
A Journey through the Inter-Cellular Interactions in the Bone Marrow in Multiple Myeloma: Implications for the Next Generation of Treatments

Abstract
:Simple Summary
Abstract
1. Introduction
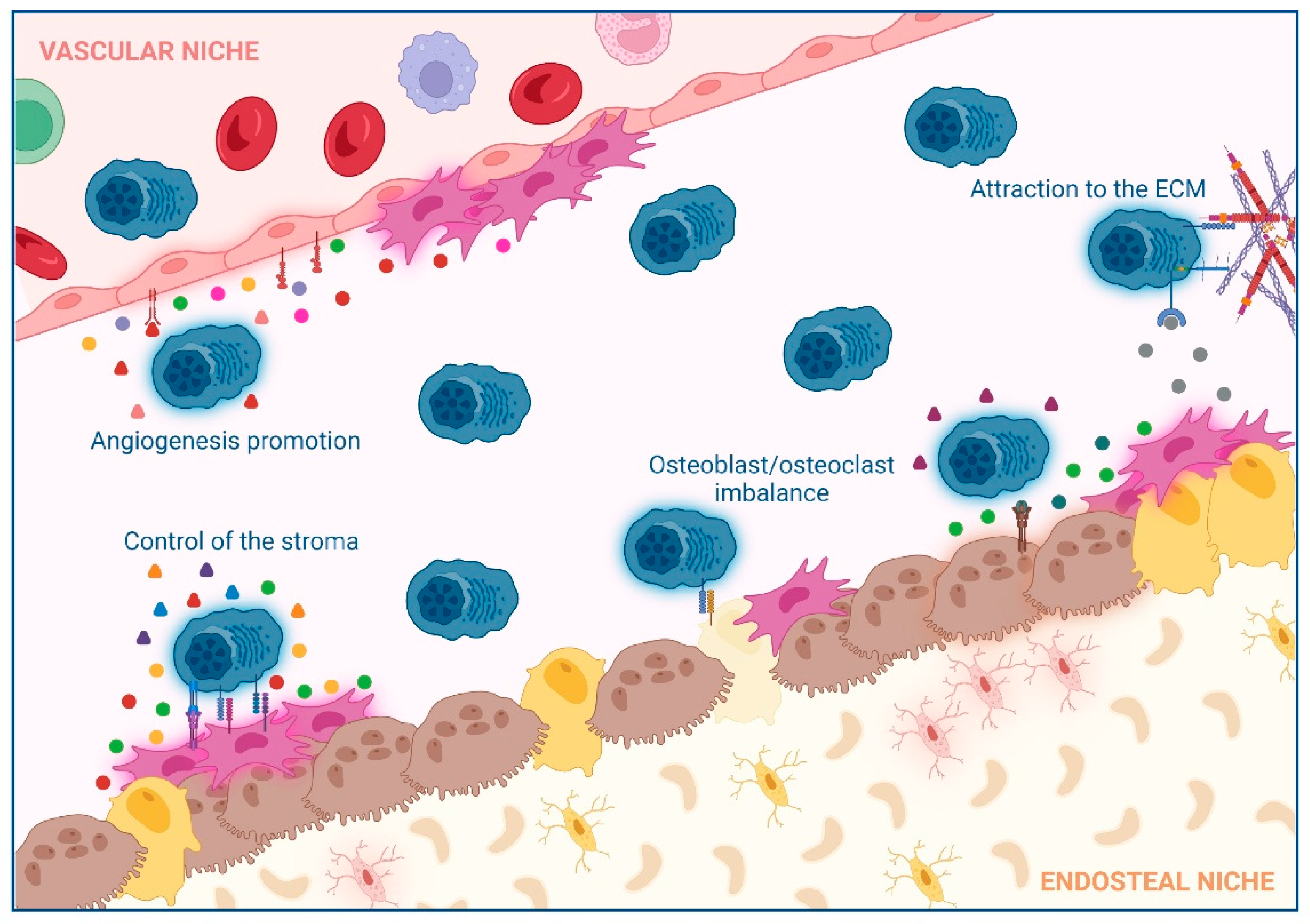
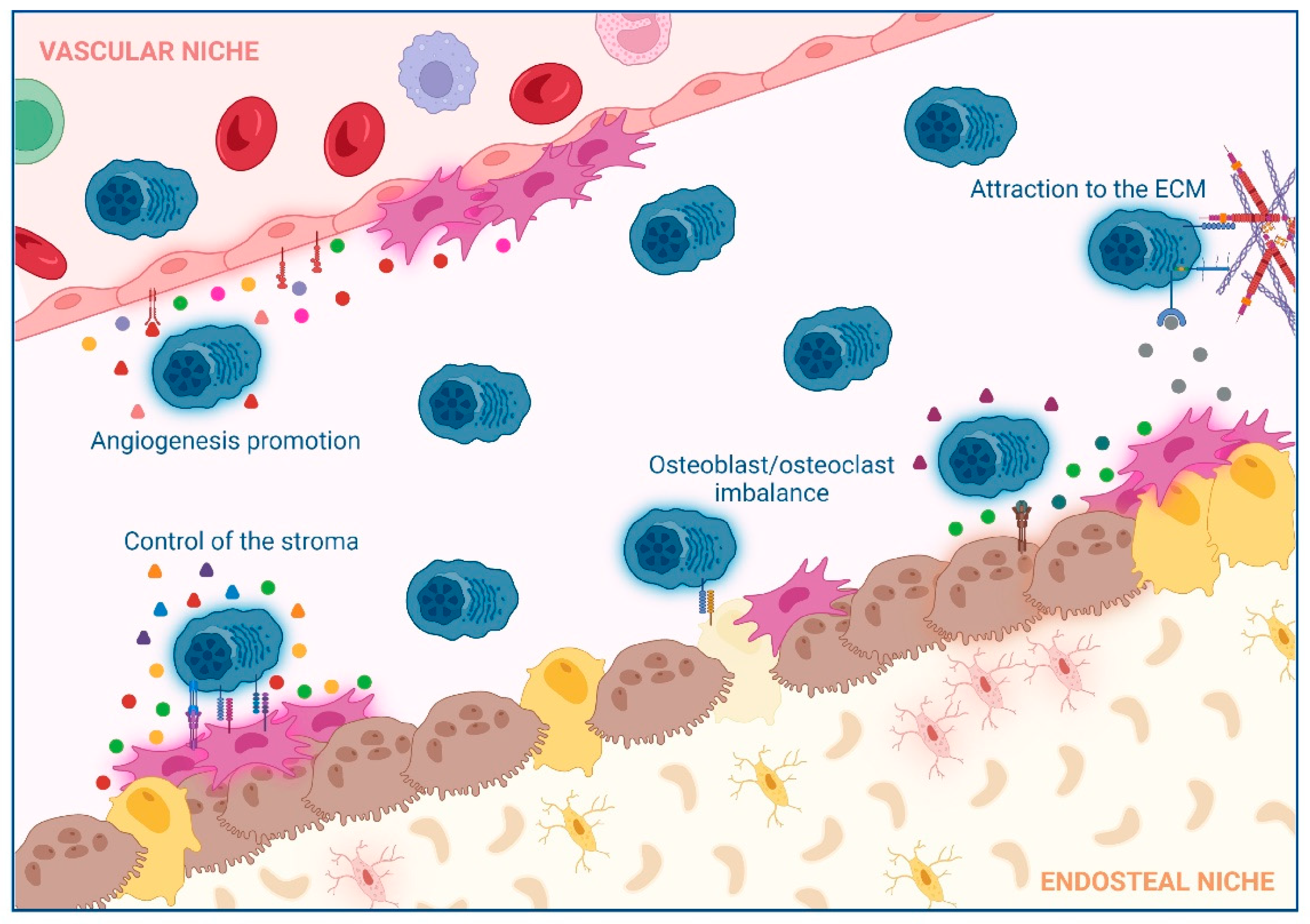
2. Impact of Interactions between Non-Hematological Cells and MM Cells in the BM
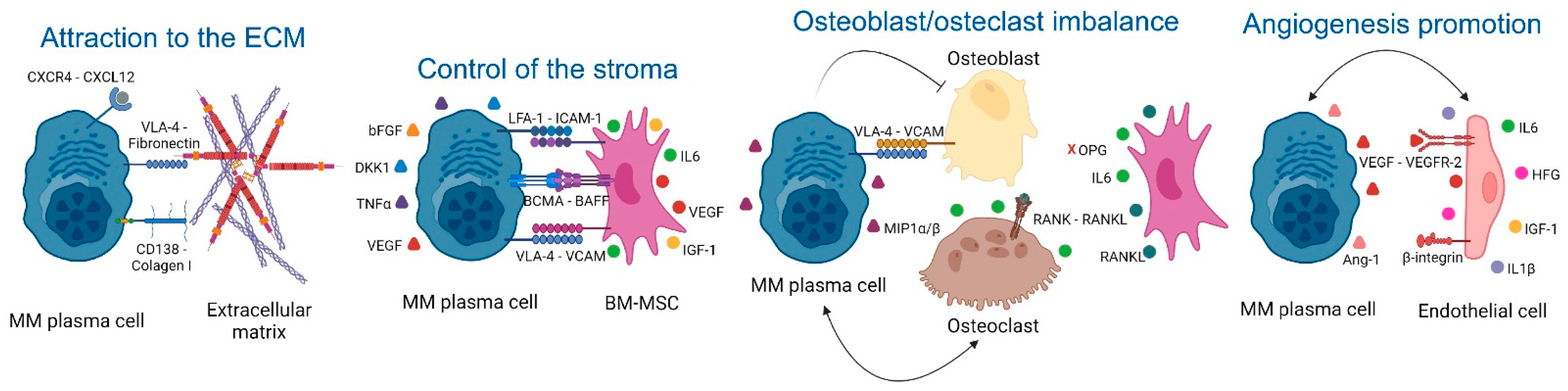
2.1. Extracellular Matrix (ECM)
2.2. Control of the Stroma by BM Mesenchymal Stromal Cells (BM-MSCs)
2.3. Osteoclast/Osteoblast Imbalance in the Endosteal Niche
2.4. Angiogenesis Promotion in the Vascular Niche
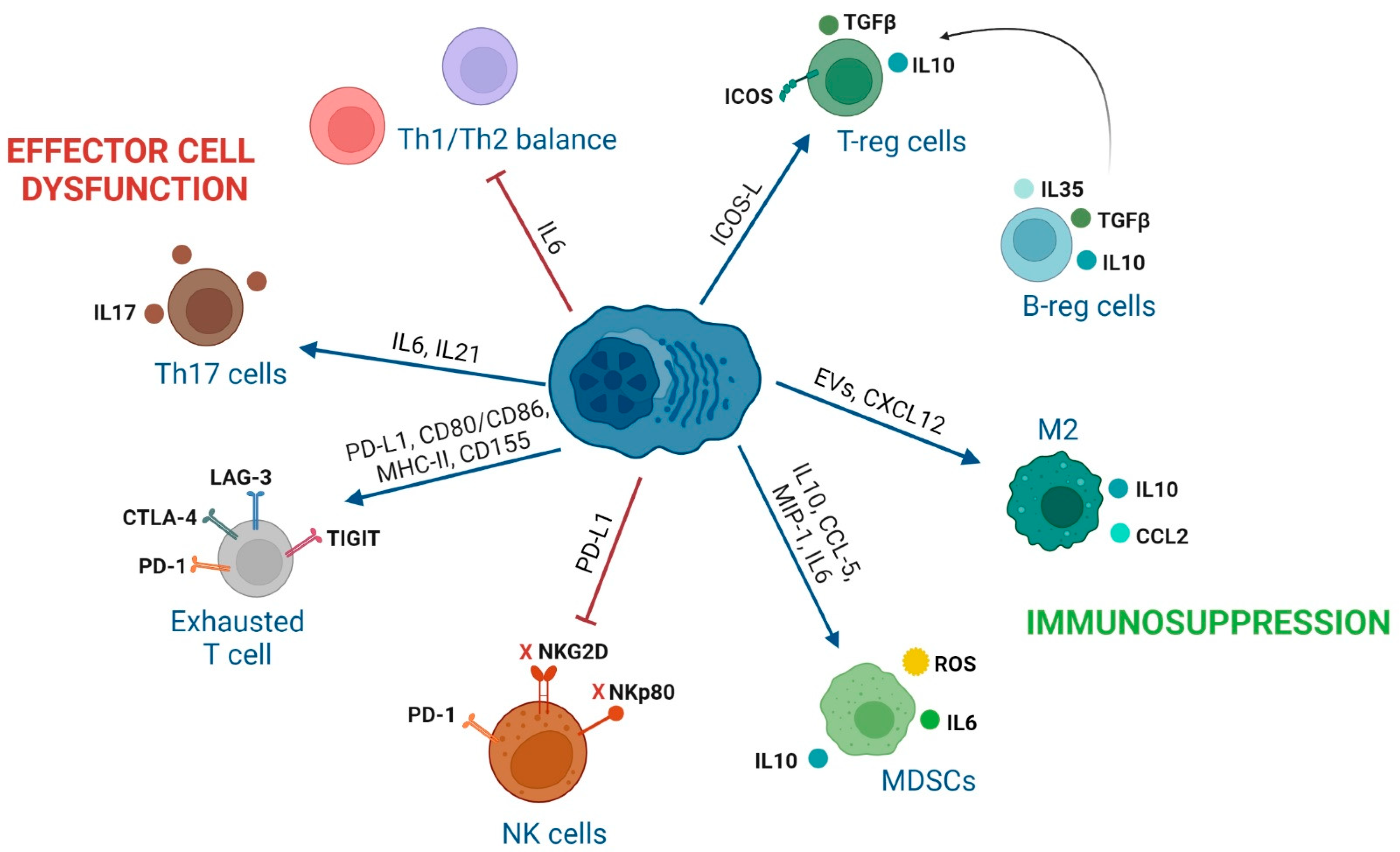
3. Impact of Interactions between Immune Cells and MM Cells in the BM of MM Patients
3.1. Effector CD8 T Lymphocytes
3.2. CD4 T Cell Subsets
3.3. The Impact of Age in T Lymphocytes in MM
3.4. NK Cells
3.5. Regulatory B Cells
3.6. Tumor-Associated-Macrophages (TAMs)
3.7. Myeloid-Derived Suppressor Cells
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Etxebeste-Mitxeltorena, M.; Del Rincón-Loza, I.; Martín-Antonio, B. Tumor Secretome to Adoptive Cellular Immunotherapy: Reduce Me Before I Make You My Partner. Front. Immunol. 2021, 12, 717850. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Ng, J.M.-K.; Wong, C.C.; Ng, E.K.W.; Yu, J. Molecular Alterations of Cancer Cell and Tumour Microenvironment in Metastatic Gastric Cancer. Oncogene 2018, 37, 4903–4920. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.-M.; Lin, W.-Y.; Shen, C.-C.; Pan, H.-C.; Keh-Bin, W.; Chen, Y.-C.; Jan, Y.-J.; Lai, D.-W.; Tang, S.-C.; Tien, H.-R.; et al. Melatonin Set out to ER Stress Signaling Thwarts Epithelial Mesenchymal Transition and Peritoneal Dissemination via Calpain-Mediated C/EBPβ and NFκB Cleavage. J. Pineal Res. 2016, 60, 142–154. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Xiao, D.; Li, G.; Ma, J.; Chen, P.; Yuan, W.; Hou, F.; Ge, J.; Zhong, M.; Tang, Y.; et al. EphA2 Promotes Epithelial-Mesenchymal Transition through the Wnt/β-Catenin Pathway in Gastric Cancer Cells. Oncogene 2014, 33, 2737–2747. [Google Scholar] [CrossRef] [Green Version]
