
Disease Modeling of Pituitary Adenoma Using Human Pluripotent Stem Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Pituitary Adenoma Pathogenesis
2.1. ACTH-Producing Adenoma
2.2. GH-Producing Adenoma
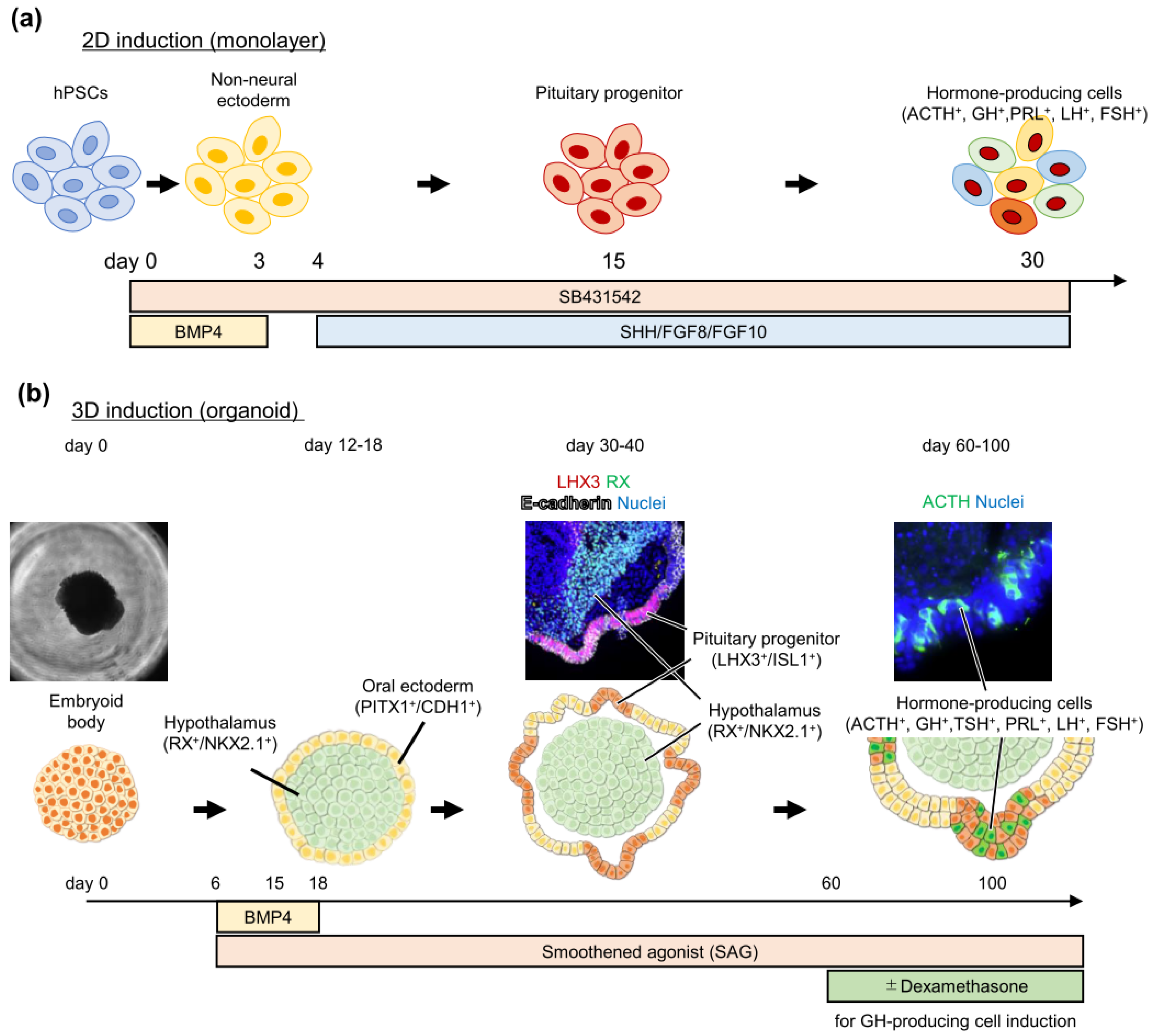
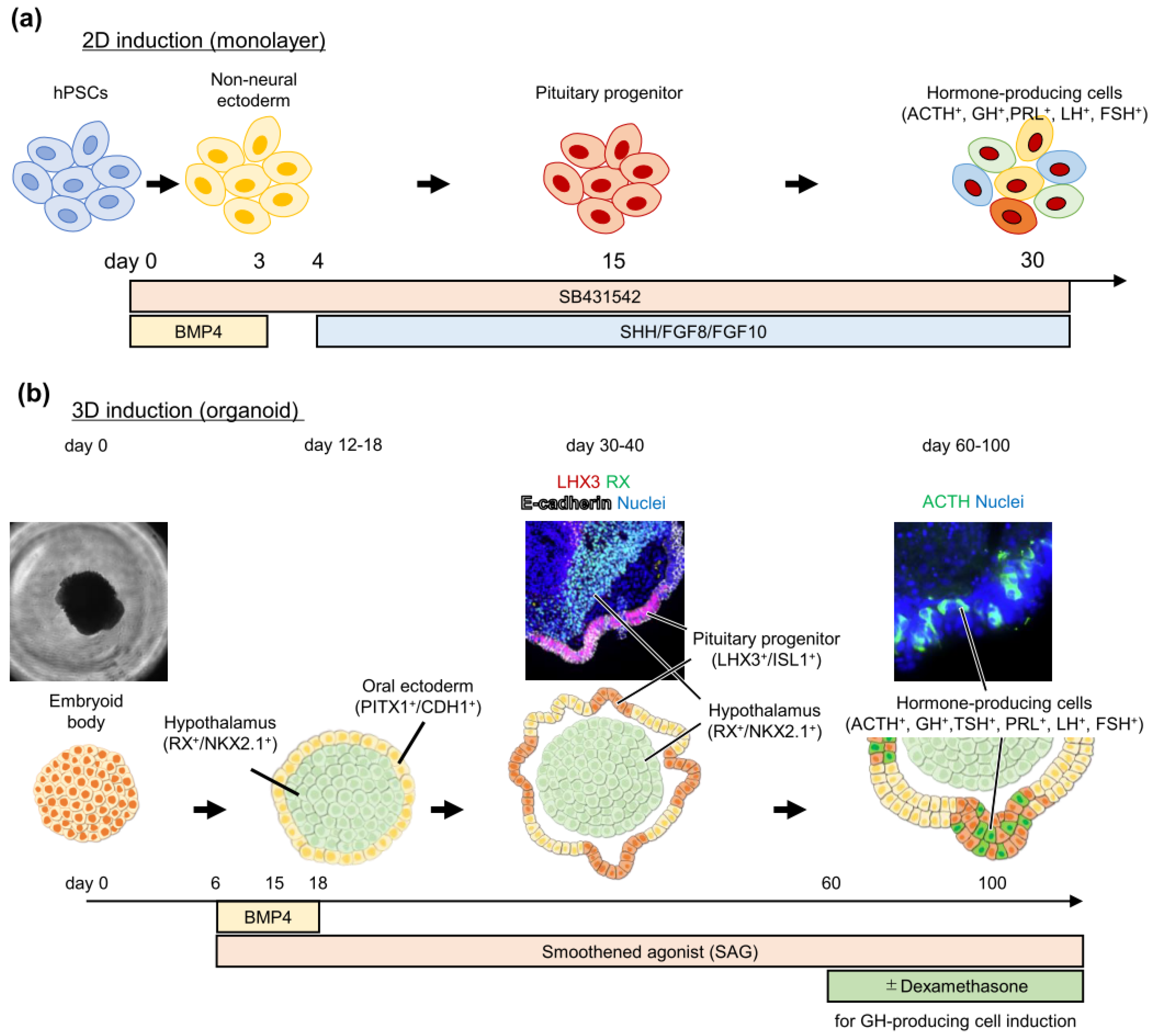
3. Pituitary Induction from Pluripotent Stem Cells
Pluripotent Stem Cell-Derived Pituitary Cell Application to Study Pituitary Diseases
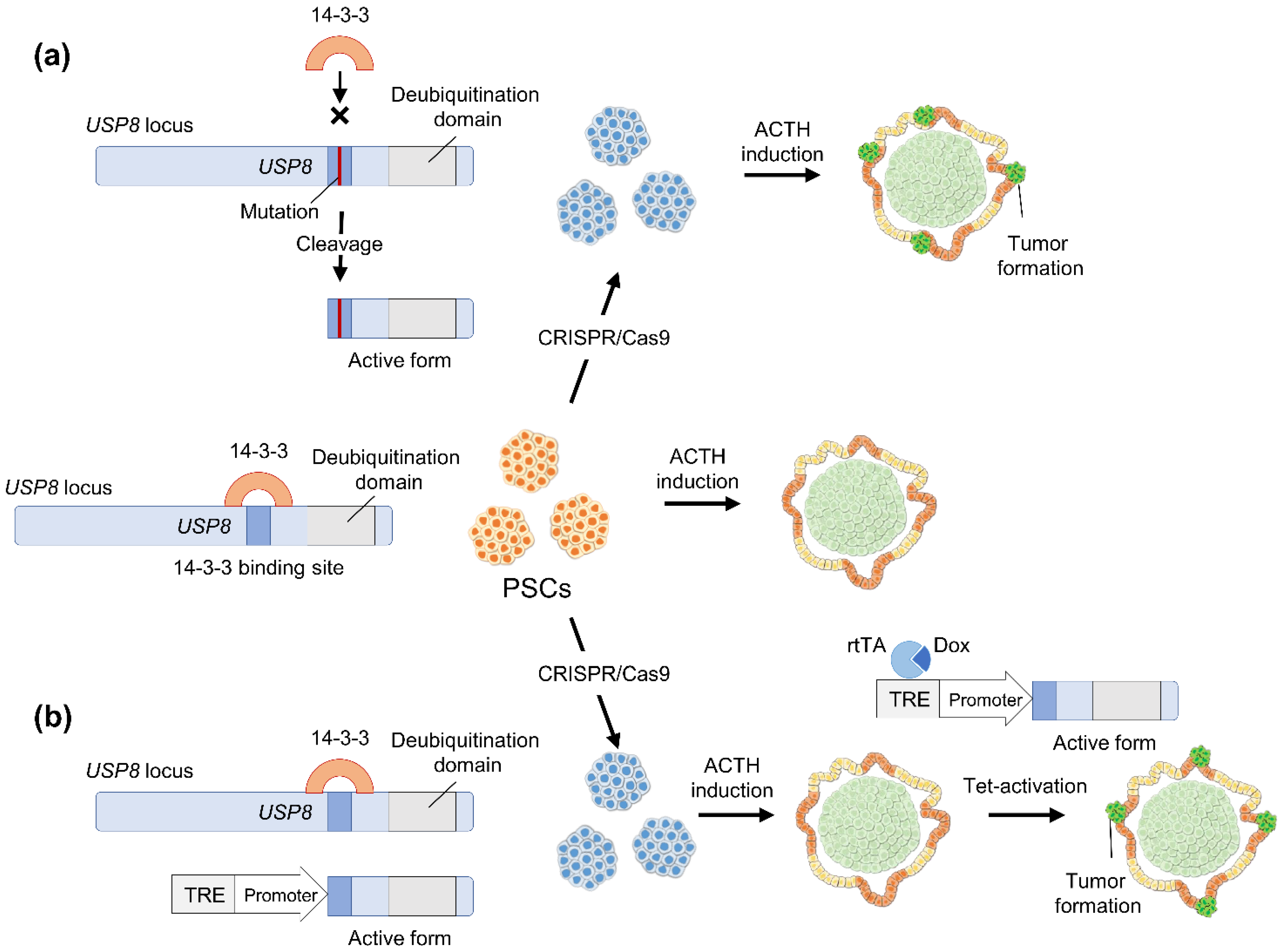
4. CRISPR/Cas9 Genome Editing
4.1. CRISPR/Cas9 Application to Study Tumor Pathophysiology
4.2. Hypothetical Strategies to Study Pituitary Adenoma Using Human Pluripotent Stem Cells
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Herman, V.; Fagin, J.; Gonsky, R.; Kovacs, K.; Melmed, S. Clonal origin of pituitary adenomas. J. Clin. Endocrinol. Metab. 1990, 71, 1427–1433. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Gittleman, H.; Liao, P.; Rouse, C.; Chen, Y.; Dowling, J.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2007-2011. Neuro Oncol. 2014, 16 (Suppl. 4), iv1–iv63. [Google Scholar] [CrossRef]
- Dallapiazza, R.F.; Oldfield, E.H.; Jane, J.A., Jr. Surgical management of Cushing’s disease. Pituitary 2015, 18, 211–216. [Google Scholar] [CrossRef]
- Amlashi, F.G.; Tritos, N.A. Thyrotropin-secreting pituitary adenomas: Epidemiology, diagnosis, and management. Endocrine 2016, 52, 427–440. [Google Scholar] [CrossRef]
- Fleseriu, M.; Biller, B.M.K.; Freda, P.U.; Gadelha, M.R.; Giustina, A.; Katznelson, L.; Molitch, M.E.; Samson, S.L.; Strasburger, C.J.; van der Lely, A.J.; et al. A Pituitary Society update to acromegaly management guidelines. Pituitary 2021, 24, 1–13. [Google Scholar] [CrossRef]
- Fleseriu, M.; Auchus, R.; Bancos, I.; Ben-Shlomo, A.; Bertherat, J.; Biermasz, N.R.; Boguszewski, C.L.; Bronstein, M.D.; Buchfelder, M.; Carmichael, J.D.; et al. Consensus on diagnosis and management of Cushing’s disease: A guideline update. Lancet Diabetes Endocrinol. 2021, 9, 847–875. [Google Scholar] [CrossRef]
- Ono, M.; Miki, N.; Kawamata, T.; Makino, R.; Amano, K.; Seki, T.; Kubo, O.; Hori, T.; Takano, K. Prospective study of high-dose cabergoline treatment of prolactinomas in 150 patients. J. Clin. Endocrinol. Metab. 2008, 93, 4721–4727. [Google Scholar] [CrossRef]
- Gadelha, M.R.; Bronstein, M.D.; Brue, T.; Coculescu, M.; Fleseriu, M.; Guitelman, M.; Pronin, V.; Raverot, G.; Shimon, I.; Lievre, K.K.; et al. Pasireotide versus continued treatment with octreotide or lanreotide in patients with inadequately controlled acromegaly (PAOLA): A randomised, phase 3 trial. Lancet Diabetes Endocrinol. 2014, 2, 875–884. [Google Scholar] [CrossRef]
