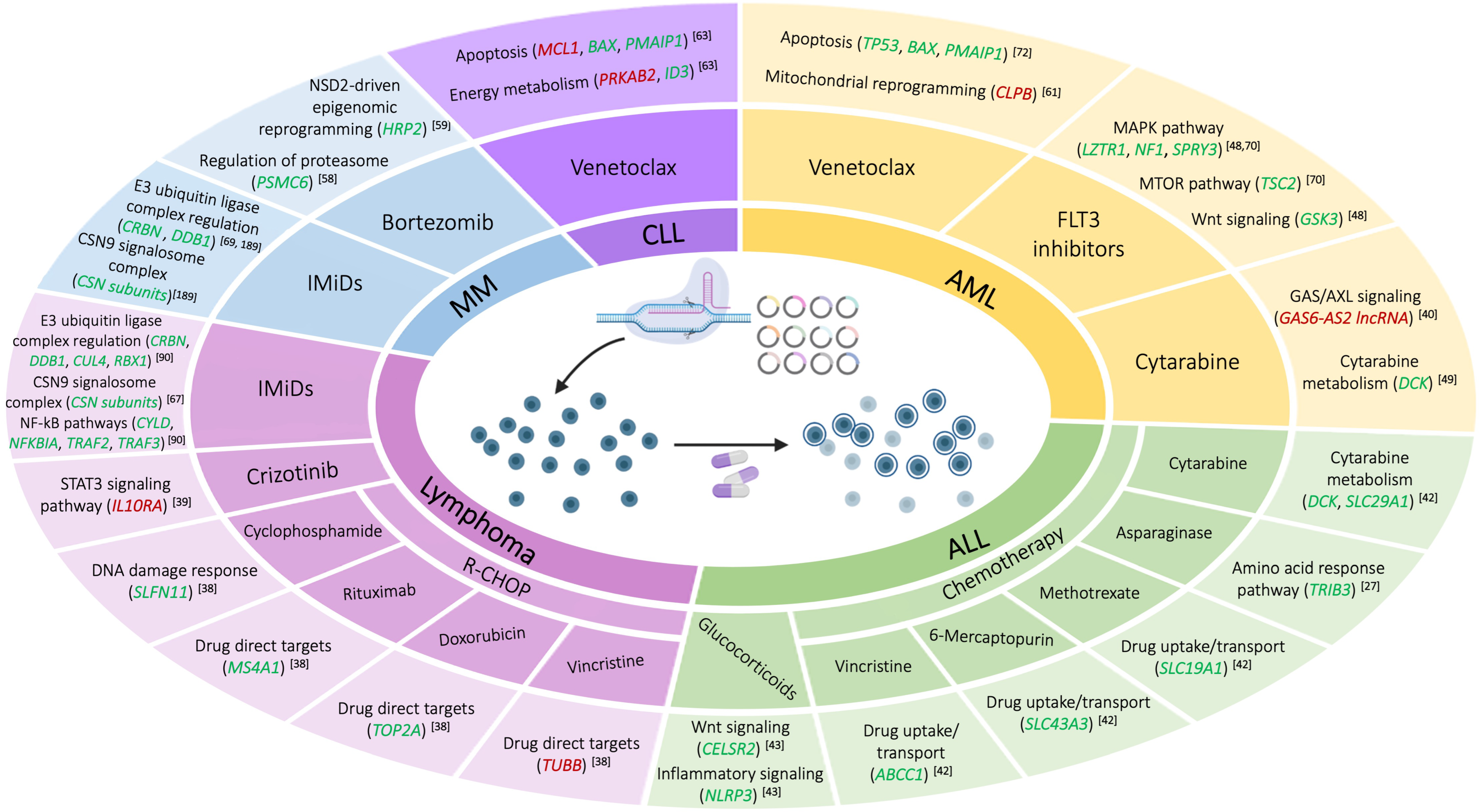
4.2. Myeloid Neoplasms
Acute Myeloid Leukemia
5. Hematologic Dependency Map through CRISPR Screens: Essential Genes and Key Regulators of Drug Sensitivity
5.1. Lymphoid Neoplasms
5.1.1. Lymphoma
5.1.2. Acute Lymphoblastic Leukemia
5.1.3. Chronic Lymphocytic Leukemia
5.1.4. Multiple Myeloma
5.2. Myeloid Neoplasms
5.2.1. Myelodysplastic Syndromes
5.2.2. Acute Myeloid Leukemia
5.2.3. Chronic Myeloid Leukemia
6. Other Applications of CRISPR Screenings
7. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Mojica, F.J.M.; Montoliu, L. On the Origin of CRISPR-Cas Technology: From Prokaryotes to Mammals. Trends Microbiol. 2016, 24, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-Guided Human Genome Engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, H.; Meng, L. CRISPR-cas9: A powerful tool towards precision medicine in cancer treatment. Acta Pharmacol. Sin. 2020, 41, 583–587. [Google Scholar] [CrossRef] [PubMed]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelsen, T.S.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-Scale CRISPR-Cas9 Knockout Screening in Human Cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moffat, J.; Grueneberg, D.A.; Yang, X.; Kim, S.Y.; Kloepfer, A.M.; Hinkle, G.; Piqani, B.; Eisenhaure, T.M.; Luo, B.; Grenier, J.K.; et al. A Lentiviral RNAi Library for Human and Mouse Genes Applied to an Arrayed Viral High-Content Screen. Cell 2006, 124, 1283–1298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuster, A.; Erasimus, H.; Fritah, S.; Nazarov, P.V.; van Dyck, E.; Niclou, S.P.; Golebiewska, A. RNAi/CRISPR Screens: From a Pool to a Valid Hit. Trends Biotechnol. 2019, 37, 38–55. [Google Scholar] [CrossRef]
- Evers, B.; Jastrzebski, K.; Heijmans, J.P.M.; Grernrum, W.; Beijersbergen, R.L.; Bernards, R. CRISPR knockout screening outperforms shRNA and CRISPRi in identifying essential genes. Nat. Biotechnol. 2016, 34, 631–633. [Google Scholar] [CrossRef]
- Munoz, D.M.; Cassiani, P.J.; Li, L.; Billy, E.; Korn, J.M.; Jones, M.D.; Golji, J.; Ruddy, D.A.; Yu, K.; McAllister, G.; et al. CRISPR Screens Provide a Comprehensive Assessment of Cancer Vulnerabilities but Generate False-Positive Hits for Highly Amplified Genomic Regions. Cancer Discov. 2016, 6, 900–913. [Google Scholar] [CrossRef] [Green Version]
- McDade, J.R.; Waxmonsky, N.C.; Swanson, L.E.; Fan, M. Practical Considerations for Using Pooled Lentiviral CRISPR Libraries. Curr. Protoc. Mol. Biol. 2016, 115, 31.5.1–31.5.13. [Google Scholar] [CrossRef]
- He, C.; Han, S.; Chang, Y.; Wu, M.; Zhao, Y.; Chen, C.; Chu, X. CRISPR screen in cancer: Status quo and future perspectives. Am. J. Cancer Res. 2021, 11, 1031–1050. [Google Scholar] [PubMed]
- Doench, J.G. Am I ready for CRISPR? A user’s guide to genetic screens. Nat. Rev. Genet. 2018, 19, 67–80. [Google Scholar] [CrossRef] [PubMed]
- Shalem, O.; Sanjana, N.E.; Zhang, F. High-throughput functional genomics using CRISPR–Cas9. Nat. Rev. Genet. 2015, 16, 299–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.; Petsalaki, E. Application of CRISPR-Cas9 Based Genome-Wide Screening Approaches to Study Cellular Signalling Mechanisms. Int. J. Mol. Sci. 2018, 19, 933. [Google Scholar] [CrossRef] [Green Version]
- Zhan, T.; Rindtorff, N.; Betge, J.; Ebert, M.P.; Boutros, M. CRISPR/Cas9 for cancer research and therapy. Semin. Cancer Biol. 2019, 55, 106–119. [Google Scholar] [CrossRef]
- Dong, M.B.; Tang, K.; Zhou, X.; Zhou, J.J.; Chen, S. Tumor immunology CRISPR screening: Present, past, and future. Trends Cancer 2022, 8, 210–225. [Google Scholar] [CrossRef]
- Hart, T.; Chandrashekhar, M.; Aregger, M.; Steinhart, Z.; Brown, K.R.; MacLeod, G.; Mis, M.; Zimmermann, M.; Fradet-Turcotte, A.; Sun, S.; et al. High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell 2015, 163, 1515–1526. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.; Lee, D.; Law, C.-T.; Zhang, M.S.; Shen, J.; Chin, D.W.-C.; Zhang, A.; Tsang, F.H.-C.; Wong, C.L.-S.; Ng, I.O.-L.; et al. Genome-wide CRISPR/Cas9 library screening identified PHGDH as a critical driver for Sorafenib resistance in HCC. Nat. Commun. 2019, 10, 4681. [Google Scholar] [CrossRef] [Green Version]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef]
- Klann, T.S.; Black, J.B.; Chellappan, M.; Safi, A.; Song, L.; Hilton, I.B.; Crawford, G.E.; Reddy, T.E.; Gersbach, C.A. CRISPR-Cas9 epigenome editing enables high-throughput screening for functional regulatory elements in the human genome. Nat. Biotechnol. 2017, 35, 561–568. [Google Scholar] [CrossRef]
- Bock, C.; Datlinger, P.; Chardon, F.; Coelho, M.A.; Dong, M.B.; Lawson, K.A.; Lu, T.; Maroc, L.; Norman, T.M.; Song, B.; et al. High-content CRISPR screening. Nat. Rev. Methods Primer 2022, 2, 1–23. [Google Scholar] [CrossRef]
- Thomsen, E.A.; Mikkelsen, J.G. CRISPR-Based Lentiviral Knockout Libraries for Functional Genomic Screening and Identification of Phenotype-Related Genes. Methods Mol. Biol. Clifton NJ 2019, 1961, 343–357. [Google Scholar]
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yau, E.H.; Rana, T.M. Next-Generation Sequencing of Genome-Wide CRISPR Screens. In Next Generation Sequencing; Head, S.R., Ordoukhanian, P., Salomon, D.R., Eds.; Methods in Molecular Biology; Springer New York: New York, NY, USA, 2018; Volume 1712, pp. 203–216. ISBN 978-1-4939-7512-9. [Google Scholar]
- Yamauchi, T.; Masuda, T.; Canver, M.C.; Seiler, M.; Semba, Y.; Shboul, M.; Al-Raqad, M.; Maeda, M.; Schoonenberg, V.A.C.; Cole, M.A.; et al. Genome-wide CRISPR-Cas9 Screen Identifies Leukemia-Specific Dependence on a Pre-mRNA Metabolic Pathway Regulated by DCPS. Cancer Cell 2018, 33, 386–400.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Birsoy, K.; Hughes, N.W.; Krupczak, K.M.; Post, Y.; Wei, J.J.; Lander, E.S.; Sabatini, D.M. Identification and characterization of essential genes in the human genome. Science 2015, 350, 1096–1101. [Google Scholar] [CrossRef] [Green Version]
- Butler, M.; van Ingen Schenau, D.S.; Yu, J.; Jenni, S.; Dobay, M.P.; Hagelaar, R.; Vervoort, B.M.T.; Tee, T.M.; Hoff, F.W.; Meijerink, J.P.; et al. BTK inhibition sensitizes acute lymphoblastic leukemia to asparaginase by suppressing the amino acid response pathway. Blood 2021, 138, 2383–2395. [Google Scholar] [CrossRef]
- Doench, J.G.; Fusi, N.; Sullender, M.; Hegde, M.; Vaimberg, E.W.; Donovan, K.F.; Smith, I.; Tothova, Z.; Wilen, C.; Orchard, R.; et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016, 34, 184–191. [Google Scholar] [CrossRef] [Green Version]
- Hart, T.; Tong, A.H.Y.; Chan, K.; Van Leeuwen, J.; Seetharaman, A.; Aregger, M.; Chandrashekhar, M.; Hustedt, N.; Seth, S.; Noonan, A.; et al. Evaluation and Design of Genome-Wide CRISPR/SpCas9 Knockout Screens. G3 GenesGenomesGenetics 2017, 7, 2719–2727. [Google Scholar] [CrossRef] [Green Version]
- Mair, B.; Tomic, J.; Masud, S.N.; Tonge, P.; Weiss, A.; Usaj, M.; Tong, A.H.Y.; Kwan, J.J.; Brown, K.R.; Titus, E.; et al. Essential Gene Profiles for Human Pluripotent Stem Cells Identify Uncharacterized Genes and Substrate Dependencies. Cell Rep. 2019, 27, 599–615.e12. [Google Scholar] [CrossRef] [Green Version]
- Sanson, K.R.; Hanna, R.E.; Hegde, M.; Donovan, K.F.; Strand, C.; Sullender, M.E.; Vaimberg, E.W.; Goodale, A.; Root, D.E.; Piccioni, F.; et al. Optimized libraries for CRISPR-Cas9 genetic screens with multiple modalities. Nat. Commun. 2018, 9, 5416. [Google Scholar] [CrossRef] [Green Version]
- Tzelepis, K.; Koike-Yusa, H.; De Braekeleer, E.; Li, Y.; Metzakopian, E.; Dovey, O.M.; Mupo, A.; Grinkevich, V.; Li, M.; Mazan, M.; et al. A CRISPR Dropout Screen Identifies Genetic Vulnerabilities and Therapeutic Targets in Acute Myeloid Leukemia. Cell Rep. 2016, 17, 1193–1205. [Google Scholar] [CrossRef] [Green Version]
- Xu, P.; Liu, Z.; Liu, Y.; Ma, H.; Xu, Y.; Bao, Y.; Zhu, S.; Cao, Z.; Wu, Z.; Zhou, Z.; et al. Genome-wide interrogation of gene functions through base editor screens empowered by barcoded sgRNAs. Nat. Biotechnol. 2021, 39, 1403–1413. [Google Scholar] [CrossRef] [PubMed]
- Konermann, S.; Brigham, M.D.; Trevino, A.E.; Joung, J.; Abudayyeh, O.O.; Barcena, C.; Hsu, P.D.; Habib, N.; Gootenberg, J.S.; Nishimasu, H.; et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 2015, 517, 583–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, L.A.; Horlbeck, M.A.; Adamson, B.; Villalta, J.E.; Chen, Y.; Whitehead, E.H.; Guimaraes, C.; Panning, B.; Ploegh, H.L.; Bassik, M.C.; et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 2014, 159, 647–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jost, M.; Chen, Y.; Gilbert, L.A.; Horlbeck, M.A.; Krenning, L.; Menchon, G.; Rai, A.; Cho, M.Y.; Stern, J.J.; Prota, A.E.; et al. Combined CRISPRi/a-Based Chemical Genetic Screens Reveal that Rigosertib Is a Microtubule-Destabilizing Agent. Mol. Cell 2017, 68, 210–223.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horlbeck, M.A.; Gilbert, L.A.; Villalta, J.E.; Adamson, B.; Pak, R.A.; Chen, Y.; Fields, A.P.; Park, C.Y.; Corn, J.E.; Kampmann, M.; et al. Compact and highly active next-generation libraries for CRISPR-mediated gene repression and activation. eLife 2016, 5, e19760. [Google Scholar] [CrossRef]