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor Microenvironment Complexity and Therapeutic Implications at a Glance. Cell Commun. Signal. 2020, 18, 59. [Google Scholar] [CrossRef] [Green Version]
- Oliver, A.J.; Lau, P.K.H.; Unsworth, A.S.; Loi, S.; Darcy, P.K.; Kershaw, M.H.; Slaney, C.Y. Tissue-Dependent Tumor Microenvironments and Their Impact on Immunotherapy Responses. Front. Immunol. 2018, 9, 70. [Google Scholar] [CrossRef] [Green Version]
- Hirata, E.; Sahai, E. Tumor Microenvironment and Differential Responses to Therapy. Cold Spring Harb. Perspect Med. 2017, 7, a026781. [Google Scholar] [CrossRef] [Green Version]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The Tumor Microenvironment at a Glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef] [Green Version]
- Bagchi, S.; Yuan, R.; Engleman, E.G. Immune Checkpoint Inhibitors for the Treatment of Cancer: Clinical Impact and Mechanisms of Response and Resistance. Annu. Rev. Pathol. 2021, 16, 223–249. [Google Scholar] [CrossRef]
- Tavora, B.; Reynolds, L.E.; Batista, S.; Demircioglu, F.; Fernandez, I.; Lechertier, T.; Lees, D.M.; Wong, P.-P.; Alexopoulou, A.; Elia, G.; et al. Endothelial-Cell FAK Targeting Sensitizes Tumours to DNA-Damaging Therapy. Nature 2014, 514, 112–116. [Google Scholar] [CrossRef] [Green Version]
- Battram, A.M.; Bachiller, M.; Martín-Antonio, B. Senescence in the Development and Response to Cancer with Immunotherapy: A Double-Edged Sword. Int. J. Mol. Sci. 2020, 21, 4346. [Google Scholar] [CrossRef]
