
Cancer Associated Fibroblast (CAF) Regulation of PDAC Parenchymal (CPC) and CSC Phenotypes Is Modulated by ECM Composition

 ,
,  , and
, and
Abstract
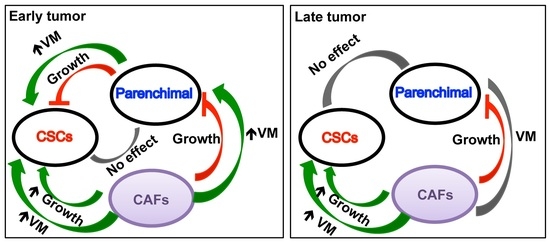
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Cancer Associated Fibroblasts (CAFs)
2.3. D Culture Models
2.4. Indirect Co-Culture
2.5. Cell Viability Measurements
2.6. Invadopodia Proteolytic Activity
2.7. Vascular Network Analysis
2.8. Statistical Analysis
3. Results
3.1. CAF Conditioned Medium (CM) Reduces CPC Growth and Increases CSC Growth on All of the Substrates
3.2. Effect of Primary CAF Conditioned Medium (CM) on CPC and CSC Invadopodia Proteolytic Activity
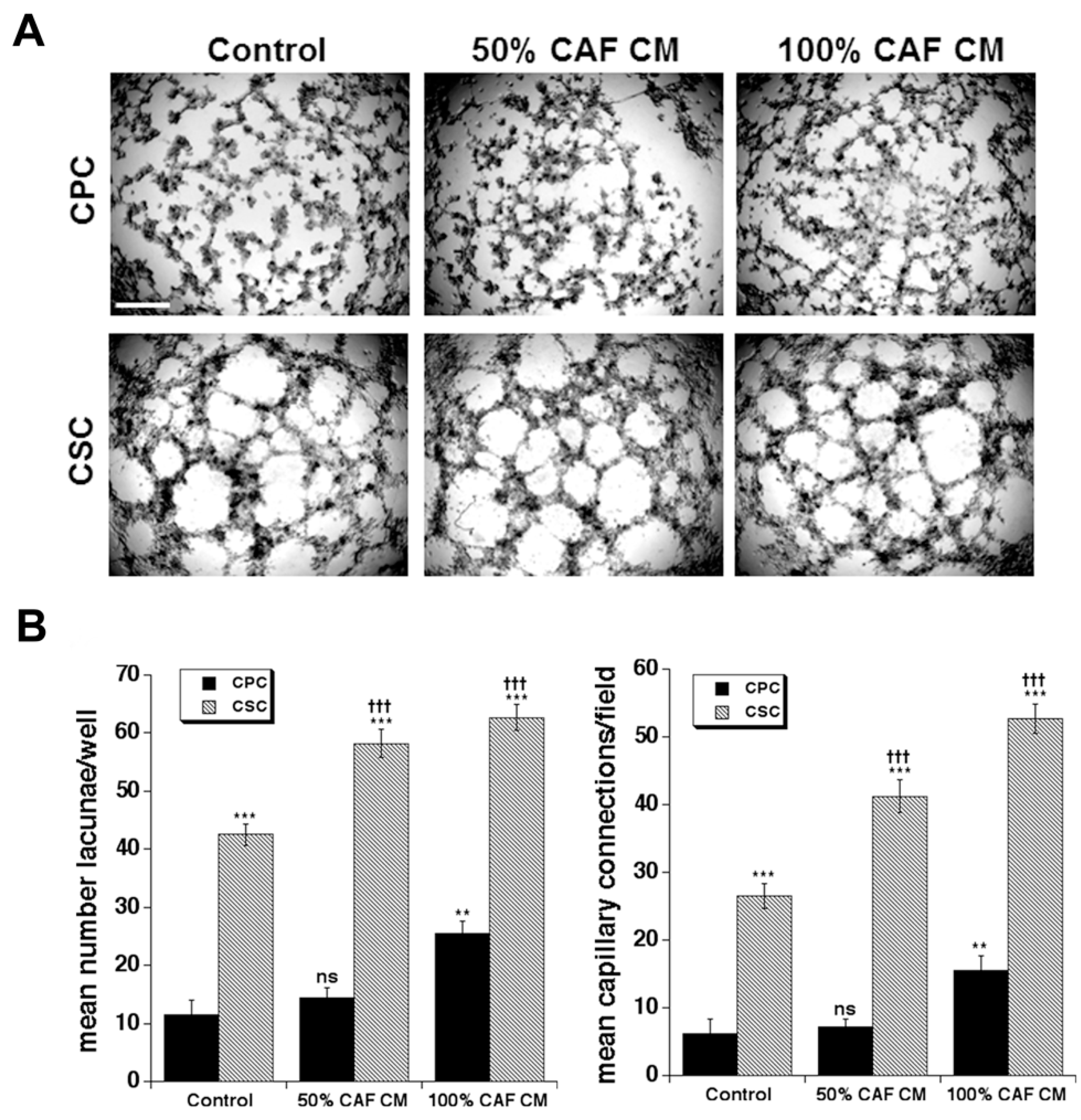
3.3. Effect of Primary CAF Conditioned Medium (CM) on CPC and CSC Vasculogenic Mimicry
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hong, S.M.; Park, J.Y.; Hruban, R.H.; Goggins, M. Molecular signatures of pancreatic cancer. Arch. Pathol. Lab. Med. 2011, 135, 716–727. [Google Scholar] [CrossRef] [PubMed]
- Di Marco, M.; Grassi, E.; Durante, S.; Vecchiarelli, S.; Palloni, A.; Macchini, M.; Casadei, R.; Ricci, C.; Panzacchi, R.; Santini, D.; et al. State of the art biological therapies in pancreatic cancer. World J. Gastrointest. Oncol. 2016, 8, 55–66. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [Green Version]
- Schober, M.; Jesenofsky, R.; Faissner, R.; Weidenauer, C.; Hagmann, W.; Michl, P.; Heuchel, R.L.; Haas, S.L.; Löhr, J.-M. Desmoplasia and chemoresistance in pancreatic cancer. Cancers 2014, 6, 2137–2154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biondani, G.; Zeeberg, K.; Greco, M.R.; Cannone, S.; Dando, I.; Pozza, E.D.; Mastrodonato, M.; Forciniti, S.; Casavola, V.; Palmieri, M.; et al. Extracellular Matrix composition modulates PDAC parenchymal and stem cell plasticity and behavior through the secretome. FEBS J. 2018, 285, 2104–2124. [Google Scholar] [CrossRef] [Green Version]
- Hadden, M.; Mittal, A.; Jaswinder Samra Zreiqat, H.; Sahni, S.; Ramaswamy, Y. Mechanically stressed cancer microenvironment: Role in pancreatic cancer progression. BBA-Rev. Cancer 2020, 1874, 188418. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, T.M.A.; Di Molfetta, D.; Greco, M.R.; Koltai, T.; Alfarouk, K.O.; Reshkin, S.J.; Cardone, R.A. Tumor Microenvironment Features and Chemoresistance in Pancreatic Ductal Adenocarcinoma: Insights into Targeting Physicochemical Barriers and Metabolism as Therapeutic Approaches. Cancers 2021, 13, 6135. [Google Scholar] [CrossRef]