- Palmer, A.C.; Chidley, C.; Sorger, P.K. A curative combination cancer therapy achieves high fractional cell killing through low cross-resistance and drug additivity. eLife 2019, 8, e50036. [Google Scholar] [CrossRef]
- Prokoph, N.; Probst, N.A.; Lee, L.C.; Monahan, J.M.; Matthews, J.D.; Liang, H.-C.; Bahnsen, K.; Montes-Mojarro, I.A.; Karaca-Atabay, E.; Sharma, G.G.; et al. IL10RA modulates crizotinib sensitivity in NPM1-ALK+ anaplastic large cell lymphoma. Blood 2020, 136, 1657–1669. [Google Scholar] [CrossRef]
- Bester, A.C.; Lee, J.D.; Chavez, A.; Lee, Y.-R.; Nachmani, D.; Vora, S.; Victor, J.; Sauvageau, M.; Monteleone, E.; Rinn, J.L.; et al. An Integrated Genome-wide CRISPRa Approach to Functionalize lncRNAs in Drug Resistance. Cell 2018, 173, 649–664.e20. [Google Scholar] [CrossRef]
- Jiang, C.; Qian, M.; Gocho, Y.; Yang, W.; Du, G.; Shen, S.; Yang, J.J.; Zhang, H. Genome-wide CRISPR/Cas9 screening identifies determinant of panobinostat sensitivity in acute lymphoblastic leukemia. Blood Adv. 2022, 6, 2496–2509. [Google Scholar] [CrossRef]
- Oshima, K.; Zhao, J.; Pérez-Durán, P.; Brown, J.A.; Patiño-Galindo, J.A.; Chu, T.; Quinn, A.; Gunning, T.; Belver, L.; Ambesi-Impiombato, A.; et al. Mutational and functional genetics mapping of chemotherapy resistance mechanisms in relapsed acute lymphoblastic leukemia. Nat. Cancer 2020, 1, 1113–1127. [Google Scholar] [CrossRef] [PubMed]
- Autry, R.J.; Paugh, S.W.; Carter, R.; Shi, L.; Liu, J.; Ferguson, D.C.; Lau, C.E.; Bonten, E.J.; Yang, W.; McCorkle, J.R.; et al. Integrative genomic analyses reveal mechanisms of glucocorticoid resistance in acute lymphoblastic leukemia. Nat. Cancer 2020, 1, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Beeharry, N.; Landrette, S.; Gayle, S.; Hernandez, M.; Grotzke, J.E.; Young, P.R.; Beckett, P.; Zhang, X.; Carter, B.Z.; Andreeff, M.; et al. LAM-003, a new drug for treatment of tyrosine kinase inhibitor-resistant FLT3-ITD-positive AML. Blood Adv. 2019, 3, 3661–3673. [Google Scholar] [CrossRef] [Green Version]
- Deb, G.; Wingelhofer, B.; Amaral, F.M.R.; Maiques-Diaz, A.; Chadwick, J.A.; Spencer, G.J.; Williams, E.L.; Leong, H.-S.; Maes, T.; Somervaille, T.C.P. Pre-clinical activity of combined LSD1 and mTORC1 inhibition in MLL-translocated acute myeloid leukaemia. Leukemia 2020, 34, 1266–1277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stavast, C.J.; van Zuijen, I.; Karkoulia, E.; Özçelik, A.; van Hoven-Beijen, A.; Leon, L.G.; Voerman, J.S.A.; Janssen, G.M.C.; van Veelen, P.A.; Burocziova, M.; et al. The tumor suppressor MIR139 is silenced by POLR2M to promote AML oncogenesis. Leukemia 2022, 36, 687–700. [Google Scholar] [CrossRef]
- Yamauchi, T.; Miyawaki, K.; Semba, Y.; Takahashi, M.; Izumi, Y.; Nogami, J.; Nakao, F.; Sugio, T.; Sasaki, K.; Pinello, L.; et al. Targeting leukemia-specific dependence on the de novo purine synthesis pathway. Leukemia 2022, 36, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Hou, P.; Wu, C.; Wang, Y.; Qi, R.; Bhavanasi, D.; Zuo, Z.; Dos Santos, C.; Chen, S.; Chen, Y.; Zheng, H.; et al. A Genome-Wide CRISPR Screen Identifies Genes Critical for Resistance to FLT3 Inhibitor AC220. Cancer Res. 2017, 77, 4402–4413. [Google Scholar] [CrossRef] [Green Version]
- Kurata, M.; Rathe, S.K.; Bailey, N.J.; Aumann, N.K.; Jones, J.M.; Veldhuijzen, G.W.; Moriarity, B.S.; Largaespada, D.A. Using genome-wide CRISPR library screening with library resistant DCK to find new sources of Ara-C drug resistance in AML. Sci. Rep. 2016, 6, 36199. [Google Scholar] [CrossRef]
- Sobh, A.; Loguinov, A.; Yazici, G.N.; Zeidan, R.S.; Tagmount, A.; Hejazi, N.S.; Hubbard, A.E.; Zhang, L.; Vulpe, C.D. Functional Profiling Identifies Determinants of Arsenic Trioxide Cellular Toxicity. Toxicol. Sci. 2019, 169, 108–121. [Google Scholar] [CrossRef]
- Constantin, D.; Widmann, C. ASH2L drives proliferation and sensitivity to bleomycin and other genotoxins in Hodgkin’s lymphoma and testicular cancer cells. Cell Death Dis. 2020, 11, 1019. [Google Scholar] [CrossRef]
- Reddy, A.; Zhang, J.; Davis, N.S.; Moffitt, A.B.; Love, C.L.; Waldrop, A.; Leppa, S.; Pasanen, A.; Meriranta, L.; Karjalainen-Lindsberg, M.-L.; et al. Genetic and Functional Drivers of Diffuse Large B Cell Lymphoma. Cell 2017, 171, 481–494.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomsen, E.A.; Rovsing, A.B.; Anderson, M.V.; Due, H.; Huang, J.; Luo, Y.; Dybkaer, K.; Mikkelsen, J.G. Identification of BLNK and BTK as mediators of rituximab-induced programmed cell death by CRISPR screens in GCB-subtype diffuse large B-cell lymphoma. Mol. Oncol. 2020, 14, 1978–1997. [Google Scholar] [CrossRef]
- Gayle, S.; Landrette, S.; Beeharry, N.; Conrad, C.; Hernandez, M.; Beckett, P.; Ferguson, S.M.; Mandelkern, T.; Zheng, M.; Xu, T.; et al. Identification of apilimod as a first-in-class PIKfyve kinase inhibitor for treatment of B-cell non-Hodgkin lymphoma. Blood 2017, 129, 1768–1778. [Google Scholar] [CrossRef]
- Cheruiyot, A.; Li, S.; Srivatsan, S.N.; Ahmed, T.; Chen, Y.; Lemacon, D.S.; Li, Y.; Yang, Z.; Wadugu, B.A.; Warner, W.A.; et al. Nonsense-mediated RNA decay is a unique vulnerability of cancer cells harboring SF3B1 or U2AF1 mutations. Cancer Res. 2021, 81, 4499–4513. [Google Scholar] [CrossRef] [PubMed]
- Sievers, Q.L.; Gasser, J.A.; Cowley, G.S.; Fischer, E.S.; Ebert, B.L. Genome-wide screen identifies cullin-RING ligase machinery required for lenalidomide-dependent CRL4CRBN activity. Blood 2018, 132, 1293–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheffer, M.; Hu, Y.; Shalem, O.; Sanjana, N.; Dhimolea, E.; Sarkar, S.; Bariteau, M.A.; Aftab, B.T.; Groen, R.W.J.; Zhang, F.; et al. Genome-Scale Crispr-Cas9 Knockout Studies Reveal Mutifactorial and Functionally Overlapping Mechanisms of Myeloma Cell Resistance to Proteasome Inhibition. Blood 2014, 124, 273. [Google Scholar] [CrossRef]
- Shi, C.-X.; Kortüm, K.M.; Zhu, Y.X.; Bruins, L.A.; Jedlowski, P.; Votruba, P.G.; Luo, M.; Stewart, R.A.; Ahmann, J.; Braggio, E.; et al. CRISPR Genome-Wide Screening Identifies Dependence on the Proteasome Subunit PSMC6 for Bortezomib Sensitivity in Multiple Myeloma. Mol. Cancer Ther. 2017, 16, 2862–2870. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zhu, X.; Dang, L.; Jiang, H.; Xie, Y.; Li, X.; Guo, J.; Wang, Y.; Peng, Z.; Wang, M.; et al. Epigenomic reprogramming via HRP2-MINA dictates response to proteasome inhibitors in multiple myeloma with t(4;14) translocation. J. Clin. Investig. 2022, 132, e149526. [Google Scholar] [CrossRef]
- Liu, Y.; Cao, Z.; Wang, Y.; Guo, Y.; Xu, P.; Yuan, P.; Liu, Z.; He, Y.; Wei, W. Genome-wide screening for functional long noncoding RNAs in human cells by Cas9 targeting of splice sites. Nat. Biotechnol. 2018, 36, 1203–1210. [Google Scholar] [CrossRef]
- Chen, X.; Glytsou, C.; Zhou, H.; Narang, S.; Reyna, D.E.; Lopez, A.; Sakellaropoulos, T.; Gong, Y.; Kloetgen, A.; Yap, Y.S.; et al. Targeting mitochondrial structure sensitizes acute myeloid leukemia to Venetoclax treatment. Cancer Discov. 2019, 9, 890–909. [Google Scholar] [CrossRef]
- Brinton, L.T.; Sher, S.; Williams, K.; Canfield, D.; Orwick, S.; Wasmuth, R.; Cempre, C.; Skinner, J.; Lehman, A.; Blachly, J.S.; et al. Cotargeting of XPO1 Enhances the Antileukemic Activity of Midostaurin and Gilteritinib in Acute Myeloid Leukemia. Cancers 2020, 12, 1574. [Google Scholar] [CrossRef] [PubMed]
- Guièze, R.; Liu, V.M.; Rosebrock, D.; Jourdain, A.A.; Hernández-Sánchez, M.; Martinez Zurita, A.; Sun, J.; Ten Hacken, E.; Baranowski, K.; Thompson, P.A.; et al. Mitochondrial Reprogramming Underlies Resistance to BCL-2 Inhibition in Lymphoid Malignancies. Cancer Cell 2019, 36, 369–384.e13. [Google Scholar] [CrossRef]
- Kazimierska, M.; Podralska, M.; Żurawek, M.; Woźniak, T.; Kasprzyk, M.E.; Sura, W.; Łosiewski, W.; Ziółkowska-Suchanek, I.; Kluiver, J.; van den Berg, A.; et al. CRISPR/Cas9 Screen for Functional MYC Binding Sites Reveals MYC-Dependent Vulnerabilities in K562 Cells. bioRxiv 2021. [Google Scholar] [CrossRef]
- Ishio, T.; Kumar, S.; Shimono, J.; Daenthanasanmak, A.; Dubois, S.; Lin, Y.; Bryant, B.; Petrus, M.N.; Bachy, E.; Huang, D.W.; et al. Genome-wide CRISPR screen identifies CDK6 as a therapeutic target in adult T-cell leukemia/lymphoma. Blood 2022, 139, 1541–1556. [Google Scholar] [CrossRef] [PubMed]
- Patil, A.; Manzano, M.; Gottwein, E. CK1α and IRF4 are essential and independent effectors of immunomodulatory drugs in primary effusion lymphoma. Blood 2018, 132, 577–586. [Google Scholar] [CrossRef]
- Patil, A.; Manzano, M.; Gottwein, E. Genome-wide CRISPR screens reveal genetic mediators of cereblon modulator toxicity in primary effusion lymphoma. Blood Adv. 2019, 3, 2105–2117. [Google Scholar] [CrossRef] [Green Version]
- Phelan, J.D.; Young, R.M.; Webster, D.E.; Roulland, S.; Wright, G.W.; Kasbekar, M.; Shaffer, A.L.; Ceribelli, M.; Wang, J.Q.; Schmitz, R.; et al. A multiprotein supercomplex controlling oncogenic signalling in lymphoma. Nature 2018, 560, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Costacurta, M.; Vervoort, S.J.; Hogg, S.J.; Martin, B.P.; Johnstone, R.W.; Shortt, J. Whole genome CRISPR screening identifies TOP2B as a potential target for IMiD sensitization in multiple myeloma. Haematologica 2021, 106, 2013–2017. [Google Scholar] [CrossRef]