- Bent, E.H.; Gilbert, L.A.; Hemann, M.T. A Senescence Secretory Switch Mediated by PI3K/AKT/MTOR Activation Controls Chemoprotective Endothelial Secretory Responses. Genes Dev. 2016, 30, 1811–1821. [Google Scholar] [CrossRef] [Green Version]
- Jeong, S.K.; Kim, J.S.; Lee, C.G.; Park, Y.-S.; Kim, S.D.; Yoon, S.O.; Han, D.H.; Lee, K.Y.; Jeong, M.H.; Jo, W.S. Tumor Associated Macrophages Provide the Survival Resistance of Tumor Cells to Hypoxic Microenvironmental Condition through IL-6 Receptor-Mediated Signals. Immunobiology 2017, 222, 55–65. [Google Scholar] [CrossRef]
- Shree, T.; Olson, O.C.; Elie, B.T.; Kester, J.C.; Garfall, A.L.; Simpson, K.; Bell-McGuinn, K.M.; Zabor, E.C.; Brogi, E.; Joyce, J.A. Macrophages and Cathepsin Proteases Blunt Chemotherapeutic Response in Breast Cancer. Genes Dev. 2011, 25, 2465–2479. [Google Scholar] [CrossRef] [Green Version]
- Dijkgraaf, E.M.; Heusinkveld, M.; Tummers, B.; Vogelpoel, L.T.C.; Goedemans, R.; Jha, V.; Nortier, J.W.R.; Welters, M.J.P.; Kroep, J.R.; van der Burg, S.H. Chemotherapy Alters Monocyte Differentiation to Favor Generation of Cancer-Supporting M2 Macrophages in the Tumor Microenvironment. Cancer Res. 2013, 73, 2480–2492. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Tang, Y.; Tan, Y.; Wei, Q.; Yu, W. Cancer-Associated Fibroblasts in Radiotherapy: Challenges and New Opportunities. Cell Commun. Signal 2019, 17, 47. [Google Scholar] [CrossRef] [Green Version]
- Tommelein, J.; De Vlieghere, E.; Verset, L.; Melsens, E.; Leenders, J.; Descamps, B.; Debucquoy, A.; Vanhove, C.; Pauwels, P.; Gespach, C.P.; et al. Radiotherapy-Activated Cancer-Associated Fibroblasts Promote Tumor Progression through Paracrine IGF1R Activation. Cancer Res. 2018, 78, 659–670. [Google Scholar] [CrossRef] [Green Version]
- Wen, S.; Hou, Y.; Fu, L.; Xi, L.; Yang, D.; Zhao, M.; Qin, Y.; Sun, K.; Teng, Y.; Liu, M. Cancer-Associated Fibroblast (CAF)-Derived IL32 Promotes Breast Cancer Cell Invasion and Metastasis via Integrin Β3-P38 MAPK Signalling. Cancer Lett. 2019, 442, 320–332. [Google Scholar] [CrossRef]