- Ferrara, B.; Pignatelli, C.; Cossutta, M.; Citro, A.; Courty, J.; Piemonti, L. The Extracellular Matrix in Pancreatic Cancer: Description of a Complex Network and Promising Therapeutic Options. Cancers 2021, 13, 4442. [Google Scholar] [CrossRef]
- Perez, V.M.; Kearney, J.F.; Yeh, J.J. The PDAC Extracellular Matrix: A Review of the ECM Protein Composition, Tumor Cell Interaction, and Therapeutic Strategies. Front. Oncol. 2021, 11, 751311. [Google Scholar] [CrossRef]
- Veenstra, V.L.; Garcia-Garijo, A.; van Laarhoven, H.W.; Bijlsma, M.F. Extracellular Influences: Molecular Subclasses and the Microenvironment in Pancreatic Cancer. Cancers 2018, 10, 34. [Google Scholar] [CrossRef] [Green Version]
- Ying, H.; Dey, P.; Yao, W.; Kimmelman, A.C.; Draetta, G.F.; Maitra, A.; De Pinho, R.A. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2016, 30, 355–385. [Google Scholar] [CrossRef] [Green Version]
- Dalla Pozza, E.; Forciniti, S.; Palmieri, M.; Dando, I. Secreted molecules inducing epithelial-to-mesenchymal transition in cancer development. Semin. Cell Dev. Biol. 2017, 78, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Whatcott, C.J.; Diep, C.H.; Jiang, P.; Watanabe, A.; LoBello, J.; Sima, C.; Hostetter, G.; Shepard, H.M.; Von Hoff, D.D.; Han, H. Desmoplasia in Primary Tumors and Metastatic Lesions of Pancreatic Cancer. Clin. Cancer Res. 2015, 21, 3561–3568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, K.; Shishodia, G.; Koul, H.K. Pancreatic cancer: Genetics, disease progression, therapeutic resistance and treatment strategies. J. Cancer Metastasis Treat. 2021, 7, 60. [Google Scholar] [CrossRef]
- Koenig, A.; Mueller, C.; Hasel, C.; Adler, G.; Menke, A. Collagen type I induces disruption of E-cadherin-mediated cell-cell contacts and promotes proliferation of pancreatic carcinoma cells. Cancer Res. 2006, 66, 4662–4671. [Google Scholar] [CrossRef] [Green Version]
- Olivares, O.; Mayers, J.R.; Gouirand, V.; Torrence, M.E.; Gicquel, T.; Borge, L.; Lac, S.; Roques, J.; Lavaut, M.; Berthezène, P.; et al. Collagen-derived proline promotes pancreatic ductal adenocarcinoma cell survival under nutrient limited conditions. Nat. Commun. 2017, 8, 16031. [Google Scholar] [CrossRef]
- Valle, S.; Martin-Hijano, L.; Alcala, S.; Alonso-Nocelo, M.; Sainz, B., Jr. The Ever-Evolving Concept of the Cancer Stem Cell in Pancreatic Cancer. Cancers 2018, 10, 33. [Google Scholar] [CrossRef] [Green Version]
- Puls, T.J.; Tan, X.; Whittington, C.F.; Voytik-Harbin, S.L. 3D collagen fibrillar microstructure guides pancreatic cancer cell phenotype and serves as a critical design parameter for phenotypic models of EMT. PLoS ONE. 2017, 12, e0188870. [Google Scholar] [CrossRef] [Green Version]