- Damnernsawad, A.; Bottomly, D.; Kurtz, S.E.; Eide, C.A.; McWeeney, S.K.; Tyner, J.W.; Nechiporuk, T. Genome-wide CRISPR screen identifies regulators of MAPK and MTOR pathways mediating sorafenib resistance in acute myeloid leukemia. Haematologica 2022, 107, 77–85. [Google Scholar] [CrossRef]
- Gallipoli, P.; Giotopoulos, G.; Tzelepis, K.; Costa, A.S.H.; Vohra, S.; Medina-Perez, P.; Basheer, F.; Marando, L.; Di Lisio, L.; Dias, J.M.L.; et al. Glutaminolysis is a metabolic dependency in FLT3ITD acute myeloid leukemia unmasked by FLT3 tyrosine kinase inhibition. Blood 2018, 131, 1639–1653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nechiporuk, T.; Kurtz, S.E.; Nikolova, O.; Liu, T.; Jones, C.L.; D’Alessandro, A.; Culp-Hill, R.; d’Almeida, A.; Joshi, S.K.; Rosenberg, M.; et al. The TP53 Apoptotic Network Is a Primary Mediator of Resistance to BCL2 Inhibition in AML Cells. Cancer Discov. 2019, 9, 910–925. [Google Scholar] [CrossRef] [PubMed]
- Romine, K.A.; Nechiporuk, T.; Bottomly, D.; Jeng, S.; McWeeney, S.K.; Kaempf, A.; Corces, M.R.; Majeti, R.; Tyner, J.W. Monocytic differentiation and AHR signaling as Primary Nodes of BET Inhibitor Response in Acute Myeloid Leukemia. Blood Cancer Discov. 2021, 2, 518–531. [Google Scholar] [CrossRef] [PubMed]
- Pacharne, S.; Dovey, O.M.; Cooper, J.L.; Gu, M.; Friedrich, M.J.; Rajan, S.S.; Barenboim, M.; Collord, G.; Vijayabaskar, M.S.; Ponstingl, H.; et al. SETBP1 overexpression acts in the place of class-defining mutations to drive FLT3-ITD-mutant AML. Blood Adv. 2021, 5, 2412–2425. [Google Scholar] [CrossRef]
- Supper, E.; Rudat, S.; Iyer, V.; Droop, A.; Wong, K.; Spinella, J.-F.; Thomas, P.; Sauvageau, G.; Adams, D.J.; Wong, C.C. Cut-like homeobox 1 (CUX1) tumor suppressor gene haploinsufficiency induces apoptosis evasion to sustain myeloid leukemia. Nat. Commun. 2021, 12, 2482. [Google Scholar] [CrossRef] [PubMed]
- Thieme, E.; Liu, T.; Bruss, N.; Roleder, C.; Lam, V.; Wang, X.; Nechiporuk, T.; Shouse, G.; Danilova, O.V.; Bottomly, D.; et al. Dual BTK/SYK inhibition with CG-806 (luxeptinib) disrupts B-cell receptor and Bcl-2 signaling networks in mantle cell lymphoma. Cell Death Dis. 2022, 13, 246. [Google Scholar] [CrossRef] [PubMed]
- Gocho, Y.; Liu, J.; Hu, J.; Yang, W.; Dharia, N.V.; Zhang, J.; Shi, H.; Du, G.; John, A.; Lin, T.-N.; et al. Network-based systems pharmacology reveals heterogeneity in LCK and BCL2 signaling and therapeutic sensitivity of T-cell acute lymphoblastic leukemia. Nat. Cancer 2021, 2, 284–299. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Takacs, G.P.; Lamba, J.K.; Vulpe, C.; Cogle, C.R. Functional Dependency Analysis Identifies Potential Druggable Targets in Acute Myeloid Leukemia. Cancers 2020, 12, 3710. [Google Scholar] [CrossRef]
- Zimmermann, M.; Murina, O.; Reijns, M.A.M.; Agathanggelou, A.; Challis, R.; Tarnauskaitė, Ž.; Muir, M.; Fluteau, A.; Aregger, M.; McEwan, A.; et al. CRISPR screens identify genomic ribonucleotides as a source of PARP-trapping lesions. Nature 2018, 559, 285–289. [Google Scholar] [CrossRef]
- Jaiswal, A.K.; Truong, H.; Tran, T.M.; Lin, T.L.; Casero, D.; Alberti, M.O.; Rao, D.S. Focused CRISPR-Cas9 genetic screening reveals USO1 as a vulnerability in B-cell acute lymphoblastic leukemia. Sci. Rep. 2021, 11, 13158. [Google Scholar] [CrossRef]
- Gabra, M.; Pastrello, C.; Machado, N.; Chow, J.T.-S.; Kotlyar, M.; Tokar, T.; Jurisica, I.; Salmena, L. Essential Gene Networks in Acute Myeloid Leukemia Identified Using a microRNA-knockout CRISPR Library Screen. bioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.; Larrue, C.; Scheidegger, N.K.; Seong, B.K.A.; Dharia, N.V.; Kuljanin, M.; Wechsler, C.S.; Kugener, G.; Robichaud, A.L.; Conway, A.S.; et al. An In Vivo CRISPR Screening Platform for Prioritizing Therapeutic Targets in AML. Cancer Discov. 2022, 12, 432–449. [Google Scholar] [CrossRef] [PubMed]
- Liss, F.; Frech, M.; Wang, Y.; Giel, G.; Fischer, S.; Simon, C.; Weber, L.M.; Nist, A.; Stiewe, T.; Neubauer, A.; et al. IRF8 Is an AML-Specific Susceptibility Factor That Regulates Signaling Pathways and Proliferation of AML Cells. Cancers 2021, 13, 764. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-H.; Vu, J.P.; Yang, C.-Y.; Sirisawad, M.; Chen, C.-T.; Dao, H.; Liu, J.; Ma, X.; Pan, C.; Cefalu, J.; et al. Glutamate-cysteine ligase catalytic subunit as a therapeutic target in acute myeloid leukemia and solid tumors. Am. J. Cancer Res. 2021, 11, 2911–2927. [Google Scholar] [PubMed]
- Lin, K.H.; Xie, A.; Rutter, J.C.; Ahn, Y.-R.; Lloyd-Cowden, J.M.; Nichols, A.G.; Soderquist, R.S.; Koves, T.R.; Muoio, D.M.; MacIver, N.J.; et al. Systematic Dissection of the Metabolic-Apoptotic Interface in AML Reveals Heme Biosynthesis to Be a Regulator of Drug Sensitivity. Cell Metab. 2019, 29, 1217–1231.e7. [Google Scholar] [CrossRef]
- Ott, C.J.; Federation, A.J.; Schwartz, L.S.; Kasar, S.; Klitgaard, J.L.; Lenci, R.; Li, Q.; Lawlor, M.; Fernandes, S.M.; Souza, A.; et al. Enhancer Architecture and Essential Core Regulatory Circuitry of Chronic Lymphocytic Leukemia. Cancer Cell 2018, 34, 982–995.e7. [Google Scholar] [CrossRef] [Green Version]
- Han, K.; Jeng, E.E.; Hess, G.T.; Morgens, D.W.; Li, A.; Bassik, M.C. Synergistic drug combinations for cancer identified in a CRISPR screen for pairwise genetic interactions. Nat. Biotechnol. 2017, 35, 463–474. [Google Scholar] [CrossRef]
- Wei, W.; Lin, Y.; Song, Z.; Xiao, W.; Chen, L.; Yin, J.; Zhou, Y.; Barta, S.K.; Petrus, M.; Waldmann, T.A.; et al. A20 and RBX1 Regulate Brentuximab Vedotin Sensitivity in Hodgkin Lymphoma Models. Clin. Cancer Res. 2020, 26, 4093–4106. [Google Scholar] [CrossRef] [Green Version]
- Song, Z.; Wei, W.; Xiao, W.; Al-Saleem, E.D.; Nejati, R.; Chen, L.; Yin, J.; Fabrizio, J.; Petrus, M.N.; Waldmann, T.A.; et al. Essential role of the linear ubiquitin chain assembly complex and TAK1 kinase in A20 mutant Hodgkin lymphoma. Proc. Natl. Acad. Sci. USA 2020, 117, 28980–28991. [Google Scholar] [CrossRef]
- Mo, Z.; Wood, S.; Namiranian, S.; Mizukoshi, R.; Weng, S.; Jang, I.S.; Fontanillo, C.; Baughman, J.M.; Silva-Torres, A.; Slade, M.; et al. Deciphering the mechanisms of CC-122 resistance in DLBCL via a genome-wide CRISPR screen. Blood Adv. 2021, 5, 2027–2039. [Google Scholar] [CrossRef]
- Bohl, S.R.; Schmalbrock, L.K.; Bauhuf, I.; Meyer, T.; Dolnik, A.; Szyska, M.; Blätte, T.J.; Knödler, S.; Röhner, L.; Miller, D.; et al. Comprehensive CRISPR-Cas9 screens identify genetic determinants of drug responsiveness in multiple myeloma. Blood Adv. 2021, 5, 2391–2402. [Google Scholar] [CrossRef]
- Shen, Y.J.; Mishima, Y.; Shi, J.; Sklavenitis-Pistofidis, R.; Redd, R.A.; Moschetta, M.; Manier, S.; Roccaro, A.M.; Sacco, A.; Tai, Y.-T.; et al. Progression signature underlies clonal evolution and dissemination of multiple myeloma. Blood 2021, 137, 2360–2372. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-Guided Platform for Sequence-Specific Control of Gene Expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Köferle, A.; Worf, K.; Breunig, C.; Baumann, V.; Herrero, J.; Wiesbeck, M.; Hutter, L.H.; Götz, M.; Fuchs, C.; Beck, S.; et al. CORALINA: A universal method for the generation of gRNA libraries for CRISPR-based screening. BMC Genomics 2016, 17, 917. [Google Scholar] [CrossRef] [Green Version]
- Heigwer, F.; Zhan, T.; Breinig, M.; Winter, J.; Brügemann, D.; Leible, S.; Boutros, M. CRISPR library designer (CLD): Software for multispecies design of single guide RNA libraries. Genome Biol. 2016, 17, 55. [Google Scholar] [CrossRef] [Green Version]
- Panda, S.K.; Boddul, S.V.; Jiménez-Andrade, G.Y.; Jiang, L.; Kasza, Z.; Fernandez-Ricaud, L.; Wermeling, F. Green listed-a CRISPR screen tool. Bioinforma. Oxf. Engl. 2017, 33, 1099–1100. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Vandenberg, C.J.; Lieschke, E.; Di Rago, L.; Scott, C.L.; Majewski, I.J. CHK2 Inhibition Provides a Strategy to Suppress Hematologic Toxicity from PARP Inhibitors. Mol. Cancer Res. MCR 2021, 19, 1350–1360. [Google Scholar] [CrossRef] [PubMed]
- Kurata, J.S.; Lin, R.-J. MicroRNA-focused CRISPR-Cas9 library screen reveals fitness-associated miRNAs. RNA 2018, 24, 966–981. [Google Scholar] [CrossRef] [Green Version]
- Jiang, F.; Liang, Y.; Wei, W.; Zou, C.; Chen, G.; Wan, Y.; Liu, Z.; Yang, Y.; Han, Z.; Zhu, J.; et al. Functional classification of prostate cancer-associated miRNAs through CRISPR/Cas9-mediated gene knockout. Mol. Med. Rep. 2020, 22, 3777–3784. [Google Scholar] [CrossRef]
- Zhu, S.; Li, W.; Liu, J.; Chen, C.-H.; Liao, Q.; Xu, P.; Xu, H.; Xiao, T.; Cao, Z.; Peng, J.; et al. Genome-scale deletion screening of human long non-coding RNAs using a paired-guide RNA CRISPR–Cas9 library. Nat. Biotechnol. 2016, 34, 1279–1286. [Google Scholar] [CrossRef]
- Pulecio, J.; Verma, N.; Mejía-Ramírez, E.; Huangfu, D.; Raya, A. CRISPR/Cas9-Based Engineering of the Epigenome. Cell Stem Cell 2017, 21, 431–447. [Google Scholar] [CrossRef] [Green Version]
- Nuñez, J.K.; Chen, J.; Pommier, G.C.; Cogan, J.Z.; Replogle, J.M.; Adriaens, C.; Ramadoss, G.N.; Shi, Q.; Hung, K.L.; Samelson, A.J.; et al. Genome-wide programmable transcriptional memory by CRISPR-based epigenome editing. Cell 2021, 184, 2503–2519.e17. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, M.; Santinha, A.J.; Platt, R.J. Moving from in vitro to in vivo CRISPR screens. Gene Genome Ed. 2021, 2, 100008. [Google Scholar] [CrossRef]