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.-C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of Carcinoma-Associated Fibroblasts and Fibrosis Induces Immunosuppression and Accelerates Pancreas Cancer with Reduced Survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [Green Version]
- Bui, J.D.; Schreiber, R.D. Cancer Immunosurveillance, Immunoediting and Inflammation: Independent or Interdependent Processes? Curr. Opin. Immunol. 2007, 19, 203–208. [Google Scholar] [CrossRef]
- Gupta, A.; Probst, H.C.; Vuong, V.; Landshammer, A.; Muth, S.; Yagita, H.; Schwendener, R.; Pruschy, M.; Knuth, A.; van den Broek, M. Radiotherapy Promotes Tumor-Specific Effector CD8+ T Cells via Dendritic Cell Activation. J. Immunol. 2012, 189, 558–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Ortíz-Maldonado, V.; Rives, S.; Castellà, M.; Alonso-Saladrigues, A.; Benítez-Ribas, D.; Caballero-Baños, M.; Baumann, T.; Cid, J.; Garcia-Rey, E.; Llanos, C.; et al. CART19-BE-01: A Multicenter Trial of ARI-0001 Cell Therapy in Patients with CD19+ Relapsed/Refractory Malignancies. Mol. Ther. 2021, 29, 636–644. [Google Scholar] [CrossRef] [PubMed]
- Perez-Amill, L.; Suñe, G.; Antoñana-Vildosola, A.; Castella, M.; Najjar, A.; Bonet, J.; Fernández-Fuentes, N.; Inogés, S.; López, A.; Bueno, C.; et al. Preclinical Development of a Humanized Chimeric Antigen Receptor against B Cell Maturation Antigen for Multiple Myeloma. Haematologica 2021, 106, 173–184. [Google Scholar] [CrossRef] [Green Version]
- Castella, M.; Fernández de Larrea, C.; Martín-Antonio, B. Immunotherapy: A Novel Era of Promising Treatments for Multiple Myeloma. Int. J. Mol. Sci. 2018, 19, 3613. [Google Scholar] [CrossRef] [Green Version]
