
Human Malignant Rhabdoid Tumor Antigens as Biomarkers and Potential Therapeutic Targets


Abstract
:Simple Summary
Abstract

1. Introduction
2. Materials and Methods
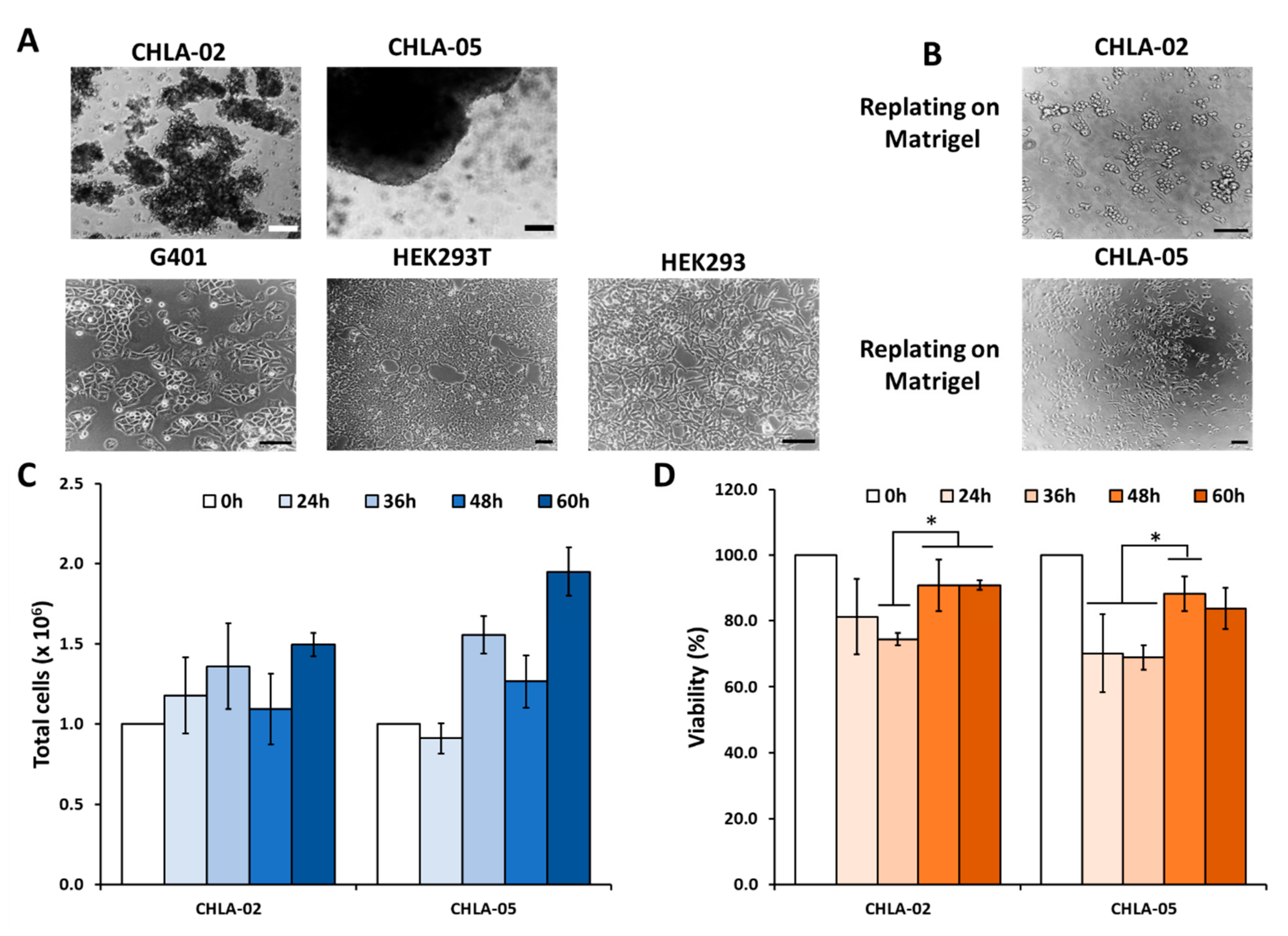
2.1. Culturing Human Malignant Rhabdoid Tumor Cells and Nonmalignant Cells
2.2. Cell Proliferation Assay and Imaging
2.3. Reverse Transcription Polymerase Chain Reaction (RT-PCR) Analysis
2.4. Immunocytochemistry
2.5. Flow Cytometry
2.6. MMP Inhibitor GM6001 Preparation and Treatment
2.7. Statistical Analysis
3. Results
3.1. AFP, MSLN, OPN, and MUC16 Are Highly Expressed Biomarkers for ATRT
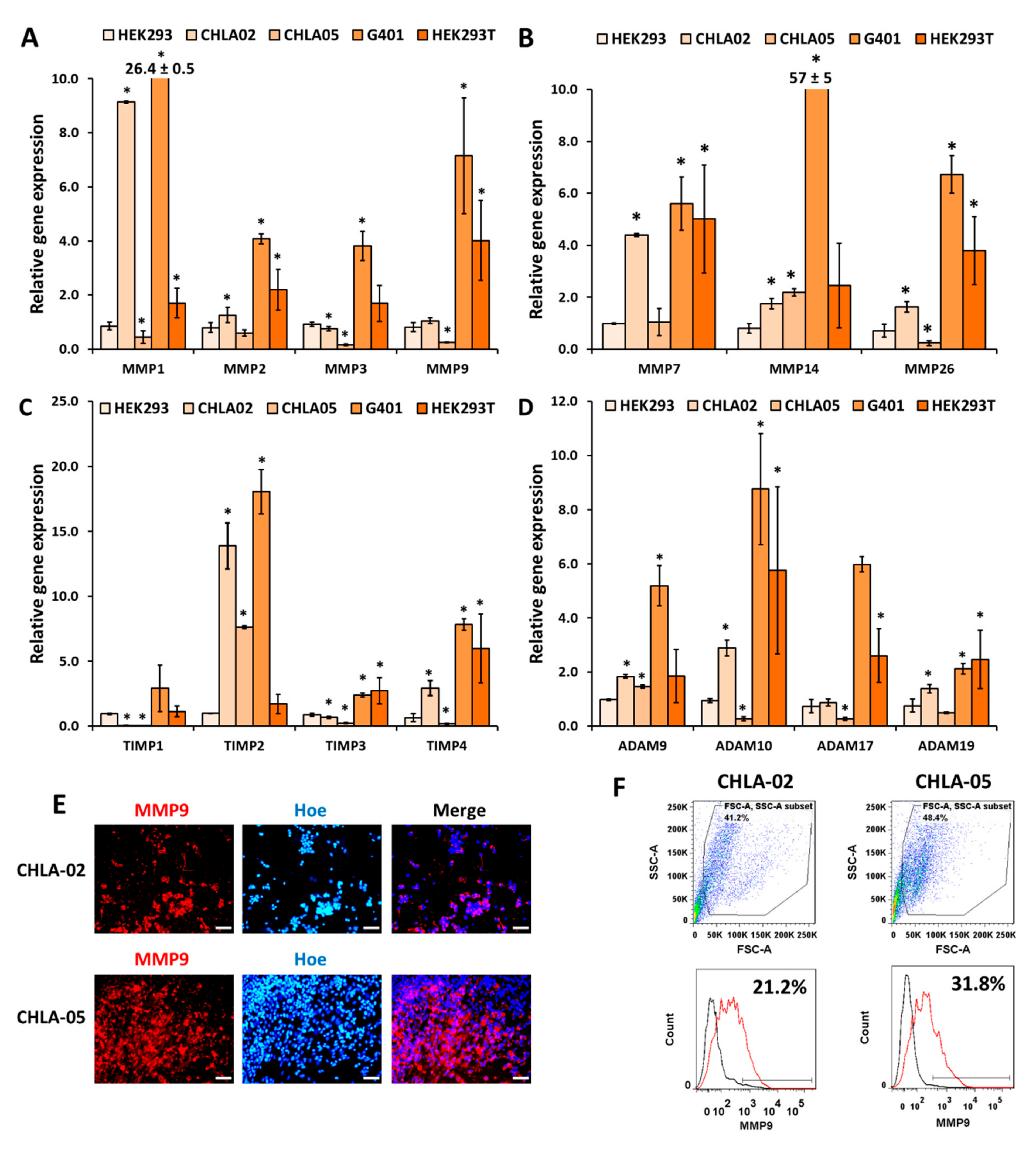
3.2. CHLA-02 Cells Expresseed Higher Levels of MMPs Than CHLA-05 Cells
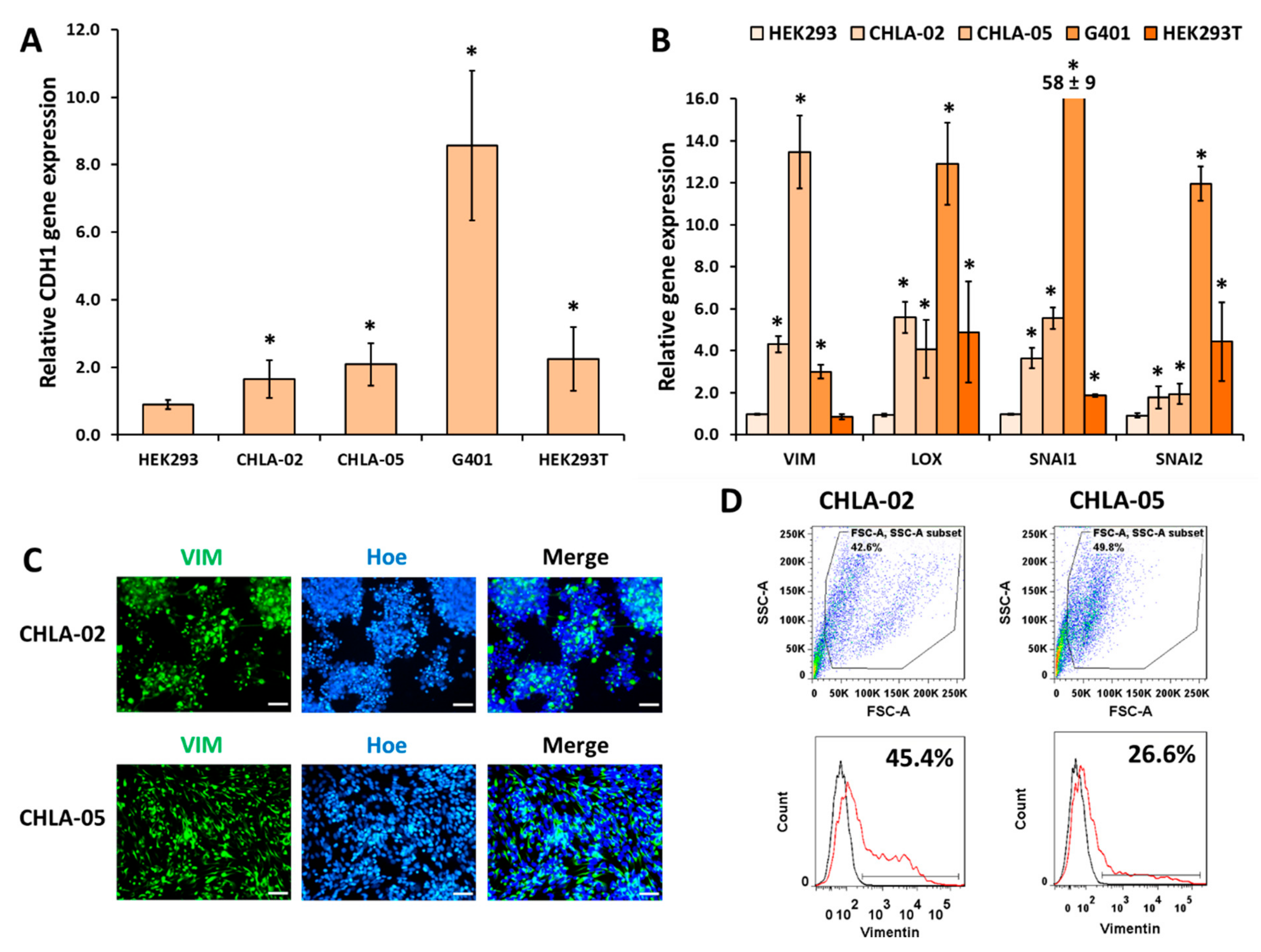
3.3. Mesenchymal Markers Were Highly Upregulated in the Malignant Rhabdoid Tumors
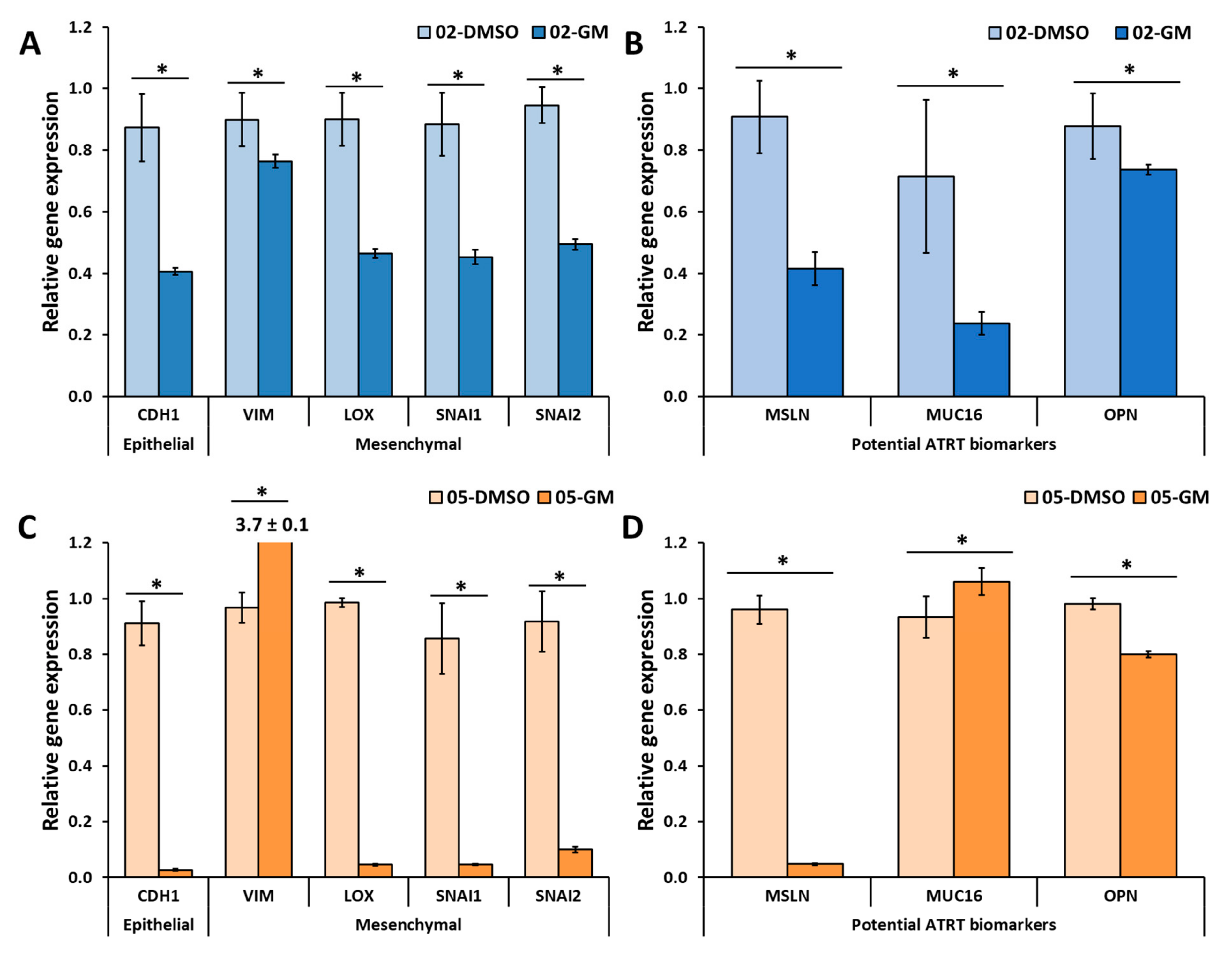
3.4. GM6001 Significantly Decreased Cell Proliferation and the Gene Expression of Several Markers in Both ATRT Cell Lines
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Von Hoff, K.; Hinkes, B.; Dannenmann-Stern, E.; von Bueren, A.O.; Warmuth-Metz, M.; Soerensen, N.; Emser, A.; Zwiener, I.; Schlegel, P.G.; Kuehl, J.; et al. Frequency, Risk-Factors and Survival of Children with Atypical Teratoid Rhabdoid Tumors (AT/RT) of the CNS Diagnosed between 1988 and 2004, and Registered to the German HIT Database. Pediatr. Blood Cancer 2011, 57, 978–985. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.; Knerlich-Lukoschus, F.; Reinhard, H.; Johann, P.D.; Sturm, D.; Sahm, F.; Bens, S.; Vogt, J.; Nemes, K.; Oyen, F.; et al. Two Molecularly Distinct Atypical Teratoid/Rhabdoid Tumors (or Tumor Components) Occurring in an Infant with Rhabdoid Tumor Predisposition Syndrome 1. Acta Neuropathol. 2019, 137, 847–850. [Google Scholar] [CrossRef] [PubMed]
- Rickert, C.H.; Paulus, W. Epidemiology of Central Nervous System Tumors in Childhood and Adolescence Based on the New WHO Classification. Child’s Nerv. Syst. 2001, 17, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Chen, Y.; de Blank, P.M.; Ondracek, A.; Farah, P.; Gittleman, H.; Wolinsky, Y.; Kruchko, C.; Cohen, M.L.; Brat, D.J.; et al. The Descriptive Epidemiology of Atypical Teratoid/Rhabdoid Tumors in the United States, 2001–2010. Neuro-Oncology 2014, 16, 1392–1399. [Google Scholar] [CrossRef] [Green Version]
- Chi, S.N.; Zimmerman, M.A.; Yao, X.; Cohen, K.J.; Burger, P.; Biegel, J.A.; Rorke-Adams, L.B.; Fisher, M.J.; Janss, A.; Mazewski, C.; et al. Intensive Multimodality Treatment for Children with Newly Diagnosed CNS Atypical Teratoid Rhabdoid Tumor. J. Clin. Oncol. 2009, 27, 385–389. [Google Scholar] [CrossRef] [Green Version]
- Lafay-Cousin, L.; Hawkins, C.; Carret, A.S.; Johnston, D.; Zelcer, S.; Wilson, B.; Jabado, N.; Scheinemann, K.; Eisenstat, D.; Fryer, C.; et al. Central Nervous System Atypical Teratoid Rhabdoid Tumours: The Canadian Paediatric Brain Tumour Consortium Experience. Eur. J. Cancer 2012, 48, 353–359. [Google Scholar] [CrossRef]
- Parham, D.M.; Weeks, D.A.; Beckwith, J.B. The Clinicopathologic Spectrum of Putative Extrarenal Rhabdoid Tumors. An Analysis of 42 Cases Studied with Immunohistochemistry or Electron Microscopy. Am. J. Surg. Pathol. 1994, 18, 1010–1029. [Google Scholar] [CrossRef]
- Biegel, J.A.; Zhou, J.Y.; Rorke, L.B.; Stenstrom, C.; Wainwright, L.M.; Fogelgren, B. Germ-Line and Acquired Mutations of INI1 in Atypical Teratoid and Rhabdoid Tumors. Cancer Res. 1999, 59, 74–79. [Google Scholar]
- Judkins, A.R.; Mauger, J.; Ht, A.; Rorke, L.B.; Biegel, J.A. Immunohistochemical Analysis of HSNF5/INI1 in Pediatric CNS Neoplasms. Am. J. Surg. Pathol. 2004, 28, 644–650. [Google Scholar] [CrossRef]
- Sullivan, L.M.; Folpe, A.L.; Pawel, B.R.; Judkins, A.R.; Biegel, J.A. Epithelioid Sarcoma Is Associated with a High Percentage of SMARCB1 Deletions. Mod. Pathol. 2013, 26, 385–392. [Google Scholar] [CrossRef]
- Hasselblatt, M.; Isken, S.; Linge, A.; Eikmeier, K.; Jeibmann, A.; Oyen, F.; Nagel, I.; Richter, J.; Bartelheim, K.; Kordes, U.; et al. High-Resolution Genomic Analysis Suggests the Absence of Recurrent Genomic Alterations Other than SMARCB1 Aberrations in Atypical Teratoid/Rhabdoid Tumors. Genes Chromosomes Cancer 2013, 52, 185–190. [Google Scholar] [CrossRef]