- Xue, W.; Chen, S.; Yin, H.; Tammela, T.; Papagiannakopoulos, T.; Joshi, N.S.; Cai, W.; Yang, G.; Bronson, R.; Crowley, D.G.; et al. CRISPR-mediated direct mutation of cancer genes in the mouse liver. Nature 2014, 514, 380–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maresch, R.; Mueller, S.; Veltkamp, C.; Öllinger, R.; Friedrich, M.; Heid, I.; Steiger, K.; Weber, J.; Engleitner, T.; Barenboim, M.; et al. Multiplexed pancreatic genome engineering and cancer induction by transfection-based CRISPR/Cas9 delivery in mice. Nat. Commun. 2016, 7, 10770. [Google Scholar] [CrossRef] [Green Version]
- Chow, R.D.; Guzman, C.D.; Wang, G.; Schmidt, F.; Youngblood, M.W.; Ye, L.; Errami, Y.; Dong, M.B.; Martinez, M.A.; Zhang, S.; et al. AAV-mediated direct in vivo CRISPR screen identifies functional suppressors in glioblastoma. Nat. Neurosci. 2017, 20, 1329–1341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, R.D.; Chen, S. Cancer CRISPR Screens In Vivo. Trends Cancer 2018, 4, 349–358. [Google Scholar] [CrossRef]
- Chen, S.; Sanjana, N.E.; Zheng, K.; Shalem, O.; Lee, K.; Shi, X.; Scott, D.A.; Song, J.; Pan, J.Q.; Weissleder, R.; et al. Genome-wide CRISPR Screen in a Mouse Model of Tumor Growth and Metastasis. Cell 2015, 160, 1246–1260. [Google Scholar] [CrossRef] [Green Version]
- Bajaj, J.; Hamilton, M.; Shima, Y.; Chambers, K.; Spinler, K.; Van Nostrand, E.L.; Yee, B.A.; Blue, S.M.; Chen, M.; Rizzeri, D.; et al. An in vivo genome-wide CRISPR screen identifies the RNA-binding protein Staufen2 as a key regulator of myeloid leukemia. Nat. Cancer 2020, 1, 410–422. [Google Scholar] [CrossRef]
- Katigbak, A.; Cencic, R.; Robert, F.; Sénécha, P.; Scuoppo, C.; Pelletier, J. A CRISPR/Cas9 Functional Screen Identifies Rare Tumor Suppressors. Sci. Rep. 2016, 6, 38968. [Google Scholar] [CrossRef]
- Colic, M.; Hart, T. Common computational tools for analyzing CRISPR screens. Emerg. Top. Life Sci. 2021, 5, 779–788. [Google Scholar] [CrossRef]
- Agrotis, A.; Ketteler, R. A new age in functional genomics using CRISPR/Cas9 in arrayed library screening. Front. Genet. 2015, 6, 300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Xu, H.; Xiao, T.; Cong, L.; Love, M.I.; Zhang, F.; Irizarry, R.A.; Liu, J.S.; Brown, M.; Liu, X.S. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 2014, 15, 554. [Google Scholar] [CrossRef] [PubMed]
- Bodapati, S.; Daley, T.P.; Lin, X.; Zou, J.; Qi, L.S. A benchmark of algorithms for the analysis of pooled CRISPR screens. Genome Biol. 2020, 21, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Köster, J.; Xu, H.; Chen, C.-H.; Xiao, T.; Liu, J.S.; Brown, M.; Liu, X.S. Quality control, modeling, and visualization of CRISPR screens with MAGeCK-VISPR. Genome Biol. 2015, 16, 281. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Wang, M.; Zhang, W.; Xiao, T.; Chen, C.-H.; Wu, A.; Wu, F.; Traugh, N.; Wang, X.; Li, Z.; et al. Integrative analysis of pooled CRISPR genetic screens using MAGeCKFlute. Nat. Protoc. 2019, 14, 756–780. [Google Scholar] [CrossRef]
- Yang, L.; Zhu, Y.; Yu, H.; Cheng, X.; Chen, S.; Chu, Y.; Huang, H.; Zhang, J.; Li, W. scMAGeCK links genotypes with multiple phenotypes in single-cell CRISPR screens. Genome Biol. 2020, 21, 19. [Google Scholar] [CrossRef] [Green Version]
- Seneviratne, A.K.; Xu, M.; Henao, J.J.A.; Fajardo, V.A.; Hao, Z.; Voisin, V.; Xu, G.W.; Hurren, R.; Kim, S.; MacLean, N.; et al. The Mitochondrial Transacylase, Tafazzin, Regulates AML Stemness by Modulating Intracellular Levels of Phospholipids. Cell Stem Cell 2019, 24, 621–636.e16. [Google Scholar] [CrossRef] [Green Version]
- Barghout, S.H.; Aman, A.; Nouri, K.; Blatman, Z.; Arevalo, K.; Thomas, G.E.; MacLean, N.; Hurren, R.; Ketela, T.; Saini, M.; et al. A genome-wide CRISPR/Cas9 screen in acute myeloid leukemia cells identifies regulators of TAK-243 sensitivity. JCI Insight 2021, 6, 141518. [Google Scholar] [CrossRef]
- Xu, J.J.; Chalk, A.M.; Nikolic, I.; Simpson, K.J.; Smeets, M.F.; Walkley, C.R. Genome-wide screening identifies cell-cycle control as a synthetic lethal pathway with SRSF2P95H mutation. Blood Adv. 2022, 6, 2092–2106. [Google Scholar] [CrossRef]
- Nie, M.; Du, L.; Ren, W.; Joung, J.; Ye, X.; Shi, X.; Ciftci, S.; Liu, D.; Wu, K.; Zhang, F.; et al. Genome-wide CRISPR screens reveal synthetic lethal interaction between CREBBP and EP300 in diffuse large B-cell lymphoma. Cell Death Dis. 2021, 12, 419. [Google Scholar] [CrossRef] [PubMed]
- Ramkumar, P.; Abarientos, A.B.; Tian, R.; Seyler, M.; Leong, J.T.; Chen, M.; Choudhry, P.; Hechler, T.; Shah, N.; Wong, S.W.; et al. CRISPR-based screens uncover determinants of immunotherapy response in multiple myeloma. Blood Adv. 2020, 4, 2899–2911. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Lee, Y.G.; Shestova, O.; Ravikumar, P.; Hayer, K.E.; Hong, S.J.; Lu, X.M.; Pajarillo, R.; Agarwal, S.; Kuramitsu, S.; et al. Impaired Death Receptor Signaling in Leukemia Causes Antigen-Independent Resistance by Inducing CAR T-cell Dysfunction. Cancer Discov. 2020, 10, 552–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koduri, V.; Duplaquet, L.; Lampson, B.L.; Wang, A.C.; Sabet, A.H.; Ishoey, M.; Paulk, J.; Teng, M.; Harris, I.S.; Endress, J.E.; et al. Targeting oncoproteins with a positive selection assay for protein degraders. Sci. Adv. 2021, 7, eabd6263. [Google Scholar] [CrossRef]
- Hart, T.; Moffat, J. BAGEL: A computational framework for identifying essential genes from pooled library screens. BMC Bioinform. 2016, 17, 164. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.; Hart, T. Improved analysis of CRISPR fitness screens and reduced off-target effects with the BAGEL2 gene essentiality classifier. Genome Med. 2021, 13, 2. [Google Scholar] [CrossRef]
- Lenoir, W.F.; Morgado, M.; DeWeirdt, P.C.; McLaughlin, M.; Griffith, A.L.; Sangree, A.K.; Feeley, M.N.; Esmaeili Anvar, N.; Kim, E.; Bertolet, L.L.; et al. Discovery of putative tumor suppressors from CRISPR screens reveals rewired lipid metabolism in acute myeloid leukemia cells. Nat. Commun. 2021, 12, 6506. [Google Scholar] [CrossRef]
- Liu, J.; Srinivasan, S.; Li, C.-Y.; Ho, I.-L.; Rose, J.; Shaheen, M.; Wang, G.; Yao, W.; Deem, A.; Bristow, C.; et al. Pooled library screening with multiplexed Cpf1 library. Nat. Commun. 2019, 10, 3144. [Google Scholar] [CrossRef] [Green Version]
- Morgens, D.W.; Deans, R.M.; Li, A.; Bassik, M.C. Systematic comparison of CRISPR/Cas9 and RNAi screens for essential genes. Nat. Biotechnol. 2016, 34, 634–636. [Google Scholar] [CrossRef] [Green Version]
- Meyers, R.M.; Bryan, J.G.; McFarland, J.M.; Weir, B.A.; Sizemore, A.E.; Xu, H.; Dharia, N.V.; Montgomery, P.G.; Cowley, G.S.; Pantel, S.; et al. Computational correction of copy number effect improves specificity of CRISPR–Cas9 essentiality screens in cancer cells. Nat. Genet. 2017, 49, 1779–1784. [Google Scholar] [CrossRef] [Green Version]
- Allen, F.; Behan, F.; Khodak, A.; Iorio, F.; Yusa, K.; Garnett, M.; Parts, L. JACKS: Joint analysis of CRISPR/Cas9 knockout screens. Genome Res. 2019, 29, 464–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spahn, P.N.; Bath, T.; Weiss, R.J.; Kim, J.; Esko, J.D.; Lewis, N.E.; Harismendy, O. PinAPL-Py: A comprehensive web-application for the analysis of CRISPR/Cas9 screens. Sci. Rep. 2017, 7, 15854. [Google Scholar] [CrossRef] [Green Version]
- Schieber, M.; Marinaccio, C.; Bolanos, L.C.; Haffey, W.D.; Greis, K.D.; Starczynowski, D.T.; Crispino, J.D. FBXO11 is a candidate tumor suppressor in the leukemic transformation of myelodysplastic syndrome. Blood Cancer J. 2020, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Akkari, Y.M.N.; Baughn, L.B.; Dubuc, A.M.; Smith, A.C.; Mallo, M.; Dal Cin, P.; Diez Campelo, M.; Gallego, M.S.; Granada Font, I.; Haase, D.T.; et al. Guiding the global evolution of cytogenetic testing for hematologic malignancies. Blood 2022, 139, 2273–2284. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, E.; Behan, F.M.; Louzada, S.; Arnol, D.; Stronach, E.A.; Yang, F.; Yusa, K.; Stegle, O.; Iorio, F.; Garnett, M.J. Structural rearrangements generate cell-specific, gene-independent CRISPR-Cas9 loss of fitness effects. Genome Biol. 2019, 20, 27. [Google Scholar] [CrossRef] [Green Version]
- Iorio, F.; Behan, F.M.; Gonçalves, E.; Bhosle, S.G.; Chen, E.; Shepherd, R.; Beaver, C.; Ansari, R.; Pooley, R.; Wilkinson, P.; et al. Unsupervised correction of gene-independent cell responses to CRISPR-Cas9 targeting. BMC Genom. 2018, 19, 604. [Google Scholar] [CrossRef] [Green Version]
- Dixit, A.; Parnas, O.; Li, B.; Chen, J.; Fulco, C.P.; Jerby-Arnon, L.; Marjanovic, N.D.; Dionne, D.; Burks, T.; Raychowdhury, R.; et al. Perturb-Seq: Dissecting Molecular Circuits with Scalable Single-Cell RNA Profiling of Pooled Genetic Screens. Cell 2016, 167, 1853–1866.e17. [Google Scholar] [CrossRef] [Green Version]
- Jaitin, D.A.; Weiner, A.; Yofe, I.; Lara-Astiaso, D.; Keren-Shaul, H.; David, E.; Salame, T.M.; Tanay, A.; van Oudenaarden, A.; Amit, I. Dissecting Immune Circuits by Linking CRISPR-Pooled Screens with Single-Cell RNA-Seq. Cell 2016, 167, 1883–1896.e15. [Google Scholar] [CrossRef] [Green Version]