- Martinez, M.; Moon, E.K. CAR T Cells for Solid Tumors: New Strategies for Finding, Infiltrating, and Surviving in the Tumor Microenvironment. Front. Immunol. 2019, 10, 128. [Google Scholar] [CrossRef] [Green Version]
- Raje, N.; Berdeja, J.; Lin, Y.; Siegel, D.; Jagannath, S.; Madduri, D.; Liedtke, M.; Rosenblatt, J.; Maus, M.V.; Turka, A.; et al. Anti-BCMA CAR T-Cell Therapy Bb2121 in Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2019, 380, 1726–1737. [Google Scholar] [CrossRef]
- García-Ortiz, A.; Rodríguez-García, Y.; Encinas, J.; Maroto-Martín, E.; Castellano, E.; Teixidó, J.; Martínez-López, J. The Role of Tumor Microenvironment in Multiple Myeloma Development and Progression. Cancers 2021, 13, 217. [Google Scholar] [CrossRef]
- Kumar, S.K.; Rajkumar, V.; Kyle, R.A.; van Duin, M.; Sonneveld, P.; Mateos, M.-V.; Gay, F.; Anderson, K.C. Multiple Myeloma. Nat. Rev. Dis. Primers. 2017, 3, 17046. [Google Scholar] [CrossRef]
- Bone Marrow Niches in Haematological Malignancies | Nature Reviews Cancer. Available online: https://www.nature.com/articles/s41568-020-0245-2 (accessed on 20 June 2022).
- Glavey, S.V.; Naba, A.; Manier, S.; Clauser, K.; Tahri, S.; Park, J.; Reagan, M.R.; Moschetta, M.; Mishima, Y.; Gambella, M.; et al. Proteomic Characterization of Human Multiple Myeloma Bone Marrow Extracellular Matrix. Leukemia 2017, 31, 2426–2434. [Google Scholar] [CrossRef]
- Landowski, T.H.; Olashaw, N.E.; Agrawal, D.; Dalton, W.S. Cell Adhesion-Mediated Drug Resistance (CAM-DR) Is Associated with Activation of NF-ΚB (RelB/P50) in Myeloma Cells. Oncogene 2003, 22, 2417–2421. [Google Scholar] [CrossRef] [Green Version]
- Targeting the Bone Marrow Microenvironment in Multiple Myeloma-Kawano-2015-Immunological Review-Wiley Online Library. Available online: https://onlinelibrary.wiley.com/doi/10.1111/imr.12233 (accessed on 23 May 2022).