- Caron, P.J.; Bevan, J.S.; Petersenn, S.; Flanagan, D.; Tabarin, A.; Prévost, G.; Maisonobe, P.; Clermont, A.; PRIMARYS Investigators. Tumor Shrinkage With Lanreotide Autogel 120 mg as Primary Therapy in Acromegaly: Results of a Prospective Multicenter Clinical Trial. J. Clin. Endocrinol. Metab. 2014, 99, 1282–1290. [Google Scholar] [CrossRef] [Green Version]
- Clayton, R.N.; Raskauskiene, D.; Reulen, R.C.; Jones, P.W. Mortality and morbidity in Cushing’s disease over 50 years in Stoke-on-Trent, UK: Audit and meta-analysis of literature. J. Clin. Endocrinol. Metab. 2011, 96, 632–642. [Google Scholar] [CrossRef] [PubMed]
- Larsson, C.; Skogseid, B.; Oberg, K.; Nakamura, Y.; Nordenskjöld, M. Multiple endocrine neoplasia type 1 gene maps to chromosome 11 and is lost in insulinoma. Nature 1988, 332, 85–87. [Google Scholar] [CrossRef] [PubMed]
- Pellegata, N.S.; Quintanilla-Martinez, L.; Siggelkow, H.; Samson, E.; Bink, K.; Höfler, H.; Fend, F.; Graw, J.; Atkinson, M.J. Germ-line mutations in p27Kip1 cause a multiple endocrine neoplasia syndrome in rats and humans. Proc. Natl. Acad. Sci. USA 2006, 103, 15558–15563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daly, A.F.; Jaffrain-Rea, M.L.; Ciccarelli, A.; Valdes-Socin, H.; Rohmer, V.; Tamburrano, G.; Borson-Chazot, C.; Estour, B.; Ciccarelli, E.; Brue, T.; et al. Clinical characterization of familial isolated pituitary adenomas. J. Clin. Endocrinol. Metab. 2006, 91, 3316–3323. [Google Scholar] [CrossRef] [Green Version]
- Yaneva, M.; Vandeva, S.; Zacharieva, S.; Daly, A.F.; Beckers, A. Genetics of Cushing’s syndrome. Neuroendocrinology 2010, 92 (Suppl. 1), 6–10. [Google Scholar] [CrossRef]
- Ma, Z.Y.; Song, Z.J.; Chen, J.H.; Wang, Y.F.; Li, S.Q.; Zhou, L.F.; Mao, Y.; Li, Y.M.; Hu, R.G.; Zhang, Z.Y.; et al. Recurrent gain-of-function USP8 mutations in Cushing’s disease. Cell Res. 2015, 25, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Reincke, M.; Sbiera, S.; Hayakawa, A.; Theodoropoulou, M.; Osswald, A.; Beuschlein, F.; Meitinger, T.; Mizuno-Yamasaki, E.; Kawaguchi, K.; Saeki, Y.; et al. Mutations in the deubiquitinase gene USP8 cause Cushing’s disease. Nat. Genet. 2015, 47, 31–38. [Google Scholar] [CrossRef]
- Perez-Rivas, L.G.; Theodoropoulou, M.; Ferraù, F.; Nusser, C.; Kawaguchi, K.; Stratakis, C.A.; Faucz, F.R.; Wildemberg, L.E.; Assié, G.; Beschorner, R.; et al. The Gene of the Ubiquitin-Specific Protease 8 Is Frequently Mutated in Adenomas Causing Cushing’s Disease. J. Clin. Endocrinol. Metab. 2015, 100, E997–E1004. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Inoshita, N.; Kawaguchi, K.; Ibrahim Ardisasmita, A.; Suzuki, H.; Fukuhara, N.; Okada, M.; Nishioka, H.; Takeuchi, Y.; Komada, M.; et al. The USP8 mutational status may predict drug susceptibility in corticotroph adenomas of Cushing’s disease. Eur. J. Endocrinol. 2016, 174, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Theodoropoulou, M.; Arzberger, T.; Gruebler, Y.; Jaffrain-Rea, M.L.; Schlegel, J.; Schaaf, L.; Petrangeli, E.; Losa, M.; Stalla, G.K.; Pagotto, U. Expression of epidermal growth factor receptor in neoplastic pituitary cells: Evidence for a role in corticotropinoma cells. J. Endocrinol. 2004, 183, 385–394. [Google Scholar] [CrossRef] [Green Version]
- Fukuoka, H.; Cooper, O.; Ben-Shlomo, A.; Mamelak, A.; Ren, S.G.; Bruyette, D.; Melmed, S. Egfr as a therapeutic target for human, canine, and mouse ACTH-secreting pituitary adenomas. J. Clin. Investig. 2011, 121, 4712–4721. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Jian, X.; Deng, S.; Ma, Z.; Shou, X.; Shen, Y.; Zhang, Q.; Song, Z.; Li, Z.; Peng, H.; et al. Identification of recurrent USP48 and BRAF mutations in Cushing’s disease. Nat. Commun. 2018, 9, 3171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sbiera, S.; Perez-Rivas, L.G.; Taranets, L.; Weigand, I.; Flitsch, J.; Graf, E.; Monoranu, C.M.; Saeger, W.; Hagel, C.; Honegger, J.; et al. Driver mutations in USP8 wild-type Cushing’s disease. Neuro Oncol. 2019, 21, 1273–1283. [Google Scholar] [CrossRef] [PubMed]
- Lidhar, K.; Korbonits, M.; Jordan, S.; Khalimova, Z.; Kaltsas, G.; Lu, X.; Clayton, R.N.; Jenkins, P.J.; Monson, J.P.; Besser, G.M.; et al. Low expression of the cell cycle inhibitor p27Kip1 in normal corticotroph cells, corticotroph tumors, and malignant pituitary tumors. J. Clin. Endocrinol. Metab. 1999, 84, 3823–3830. [Google Scholar] [CrossRef]
- Jordan, S.; Lidhar, K.; Korbonits, M.; Lowe, D.G.; Grossman, A.B. Cyclin D and cyclin E expression in normal and adenomatous pituitary. Eur. J. Endocrinol. 2000, 143, R1–R6. [Google Scholar] [CrossRef] [Green Version]
- Roussel-Gervais, A.; Couture, C.; Langlais, D.; Takayasu, S.; Balsalobre, A.; Rueda, B.R.; Zukerberg, L.R.; Figarella-Branger, D.; Brue, T.; Drouin, J. The Cables1 Gene in Glucocorticoid Regulation of Pituitary Corticotrope Growth and Cushing Disease. J. Clin. Endocrinol. Metab. 2016, 101, 513–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melmed, S. Acromegaly. N. Engl. J. Med. 2006, 355, 2558–2573. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, R.; Izawa, M.; Fukuoka, H.; Iguchi, G.; Odake, Y.; Yoshida, K.; Bando, H.; Suda, K.; Nishizawa, H.; Takahashi, M.; et al. Genetic and clinical characteristics of japanese patients with sporadic somatotropinoma. Endocr. J. 2016, 63, 953–963. [Google Scholar] [CrossRef] [Green Version]