- Calderaro, J.; Moroch, J.; Pierron, G.; Pedeutour, F.; Grison, C.; Maillé, P.; Soyeux, P.; de la Taille, A.; Couturier, J.; Vieillefond, A.; et al. SMARCB1/INI1 Inactivation in Renal Medullary Carcinoma. Histopathology 2012, 61, 428–435. [Google Scholar] [CrossRef] [Green Version]
- Mobley, B.C.; McKenney, J.K.; Bangs, C.D.; Callahan, K.; Yeom, K.W.; Schneppenheim, R.; Hayden, M.G.; Cherry, A.M.; Gokden, M.; Edwards, M.S.B.; et al. Loss of SMARCB1/INI1 Expression in Poorly Differentiated Chordomas. Acta Neuropathol. 2010, 120, 745–753. [Google Scholar] [CrossRef]
- Yang, Y.-P.; Nguyen, P.N.N.; Ma, H.-I.; Ho, W.-J.; Chen, Y.-W.; Chien, Y.; Yarmishyn, A.A.; Huang, P.-I.; Lo, W.-L.; Wang, C.-Y.; et al. Tumor Mesenchymal Stromal Cells Regulate Cell Migration of Atypical Teratoid Rhabdoid Tumor through Exosome-Mediated MiR155/SMARCA4 Pathway. Cancers 2019, 11, 720. [Google Scholar] [CrossRef] [Green Version]
- Strobeck, M.W.; DeCristofaro, M.F.; Banine, F.; Weissman, B.E.; Sherman, L.S.; Knudsen, E.S. The BRG-1 Subunit of the SWI/SNF Complex Regulates CD44 Expression. J. Biol. Chem. 2001, 276, 9273–9278. [Google Scholar] [CrossRef] [Green Version]
- Hsu, K.-H.; Tsai, H.-W.; Lin, P.-W.; Hsu, Y.-S.; Shan, Y.-S.; Lu, P.-J. Clinical Implication and Mitotic Effect of CD44 Cleavage in Relation to Osteopontin/CD44 Interaction and Dysregulated Cell Cycle Protein in Gastrointestinal Stromal Tumor. Ann. Surg. Oncol. 2010, 17, 2199–2212. [Google Scholar] [CrossRef]
- Schenkel, A.R.; Mamdouh, Z.; Chen, X.; Liebman, R.M.; Muller, W.A. CD99 Plays a Major Role in the Migration of Monocytes through Endothelial Junctions. Nat. Immunol. 2002, 3, 143–150. [Google Scholar] [CrossRef]
- Yang, C.-S.; Jan, Y.-J.; Wang, J.; Shen, C.-C.; Chen, C.C.C.; Chen, M. Spinal Atypical Teratoid/Rhabdoid Tumor in a 7-Year-Old Boy. Neuropathology 2007, 27, 139–144. [Google Scholar] [CrossRef]
- Las Heras, F.; Pritzker, K.P.H. Adult Variant of Atypical Teratoid/Rhabdoid Tumor: Immunohistochemical and Ultrastructural Confirmation of a Rare Tumor in the Sella Tursica. Pathol. Res. Pract. 2010, 206, 788–791. [Google Scholar] [CrossRef]
- Park, H.G.; Yoon, J.H.; Kim, S.H.; Cho, K.H.; Park, H.J.; Kim, S.H.; Kim, E.H. Adult-Onset Sellar and Suprasellar Atypical Teratoid Rhabdoid Tumor Treated with a Multimodal Approach: A Case Report. Brain Tumor Res. Treat. 2014, 2, 108–113. [Google Scholar] [CrossRef] [Green Version]
- Al-Hussaini, M.; Dissi, N.; Souki, C.; Amayiri, N. Atypical Teratoid/Rhabdoid Tumor, an Immunohistochemical Study of Potential Diagnostic and Prognostic Markers. Neuropathology 2016, 36, 17–26. [Google Scholar] [CrossRef]
- Acevedo, H.F.; Tong, J.Y.; Hartsock, R.J. Human Chorionic Gonadotropin-Beta Subunit Gene Expression in Cultured Human Fetal and Cancer Cells of Different Types and Origins. Cancer 1995, 76, 1467–1475. [Google Scholar] [CrossRef]
- García-García, A.G.; Polo-Hernández, E.; Tabernero, A.; Medina, J.M. Alpha-Fetoprotein (AFP) Modulates the Effect of Serum Albumin on Brain Development by Restraining the Neurotrophic Effect of Oleic Acid. Brain Res. 2015, 1624, 45–58. [Google Scholar] [CrossRef] [Green Version]
- Rizzo, A.; Galgano, M.; Mutinati, M.; Sciorsci, R.L. Alpha-Fetoprotein in Animal Reproduction. Res. Vet. Sci. 2019, 123, 281–285. [Google Scholar] [CrossRef]
- Okunaka, T.; Kato, H.; Konaka, C.; Yamamoto, H.; Furukawa, K. Primary Lung Cancer Producing Alpha-Fetoprotein. Ann. Thorac. Surg. 1992, 53, 151–152. [Google Scholar] [CrossRef]
- Van der Gaast, A.; Hoekstra, J.W.; Croles, J.J.; Splinter, T.A.W. Elevated Serum Tumor Markers in Patients with Testicular Cancer after Induction Chemotherapy Due to a Reservoir of Markers in Cystic Differentiated Mature Teratoma. J. Urol. 1991, 145, 829–831. [Google Scholar] [CrossRef]
- Box, J.K.; Paquet, N.; Adams, M.N.; Boucher, D.; Bolderson, E.; O’Byrne, K.J.; Richard, D.J. Nucleophosmin: From Structure and Function to Disease Development. BMC Mol. Biol. 2016, 17, 19. [Google Scholar] [CrossRef] [Green Version]