- Mimitou, E.P.; Cheng, A.; Montalbano, A.; Hao, S.; Stoeckius, M.; Legut, M.; Roush, T.; Herrera, A.; Papalexi, E.; Ouyang, Z.; et al. Multiplexed detection of proteins, transcriptomes, clonotypes and CRISPR perturbations in single cells. Nat. Methods 2019, 16, 409–412. [Google Scholar] [CrossRef]
- Yang, L.; Chan, A.K.N.; Miyashita, K.; Delaney, C.D.; Wang, X.; Li, H.; Pokharel, S.P.; Li, S.; Li, M.; Xu, X.; et al. High-resolution characterization of gene function using single-cell CRISPR tiling screen. Nat. Commun. 2021, 12, 4063. [Google Scholar] [CrossRef]
- Lopes, R.; Sprouffske, K.; Sheng, C.; Uijttewaal, E.C.H.; Wesdorp, A.E.; Dahinden, J.; Wengert, S.; Diaz-Miyar, J.; Yildiz, U.; Bleu, M.; et al. Systematic dissection of transcriptional regulatory networks by genome-scale and single-cell CRISPR screens. Sci. Adv. 2021, 7, eabf5733. [Google Scholar] [CrossRef] [PubMed]
- Frangieh, C.J.; Melms, J.C.; Thakore, P.I.; Geiger-Schuller, K.R.; Ho, P.; Luoma, A.M.; Cleary, B.; Jerby-Arnon, L.; Malu, S.; Cuoco, M.S.; et al. Multimodal pooled Perturb-CITE-seq screens in patient models define mechanisms of cancer immune evasion. Nat. Genet. 2021, 53, 332–341. [Google Scholar] [CrossRef] [PubMed]
- Datlinger, P.; Rendeiro, A.F.; Schmidl, C.; Krausgruber, T.; Traxler, P.; Klughammer, J.; Schuster, L.C.; Kuchler, A.; Alpar, D.; Bock, C. Pooled CRISPR screening with single-cell transcriptome readout. Nat. Methods 2017, 14, 297–301. [Google Scholar] [CrossRef] [Green Version]
- Norman, T.M.; Horlbeck, M.A.; Replogle, J.M.; Ge, A.Y.; Xu, A.; Jost, M.; Gilbert, L.A.; Weissman, J.S. Exploring genetic interaction manifolds constructed from rich single-cell phenotypes. Science 2019, 365, 786–793. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Chemparathy, A.; La Russa, M.; Daley, T.; Qi, L.S. Computational Methods for Analysis of Large-Scale CRISPR Screens. Annu. Rev. Biomed. Data Sci. 2020, 3, 137–162. [Google Scholar] [CrossRef]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M.; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated analysis of multimodal single-cell data. Cell 2021, 184, 3573–3587.e29. [Google Scholar] [CrossRef]
- Papalexi, E.; Mimitou, E.P.; Butler, A.W.; Foster, S.; Bracken, B.; Mauck, W.M.; Wessels, H.-H.; Hao, Y.; Yeung, B.Z.; Smibert, P.; et al. Characterizing the molecular regulation of inhibitory immune checkpoints with multimodal single-cell screens. Nat. Genet. 2021, 53, 322–331. [Google Scholar] [CrossRef]
- Dempster, J.M.; Pacini, C.; Pantel, S.; Behan, F.M.; Green, T.; Krill-Burger, J.; Beaver, C.M.; Younger, S.T.; Zhivich, V.; Najgebauer, H.; et al. Agreement between two large pan-cancer CRISPR-Cas9 gene dependency data sets. Nat. Commun. 2019, 10, 5817. [Google Scholar] [CrossRef] [Green Version]
- Pacini, C.; Dempster, J.M.; Boyle, I.; Gonçalves, E.; Najgebauer, H.; Karakoc, E.; van der Meer, D.; Barthorpe, A.; Lightfoot, H.; Jaaks, P.; et al. Integrated cross-study datasets of genetic dependencies in cancer. Nat. Commun. 2021, 12, 1661. [Google Scholar] [CrossRef]
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Pantel, S.; Krill-Burger, J.M.; et al. Defining a Cancer Dependency Map. Cell 2017, 170, 564–576.e16. [Google Scholar] [CrossRef] [Green Version]
- Behan, F.M.; Iorio, F.; Picco, G.; Gonçalves, E.; Beaver, C.M.; Migliardi, G.; Santos, R.; Rao, Y.; Sassi, F.; Pinnelli, M.; et al. Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature 2019, 568, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Onishi, I.; Yamamoto, K.; Kinowaki, Y.; Kitagawa, M.; Kurata, M. To Discover the Efficient and Novel Drug Targets in Human Cancers Using CRISPR/Cas Screening and Databases. Int. J. Mol. Sci. 2021, 22, 12322. [Google Scholar] [CrossRef] [PubMed]
- Dempster, J.M.; Boyle, I.; Vazquez, F.; Root, D.E.; Boehm, J.S.; Hahn, W.C.; Tsherniak, A.; McFarland, J.M. Chronos: A cell population dynamics model of CRISPR experiments that improves inference of gene fitness effects. Genome Biol. 2021, 22, 343. [Google Scholar] [CrossRef] [PubMed]
- Rauscher, B.; Heigwer, F.; Breinig, M.; Winter, J.; Boutros, M. GenomeCRISPR—A database for high-throughput CRISPR/Cas9 screens. Nucleic Acids Res. 2017, 45, D679–D686. [Google Scholar] [CrossRef] [Green Version]
- Lenoir, W.F.; Lim, T.L.; Hart, T. PICKLES: The database of pooled in-vitro CRISPR knockout library essentiality screens. Nucleic Acids Res. 2018, 46, D776–D780. [Google Scholar] [CrossRef] [Green Version]
- Oughtred, R.; Rust, J.; Chang, C.; Breitkreutz, B.; Stark, C.; Willems, A.; Boucher, L.; Leung, G.; Kolas, N.; Zhang, F.; et al. The BioGRID database: A comprehensive biomedical resource of curated protein, genetic, and chemical interactions. Protein Sci. 2021, 30, 187–200. [Google Scholar] [CrossRef]
- Choi, A.; Jang, I.; Han, H.; Kim, M.-S.; Choi, J.; Lee, J.; Cho, S.-Y.; Jun, Y.; Lee, C.; Kim, J.; et al. iCSDB: An integrated database of CRISPR screens. Nucleic Acids Res. 2021, 49, D956–D961. [Google Scholar] [CrossRef]
- Armitage, J.O.; Gascoyne, R.D.; Lunning, M.A.; Cavalli, F. Non-Hodgkin lymphoma. Lancet Lond. Engl. 2017, 390, 298–310. [Google Scholar] [CrossRef]
- Sun, R.; Medeiros, L.J.; Young, K.H. Diagnostic and predictive biomarkers for lymphoma diagnosis and treatment in the era of precision medicine. Mod. Pathol. 2016, 29, 1118–1142. [Google Scholar] [CrossRef] [Green Version]
- He, M.Y.; Kridel, R. Treatment resistance in diffuse large B-cell lymphoma. Leukemia 2021, 35, 2151–2165. [Google Scholar] [CrossRef]
- Liu, Y.; Barta, S.K. Diffuse large B-cell lymphoma: 2019 update on diagnosis, risk stratification, and treatment. Am. J. Hematol. 2019, 94, 604–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ioannou, N.; Jain, K.; Ramsay, A.G. Immunomodulatory Drugs for the Treatment of B Cell Malignancies. Int. J. Mol. Sci. 2021, 22, 8572. [Google Scholar] [CrossRef]
- Grover, N.S.; Park, S.I. Novel Targeted Agents in Hodgkin and Non-Hodgkin Lymphoma Therapy. Pharmaceuticals 2015, 8, 607–636. [Google Scholar] [CrossRef] [PubMed]
- Mossé, Y.P.; Voss, S.D.; Lim, M.S.; Rolland, D.; Minard, C.G.; Fox, E.; Adamson, P.; Wilner, K.; Blaney, S.M.; Weigel, B.J. Targeting ALK With Crizotinib in Pediatric Anaplastic Large Cell Lymphoma and Inflammatory Myofibroblastic Tumor: A Children’s Oncology Group Study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2017, 35, 3215–3221. [Google Scholar] [CrossRef] [PubMed]
- Brugières, L.; Houot, R.; Cozic, N.; De La Fouchardière, C.; Morschhauser, F.; Brice, P.; Arakelyan Laboure, N.; Auvrignon, A.; Aladjidi, N.; Kolb, B.; et al. Crizotinib in Advanced ALK+ Anaplastic Large Cell Lymphoma in Children and Adults: Results of the Acs© Phase II Trial. Blood 2017, 130, 2831. [Google Scholar]
- Bhat, M.; Robichaud, N.; Hulea, L.; Sonenberg, N.; Pelletier, J.; Topisirovic, I. Targeting the translation machinery in cancer. Nat. Rev. Drug Discov. 2015, 14, 261–278. [Google Scholar] [CrossRef] [PubMed]
- Sanghvi, V.R.; Mohan, P.; Singh, K.; Cao, L.; Berishaj, M.; Wolfe, A.L.; Schatz, J.H.; Lailler, N.; de Stanchina, E.; Viale, A.; et al. NRF2 Activation Confers Resistance to eIF4A Inhibitors in Cancer Therapy. Cancers 2021, 13, 639. [Google Scholar] [CrossRef]
- Bhojwani, D.; Pui, C.-H. Relapsed childhood acute lymphoblastic leukaemia. Lancet Oncol. 2013, 14, e205–e217. [Google Scholar] [CrossRef]
- Hsiao, M.H.; Yu, A.L.; Yeargin, J.; Ku, D.; Haas, M. Nonhereditary p53 mutations in T-cell acute lymphoblastic leukemia are associated with the relapse phase. Blood 1994, 83, 2922–2930. [Google Scholar] [CrossRef] [Green Version]
- Tzoneva, G.; Perez-Garcia, A.; Carpenter, Z.; Khiabanian, H.; Tosello, V.; Allegretta, M.; Paietta, E.; Racevskis, J.; Rowe, J.M.; Tallman, M.S.; et al. Activating mutations in the NT5C2 nucleotidase gene drive chemotherapy resistance in relapsed ALL. Nat. Med. 2013, 19, 368–371. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Li, H.; Bai, Y.; Kirschner-Schwabe, R.; Yang, J.J.; Chen, Y.; Lu, G.; Tzoneva, G.; Ma, X.; Wu, T.; et al. Negative feedback-defective PRPS1 mutants drive thiopurine resistance in relapsed childhood ALL. Nat. Med. 2015, 21, 563–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullighan, C.G.; Zhang, J.; Kasper, L.H.; Lerach, S.; Payne-Turner, D.; Phillips, L.A.; Heatley, S.L.; Holmfeldt, L.; Collins-Underwood, J.R.; Ma, J.; et al. CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature 2011, 471, 235–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inaba, H.; Pui, C.-H. Glucocorticoid use in acute lymphoblastic leukemia: Comparison of prednisone and dexamethasone. Lancet Oncol. 2010, 11, 1096–1106. [Google Scholar] [CrossRef] [Green Version]
- Paugh, S.W.; Bonten, E.J.; Savic, D.; Ramsey, L.B.; Thierfelder, W.E.; Gurung, P.; Malireddi, R.K.S.; Actis, M.; Mayasundari, A.; Min, J.; et al. NALP3 inflammasome upregulation and CASP1 cleavage of the glucocorticoid receptor cause glucocorticoid resistance in leukemia cells. Nat. Genet. 2015, 47, 607–614. [Google Scholar] [CrossRef] [Green Version]
- Pottier, N.; Yang, W.; Assem, M.; Panetta, J.C.; Pei, D.; Paugh, S.W.; Cheng, C.; Den Boer, M.L.; Relling, M.V.; Pieters, R.; et al. The SWI/SNF chromatin-remodeling complex and glucocorticoid resistance in acute lymphoblastic leukemia. J. Natl. Cancer Inst. 2008, 100, 1792–1803. [Google Scholar] [CrossRef] [Green Version]