- Kouroukis, T.C.; Baldassarre, F.G.; Haynes, A.E.; Imrie, K.; Reece, D.E.; Cheung, M.C. Bortezomib in Multiple Myeloma: Systematic Review and Clinical Considerations. Curr. Oncol. 2014, 21, 573–603. [Google Scholar] [CrossRef] [Green Version]
- Ghobrial, I.M.; Liu, C.-J.; Zavidij, O.; Azab, A.K.; Baz, R.; Laubach, J.P.; Mishima, Y.; Armand, P.; Munshi, N.C.; Basile, F.; et al. Phase I/II Trial of the CXCR4 Inhibitor Plerixafor in Combination with Bortezomib as a Chemosensitization Strategy in Relapsed/Refractory Multiple Myeloma. Am. J. Hematol. 2019, 94, 1244–1253. [Google Scholar] [CrossRef]
- Ghobrial, I.M. Myeloma as a Model for the Process of Metastasis: Implications for Therapy. Blood 2012, 120, 20–30. [Google Scholar] [CrossRef] [Green Version]
- Méndez-Ferrer, S.; Michurina, T.V.; Ferraro, F.; Mazloom, A.R.; MacArthur, B.D.; Lira, S.A.; Scadden, D.T.; Ma’ayan, A.; Enikolopov, G.N.; Frenette, P.S. Mesenchymal and Haematopoietic Stem Cells Form a Unique Bone Marrow Niche. Nature 2010, 466, 829–834. [Google Scholar] [CrossRef]
- Dazzi, F.; Ramasamy, R.; Glennie, S.; Jones, S.P.; Roberts, I. The Role of Mesenchymal Stem Cells in Haemopoiesis. Blood Rev. 2006, 20, 161–171. [Google Scholar] [CrossRef]
- Hideshima, T.; Mitsiades, C.; Tonon, G.; Richardson, P.G.; Anderson, K.C. Understanding Multiple Myeloma Pathogenesis in the Bone Marrow to Identify New Therapeutic Targets. Nat. Rev. Cancer 2007, 7, 585–598. [Google Scholar] [CrossRef]
- Asosingh, K.; Vankerkhove, V.; Riet, I.V.; Camp, B.V.; Vanderkerken, K. Selective in Vivo Growth of Lymphocyte Function- Associated Antigen-1–Positive Murine Myeloma Cells: Involvement of Function-Associated Antigen-1–Mediated Homotypic Cell-Cell Adhesion. Exp. Hematol. 2003, 31, 48–55. [Google Scholar] [CrossRef]
- Schmidmaier, R.; Mandl-Weber, S.; Gaul, L.; Baumann, P.; Bumeder, I.; Straka, C.; Emmerich, B. Inhibition of Lymphocyte Function Associated Antigen 1 by LFA878 Induces Apoptosis in Multiple Myeloma Cells and Is Associated with Downregulation of the Focal Adhesion Kinase/Phosphatidylinositol 3 Kinase/Akt Pathway. Int. J. Oncol. 2007, 31, 969–976. [Google Scholar] [CrossRef] [Green Version]
- Harmer, D.; Falank, C.; Reagan, M.R. Interleukin-6 Interweaves the Bone Marrow Microenvironment, Bone Loss, and Multiple Myeloma. Front. Endocrinol. 2019, 9, 788. [Google Scholar] [CrossRef] [Green Version]
- Hideshima, T.; Bergsagel, P.L.; Kuehl, W.M.; Anderson, K.C. Advances in Biology of Multiple Myeloma: Clinical Applications. Blood 2004, 104, 607–618. [Google Scholar] [CrossRef] [Green Version]
- Orlowski, R.Z.; Gercheva, L.; Williams, C.; Sutherland, H.; Robak, T.; Masszi, T.; Goranova-Marinova, V.; Dimopoulos, M.A.; Cavenagh, J.D.; Špička, I.; et al. A Phase 2, Randomized, Double-Blind, Placebo-Controlled Study of Siltuximab (Anti-IL-6 MAb) and Bortezomib versus Bortezomib Alone in Patients with Relapsed or Refractory Multiple Myeloma. Am. J. Hematol. 2015, 90, 42–49. [Google Scholar] [CrossRef] [Green Version]
- Matsuyama, Y.; Nagashima, T.; Honne, K.; Kamata, Y.; Iwamoto, M.; Okazaki, H.; Sato, K.; Ozawa, K.; Minota, S. Successful Treatment of a Patient with Rheumatoid Arthritis and IgA-κ Multiple Myeloma with Tocilizumab. Intern. Med. 2011, 50, 639–642. [Google Scholar] [CrossRef] [Green Version]
- Corre, J.; Mahtouk, K.; Attal, M.; Gadelorge, M.; Huynh, A.; Fleury-Cappellesso, S.; Danho, C.; Laharrague, P.; Klein, B.; Rème, T.; et al. Bone Marrow Mesenchymal Stem Cells Are Abnormal in Multiple Myeloma. Leukemia 2007, 21, 1079–1088. [Google Scholar] [CrossRef] [PubMed]
- Zdzisińska, B.; Bojarska-Junak, A.; Dmoszyńska, A.; Kandefer-Szerszeń, M. Abnormal Cytokine Production by Bone Marrow Stromal Cells of Multiple Myeloma Patients in Response to RPMI8226 Myeloma Cells. Arch. Immunol. Ther. Exp. 2008, 56, 207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, M.; Yuan, H.; Peng, H.; Liu, S.; Xiao, X.; Wang, Z.; Zhang, G.; Xiao, H. Mesenchymal Stem Cells Inhibit the Effects of Dexamethasone in Multiple Myeloma Cells. Stem Cells Int. 2022, 2022, e4855517. [Google Scholar] [CrossRef] [PubMed]
- BAFF and APRIL Protect Myeloma Cells from Apoptosis Induced by Interleukin 6 Deprivation and Dexamethasone | Blood | American Society of Hematology. Available online: https://ashpublications.org/blood/article/103/8/3148/18054/BAFF-and-APRIL-protect-myeloma-cells-from (accessed on 23 May 2022).