- Phi, J.H.; Sun, C.; Lee, S.-H.; Lee, S.; Park, I.; Choi, S.A.; Park, S.-H.; Lee, J.Y.; Wang, K.; Kim, S.; et al. NPM1 as a Potential Therapeutic Target for Atypical Teratoid/Rhabdoid Tumors. BMC Cancer 2019, 19, 848. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.-I.I.; Kao, C.-L.L.; Lee, Y.-Y.Y.; Chiou, G.-Y.Y.; Tai, L.-K.K.; Lu, K.-H.H.; Huang, C.-S.S.; Chen, Y.-W.W.; Chiou, S.-H.H.; Cheng, I.-C.C.; et al. Differential Expression Profiling between Atypical Teratoid/Rhabdoid and Medulloblastoma Tumor in Vitro and in Vivo Using Microarray Analysis. Child’s Nerv. Syst. 2010, 26, 293–303. [Google Scholar] [CrossRef]
- Son, M.J.; Woolard, K.; Nam, D.H.; Lee, J.; Fine, H.A. SSEA-1 Is an Enrichment Marker for Tumor-Initiating Cells in Human Glioblastoma. Cell Stem Cell 2009, 4, 440–452. [Google Scholar] [CrossRef] [Green Version]
- Hollingsworth, M.A.; Swanson, B.J. Mucins in Cancer: Protection and Control of the Cell Surface. Nat. Rev. Cancer 2004, 4, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Tan, Y.; Li, S.; Zhou, J.; Wang, Q.; Wang, Y.; Xie, Y.; Chen, L.; Li, J.; Fang, C.; et al. Genomic Landscapes by Multiregion Sequencing Combined with Circulation Tumor DNA Detection Contribute to Molecular Diagnosis in Glioblastomas. Aging 2019, 11, 11224–11243. [Google Scholar] [CrossRef] [PubMed]
- Robbins, C.J.; Bou-Dargham, M.J.; Sanchez, K.; Rosen, M.C.; Sang, Q.-X.A. Decoding Somatic Driver Gene Mutations and Affected Signaling Pathways in Human Medulloblastoma Subgroups. J. Cancer 2018, 9, 4596–4610. [Google Scholar] [CrossRef] [PubMed]
- Bast, R.C.; Badgwell, D.; Lu, Z.; Marquez, R.; Rosen, D.; Liu, J.; Baggerly, K.A.; Atkinson, E.N.; Skates, S.; Zhang, Z.; et al. New Tumor Markers: CA125 and Beyond. Int. J. Gynecol. Cancer 2005, 15 (Suppl. 3), 274–281. [Google Scholar] [CrossRef]
- Lakshmanan, I.; Ponnusamy, M.P.; Das, S.; Chakraborty, S.; Haridas, D.; Mukhopadhyay, P.; Lele, S.M.; Batra, S.K. MUC16 Induced Rapid G2/M Transition via Interactions with JAK2 for Increased Proliferation and Anti-Apoptosis in Breast Cancer Cells. Oncogene 2012, 31, 805–817. [Google Scholar] [CrossRef] [Green Version]
- Cwik, G.; Wallner, G.; Skoczylas, T.; Ciechanski, A.; Zinkiewicz, K. Cancer Antigens 19-9 and 125 in the Differential Diagnosis of Pancreatic Mass Lesions. Arch. Surg. 2006, 141, 968–973, discussion 974. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Pasche, B.; Zhang, W.; Chen, K. Association of MUC16 Mutation With Tumor Mutation Load and Outcomes in Patients With Gastric Cancer. JAMA Oncol. 2018, 4, 1691–1698. [Google Scholar] [CrossRef]
- Guo, J.; Zhou, S.; Rao, N.P.; Pez, G.H. Pleomorphic Malignant Fibrous Histiocytoma/Undifferentiated High-Grade Pleomorphic Sarcoma of the Scrotum in a Patient Presenting as Fournier Gangrene: A Case Report. Appl. Immunohistochem. Mol. Morphol. 2010, 18, 473–478. [Google Scholar] [CrossRef]
- Das, S.; Rachagani, S.; Torres-Gonzalez, M.P.; Lakshmanan, I.; Majhi, P.D.; Smith, L.M.; Wagner, K.-U.; Batra, S.K. Carboxyl-Terminal Domain of MUC16 Imparts Tumorigenic and Metastatic Functions through Nuclear Translocation of JAK2 to Pancreatic Cancer Cells. Oncotarget 2015, 6, 5772–5787. [Google Scholar] [CrossRef] [Green Version]
- Haridas, D.; Ponnusamy, M.P.; Chugh, S.; Lakshmanan, I.; Seshacharyulu, P.; Batra, S.K. MUC16: Molecular Analysis and Its Functional Implications in Benign and Malignant Conditions. FASEB J. 2014, 28, 4183–4199. [Google Scholar] [CrossRef]
- Akita, K.; Tanaka, M.; Tanida, S.; Mori, Y.; Toda, M.; Nakada, H. CA125/MUC16 Interacts with Src Family Kinases, and over-Expression of Its C-Terminal Fragment in Human Epithelial Cancer Cells Reduces Cell-Cell Adhesion. Eur. J. Cell Biol. 2013, 92, 257–263. [Google Scholar] [CrossRef]
- Thériault, C.; Pinard, M.; Comamala, M.; Migneault, M.; Beaudin, J.; Matte, I.; Boivin, M.; Piché, A.; Rancourt, C. MUC16 (CA125) Regulates Epithelial Ovarian Cancer Cell Growth, Tumorigenesis and Metastasis. Gynecol. Oncol. 2011, 121, 434–443. [Google Scholar] [CrossRef]
- Gubbels, J.A.A.; Belisle, J.; Onda, M.; Rancourt, C.; Migneault, M.; Ho, M.; Bera, T.K.; Connor, J.; Sathyanarayana, B.K.; Lee, B.; et al. Mesothelin-MUC16 Binding Is a High Affinity, N-Glycan Dependent Interaction That Facilitates Peritoneal Metastasis of Ovarian Tumors. Mol. Cancer 2006, 5, 50. [Google Scholar] [CrossRef] [Green Version]
- Rump, A.; Morikawa, Y.; Tanaka, M.; Minami, S.; Umesaki, N.; Takeuchi, M.; Miyajima, A. Binding of Ovarian Cancer Antigen CA125/MUC16 to Mesothelin Mediates Cell Adhesion. J. Biol. Chem. 2004, 279, 9190–9198. [Google Scholar] [CrossRef] [Green Version]