- Forciniti, S.; Dalla Pozza, E.; Greco, M.R.; Carvalho, T.M.A.; Rolando, B.; Ambrosini, G.; Carmona-Carmona, C.A.; Pacchiana, R.; Di Molfetta, D.; Donadelli, M.; et al. Extracellular Matrix Composition Modulates the Responsiveness of Differentiated and Stem Pancreatic Cancer Cells to Lipophilic Derivate of Gemcitabine. Int. J. Mol. Sci. 2021, 22, 29. [Google Scholar] [CrossRef]
- Phillips, P. Pancreatic stellate cells and fibrosis. Chapter 3. In Pancreatic Cancer and Tumor Microenvironment; Grippo, P.J., Munshi, H.G., Eds.; Transworld Research Network: Trivandrum, India, 2012. [Google Scholar] [PubMed]
- Moir, J.A.; Mann, J.; White, S.A. The role of pancreatic stellate cells in pancreatic cancer. Surg. Oncol. 2015, 24, 232–238. [Google Scholar] [CrossRef]
- von Ahrens, D.; Bhagat, T.D.; Nagrath, D.; Maitra, A.; Verma, A. The role of stromal cancer-associated fibroblasts in pancreatic cancer. J. Hematol. Oncol. 2017, 10, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarroll, J.A.; Naim, S.; Sharbeen, G.; Russia, N.; Lee, J.; Kavallaris, M.; Goldstein, D.; Phillips, P.A. Role of pancreatic stellate cells in chemoresistance in pancreatic cancer. Front. Physiol. 2014, 5, 141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolm, L.; Cigolla, S.; Wittel, U.A.; Hopt, U.T.; Keck, T.; Rades, D.; Bronsert, P.; Wellner, U.F. The Role of Fibroblasts in Pancreatic Cancer: Extracellular Matrix Versus Paracrine Factors. Transl. Oncol. 2017, 10, 578–588. [Google Scholar] [CrossRef] [PubMed]
- Shan, T.; Chen, S.; Chen, X.; Lin, W.R.; Li, W.; Ma, J.; Wu, T.; Cui, X.; Ji, H.; Li, Y.; et al. Cancer-associated fibroblasts enhance pancreatic cancer cell invasion by remodeling the metabolic conversion mechanism. Oncol. Rep. 2017, 37, 1971–1979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.P.; Liu, S.L.; Xu, J.F.; Cao, S.G.; Li, Y.; Zhou, Y.B. Pancreatic stellate cells increase pancreatic cancer cells invasion through the hepatocyte growth factor /c-Met/survivin regulated by P53/P21. Exp. Cell Res. 2017, 357, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Hwang, R.F.; Moore, T.; Arumugam, T.; Ramachandran, V.; Amos, K.D.; Rivera, A.; Ji, B.; Evans, D.B.; Logsdon, C.D. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008, 68, 918–926. [Google Scholar] [CrossRef] [Green Version]
- Sherman, M.H.; Yu, R.T.; Tseng, T.W.; Sousa, C.M.; Liu, S.; Truitt, M.L.; He, N.; Ding, N.; Liddle, C.; Atkins, A.R.; et al. Stromal cues regulate the pancreatic cancer epigenome and metabolome. Proc. Natl. Acad. Sci. USA 2017, 114, 1129–1134. [Google Scholar] [CrossRef] [Green Version]