- Expression of BCMA, TACI, and BAFF-R in Multiple Myeloma: A Mechanism for Growth and Survival | Blood | American Society of Hematology. Available online: https://ashpublications.org/blood/article/103/2/689/17829/Expression-of-BCMA-TACI-and-BAFF-R-in-multiple (accessed on 23 May 2022).
- APRIL and BCMA Promote Human Multiple Myeloma Growth and Immunosuppression in the Bone Marrow Microenvironment | Blood | American Society of Hematology. Available online: https://ashpublications.org/blood/article/127/25/3225/35206/APRIL-and-BCMA-promote-human-multiple-myeloma (accessed on 23 May 2022).
- Lee, L.; Draper, B.; Chaplin, N.; Philip, B.; Chin, M.; Galas-Filipowicz, D.; Onuoha, S.; Thomas, S.; Baldan, V.; Bughda, R.; et al. An APRIL-Based Chimeric Antigen Receptor for Dual Targeting of BCMA and TACI in Multiple Myeloma. Blood 2018, 131, 746–758. [Google Scholar] [CrossRef] [PubMed]
- Holthof, L.C.; van der Schans, J.J.; Katsarou, A.; Poels, R.; Gelderloos, A.T.; Drent, E.; van Hal-van Veen, S.E.; Li, F.; Zweegman, S.; van de Donk, N.W.C.J.; et al. Bone Marrow Mesenchymal Stromal Cells Can Render Multiple Myeloma Cells Resistant to Cytotoxic Machinery of CAR T Cells through Inhibition of Apoptosis. Clin. Cancer Res. 2021, 27, 3793–3803. [Google Scholar] [CrossRef]
- The Role of Tumor Necrosis Factor α in the Pathophysiology of Human Multiple Myeloma: Therapeutic Applications | Oncogene. Available online: https://www.nature.com/articles/1204623 (accessed on 23 May 2022).
- Tsubaki, M.; Komai, M.; Itoh, T.; Imano, M.; Sakamoto, K.; Shimaoka, H.; Ogawa, N.; Mashimo, K.; Fujiwara, D.; Takeda, T.; et al. Inhibition of the Tumour Necrosis Factor-Alpha Autocrine Loop Enhances the Sensitivity of Multiple Myeloma Cells to Anticancer Drugs. Eur. J. Cancer 2013, 49, 3708–3717. [Google Scholar] [CrossRef]
- Calip, G.S.; Lee, W.-J.; Lee, T.A.; Schumock, G.T.; Chiu, B.C.-H. Tumor Necrosis Factor-Alpha Inhibitor Medications for Inflammatory Conditions and Incidence of Multiple Myeloma. Blood 2015, 126, 2954. [Google Scholar] [CrossRef]
- Sebba, A. Tocilizumab: The First Interleukin-6-Receptor Inhibitor. Am. J. Health Syst. Pharm. 2008, 65, 1413–1418. [Google Scholar] [CrossRef]
- Iyer, S.P.; Beck, J.T.; Stewart, A.K.; Shah, J.; Kelly, K.R.; Isaacs, R.; Bilic, S.; Sen, S.; Munshi, N.C. A Phase IB Multicentre Dose-Determination Study of BHQ880 in Combination with Anti-Myeloma Therapy and Zoledronic Acid in Patients with Relapsed or Refractory Multiple Myeloma and Prior Skeletal-Related Events. Br. J. Haematol. 2014, 167, 366–375. [Google Scholar] [CrossRef]
- Raje, N.S.; Moreau, P.; Terpos, E.; Benboubker, L.; Grząśko, N.; Holstein, S.A.; Oriol, A.; Huang, S.-Y.; Beksac, M.; Kuliczkowski, K.; et al. Phase 2 Study of Tabalumab, a Human Anti-B-Cell Activating Factor Antibody, with Bortezomib and Dexamethasone in Patients with Previously Treated Multiple Myeloma. Br. J. Haematol. 2017, 176, 783–795. [Google Scholar] [CrossRef]
- Roodman, G.D. Osteoblast Function in Myeloma. Bone 2011, 48, 135–140. [Google Scholar] [CrossRef]
- Gau, Y.-C.; Yeh, T.-J.; Hsu, C.-M.; Hsiao, S.Y.; Hsiao, H.-H. Pathogenesis and Treatment of Myeloma-Related Bone Disease. Int. J. Mol. Sci. 2022, 23, 3112. [Google Scholar] [CrossRef]
- Valentin-Opran, A.; Charhon, S.A.; Meunier, P.J.; Edouard, C.M.; Arlot, M.E. Quantitative Histology of Myeloma-Induced Bone Changes. Br. J. Haematol. 1982, 52, 601–610. [Google Scholar] [CrossRef]
- Choi, S.J.; Oba, Y.; Gazitt, Y.; Alsina, M.; Cruz, J.; Anderson, J.; Roodman, G.D. Antisense Inhibition of Macrophage Inflammatory Protein 1-α Blocks Bone Destruction in a Model of Myeloma Bone Disease. J. Clin. Investig. 2001, 108, 1833–1841. [Google Scholar] [CrossRef]
- Abe, M.; Hiura, K.; Wilde, J.; Moriyama, K.; Hashimoto, T.; Ozaki, S.; Wakatsuki, S.; Kosaka, M.; Kido, S.; Inoue, D.; et al. Role for Macrophage Inflammatory Protein (MIP)-1alpha and MIP-1beta in the Development of Osteolytic Lesions in Multiple Myeloma. Blood 2002, 100, 2195–2202. [Google Scholar] [CrossRef]
- Macrophage Inflammatory Protein-1α Is an Osteoclastogenic Factor in Myeloma That Is Independent of Receptor Activator of Nuclear Factor ΚB Ligand | Blood | American Society of Hematology. Available online: https://ashpublications.org/blood/article/97/11/3349/107426/Macrophage-inflammatory-protein-1-is-an (accessed on 23 May 2022).