- Yonezawa, S.; Goto, M.; Yamada, N.; Higashi, M.; Nomoto, M. Expression Profiles of MUC1, MUC2, and MUC4 Mucins in Human Neoplasms and Their Relationship with Biological Behavior. Proteomics 2008, 8, 3329–3341. [Google Scholar] [CrossRef]
- Bera, T.K.; Pastan, I. Mesothelin Is Not Required for Normal Mouse Development or Reproduction. Mol. Cell. Biol. 2000, 20, 2902–2906. [Google Scholar] [CrossRef] [Green Version]
- Chang, K.; Pastan, I. Molecular Cloning of Mesothelin, a Differentiation Antigen Present on Mesothelium, Mesotheliomas, and Ovarian Cancers. Proc. Natl. Acad. Sci. USA 1996, 93, 136–140. [Google Scholar] [CrossRef] [Green Version]
- Thomas, A.; Chen, Y.; Steinberg, S.M.; Luo, J.; Pack, S.; Raffeld, M.; Abdullaev, Z.; Alewine, C.; Rajan, A.; Giaccone, G.; et al. High Mesothelin Expression in Advanced Lung Adenocarcinoma Is Associated with KRAS Mutations and a Poor Prognosis. Oncotarget 2015, 6, 11694–11703. [Google Scholar] [CrossRef] [Green Version]
- Argani, P.; Iacobuzio-Donahue, C.; Ryu, B.; Rosty, C.; Goggins, M.; Wilentz, R.E.; Murugesan, S.R.; Leach, S.D.; Jaffee, E.; Yeo, C.J.; et al. Mesothelin Is Overexpressed in the Vast Majority of Ductal Adenocarcinomas of the Pancreas: Identification of a New Pancreatic Cancer Marker by Serial Analysis of Gene Expression (SAGE). Clin. Cancer Res. 2001, 7, 3862–3868. [Google Scholar]
- Hassan, R.; Laszik, Z.G.; Lerner, M.; Raffeld, M.; Postier, R.; Brackett, D. Mesothelin Is Overexpressed in Pancreaticobiliary Adenocarcinomas but Not in Normal Pancreas and Chronic Pancreatitis. Am. J. Clin. Pathol. 2005, 124, 838–845. [Google Scholar] [CrossRef]
- Hassan, R.; Kreitman, R.J.; Pastan, I.; Willingham, M.C. Localization of Mesothelin in Epithelial Ovarian Cancer. Appl. Immunohistochem. Mol. Morphol. 2005, 13, 243–247. [Google Scholar] [CrossRef]
- Parinyanitikul, N.; Blumenschein, G.R.; Wu, Y.; Lei, X.; Chavez-Macgregor, M.; Smart, M.; Gonzalez-Angulo, A.M. Mesothelin Expression and Survival Outcomes in Triple Receptor Negative Breast Cancer. Clin. Breast Cancer 2013, 13, 378–384. [Google Scholar] [CrossRef] [Green Version]
- Johnson, M.D.; Vito, F.; O’Connell, M.J. Mesothelin Expression in the Leptomeninges and Meningiomas. J. Histochem. Cytochem. 2008, 56, 579–585. [Google Scholar] [CrossRef] [Green Version]
- Tozbikian, G.; Brogi, E.; Kadota, K.; Catalano, J.; Akram, M.; Patil, S.; Ho, A.Y.; Reis-Filho, J.S.; Weigelt, B.; Norton, L.; et al. Mesothelin Expression in Triple Negative Breast Carcinomas Correlates Significantly with Basal-like Phenotype, Distant Metastases and Decreased Survival. PLoS ONE 2014, 9, e114900. [Google Scholar] [CrossRef] [Green Version]
- Roycik, M.D.; Myers, J.S.; Newcomer, R.G.; Sang, Q.-X.A. Matrix Metalloproteinase Inhibition in Atherosclerosis and Stroke. Curr. Mol. Med. 2013, 13, 1299–1313. [Google Scholar] [CrossRef]
- Hu, J.; Van den Steen, P.E.; Sang, Q.-X.A.; Opdenakker, G. Matrix Metalloproteinase Inhibitors as Therapy for Inflammatory and Vascular Diseases. Nat. Rev. Drug Discov. 2007, 6, 480–498. [Google Scholar] [CrossRef]
- Roycik, M.D.; Fang, X.; Sang, Q.-X. A Fresh Prospect of Extracellular Matrix Hydrolytic Enzymes and Their Substrates. Curr. Pharm. Des. 2009, 15, 1295–1308. [Google Scholar] [CrossRef]
- Qi, B.; Newcomer, R.G.; Sang, Q.-X.A. ADAM19/Adamalysin 19 Structure, Function, and Role as a Putative Target in Tumors and Inflammatory Diseases. Curr. Pharm. Des. 2009, 15, 2336–2348. [Google Scholar] [CrossRef]
- Sang, Q.-X.A.; Jin, Y.; Newcomer, R.G.; Monroe, S.C.; Fang, X.; Hurst, D.R.; Lee, S.; Cao, Q.; Schwartz, M.A. Matrix Metalloproteinase Inhibitors as Prospective Agents for the Prevention and Treatment of Cardiovascular and Neoplastic Diseases. Curr. Top. Med. Chem. 2006, 6, 289–316. [Google Scholar] [CrossRef]
- Johann, P.D.; Erkek, S.; Zapatka, M.; Kerl, K.; Buchhalter, I.; Hovestadt, V.; Jones, D.T.W.; Sturm, D.; Hermann, C.; Segura Wang, M.; et al. Atypical Teratoid/Rhabdoid Tumors Are Comprised of Three Epigenetic Subgroups with Distinct Enhancer Landscapes. Cancer Cell 2016, 29, 379–393. [Google Scholar] [CrossRef] [Green Version]