- Mendoza, V.; Sosa, E.; Espinosa-de-los-Monteros, A.L.; Salcedo, M.; Guinto, G.; Cheng, S.; Sandoval, C.; Mercado, M. Gspα mutations in Mexican patients with acromegaly: Potential impact on long term prognosis. Growth Horm. IGF Res. 2005, 15, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Landis, C.A.; Masters, S.B.; Spada, A.; Pace, A.M.; Bourne, H.R.; Vallar, L. GTPase inhibiting mutations activate the α chain of Gs and stimulate adenylyl cyclase in human pituitary tumours. Nature 1989, 340, 692–696. [Google Scholar] [CrossRef] [PubMed]
- Freda, P.; Chung, W.; Matsuoka, N.; Walsh, J.; Kanibir, M.N.; Kleinman, G.; Wang, Y.; Bruce, J.; Post, K. Analysis of GNAS mutations in 60 growth hormone secreting pituitary tumors: Correlation with clinical and pathological characteristics and surgical outcome based on highly sensitive GH and IGF-I criteria for remission. Pituitary 2007, 10, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Vallar, L.; Spada, A.; Giannattasio, G. Altered Gs and adenylate cyclase activity in human GH-secreting pituitary adenomas. Nature 1987, 330, 566–568. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekharappa, S.C.; Guru, S.C.; Manickam, P.; Olufemi, S.E.; Collins, F.S.; Emmert-Buck, M.R.; Debelenko, L.V.; Zhuang, Z.; Lubensky, I.A.; Liotta, L.A.; et al. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science 1997, 276, 404–407. [Google Scholar] [CrossRef] [PubMed]
- Kirschner, L.S.; Carney, J.A.; Pack, S.D.; Taymans, S.E.; Giatzakis, C.; Cho, Y.S.; Cho-Chung, Y.S.; Stratakis, C.A. Mutations of the gene encoding the protein kinase A type I-alpha regulatory subunit in patients with the Carney complex. Nat. Genet. 2000, 26, 89–92. [Google Scholar] [CrossRef]
- Vierimaa, O.; Georgitsi, M.; Lehtonen, R.; Vahteristo, P.; Kokko, A.; Raitila, A.; Tuppurainen, K.; Ebeling, T.M.L.; Salmela, P.I.; Paschke, R.; et al. Pituitary Adenoma Predisposition Caused by Germline Mutations in the AIP Gene. Science 2006, 312, 1228–1230. [Google Scholar] [CrossRef] [PubMed]
- Trivellin, G.; Daly, A.F.; Faucz, F.R.; Yuan, B.; Rostomyan, L.; Larco, D.O.; Schernthaner-Reiter, M.H.; Szarek, E.; Leal, L.F.; Caberg, J.H.; et al. Gigantism and acromegaly due to Xq26 microduplications and GPR101 mutation. N. Engl. J. Med. 2014, 371, 2363–2374. [Google Scholar] [CrossRef]
- Bogusławska, A.; Korbonits, M. Genetics of Acromegaly and Gigantism. J. Clin. Med. 2021, 10, 1377. [Google Scholar] [CrossRef]
- Valimaki, N.; Demir, H.; Pitkanen, E.; Kaasinen, E.; Karppinen, A.; Kivipelto, L.; Schalin-Jantti, C.; Aaltonen, L.A.; Karhu, A. Whole-Genome Sequencing of Growth Hormone (GH)-Secreting Pituitary Adenomas. J. Clin. Endocrinol. Metab. 2015, 100, 3918–3927. [Google Scholar] [CrossRef] [Green Version]