- Dalla Pozza, E.; Dando, I.; Biondani, G.; Brandi, J.; Costanzo, C.; Zoratti, E.; Fassan, M.; Boschi, F.; Melisi, D.; Cecconi, D.; et al. Pancreatic ductal adenocarcinoma cell lines display a plastic ability to bidirectionally convert into cancer stem cells. Int. J. Oncol. 2014, 46, 1099–1108. [Google Scholar] [CrossRef] [Green Version]
- Deer, E.L.; Gonzalez-Hernandez, J.; Coursen, J.D.; Shea, J.E.; Ngatia, J.; Scaife, C.L.; Firpo, M.A.; Mulvihill, S.J. Phenotype and genotype of pancreatic cancer cell lines. Pancreas 2010, 39, 425–435. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Han, D.; Min, H.; Jin, J.; Yi, E.C. Comparative proteomic profiling of pancreatic ductal adenocarcinoma cell lines. Mol. Cells 2014, 37, 888–898. [Google Scholar] [CrossRef] [Green Version]
- Leca, J.; Martinez, S.; Lac, S.; Nigri, J.; Secq, V.; Rubis, M.; Bressy, C.; Sergé, A.; Lavaut, M.N.; Dusetti, N.; et al. Cancer-associated fibroblast-derived annexin A6+ extracellular vesicles support pancreatic cancer aggressiveness. J. Clin. Investig. 2016, 126, 4140–4156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeeberg, K.; Cardone, R.A.; Greco, M.R.; Saccomano, M.; Nohr-Nielsen, A.; Alves, F.; Pedersen, S.F.; Reshkin, S.J. Assessment of different 3D culture systems to study tumor phenotype and chemosensitivity in pancreatic ductal adenocarcinoma. Int. J. Oncol. 2016, 49, 243–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valenti, G.; Quinn, H.M.; Heynen, G.; Lan, L.; Holland, J.D.; Vogel, R.; Wulf-Goldenberg, A.; Birchmeier, W. Cancer Stem Cells Regulate Cancer-Associated Fibroblasts via Activation of Hedgehog Signaling in Mammary Gland Tumors. Cancer Res. 2017, 77, 2134–2147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, S.; Chen, J.; Yao, H.; Liu, J.; Yu, S.; Lao, L.; Wang, M.; Luo, M.; Xing, Y.; Chen, F.; et al. CD10(+)GPR77(+) Cancer-Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness. Cell 2018, 172, 841–856.e16. [Google Scholar] [CrossRef]
- Busco, G.; Cardone, R.A.; Greco, M.R.; Bellizzi, A.; Colella, M.; Antelmi, E.; Mancini, M.T.; Dell’Aquila, M.E.; Casavola, V.; Paradiso, A.; et al. NHE1 promotes invadopodial ECM proteolysis through acidification of the peri-invadopodial space. FASEB J. 2010, 24, 3903–3915. [Google Scholar] [CrossRef]
- Chu, G.C.; Kimmelman, A.C.; Hezel, A.F.; De Pinho, R.A. Stromal biology of pancreatic cancer. J. Cell Biochem. 2007, 101, 887–907. [Google Scholar] [CrossRef]