- Ho, B.; Johann, P.D.; Grabovska, Y.; De Dieu Andrianteranagna, M.J.; Yao, F.; Frühwald, M.; Hasselblatt, M.; Bourdeaut, F.; Williamson, D.; Huang, A.; et al. Molecular Subgrouping of Atypical Teratoid/Rhabdoid Tumors—A Reinvestigation and Current Consensus. Neuro-Oncology 2020, 22, 613–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosco, D.B.; Roycik, M.D.; Jin, Y.; Schwartz, M.A.; Lively, T.J.; Zorio, D.A.R.; Sang, Q.X.A. A New Synthetic Matrix Metalloproteinase Inhibitor Reduces Human Mesenchymal Stem Cell Adipogenesis. PLoS ONE 2017, 12, e0172925. [Google Scholar] [CrossRef] [PubMed]
- Russell, M.D.; Young, A.M.H.; Karri, S.K. Biomarkers of Pediatric Brain Tumors. Front. Pediatr. 2013, 1, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardoso, L.C.; Soares, R.d.S.; Laurentino, T.D.S.; Lerario, A.M.; Marie, S.K.N.; Oba-Shinjo, S.M. CD99 Expression in Glioblastoma Molecular Subtypes and Role in Migration and Invasion. Int. J. Mol. Sci. 2019, 20, 1137. [Google Scholar] [CrossRef] [Green Version]
- Sherman, L.; Sleeman, J.; Dall, P.; Hekele, A.; Moll, J.; Ponta, H.; Herrlich, P. The CD44 Proteins in Embryonic Development and in Cancer. Curr. Top. Microbiol. Immunol. 1996, 213 Pt 1, 249–269. [Google Scholar] [CrossRef]
- Servais, E.L.; Colovos, C.; Rodriguez, L.; Bograd, A.J.; Nitadori, J.; Sima, C.; Rusch, V.W.; Sadelain, M.; Adusumilli, P.S. Mesothelin Overexpression Promotes Mesothelioma Cell Invasion and MMP-9 Secretion in an Orthotopic Mouse Model and in Epithelioid Pleural Mesothelioma Patients. Clin. Cancer Res. 2012, 18, 2478–2489. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.Z.; Poore, B.; Alt, J.; Price, A.; Allen, S.J.; Hanaford, A.R.; Kaur, H.; Orr, B.A.; Slusher, B.S.; Eberhart, C.G.; et al. Unbiased Metabolic Profiling Predicts Sensitivity of High MYC-Expressing Atypical Teratoid/Rhabdoid Tumors to Glutamine Inhibition with 6-Diazo-5-Oxo-L-Norleucine. Clin. Cancer Res. 2019, 25, 5925–5936. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.Y.; Cha, J.; Kim, S.K.; Park, J.H.; Song, K.H.; Kim, P.; Kim, M.-Y. C-MYC Drives Breast Cancer Metastasis to the Brain, but Promotes Synthetic Lethality with TRAIL. Mol. Cancer Res. 2019, 17, 544–554. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Le, A. Glutamine Metabolism in Cancer. Adv. Exp. Med. Biol. 2018, 1063, 13–32. [Google Scholar] [CrossRef]
- Pietras, A.; Katz, A.M.; Ekström, E.J.; Wee, B.; Halliday, J.J.; Pitter, K.L.; Werbeck, J.L.; Amankulor, N.M.; Huse, J.T.; Holland, E.C. Osteopontin-CD44 Signaling in the Glioma Perivascular Niche Enhances Cancer Stem Cell Phenotypes and Promotes Aggressive Tumor Growth. Cell Stem Cell 2014, 14, 357–369. [Google Scholar] [CrossRef] [Green Version]
- Perez, B.H.; Gipson, I.K. Focus on Molecules: Human Mucin MUC16. Exp. Eye Res. 2008, 87, 400–401. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.-H.H.; Hung, W.-C.C.; Wang, P.; Paul, C.; Konstantopoulos, K. Mesothelin Binding to CA125/MUC16 Promotes Pancreatic Cancer Cell Motility and Invasion via MMP-7 Activation. Sci. Rep. 2013, 3, 1870. [Google Scholar] [CrossRef] [Green Version]
- Valacca, C.; Tassone, E.; Mignatti, P. TIMP-2 Interaction with MT1-MMP Activates the AKT Pathway and Protects Tumor Cells from Apoptosis. PLoS ONE 2015, 10, e0136797. [Google Scholar] [CrossRef]
- D’Alessio, S.; Ferrari, G.; Cinnante, K.; Scheerer, W.; Galloway, A.C.; Roses, D.F.; Rozanov, D.V.; Remacle, A.G.; Oh, E.-S.; Shiryaev, S.A.; et al. Tissue Inhibitor of Metalloproteinases-2 Binding to Membrane-Type 1 Matrix Metalloproteinase Induces MAPK Activation and Cell Growth by a Non-Proteolytic Mechanism. J. Biol. Chem. 2008, 283, 87–99. [Google Scholar] [CrossRef]
- Johnson, M.D.; Vito, F.; Xu, H.H.; Xu, H.H. MUC16 Expression and Risk of Adenocarcinoma Metastases to Peritoneum, Pleura, Leptomeninges, and Brain. Appl. Immunohistochem. Mol. Morphol. 2010, 18, 250–253. [Google Scholar] [CrossRef]
- Holmberg, J.; He, X.; Peredo, I.; Orrego, A.; Hesselager, G.; Ericsson, C.; Hovatta, O.; Oba-Shinjo, S.M.; Marie, S.K.N.; Nistér, M.; et al. Activation of Neural and Pluripotent Stem Cell Signatures Correlates with Increased Malignancy in Human Glioma. PLoS ONE 2011, 6, e18454. [Google Scholar] [CrossRef] [Green Version]
- Yin, X.; Li, Y.-W.; Jin, J.-J.; Zhou, Y.; Ren, Z.-G.; Qiu, S.-J.; Zhang, B.-H. The Clinical and Prognostic Implications of Pluripotent Stem Cell Gene Expression in Hepatocellular Carcinoma. Oncol. Lett. 2013, 5, 1155–1162. [Google Scholar] [CrossRef] [Green Version]