- Neou, M.; Villa, C.; Armignacco, R.; Jouinot, A.; Raffin-Sanson, M.-L.; Septier, A.; Letourneur, F.; Diry, S.; Diedisheim, M.; Izac, B.; et al. Pangenomic Classification of Pituitary Neuroendocrine Tumors. Cancer Cell 2020, 37, 123–134.e125. [Google Scholar] [CrossRef] [PubMed]
- Occhi, G.; Losa, M.; Albiger, N.; Trivellin, G.; Regazzo, D.; Scanarini, M.; Monteserin-Garcia, J.L.; Frohlich, B.; Ferasin, S.; Terreni, M.R. The glucose-dependent insulinotropic polypeptide receptor is overexpressed amongst GNAS1 mutation-negative somatotropinomas and drives growth hormone (GH)-promoter activity in GH3 cells. J. Neuroendocrinol. 2011, 23, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Umahara, M.; Okada, S.; Ohshima, K.; Mori, M. Glucose-dependent insulinotropic polypeptide induced growth hormone secretion in acromegaly. Endocr. J. 2003, 50, 643–650. [Google Scholar] [CrossRef] [Green Version]
- Volz, A.; Goke, R.; Lankat-Buttgereit, B.; Fehmann, H.C.; Bode, H.P.; Goke, B. Molecular cloning, functional expression, and signal transduction of the GIP-receptor cloned from a human insulinoma. FEBS Lett. 1995, 373, 23–29. [Google Scholar] [CrossRef] [Green Version]
- Gremlich, S.; Porret, A.; Hani, E.H.; Cherif, D.; Vionnet, N.; Froguel, P.; Thorens, B. Cloning, functional expression, and chromosomal localization of the human pancreatic islet glucose-dependent insulinotropic polypeptide receptor. Diabetes 1995, 44, 1202–1208. [Google Scholar] [CrossRef] [PubMed]
- Regazzo, D.; Losa, M.; Albiger, N.M.; Terreni, M.R.; Vazza, G.; Ceccato, F.; Emanuelli, E.; Denaro, L.; Scaroni, C.; Occhi, G. The GIP/GIPR axis is functionally linked to gh-secretion increase in a significant proportion of gsp− somatotropinomas. Eur. J. Endocrinol. 2017, 176, 543–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic Stem Cell Lines Derived from Human Blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chambers, S.M.; Fasano, C.A.; Papapetrou, E.P.; Tomishima, M.; Sadelain, M.; Studer, L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 2009, 27, 275–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lancaster, M.A.; Renner, M.; Martin, C.-A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, S.; Ito, I.; Nagasaki, T.; Yamamoto, Y.; Konishi, S.; Korogi, Y.; Matsumoto, H.; Muro, S.; Hirai, T.; Funato, M.; et al. Generation of Alveolar Epithelial Spheroids via Isolated Progenitor Cells from Human Pluripotent Stem Cells. Stem Cell Rep. 2014, 3, 394–403. [Google Scholar] [CrossRef] [Green Version]