- Foà, R.; Vitale, A.; Vignetti, M.; Meloni, G.; Guarini, A.; De Propris, M.S.; Elia, L.; Paoloni, F.; Fazi, P.; Cimino, G.; et al. Dasatinib as first-line treatment for adult patients with Philadelphia chromosome–positive acute lymphoblastic leukemia. Blood 2011, 118, 6521–6528. [Google Scholar] [CrossRef] [Green Version]
- Vilas-Zornoza, A.; Agirre, X.; Abizanda, G.; Moreno, C.; Segura, V.; De Martino Rodriguez, A.; José-Eneriz, E.S.; Miranda, E.; Martín-Subero, J.I.; Garate, L.; et al. Preclinical activity of LBH589 alone or in combination with chemotherapy in a xenogeneic mouse model of human acute lymphoblastic leukemia. Leukemia 2012, 26, 1517–1526. [Google Scholar] [CrossRef]
- Garrido Castro, P.; van Roon, E.H.J.; Pinhanços, S.S.; Trentin, L.; Schneider, P.; Kerstjens, M.; Te Kronnie, G.; Heidenreich, O.; Pieters, R.; Stam, R.W. The HDAC inhibitor panobinostat (LBH589) exerts in vivo anti-leukaemic activity against MLL-rearranged acute lymphoblastic leukaemia and involves the RNF20/RNF40/WAC-H2B ubiquitination axis. Leukemia 2018, 32, 323–331. [Google Scholar] [CrossRef]
- Hallek, M. Chronic lymphocytic leukemia: 2020 update on diagnosis, risk stratification and treatment. Am. J. Hematol. 2019, 94, 1266–1287. [Google Scholar] [CrossRef] [Green Version]
- Kipps, T.J.; Choi, M.Y. Targeted Therapy in Chronic Lymphocytic Leukemia. Cancer J. 2019, 25, 378–385. [Google Scholar] [CrossRef]
- Pérez-Carretero, C.; González-Gascón-Y-Marín, I.; Rodríguez-Vicente, A.E.; Quijada-Álamo, M.; Hernández-Rivas, J.-Á.; Hernández-Sánchez, M.; Hernández-Rivas, J.M. The Evolving Landscape of Chronic Lymphocytic Leukemia on Diagnosis, Prognosis and Treatment. Diagn. Basel Switz. 2021, 11, 853. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.A.; Tam, C.; Lew, T.E.; Juneja, S.; Juneja, M.; Westerman, D.; Wall, M.; Lade, S.; Gorelik, A.; Huang, D.C.S.; et al. Clinicopathological features and outcomes of progression of CLL on the BCL2 inhibitor venetoclax. Blood 2017, 129, 3362–3370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyle, R.A.; Rajkumar, S.V. Multiple myeloma. Blood 2008, 111, 2962–2972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, J.J.; Orlowski, R.Z. Proteasome inhibitors in the treatment of multiple myeloma. Leukemia 2009, 23, 1964–1979. [Google Scholar] [CrossRef] [Green Version]
- Acosta-Alvear, D.; Cho, M.Y.; Wild, T.; Buchholz, T.J.; Lerner, A.G.; Simakova, O.; Hahn, J.; Korde, N.; Landgren, O.; Maric, I.; et al. Paradoxical resistance of multiple myeloma to proteasome inhibitors by decreased levels of 19S proteasomal subunits. eLife 2015, 4, e08153. [Google Scholar] [CrossRef]
- Franke, N.E.; Niewerth, D.; Assaraf, Y.G.; van Meerloo, J.; Vojtekova, K.; van Zantwijk, C.H.; Zweegman, S.; Chan, E.T.; Kirk, C.J.; Geerke, D.P.; et al. Impaired bortezomib binding to mutant β5 subunit of the proteasome is the underlying basis for bortezomib resistance in leukemia cells. Leukemia 2012, 26, 757–768. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.K.; Rajkumar, S.V.; Dispenzieri, A.; Lacy, M.Q.; Hayman, S.R.; Buadi, F.K.; Zeldenrust, S.R.; Dingli, D.; Russell, S.J.; Lust, J.A.; et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood 2008, 111, 2516–2520. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Song, T.; Zhou, W.; Xing, L.; Wang, S.; Ho, M.; Peng, Z.; Tai, Y.-T.; Hideshima, T.; Anderson, K.C.; et al. A genome-scale CRISPR-Cas9 screening in myeloma cells identifies regulators of immunomodulatory drug sensitivity. Leukemia 2019, 33, 171–180. [Google Scholar] [CrossRef]
- Shirasaki, R.; Matthews, G.M.; Gandolfi, S.; de Matos Simoes, R.; Buckley, D.L.; Raja Vora, J.; Sievers, Q.L.; Brüggenthies, J.B.; Dashevsky, O.; Poarch, H.; et al. Functional Genomics Identify Distinct and Overlapping Genes Mediating Resistance to Different Classes of Heterobifunctional Degraders of Oncoproteins. Cell Rep. 2021, 34, 108532. [Google Scholar] [CrossRef]
- Anderson, K.C. Progress and Paradigms in Multiple Myeloma. Clin. Cancer Res. 2016, 22, 5419–5427. [Google Scholar] [CrossRef] [Green Version]
- Shallis, R.M.; Wang, R.; Davidoff, A.; Ma, X.; Zeidan, A.M. Epidemiology of acute myeloid leukemia: Recent progress and enduring challenges. Blood Rev. 2019, 36, 70–87. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurnari, C.; Pagliuca, S.; Visconte, V. Deciphering the Therapeutic Resistance in Acute Myeloid Leukemia. Int. J. Mol. Sci. 2020, 21, 8505. [Google Scholar] [CrossRef]
- Sakamoto, K.M.; Grant, S.; Saleiro, D.; Crispino, J.D.; Hijiya, N.; Giles, F.; Platanias, L.; Eklund, E.A. Targeting novel signaling pathways for resistant acute myeloid leukemia. Mol. Genet. Metab. 2015, 114, 397–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rathe, S.K.; Moriarity, B.S.; Stoltenberg, C.B.; Kurata, M.; Aumann, N.K.; Rahrmann, E.P.; Bailey, N.J.; Melrose, E.G.; Beckmann, D.A.; Liska, C.R.; et al. Using RNA-seq and targeted nucleases to identify mechanisms of drug resistance in acute myeloid leukemia. Sci. Rep. 2014, 4, 6048. [Google Scholar] [CrossRef] [Green Version]
- K Bhanumathy, K.; Balagopal, A.; Vizeacoumar, F.S.; Vizeacoumar, F.J.; Freywald, A.; Giambra, V. Protein Tyrosine Kinases: Their Roles and Their Targeting in Leukemia. Cancers 2021, 13, 184. [Google Scholar] [CrossRef] [PubMed]
- Scholl, S.; Fleischmann, M.; Schnetzke, U.; Heidel, F.H. Molecular Mechanisms of Resistance to FLT3 Inhibitors in Acute Myeloid Leukemia: Ongoing Challenges and Future Treatments. Cells 2020, 9, 2493. [Google Scholar] [CrossRef]
- Morales, M.L.; Arenas, A.; Ortiz-Ruiz, A.; Leivas, A.; Rapado, I.; Rodríguez-García, A.; Castro, N.; Zagorac, I.; Quintela-Fandino, M.; Gómez-López, G.; et al. MEK inhibition enhances the response to tyrosine kinase inhibitors in acute myeloid leukemia. Sci. Rep. 2019, 9, 18630. [Google Scholar] [CrossRef] [Green Version]
- Burnett, A.; Stone, R. AML: New Drugs but New Challenges. Clin. Lymphoma Myeloma Leuk. 2020, 20, 341–350. [Google Scholar] [CrossRef]
- Grieselhuber, N.R.; Mims, A.S. Novel Targeted Therapeutics in Acute Myeloid Leukemia: An Embarrassment of Riches. Curr. Hematol. Malig. Rep. 2021, 16, 192–206. [Google Scholar] [CrossRef]
- Surka, C.; Jin, L.; Mbong, N.; Lu, C.-C.; Jang, I.S.; Rychak, E.; Mendy, D.; Clayton, T.; Tindall, E.; Hsu, C.; et al. CC-90009, a novel cereblon E3 ligase modulator, targets acute myeloid leukemia blasts and leukemia stem cells. Blood 2021, 137, 661–677. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.-C.; Zheng, S.; Wang, L.-J.; Iskra, B.S.; Rao, M.K.; Houghton, P.J.; Huang, Y.; Chen, Y. Predicting and characterizing a cancer dependency map of tumors with deep learning. Sci. Adv. 2021, 7, eabh1275. [Google Scholar] [CrossRef] [PubMed]
- Blanck, M.; Budnik-Zawilska, M.B.; Lenger, S.R.; McGonigle, J.E.; Martin, G.R.A.; le Sage, C.; Lawo, S.; Pemberton, H.N.; Tiwana, G.S.; Sorrell, D.A.; et al. A Flexible, Pooled CRISPR Library for Drug Development Screens. CRISPR J. 2020, 3, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Wilke, A.C.; Doebele, C.; Zindel, A.; Lee, K.S.; Rieke, S.A.; Ceribelli, M.; Comoglio, F.; Phelan, J.D.; Wang, J.Q.; Pikman, Y.; et al. SHMT2 inhibition disrupts the TCF3 transcriptional survival program in Burkitt lymphoma. Blood 2022, 139, 538–553. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Medeiros, L.J.; Li, Y.; Li, J.; Young, K.H. Genetic alterations and their clinical implications in DLBCL. Nat. Rev. Clin. Oncol. 2019, 16, 634–652. [Google Scholar] [CrossRef]
- Sakihama, S.; Karube, K. Genetic Alterations in Adult T-Cell Leukemia/Lymphoma: Novel Discoveries with Clinical and Biological Significance. Cancers 2022, 14, 2394. [Google Scholar] [CrossRef]
- Borchmann, S.; Engert, A. The genetics of Hodgkin lymphoma: An overview and clinical implications. Curr. Opin. Oncol. 2017, 29, 307–314. [Google Scholar] [CrossRef]
- Juskevicius, D.; Jucker, D.; Klingbiel, D.; Mamot, C.; Dirnhofer, S.; Tzankov, A. Mutations of CREBBP and SOCS1 are independent prognostic factors in diffuse large B cell lymphoma: Mutational analysis of the SAKK 38/07 prospective clinical trial cohort. J. Hematol. Oncol.J Hematol Oncol 2017, 10, 70. [Google Scholar] [CrossRef] [Green Version]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [Green Version]
- Boxer, L.M.; Dang, C.V. Translocations involving c-myc and c-myc function. Oncogene 2001, 20, 5595–5610. [Google Scholar] [CrossRef] [Green Version]
- Cucco, F.; Barrans, S.; Sha, C.; Clipson, A.; Crouch, S.; Dobson, R.; Chen, Z.; Thompson, J.S.; Care, M.A.; Cummin, T.; et al. Distinct genetic changes reveal evolutionary history and heterogeneous molecular grade of DLBCL with MYC/BCL2 double-hit. Leukemia 2020, 34, 1329–1341. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, R.; Hansmann, M.-L.; Bohle, V.; Martin-Subero, J.I.; Hartmann, S.; Mechtersheimer, G.; Klapper, W.; Vater, I.; Giefing, M.; Gesk, S.; et al. TNFAIP3 (A20) is a tumor suppressor gene in Hodgkin lymphoma and primary mediastinal B cell lymphoma. J. Exp. Med. 2009, 206, 981–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aalipour, A.; Advani, R.H. Bruton’s tyrosine kinase inhibitors and their clinical potential in the treatment of B-cell malignancies: Focus on ibrutinib. Ther. Adv. Hematol. 2014, 5, 121–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alinari, L.; Quinion, C.; Blum, K.A. Bruton’s tyrosine kinase inhibitors in B-cell non-Hodgkin’s lymphomas. Clin. Pharmacol. Ther. 2015, 97, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Tong, K.I.; Yoon, S.; Isaev, K.; Bakhtiari, M.; Lackraj, T.; He, M.Y.; Joynt, J.; Silva, A.; Xu, M.C.; Privé, G.G.; et al. Combined EZH2 Inhibition and IKAROS Degradation Leads to Enhanced Antitumor Activity in Diffuse Large B-cell Lymphoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 5401–5414. [Google Scholar] [CrossRef]