- Saigusa, S.; Tanaka, K.; Toiyama, Y.; Yokoe, T.; Okugawa, Y.; Ioue, Y.; Miki, C.; Kusunoki, M. Correlation of CD133, OCT4, and SOX2 in Rectal Cancer and Their Association with Distant Recurrence after Chemoradiotherapy. Ann. Surg. Oncol. 2009, 16, 3488–3498. [Google Scholar] [CrossRef]
- Lu, Y.; Zhu, H.; Shan, H.; Lu, J.; Chang, X.; Li, X.; Lu, J.; Fan, X.; Zhu, S.; Wang, Y.; et al. Knockdown of Oct4 and Nanog Expression Inhibits the Stemness of Pancreatic Cancer Cells. Cancer Lett. 2013, 340, 113–123. [Google Scholar] [CrossRef]
- Kohashi, K.; Yamada, Y.; Hotokebuchi, Y.; Yamamoto, H.; Taguchi, T.; Iwamoto, Y.; Oda, Y. ERG and SALL4 Expressions in SMARCB1/INI1-Deficient Tumors: A Useful Tool for Distinguishing Epithelioid Sarcoma from Malignant Rhabdoid Tumor. Hum. Pathol. 2015, 46, 225–230. [Google Scholar] [CrossRef]
- Miettinen, M.; Wang, Z.; McCue, P.A.; Sarlomo-Rikala, M.; Rys, J.; Biernat, W.; Lasota, J.; Lee, Y.-S. SALL4 Expression in Germ Cell and Non-Germ Cell Tumors: A Systematic Immunohistochemical Study of 3215 Cases. Am. J. Surg. Pathol. 2014, 38, 410–420. [Google Scholar] [CrossRef] [Green Version]
- Venneti, S.; Le, P.; Martinez, D.; Xie, S.X.; Sullivan, L.M.; Rorke-Adams, L.B.; Pawel, B.; Judkins, A.R. Malignant Rhabdoid Tumors Express Stem Cell Factors, Which Relate To the Expression of EZH2 and Id Proteins. Am. J. Surg. Pathol. 2011, 35, 1463–1472. [Google Scholar] [CrossRef]
- Deisch, J.; Raisanen, J.; Rakheja, D. Immunohistochemical Expression of Embryonic Stem Cell Markers in Malignant Rhabdoid Tumors. Pediatr. Dev. Pathol. 2011, 14, 353–359. [Google Scholar] [CrossRef]
- Hodgson, J.G.; Yeh, R.-F.; Ray, A.; Wang, N.J.; Smirnov, I.; Yu, M.; Hariono, S.; Silber, J.; Feiler, H.S.; Gray, J.W.; et al. Comparative Analyses of Gene Copy Number and MRNA Expression in Glioblastoma Multiforme Tumors and Xenografts. Neuro-Oncology 2009, 11, 477–487. [Google Scholar] [CrossRef] [Green Version]
- Blackburn, J.S.; Brinckerhoff, C.E. Matrix Metalloproteinase-1 and Thrombin Differentially Activate Gene Expression in Endothelial Cells via PAR-1 and Promote Angiogenesis. Am. J. Pathol. 2008, 173, 1736–1746. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Li, W.; Li, Y.; Yang, H.; Xu, H.; Liang, S.; Zhang, L.; Li, Y. Expression of Matrix Metalloproteinase-26 Promotes Human Glioma U251 Cell Invasion in Vitro and in Vivo. Oncol. Rep. 2010, 23, 69–78. [Google Scholar] [CrossRef]
- Guo, J.-G.; Guo, C.-C.; He, Z.-Q.; Cai, X.-Y.; Mou, Y.-G. High MMP-26 Expression in Glioma Is Correlated with Poor Clinical Outcome of Patients. Oncol. Lett. 2018, 16, 2237–2242. [Google Scholar] [CrossRef] [Green Version]
- Hu, Q.; Yan, C.; Xu, C.; Yan, H.; Qing, L.; Pu, Y.; He, Z.; Li, X. Matrilysin-2 Expression in Colorectal Cancer Is Associated with Overall Survival of Patients. Tumour Biol. 2014, 35, 3569–3574. [Google Scholar] [CrossRef]
- Yamamoto, H.; Vinitketkumnuen, A.; Adachi, Y.; Taniguchi, H.; Hirata, T.; Miyamoto, N.; Nosho, K.; Imsumran, A.; Fujita, M.; Hosokawa, M.; et al. Association of Matrilysin-2 (MMP-26) Expression with Tumor Progression and Activation of MMP-9 in Esophageal Squamous Cell Carcinoma. Carcinogenesis 2004, 25, 2353–2360. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.G.; Xiao, A.Z.; Newcomer, R.G.; Park, H.I.; Kang, T.; Chung, L.W.K.; Swanson, M.G.; Zhau, H.E.; Kurhanewicz, J.; Sang, Q.X.A. Activation of Pro-Gelatinase B by Endometase/Matrilysin-2 Promotes Invasion of Human Prostate Cancer Cells. J. Biol. Chem. 2003, 278, 15056–15064. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Cao, Y.-J.; Zhao, Y.-G.; Sang, Q.-X.A.; Duan, E.-K. Expression of Matrix Metalloproteinase-26 and Tissue Inhibitor of Metalloproteinase-4 in Human Normal Cytotrophoblast Cells and a Choriocarcinoma Cell Line, JEG-3. Mol. Hum. Reprod. 2002, 8, 659–666. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Desai, K.K.; Iczkowski, K.A.; Newcomer, R.G.; Wu, K.J.; Zhao, Y.-G.; Tan, W.W.; Roycik, M.D.; Sang, Q.-X.A. Coordinated Peak Expression of MMP-26 and TIMP-4 in Preinvasive Human Prostate Tumor. Cell Res. 2006, 16, 750–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheau, C.; Badarau, I.A.; Costache, R.; Caruntu, C.; Mihai, G.L.; Didilescu, A.C.; Constantin, C.; Neagu, M. The Role of Matrix Metalloproteinases in the Epithelial-Mesenchymal Transition of Hepatocellular Carcinoma. Anal. Cell. Pathol. 2019, 2019, 9423907. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Liu, J.; Huang, Y.; Liu, D.; Zhang, G.; Kan, H. MicroRNA-489 Plays an Anti-Metastatic Role in Human Hepatocellular Carcinoma by Targeting Matrix Metalloproteinase-7. Transl. Oncol. 2017, 10, 211–220. [Google Scholar] [CrossRef] [PubMed]