- Burridge, P.W.; Matsa, E.; Shukla, P.; Lin, Z.C.; Churko, J.M.; Ebert, A.D.; Lan, F.; Diecke, S.; Huber, B.; Mordwinkin, N.M.; et al. Chemically defined generation of human cardiomyocytes. Nat. Methods 2014, 11, 855–860. [Google Scholar] [CrossRef] [Green Version]
- Spence, J.R.; Mayhew, C.N.; Rankin, S.A.; Kuhar, M.F.; Vallance, J.E.; Tolle, K.; Hoskins, E.E.; Kalinichenko, V.V.; Wells, S.I.; Zorn, A.M.; et al. Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature 2011, 470, 105–109. [Google Scholar] [CrossRef] [Green Version]
- Crespo, M.; Vilar, E.; Tsai, S.-Y.; Chang, K.; Amin, S.; Srinivasan, T.; Zhang, T.; Pipalia, N.H.; Chen, H.J.; Witherspoon, M.; et al. Colonic organoids derived from human induced pluripotent stem cells for modeling colorectal cancer and drug testing. Nat. Med. 2017, 23, 878–884. [Google Scholar] [CrossRef]
- Hosoyama, T.; McGivern, J.V.; Van Dyke, J.M.; Ebert, A.D.; Suzuki, M. Derivation of myogenic progenitors directly from human pluripotent stem cells using a sphere-based culture. Stem Cells Transl. Med. 2014, 3, 564–574. [Google Scholar] [CrossRef]
- Oldershaw, R.A.; Baxter, M.A.; Lowe, E.T.; Bates, N.; Grady, L.M.; Soncin, F.; Brison, D.R.; Hardingham, T.E.; Kimber, S.J. Directed differentiation of human embryonic stem cells toward chondrocytes. Nat. Biotechnol. 2010, 28, 1187–1194. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, R.; Takahashi, Y. Human pituitary development and application of iPSCs for pituitary disease. Cell. Mol. Life Sci. 2020, 78, 2069–2079. [Google Scholar] [CrossRef]
- Zhu, X.; Gleiberman, A.S.; Rosenfeld, M.G. Molecular Physiology of Pituitary Development: Signaling and Transcriptional Networks. Physiol. Rev. 2007, 87, 933–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suga, H.; Kadoshima, T.; Minaguchi, M.; Ohgushi, M.; Soen, M.; Nakano, T.; Takata, N.; Wataya, T.; Muguruma, K.; Miyoshi, H.; et al. Self-formation of functional adenohypophysis in three-dimensional culture. Nature 2011, 480, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Wataya, T.; Ando, S.; Muguruma, K.; Ikeda, H.; Watanabe, K.; Eiraku, M.; Kawada, M.; Takahashi, J.; Hashimoto, N.; Sasai, Y. Minimization of exogenous signals in ES cell culture induces rostral hypothalamic differentiation. Proc. Natl. Acad. Sci. USA 2008, 105, 11796–11801. [Google Scholar] [CrossRef] [Green Version]
- Dincer, Z.; Piao, J.; Niu, L.; Ganat, Y.; Kriks, S.; Zimmer, B.; Shi, S.H.; Tabar, V.; Studer, L. Specification of functional cranial placode derivatives from human pluripotent stem cells. Cell Rep. 2013, 5, 1387–1402. [Google Scholar] [CrossRef] [Green Version]