- Gayle, S.; Landrette, S.; Beeharry, N.; Conrad, C.; Hernandez, M.; Beckett, P.; Ferguson, S.M.; Xu, T.; Rothberg, J.; Lichenstein, H. B-cell non-Hodgkin lymphoma: Selective vulnerability to PIKFYVE inhibition. Autophagy 2017, 13, 1082–1083. [Google Scholar] [CrossRef] [Green Version]
- Dharia, N.V.; Kugener, G.; Guenther, L.M.; Malone, C.F.; Durbin, A.D.; Hong, A.L.; Howard, T.P.; Bandopadhayay, P.; Wechsler, C.S.; Fung, I.; et al. A first-generation pediatric cancer dependency map. Nat. Genet. 2021, 53, 529–538. [Google Scholar] [CrossRef]
- Chan, L.N.; Chen, Z.; Braas, D.; Lee, J.-W.; Xiao, G.; Geng, H.; Cosgun, K.N.; Hurtz, C.; Shojaee, S.; Cazzaniga, V.; et al. Metabolic gatekeeper function of B-lymphoid transcription factors. Nature 2017, 542, 479–483. [Google Scholar] [CrossRef]
- Guièze, R.; Wu, C.J. Genomic and epigenomic heterogeneity in chronic lymphocytic leukemia. Blood 2015, 126, 445–453. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Vicente, A.E.; Bikos, V.; Hernández-Sánchez, M.; Malcikova, J.; Hernández-Rivas, J.-M.; Pospisilova, S. Next-generation sequencing in chronic lymphocytic leukemia: Recent findings and new horizons. Oncotarget 2017, 8, 71234–71248. [Google Scholar] [CrossRef] [Green Version]
- Quijada-Álamo, M.; Pérez-Carretero, C.; Hernández-Sánchez, M.; Rodríguez-Vicente, A.-E.; Herrero, A.-B.; Hernández-Sánchez, J.-M.; Martín-Izquierdo, M.; Santos-Mínguez, S.; Del Rey, M.; González, T.; et al. Dissecting the role of TP53 alterations in del(11q) chronic lymphocytic leukemia. Clin. Transl. Med. 2021, 11, e304. [Google Scholar] [CrossRef] [PubMed]
- Quijada-Álamo, M.; Hernández-Sánchez, M.; Rodríguez-Vicente, A.-E.; Pérez-Carretero, C.; Rodríguez-Sánchez, A.; Martín-Izquierdo, M.; Alonso-Pérez, V.; García-Tuñón, I.; Bastida, J.M.; Vidal-Manceñido, M.J.; et al. Biological significance of monoallelic and biallelic BIRC3 loss in del(11q) chronic lymphocytic leukemia progression. Blood Cancer J. 2021, 11, 127. [Google Scholar] [CrossRef] [PubMed]
- Quijada-Álamo, M.; Hernández-Sánchez, M.; Alonso-Pérez, V.; Rodríguez-Vicente, A.E.; García-Tuñón, I.; Martín-Izquierdo, M.; Hernández-Sánchez, J.M.; Herrero, A.B.; Bastida, J.M.; San Segundo, L.; et al. CRISPR/Cas9-generated models uncover therapeutic vulnerabilities of del(11q) CLL cells to dual BCR and PARP inhibition. Leukemia 2020, 34, 1599–1612. [Google Scholar] [CrossRef]
- Ten Hacken, E.; Clement, K.; Li, S.; Hernández-Sánchez, M.; Redd, R.; Wang, S.; Ruff, D.; Gruber, M.; Baranowski, K.; Jacob, J.; et al. High throughput single-cell detection of multiplex CRISPR-edited gene modifications. Genome Biol. 2020, 21, 266. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Knittel, G.; Rehkämper, T.; Korovkina, D.; Liedgens, P.; Fritz, C.; Torgovnick, A.; Al-Baldawi, Y.; Al-Maarri, M.; Cun, Y.; Fedorchenko, O.; et al. Two mouse models reveal an actionable PARP1 dependence in aggressive chronic lymphocytic leukemia. Nat. Commun. 2017, 8, 153. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.A.; Mavrommatis, K.; Wardell, C.P.; Ashby, T.C.; Bauer, M.; Davies, F.E.; Rosenthal, A.; Wang, H.; Qu, P.; Hoering, A.; et al. Identification of novel mutational drivers reveals oncogene dependencies in multiple myeloma. Blood 2018, 132, 587–597. [Google Scholar] [CrossRef]
- Walker, B.A.; Boyle, E.M.; Wardell, C.P.; Murison, A.; Begum, D.B.; Dahir, N.M.; Proszek, P.Z.; Johnson, D.C.; Kaiser, M.F.; Melchor, L.; et al. Mutational Spectrum, Copy Number Changes, and Outcome: Results of a Sequencing Study of Patients with Newly Diagnosed Myeloma. J. Clin. Oncol. 2015, 33, 3911–3920. [Google Scholar] [CrossRef]
- García-Ortiz, A.; Rodríguez-García, Y.; Encinas, J.; Maroto-Martín, E.; Castellano, E.; Teixidó, J.; Martínez-López, J. The Role of Tumor Microenvironment in Multiple Myeloma Development and Progression. Cancers 2021, 13, 217. [Google Scholar] [CrossRef]
- Cazzola, M. Myelodysplastic Syndromes. N. Engl. J. Med. 2020, 383, 1358–1374. [Google Scholar] [CrossRef]
- Platzbecker, U. Treatment of MDS. Blood 2019, 133, 1096–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, R.N.; Steeples, V.; Singh, S.; Sanchi, A.; Boultwood, J.; Pellagatti, A. Splicing factor mutations in the myelodysplastic syndromes: Target genes and therapeutic approaches. Adv. Biol. Regul. 2018, 67, 13–29. [Google Scholar] [CrossRef] [PubMed]
- Navada, S.C.; Fruchtman, S.M.; Odchimar-Reissig, R.; Demakos, E.P.; Petrone, M.E.; Zbyszewski, P.S.; Holland, J.F.; Silverman, L.R. A phase 1/2 study of rigosertib in patients with myelodysplastic syndromes (MDS) and MDS progressed to acute myeloid leukemia. Leuk. Res. 2018, 64, 10–16. [Google Scholar] [CrossRef]
- Wang, E.; Zhou, H.; Nadorp, B.; Cayanan, G.; Chen, X.; Yeaton, A.H.; Nomikou, S.; Witkowski, M.T.; Narang, S.; Kloetgen, A.; et al. Surface antigen-guided CRISPR screens identify regulators of myeloid leukemia differentiation. Cell Stem Cell 2021, 28, 718–731.e6. [Google Scholar] [CrossRef]
- Stavropoulou, V.; Peters, A.H.F.M.; Schwaller, J. Aggressive leukemia driven by MLL-AF9. Mol. Cell. Oncol. 2018, 5, e1241854. [Google Scholar] [CrossRef] [Green Version]
- Au, Y.Z.; Gu, M.; De Braekeleer, E.; Gozdecka, M.; Aspris, D.; Tarumoto, Y.; Cooper, J.; Yu, J.; Ong, S.H.; Chen, X.; et al. KAT7 is a genetic vulnerability of acute myeloid leukemias driven by MLL rearrangements. Leukemia 2021, 35, 1012–1022. [Google Scholar] [CrossRef]
- Zhou, J.-D.; Yao, D.-M.; Li, X.-X.; Zhang, T.-J.; Zhang, W.; Ma, J.-C.; Guo, H.; Deng, Z.-Q.; Lin, J.; Qian, J. KRAS overexpression independent of RAS mutations confers an adverse prognosis in cytogenetically normal acute myeloid leukemia. Oncotarget 2017, 8, 66087–66097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Yu, H.; Hughes, N.W.; Liu, B.; Kendirli, A.; Klein, K.; Chen, W.W.; Lander, E.S.; Sabatini, D.M. Gene Essentiality Profiling Reveals Gene Networks and Synthetic Lethal Interactions with Oncogenic Ras. Cell 2017, 168, 890–903.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, R.; Salinas-Illarena, A.; Baldauf, H.-M. New strategies to treat AML: Novel insights into AML survival pathways and combination therapies. Leukemia 2021, 35, 299–311. [Google Scholar] [CrossRef]
- Brinton, L.T.; Zhang, P.; Williams, K.; Canfield, D.; Orwick, S.; Sher, S.; Wasmuth, R.; Beaver, L.; Cempre, C.; Skinner, J.; et al. Synergistic effect of BCL2 and FLT3 co-inhibition in acute myeloid leukemia. J. Hematol. Oncol.J Hematol Oncol 2020, 13, 139. [Google Scholar] [CrossRef]
- Fiskus, W.; Mill, C.P.; Nabet, B.; Perera, D.; Birdwell, C.; Manshouri, T.; Lara, B.; Kadia, T.M.; DiNardo, C.; Takahashi, K.; et al. Superior efficacy of co-targeting GFI1/KDM1A and BRD4 against AML and post-MPN secondary AML cells. Blood Cancer J. 2021, 11, 98. [Google Scholar] [CrossRef] [PubMed]
- Druker, B.J.; Tamura, S.; Buchdunger, E.; Ohno, S.; Segal, G.M.; Fanning, S.; Zimmermann, J.; Lydon, N.B. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat. Med. 1996, 2, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Saußele, S.; Silver, R.T. Management of chronic myeloid leukemia in blast crisis. Ann. Hematol. 2015, 94 (Suppl. S2), S159–S165. [Google Scholar] [CrossRef] [PubMed]
- Alves, R.; Gonçalves, A.C.; Rutella, S.; Almeida, A.M.; De Las Rivas, J.; Trougakos, I.P.; Sarmento Ribeiro, A.B. Resistance to Tyrosine Kinase Inhibitors in Chronic Myeloid Leukemia—From Molecular Mechanisms to Clinical Relevance. Cancers 2021, 13, 4820. [Google Scholar] [CrossRef] [PubMed]
- El Eit, R.M.; Iskandarani, A.N.; Saliba, J.L.; Jabbour, M.N.; Mahfouz, R.A.; Bitar, N.M.A.; Ayoubi, H.R.E.; Zaatari, G.S.; Mahon, F.-X.; De Thé, H.B.; et al. Effective targeting of chronic myeloid leukemia initiating activity with the combination of arsenic trioxide and interferon alpha: Arsenic and interferon targets CML initiating cells activity. Int. J. Cancer 2014, 134, 988–996. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Gong, L.; Su, D.; Jin, Y.; Guo, C.; Yue, M.; Yao, S.; Qin, Z.; Ye, Y.; Tang, Y.; et al. Cullin5 deficiency promotes small-cell lung cancer metastasis by stabilizing integrin β1. J. Clin. Investig. 2019, 129, 972–987. [Google Scholar] [CrossRef]
- Marinaccio, C.; Suraneni, P.; Celik, H.; Volk, A.; Wen, Q.J.; Ling, T.; Bulic, M.; Lasho, T.; Koche, R.P.; Famulare, C.A.; et al. LKB1/STK11 Is a Tumor Suppressor in the Progression of Myeloproliferative Neoplasms. Cancer Discov. 2021, 11, 1398–1410. [Google Scholar] [CrossRef]
- Mintzer, D.; Bagg, A. Clinical syndromes of transformation in clonal hematologic disorders. Am. J. Med. 2001, 111, 480–488. [Google Scholar] [CrossRef]