- McDonnell, S.; Navre, M.; Coffey, R.J.; Matrisian, L.M. Expression and Localization of the Matrix Metalloproteinase Pump-1 (MMP-7) in Human Gastric and Colon Carcinomas. Mol. Carcinog. 1991, 4, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Nakano, A.; Tani, E.; Miyazaki, K.; Yamamoto, Y.; Furuyama, J. Matrix Metalloproteinases and Tissue Inhibitors of Metalloproteinases in Human Gliomas. J. Neurosurg. 1995, 83, 298–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brew, K.; Nagase, H. The Tissue Inhibitors of Metalloproteinases (TIMPs): An Ancient Family with Structural and Functional Diversity. Biochim. Biophys. Acta 2010, 1803, 55–71. [Google Scholar] [CrossRef] [Green Version]
- Muller, M.; Hubbard, S.L.; Fukuyama, K.; Dirks, P.; Matsuzawa, K.; Rutka, J.T. Characterization of a Pineal Region Malignant Rhabdoid Tumor. Towards Understanding Brain Tumor Cell Invasion. Pediatr. Neurosurg. 1995, 22, 204–209. [Google Scholar] [CrossRef]
- Rorive, S.; Lopez, X.M.; Maris, C.; Trepant, A.-L.; Sauvage, S.; Sadeghi, N.; Roland, I.; Decaestecker, C.; Salmon, I. TIMP-4 and CD63: New Prognostic Biomarkers in Human Astrocytomas. Mod. Pathol. 2010, 23, 1418–1428. [Google Scholar] [CrossRef] [Green Version]
- Lizarraga, F.; Espinosa, M.; Ceballos-Cancino, G.; Vazquez-Santillan, K.; Bahena-Ocampo, I.; Schwarz-Cruz y Celis, A.; Vega-Gordillo, M.; Garcia Lopez, P.; Maldonado, V.; Melendez-Zajgla, J. Tissue Inhibitor of Metalloproteinases-4 (TIMP-4) Regulates Stemness in Cervical Cancer Cells. Mol. Carcinog. 2016, 55, 1952–1961. [Google Scholar] [CrossRef]
- Boufraqech, M.; Zhang, L.; Nilubol, N.; Sadowski, S.M.; Kotian, S.; Quezado, M.; Kebebew, E. Lysyl Oxidase (LOX) Transcriptionally Regulates SNAI2 Expression and TIMP4 Secretion in Human Cancers. Clin. Cancer Res. 2016, 22, 4491–4504. [Google Scholar] [CrossRef] [Green Version]
- Zigrino, P.; Nischt, R.; Mauch, C. The Disintegrin-like and Cysteine-Rich Domains of ADAM-9 Mediate Interactions between Melanoma Cells and Fibroblasts. J. Biol. Chem. 2011, 286, 6801–6807. [Google Scholar] [CrossRef] [Green Version]
- O’Shea, C.; McKie, N.; Buggy, Y.; Duggan, C.; Hill, A.D.K.; McDermott, E.; O’Higgins, N.; Duffy, M.J. Expression of ADAM-9 MRNA and Protein in Human Breast Cancer. Int. J. Cancer 2003, 105, 754–761. [Google Scholar] [CrossRef]
- Micocci, K.C.; Martin, A.C.B.M.; de Freitas Montenegro, C.; Durante, A.C.; Pouliot, N.; Cominetti, M.R.; Selistre-de-Araujo, H.S. ADAM9 Silencing Inhibits Breast Tumor Cell Invasion in Vitro. Biochimie 2013, 95, 1371–1378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, X.; Wang, Y.; Zhang, C.; Liu, L.; Yang, S.; Wang, Y.; Liu, X.; Qian, Z.; Fang, S.; Qiao, H.; et al. ADAM9 Expression Is Associate with Glioma Tumor Grade and Histological Type, and Acts as a Prognostic Factor in Lower-Grade Gliomas. Int. J. Mol. Sci. 2016, 17, 1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, S.; Zemp, F.J.; Senger, D.; Robbins, S.M.; Yong, V.W. ADAM-9 Is a Novel Mediator of Tenascin-C-Stimulated Invasiveness of Brain Tumor-Initiating Cells. Neuro-Oncology 2015, 17, 1095–1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Wang, S.; Yuan, A.; Yuan, X.; Liu, B. MicroRNA-140 Represses Glioma Growth and Metastasis by Directly Targeting ADAM9. Oncol. Rep. 2016, 36, 2329–2338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zigrino, P.; Mauch, C.; Fox, J.W.; Nischt, R. Adam-9 Expression and Regulation in Human Skin Melanoma and Melanoma Cell Lines. Int. J. Cancer 2005, 116, 853–859. [Google Scholar] [CrossRef]
- Hsia, H.-E.; Tüshaus, J.; Brummer, T.; Zheng, Y.; Scilabra, S.D.; Lichtenthaler, S.F. Functions of “A Disintegrin and Metalloproteases (ADAMs)” in the Mammalian Nervous System. Cell. Mol. Life Sci. 2019, 76, 3055–3081. [Google Scholar] [CrossRef]