- Zimmer, B.; Piao, J.; Ramnarine, K.; Tomishima, M.J.; Tabar, V.; Studer, L. Derivation of Diverse Hormone-Releasing Pituitary Cells from Human Pluripotent Stem Cells. Stem Cell Rep. 2016, 6, 858–872. [Google Scholar] [CrossRef] [Green Version]
- Ozone, C.; Suga, H.; Eiraku, M.; Kadoshima, T.; Yonemura, S.; Takata, N.; Oiso, Y.; Tsuji, T.; Sasai, Y. Functional anterior pituitary generated in self-organizing culture of human embryonic stem cells. Nat. Commun. 2016, 7, 10351. [Google Scholar] [CrossRef] [Green Version]
- Kasai, T.; Suga, H.; Sakakibara, M.; Ozone, C.; Matsumoto, R.; Kano, M.; Mitsumoto, K.; Ogawa, K.; Kodani, Y.; Nagasaki, H.; et al. Hypothalamic Contribution to Pituitary Functions Is Recapitulated In Vitro Using 3D-Cultured Human iPS Cells. Cell Rep. 2020, 30, 18–24.e15. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, R.; Suga, H.; Aoi, T.; Bando, H.; Fukuoka, H.; Iguchi, G.; Narumi, S.; Hasegawa, T.; Muguruma, K.; Ogawa, W.; et al. Congenital pituitary hypoplasia model demonstrates hypothalamic OTX2 regulation of pituitary progenitor cells. J. Clin. Invest 2020, 130, 641–654. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Iguchi, G.; Takeno, R.; Okimura, Y.; Sano, T.; Takahashi, M.; Nishizawa, H.; Handayaningshi, A.E.; Fukuoka, H.; Tobita, M.; et al. Adult combined GH, prolactin, and TSH deficiency associated with circulating PIT-1 antibody in humans. J. Clin. Investig. 2011, 121, 113–119. [Google Scholar] [CrossRef] [Green Version]
- Bando, H.; Iguchi, G.; Fukuoka, H.; Yamamoto, M.; Hidaka-Takeno, R.; Okimura, Y.; Matsumoto, R.; Suda, K.; Nishizawa, H.; Takahashi, M.; et al. Involvement of PIT-1-reactive cytotoxic T lymphocytes in anti-PIT-1 antibody syndrome. J. Clin. Endocrinol. Metab. 2014, 99, E1744–E1749. [Google Scholar] [CrossRef] [Green Version]
- Kanie, K.; Bando, H.; Iguchi, G.; Muguruma, K.; Matsumoto, R.; Hidaka-Takeno, R.; Okimura, Y.; Yamamoto, M.; Fujita, Y.; Fukuoka, H.; et al. Pathogenesis of anti-PIT-1 antibody syndrome: PIT-1 presentation by HLA Class I on Anterior Pituitary Cells. J. Endocr. Soc. 2019, 3, 1969–1978. [Google Scholar] [CrossRef] [PubMed]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR provides acquired resistance against viruses in prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable Dual-RNA-guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Tuveson, D.; Clevers, H. Cancer modeling meets human organoid technology. Science 2019, 364, 952–955. [Google Scholar] [CrossRef]
- Ogawa, J.; Pao, G.M.; Shokhirev, M.N.; Verma, I.M. Glioblastoma Model Using Human Cerebral Organoids. Cell Rep. 2018, 23, 1220–1229. [Google Scholar] [CrossRef] [Green Version]