- Menssen, A.J.; Walter, M.J. Genetics of progression from MDS to secondary leukemia. Blood 2020, 136, 50–60. [Google Scholar] [CrossRef]
- Mo, A.; Chang, L.; Duns, G.; Ibrahim, R.; Docking, R.; Umlandt, P.; Parker, J.; Karsan, A. Loss of fbxo11 functions drives acute myeloid leukemia. Exp. Hematol. 2019, 76, S78. [Google Scholar] [CrossRef]
- Rampal, R.; Ahn, J.; Abdel-Wahab, O.; Nahas, M.; Wang, K.; Lipson, D.; Otto, G.A.; Yelensky, R.; Hricik, T.; McKenney, A.S.; et al. Genomic and functional analysis of leukemic transformation of myeloproliferative neoplasms. Proc. Natl. Acad. Sci. USA 2014, 111, E5401–E5410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Courtier, F.; Carbuccia, N.; Garnier, S.; Guille, A.; Adélaïde, J.; Cervera, N.; Gelsi-Boyer, V.; Mozziconacci, M.-J.; Rey, J.; Vey, N.; et al. Genomic analysis of myeloproliferative neoplasms in chronic and acute phases. Haematologica 2017, 102, e11–e14. [Google Scholar] [CrossRef] [PubMed]
- Mannelli, F. Acute Myeloid Leukemia Evolving from Myeloproliferative Neoplasms: Many Sides of a Challenging Disease. J. Clin. Med. 2021, 10, 436. [Google Scholar] [CrossRef] [PubMed]
- Krump, N.A.; You, J. Molecular mechanisms of viral oncogenesis in humans. Nat. Rev. Microbiol. 2018, 16, 684–698. [Google Scholar] [CrossRef]
- Brady, G.; MacArthur, G.J.; Farrell, P.J. Epstein–Barr virus and Burkitt lymphoma. J. Clin. Pathol. 2007, 60, 1397–1402. [Google Scholar] [CrossRef]
- Ma, Y.; Walsh, M.J.; Bernhardt, K.; Ashbaugh, C.W.; Trudeau, S.J.; Ashbaugh, I.Y.; Jiang, S.; Jiang, C.; Zhao, B.; Root, D.E.; et al. CRISPR/Cas9 Screens Reveal Epstein-Barr Virus-Transformed B Cell Host Dependency Factors. Cell Host Microbe 2017, 21, 580–591.e7. [Google Scholar] [CrossRef] [Green Version]
- Guo, R.; Zhang, Y.; Teng, M.; Jiang, C.; Schineller, M.; Zhao, B.; Doench, J.G.; O’Reilly, R.J.; Cesarman, E.; Giulino-Roth, L.; et al. DNA methylation enzymes and PRC1 restrict B-cell Epstein-Barr virus oncoprotein expression. Nat. Microbiol. 2020, 5, 1051–1063. [Google Scholar] [CrossRef]
- Manzano, M.; Patil, A.; Waldrop, A.; Dave, S.S.; Behdad, A.; Gottwein, E. Gene essentiality landscape and druggable oncogenic dependencies in herpesviral primary effusion lymphoma. Nat. Commun. 2018, 9, 3263. [Google Scholar] [CrossRef]
- Miri, S.M.; Tafsiri, E.; Cho, W.C.S.; Ghaemi, A. CRISPR-Cas, a robust gene-editing technology in the era of modern cancer immunotherapy. Cancer Cell Int. 2020, 20, 456. [Google Scholar] [CrossRef]
- Dimitri, A.; Herbst, F.; Fraietta, J.A. Engineering the next-generation of CAR T-cells with CRISPR-Cas9 gene editing. Mol. Cancer 2022, 21, 78. [Google Scholar] [CrossRef]
- Im, A.; Pavletic, S.Z. Immunotherapy in hematologic malignancies: Past, present, and future. J. Hematol. Oncol. 2017, 10, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salik, B.; Smyth, M.J.; Nakamura, K. Targeting immune checkpoints in hematological malignancies. J. Hematol. Oncol. 2020, 13, 111. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef]
- Zhang, J.-P.; Song, Z.; Wang, H.-B.; Lang, L.; Yang, Y.-Z.; Xiao, W.; Webster, D.E.; Wei, W.; Barta, S.K.; Kadin, M.E.; et al. A novel model of controlling PD-L1 expression in ALK+ anaplastic large cell lymphoma revealed by CRISPR screening. Blood 2019, 134, 171–185. [Google Scholar] [CrossRef]
- Schuster, S.J. Bispecific antibodies for the treatment of lymphomas: Promises and challenges. Hematol. Oncol. 2021, 39 (Suppl. S1), 113–116. [Google Scholar] [CrossRef]
- Decker, C.E.; Young, T.; Pasnikowski, E.; Chiu, J.; Song, H.; Wei, Y.; Thurston, G.; Daly, C. Genome-scale CRISPR activation screen uncovers tumor-intrinsic modulators of CD3 bispecific antibody efficacy. Sci. Rep. 2019, 9, 20068. [Google Scholar] [CrossRef] [PubMed]
- Vitale, C.; Strati, P. CAR T-Cell Therapy for B-Cell non-Hodgkin Lymphoma and Chronic Lymphocytic Leukemia: Clinical Trials and Real-World Experiences. Front. Oncol. 2020, 10, 849. [Google Scholar] [CrossRef] [PubMed]
- Dufva, O.; Koski, J.; Maliniemi, P.; Ianevski, A.; Klievink, J.; Leitner, J.; Pölönen, P.; Hohtari, H.; Saeed, K.; Hannunen, T.; et al. Integrated drug profiling and CRISPR screening identify essential pathways for CAR T-cell cytotoxicity. Blood 2020, 135, 597–609. [Google Scholar] [CrossRef] [Green Version]
- Upadhyay, R.; Boiarsky, J.A.; Pantsulaia, G.; Svensson-Arvelund, J.; Lin, M.J.; Wroblewska, A.; Bhalla, S.; Scholler, N.; Bot, A.; Rossi, J.M.; et al. A Critical Role for Fas-Mediated Off-Target Tumor Killing in T-cell Immunotherapy. Cancer Discov. 2021, 11, 599–613. [Google Scholar] [CrossRef]
- Yan, X.; Chen, D.; Wang, Y.; Guo, Y.; Tong, C.; Wei, J.; Zhang, Y.; Wu, Z.; Han, W. Identification of NOXA as a pivotal regulator of resistance to CAR T-cell therapy in B-cell malignancies. Signal Transduct. Target. Ther. 2022, 7, 98. [Google Scholar] [CrossRef]
- Raje, N.; Berdeja, J.; Lin, Y.; Siegel, D.; Jagannath, S.; Madduri, D.; Liedtke, M.; Rosenblatt, J.; Maus, M.V.; Turka, A.; et al. Anti-BCMA CAR T-Cell Therapy bb2121 in Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2019, 380, 1726–1737. [Google Scholar] [CrossRef] [PubMed]
- Brudno, J.N.; Maric, I.; Hartman, S.D.; Rose, J.J.; Wang, M.; Lam, N.; Stetler-Stevenson, M.; Salem, D.; Yuan, C.; Pavletic, S.; et al. T Cells Genetically Modified to Express an Anti-B-Cell Maturation Antigen Chimeric Antigen Receptor Cause Remissions of Poor-Prognosis Relapsed Multiple Myeloma. J. Clin. Oncol. 2018, 36, 2267–2280. [Google Scholar] [CrossRef] [PubMed]
- Soares, F.; Chen, B.; Lee, J.B.; Ahmed, M.; Ly, D.; Tin, E.; Kang, H.; Zeng, Y.; Akhtar, N.; Minden, M.D.; et al. CRISPR screen identifies genes that sensitize AML cells to double-negative T-cell therapy. Blood 2021, 137, 2171–2181. [Google Scholar] [CrossRef] [PubMed]
| Name Algorithm | Brief Description | Original Purpose | Software | CN Correction | Guide Inefficiencies | Visualization Tools | Type of Library in Hematology | Applications in Hematological Neoplasms |
|---|---|---|---|---|---|---|---|---|
| MAGeCK [114,116,117,118] | Binomial model method that prioritizes sgRNA, genes and pathways. MAGeCK–VISPR and MAGeCK–Flute are updated versions that provide advantages such as QC analysis, visualization or CN correction while scMAGeCK is a version specifically adapted for single-cell sequencing data. | CRISPRko | Python, R | Yes | Yes | Yes | CRISPRko, CRISPRa, CRISPRi | AML [25,32,47,62,70,71,72,74,81,119,120,121], NHL [39,52,53,65,67,76,90,97,122], HL [51], CLL [79], MM [91,123], ALL [27,41,42,43,124], MDS [55] |
| STARS [28] | A method based on a gene-ranking system that calculates gene scores using a binomial model. | All CRISPR screens | Python | No | No | No | CRISPRko | ALL [125], LLC [63], MM [56] |
| BAGEL [126,127] | Supervised learning method for analyzing CRISPR knockout screens which uses the fold changes of all gRNAs targeting all genes and core essential and nonessential gene lists to estimate an essentiality factor. | CRISPRko | Python | No | No | No | CRISPRko | AML [75,128], CML [129] |
| casTLE [130] | Maximum Likelihood Estimator that combines measurements from multiple targeting reagents to estimate a maximum essentiality effect size and a p-value. | CRISPRko, CRISPRi, CRISPRa and RNAi | Python | No | No | Yes | CRISPRko | CML [87] |
| CERES [131] | A method that estimates gene dependency levels in multiple CRISPR essentiality screens while correcting the CN specific effect. | CRISPRko in multiple screens | R | Yes | Yes | No | CRISPRko | AML [128] |
| JACKS [132] | Bayesian method that models gRNA efficacies in multiple screens performed with the same sgRNA library. | CRISPRko in multiple screens | Python | No | Yes | No | CRISPRko | AML [128] |
| PinAPL-Py [133] | A method that develops a full automated workflow for CRISPR screening analysis. | All CRISPR screens | Web-based (http://pinapl-py.ucsd.edu, accessed on 25 June 2022) | No | No | Yes | CRISPRko | MDS [134] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ancos-Pintado, R.; Bragado-García, I.; Morales, M.L.; García-Vicente, R.; Arroyo-Barea, A.; Rodríguez-García, A.; Martínez-López, J.; Linares, M.; Hernández-Sánchez, M. High-Throughput CRISPR Screening in Hematological Neoplasms. Cancers 2022, 14, 3612. https://doi.org/10.3390/cancers14153612
Ancos-Pintado R, Bragado-García I, Morales ML, García-Vicente R, Arroyo-Barea A, Rodríguez-García A, Martínez-López J, Linares M, Hernández-Sánchez M. High-Throughput CRISPR Screening in Hematological Neoplasms. Cancers. 2022; 14(15):3612. https://doi.org/10.3390/cancers14153612
Chicago/Turabian StyleAncos-Pintado, Raquel, Irene Bragado-García, María Luz Morales, Roberto García-Vicente, Andrés Arroyo-Barea, Alba Rodríguez-García, Joaquín Martínez-López, María Linares, and María Hernández-Sánchez. 2022. "High-Throughput CRISPR Screening in Hematological Neoplasms" Cancers 14, no. 15: 3612. https://doi.org/10.3390/cancers14153612
APA StyleAncos-Pintado, R., Bragado-García, I., Morales, M. L., García-Vicente, R., Arroyo-Barea, A., Rodríguez-García, A., Martínez-López, J., Linares, M., & Hernández-Sánchez, M. (2022). High-Throughput CRISPR Screening in Hematological Neoplasms. Cancers, 14(15), 3612. https://doi.org/10.3390/cancers14153612