- Weber, S.; Saftig, P. Ectodomain Shedding and ADAMs in Development. Development 2012, 139, 3693–3709. [Google Scholar] [CrossRef] [Green Version]
- Venkatesh, H.S.; Tam, L.T.; Woo, P.J.; Lennon, J.; Nagaraja, S.; Gillespie, S.M.; Ni, J.; Duveau, D.Y.; Morris, P.J.; Zhao, J.J.; et al. Targeting Neuronal Activity-Regulated Neuroligin-3 Dependency in High-Grade Glioma. Nature 2017, 549, 533–537. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Qin, X.-P.; Zhuang, Y.; Zhang, Y.; Liao, H.-B.; Tang, J.-C.; Pan, M.-X.; Zeng, F.-F.; Lei, Y.; Lei, R.-X.; et al. Glioblastoma Recurrence Correlates with NLGN3 Levels. Cancer Med. 2018, 7, 2848–2859. [Google Scholar] [CrossRef]
- Mullooly, M.; McGowan, P.M.; Sukor, S.U.; Madden, S.F.; McDermott, E.; Crown, J.; O’Donovan, N.; Duffy, M.J. ADAMs as New Therapeutic Targets for Triple-Negative Breast Cancer. J. Clin. Oncol. 2011, 29, 1062. [Google Scholar] [CrossRef]
- Liu, F.; Zhuang, L.; Wu, R.; Li, D. MiR-365 Inhibits Cell Invasion and Migration of Triple Negative Breast Cancer through ADAM10. J. Buon 2019, 24, 1905–1912. [Google Scholar]
- Chen, T.; You, Y.; Jiang, H.; Wang, Z.Z. Epithelial-Mesenchymal Transition (EMT): A Biological Process in the Development, Stem Cell Differentiation, and Tumorigenesis. J. Cell. Physiol. 2017, 232, 3261–3272. [Google Scholar] [CrossRef]
- Pastushenko, I.; Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Donaher, J.L.; Das, S.; Li, X.; Reinhardt, F.; Krall, J.A.; Lambert, A.W.; Thiru, P.; Keys, H.R.; Khan, M.; et al. Genome-Wide CRISPR Screen Identifies PRC2 and KMT2D-COMPASS as Regulators of Distinct EMT Trajectories That Contribute Differentially to Metastasis. Nat. Cell Biol. 2022, 24, 554–564. [Google Scholar] [CrossRef]
- Du, B.; Shim, J.S. Targeting Epithelial-Mesenchymal Transition (EMT) to Overcome Drug Resistance in Cancer. Molecules 2016, 21, 965. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Zheng, J.; Yu, J.; Wu, Y.; Guo, J.; Xu, Z.; Sun, X. Knockdown of MMP-1 Inhibits the Progression of Colorectal Cancer by Suppressing the PI3K/Akt/C-myc Signaling Pathway and EMT. Oncol. Rep. 2020, 43, 1103–1112. [Google Scholar] [CrossRef]
- Terada, Y.; Jo, N.; Arakawa, Y.; Sakakura, M.; Yamada, Y.Y.; Ukai, T.; Kabata, M.; Mitsunaga, K.; Mineharu, Y.; Ohta, S.; et al. Human Pluripotent Stem Cell-Derived Tumor Model Uncovers the Embryonic Stem Cell Signature as a Key Driver in Atypical Teratoid/Rhabdoid Tumor. Cell Rep. 2019, 26, 2608–2621. [Google Scholar] [CrossRef] [Green Version]
- Vincent, P.H.; Benedikz, E.; Uhlén, P.; Hovatta, O.; Sundström, E. Expression of Pluripotency Markers in Nonpluripotent Human Neural Stem and Progenitor Cells. Stem Cells Dev. 2017, 26, 876–887. [Google Scholar] [CrossRef] [Green Version]
- Koçak, N.; Dönmez, H.; Yildirim, İ.H. Effects of Melatonin on Apoptosis and Cell Differentiation in MCF-7 Derived Cancer Stem Cells. Cell. Mol. Biol. 2018, 64, 56–61. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Gene | Name | Location |
|---|---|---|---|
| ATRT | OPN | Osteopontin | Secreted |
| NPM1 | Nucleophosmin 1 | Intracellular | |
| Common cancer biomarkers | MUC16 | Mucin-16 | Membrane, secreted |
| CD44 | Homing cell adhesion molecule | Intracellular, membrane, secreted | |
| AFP | Alpha-fetoprotein | Secreted | |
| MSLN | Mesothelin | Intracellular, membrane | |
| CD99 | Single-chain type-1 glycoprotein | Intracellular, membrane | |
| CGB3 | Chorionic gonadotropin β-3 | Secreted | |
| Embryonic | SSEA1 | Fucosyltransferase 4 | Membrane |
| OCT4 | Octamer-binding transcription factor 4 | Intracellular | |
| Neuronal | ENO2 | Neuron-specific enolase | Intracellular |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hua, T.; Zeng, Z.; Chen, J.; Xue, Y.; Li, Y.; Sang, Q. Human Malignant Rhabdoid Tumor Antigens as Biomarkers and Potential Therapeutic Targets. Cancers 2022, 14, 3685. https://doi.org/10.3390/cancers14153685
Hua T, Zeng Z, Chen J, Xue Y, Li Y, Sang Q. Human Malignant Rhabdoid Tumor Antigens as Biomarkers and Potential Therapeutic Targets. Cancers. 2022; 14(15):3685. https://doi.org/10.3390/cancers14153685
Chicago/Turabian StyleHua, Timothy, Ziwei Zeng, Junji Chen, Yu Xue, Yan Li, and Qingxiang Sang. 2022. "Human Malignant Rhabdoid Tumor Antigens as Biomarkers and Potential Therapeutic Targets" Cancers 14, no. 15: 3685. https://doi.org/10.3390/cancers14153685