- Bian, S.; Repic, M.; Guo, Z.; Kavirayani, A.; Burkard, T.; Bagley, J.A.; Krauditsch, C.; Knoblich, J.A. Genetically engineered cerebral organoids model brain tumor formation. Nat. Methods 2018, 15, 631–639. [Google Scholar] [CrossRef]
- Matano, M.; Date, S.; Shimokawa, M.; Takano, A.; Fujii, M.; Ohta, Y.; Watanabe, T.; Kanai, T.; Sato, T. Modeling colorectal cancer using CRISPR-Cas9-mediated engineering of human intestinal organoids. Nat. Med. 2015, 21, 256–262. [Google Scholar] [CrossRef]
- Drost, J.; van Jaarsveld, R.H.; Ponsioen, B.; Zimberlin, C.; van Boxtel, R.; Buijs, A.; Sachs, N.; Overmeer, R.M.; Offerhaus, G.J.; Begthel, H.; et al. Sequential cancer mutations in cultured human intestinal stem cells. Nature 2015, 521, 43–47. [Google Scholar] [CrossRef]
- Artegiani, B.; van Voorthuijsen, L.; Lindeboom, R.G.H.; Seinstra, D.; Heo, I.; Tapia, P.; López-Iglesias, C.; Postrach, D.; Dayton, T.; Oka, R.; et al. Probing the Tumor Suppressor Function of BAP1 in CRISPR-Engineered Human Liver Organoids. Cell Stem Cell 2019, 24, 927–943.e926. [Google Scholar] [CrossRef]
- Seino, T.; Kawasaki, S.; Shimokawa, M.; Tamagawa, H.; Toshimitsu, K.; Fujii, M.; Ohta, Y.; Matano, M.; Nanki, K.; Kawasaki, K.; et al. Human Pancreatic Tumor Organoids Reveal Loss of Stem Cell Niche Factor Dependence during Disease Progression. Cell Stem Cell 2018, 22, 454–467.e456. [Google Scholar] [CrossRef] [Green Version]
- Dekkers, J.F.; Whittle, J.R.; Vaillant, F.; Chen, H.R.; Dawson, C.; Liu, K.; Geurts, M.H.; Herold, M.J.; Clevers, H.; Lindeman, G.J.; et al. Modeling Breast Cancer Using CRISPR-Cas9-Mediated Engineering of Human Breast Organoids. J. Natl. Cancer Inst. 2020, 112, 540–544. [Google Scholar] [CrossRef]
- Cox, B.; Laporte, E.; Vennekens, A.; Kobayashi, H.; Nys, C.; Van Zundert, I.; Uji, I.H.; Vercauteren Drubbel, A.; Beck, B.; Roose, H.; et al. Organoids from pituitary as a novel research model toward pituitary stem cell exploration. J. Endocrinol. 2019, 240, 287–308. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matsumoto, R.; Suga, H.; Arima, H.; Yamamoto, T. Disease Modeling of Pituitary Adenoma Using Human Pluripotent Stem Cells. Cancers 2022, 14, 3660. https://doi.org/10.3390/cancers14153660
Matsumoto R, Suga H, Arima H, Yamamoto T. Disease Modeling of Pituitary Adenoma Using Human Pluripotent Stem Cells. Cancers. 2022; 14(15):3660. https://doi.org/10.3390/cancers14153660
Chicago/Turabian StyleMatsumoto, Ryusaku, Hidetaka Suga, Hiroshi Arima, and Takuya Yamamoto. 2022. "Disease Modeling of Pituitary Adenoma Using Human Pluripotent Stem Cells" Cancers 14, no. 15: 3660. https://doi.org/10.3390/cancers14153660