- Shields, M.A.; Dangi-Garimella, S.; Krantz, S.B.; Bentrem, D.J.; Munshi, H.G. Pancreatic cancer cells respond to type I collagen by inducing snail expression to promote membrane type 1 matrix metalloproteinase-dependent collagen invasion. J. Biol. Chem. 2011, 286, 10495–10504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhim, A.D.; Mirek, E.T.; Aiello, N.M.; Maitra, A.; Bailey, J.M.; McAllister, F.; Reichert, M.; Beatty, G.L.; Rustgi, A.K.; Vonderheide, R.H.; et al. EMT and dissemination precede pancreatic tumor formation. Cell 2012, 148, 349–361. [Google Scholar] [CrossRef] [Green Version]
- Cardone, R.A.; Greco, M.R.; Zeeberg, K.; Zaccagnino, A.; Saccomano, M.; Bellizzi, A.; Bruns, P.; Menga, M.; Pilarsky, C.; Schwab, A.; et al. A Novel NHE1-Centered Signaling Cassette Drives Epidermal Growth Factor Receptor–Dependent Pancreatic Tumor Metastasis and Is a Target for Combination Therapy. Neoplasia 2015, 17, 155–166. [Google Scholar] [CrossRef] [Green Version]
- Mostafa, M.E.; Erbarut-Seven, I.; Pehlivanoglu, B.; Adsay, V. Pathologic classification of “pancreatic cancers”: Current concepts and challenges. Chin. Clin. Oncol. 2017, 6, 59. [Google Scholar] [CrossRef]
- Zhao, Y.; Bao, Q.; Renner, A.; Camaj, P.; Eichhorn, M.; Ischenko, I.; Angele, M.; Kleespies, A.; Jauch, K.W.; Bruns, C. Cancer stem cells and angiogenesis. Int. J. Dev. Biol. 2011, 55, 477–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cannone, S.; Greco, M.R.; Carvalho, T.M.A.; Guizouarn, H.; Soriani, O.; Di Molfetta, D.; Tomasini, R.; Zeeberg, K.; Reshkin, S.J.; Cardone, R.A. Cancer Associated Fibroblast (CAF) Regulation of PDAC Parenchymal (CPC) and CSC Phenotypes Is Modulated by ECM Composition. Cancers 2022, 14, 3737. https://doi.org/10.3390/cancers14153737
Cannone S, Greco MR, Carvalho TMA, Guizouarn H, Soriani O, Di Molfetta D, Tomasini R, Zeeberg K, Reshkin SJ, Cardone RA. Cancer Associated Fibroblast (CAF) Regulation of PDAC Parenchymal (CPC) and CSC Phenotypes Is Modulated by ECM Composition. Cancers. 2022; 14(15):3737. https://doi.org/10.3390/cancers14153737
Chicago/Turabian StyleCannone, Stefania, Maria Raffaella Greco, Tiago M. A. Carvalho, Helene Guizouarn, Olivier Soriani, Daria Di Molfetta, Richard Tomasini, Katrine Zeeberg, Stephan Joel Reshkin, and Rosa Angela Cardone. 2022. "Cancer Associated Fibroblast (CAF) Regulation of PDAC Parenchymal (CPC) and CSC Phenotypes Is Modulated by ECM Composition" Cancers 14, no. 15: 3737. https://doi.org/10.3390/cancers14153737
APA StyleCannone, S., Greco, M. R., Carvalho, T. M. A., Guizouarn, H., Soriani, O., Di Molfetta, D., Tomasini, R., Zeeberg, K., Reshkin, S. J., & Cardone, R. A. (2022). Cancer Associated Fibroblast (CAF) Regulation of PDAC Parenchymal (CPC) and CSC Phenotypes Is Modulated by ECM Composition. Cancers, 14(15), 3737. https://doi.org/10.3390/cancers14153737