- Notch-Directed Microenvironment Reprogramming in Myeloma: A Single Path to Multiple Outcomes | Leukemia. Available online: https://www.nature.com/articles/leu20136 (accessed on 23 May 2022).
- Boyce, B.F.; Xing, L. Functions of RANKL/RANK/OPG in Bone Modeling and Remodeling. Arch. Biochem. Biophys. 2008, 473, 139–146. [Google Scholar] [CrossRef] [Green Version]
- Michigami, T.; Shimizu, N.; Williams, P.J.; Niewolna, M.; Dallas, S.L.; Mundy, G.R.; Yoneda, T. Cell-Cell Contact between Marrow Stromal Cells and Myeloma Cells via VCAM-1 and Alpha (4) Beta (1)-Integrin Enhances Production of Osteoclast-Stimulating Activity. Blood 2000, 96, 1953–1960. [Google Scholar] [CrossRef]
- Terpos, E.; Raje, N.; Croucher, P.; Garcia-Sanz, R.; Leleu, X.; Pasteiner, W.; Wang, Y.; Glennane, A.; Canon, J.; Pawlyn, C. Denosumab Compared with Zoledronic Acid on PFS in Multiple Myeloma: Exploratory Results of an International Phase 3 Study. Blood Adv. 2021, 5, 725–736. [Google Scholar] [CrossRef]
- Huang, S.-Y.; Yoon, S.-S.; Shimizu, K.; Chng, W.J.; Chang, C.-S.; Wong, R.S.-M.; Gao, S.; Wang, Y.; Gordon, S.W.; Glennane, A.; et al. Denosumab Versus Zoledronic Acid in Bone Disease Treatment of Newly Diagnosed Multiple Myeloma: An International, Double-Blind, Randomized Controlled Phase 3 Study—Asian Subgroup Analysis. Adv. Ther. 2020, 37, 3404–3416. [Google Scholar] [CrossRef]
- Hanley, D.A.; Adachi, J.D.; Bell, A.; Brown, V. Denosumab: Mechanism of Action and Clinical Outcomes. Int. J. Clin. Pract. 2012, 66, 1139–1146. [Google Scholar] [CrossRef] [Green Version]
- Myeloma Cells Block RUNX2/CBFA1 Activity in Human Bone Marrow Osteoblast Progenitors and Inhibit Osteoblast Formation and Differentiation | Blood | American Society of Hematology. Available online: https://ashpublications.org/blood/article/106/7/2472/21687/Myeloma-cells-block-RUNX2-CBFA1-activity-in-human (accessed on 23 May 2022).
- Osteoprotegerin Is Bound, Internalized, and Degraded by Multiple Myeloma Cells | Blood | American Society of Hematology. Available online: https://ashpublications.org/blood/article/100/8/3002/106454/Osteoprotegerin-is-bound-internalized-and-degraded (accessed on 23 May 2022).
- Mori, Y.; Shimizu, N.; Dallas, M.; Niewolna, M.; Story, B.; Williams, P.J.; Mundy, G.R.; Yoneda, T. Anti-A4 Integrin Antibody Suppresses the Development of Multiple Myeloma and Associated Osteoclastic Osteolysis. Blood 2004, 104, 2149–2154. [Google Scholar] [CrossRef]
- Padmanabhan, S.; Beck, J.T.; Kelly, K.R.; Munshi, N.C.; Dzik-Jurasz, A.; Gangolli, E.; Ettenberg, S.; Miner, K.; Bilic, S.; Whyte, W.; et al. A Phase I/II Study of BHQ880, a Novel Osteoblat Activating, Anti-DKK1 Human Monoclonal Antibody, in Relapsed and Refractory Multiple Myeloma (MM) Patients Treated with Zoledronic Acid (Zol) and Anti-Myeloma Therapy (MM Tx). Blood 2009, 114, 750. [Google Scholar] [CrossRef]
- Vacca, A.; Ria, R.; Reale, A.; Ribatti, D. Angiogenesis in Multiple Myeloma. Angiogenes. Lymphangiogenes. Clin. Implic. 2014, 99, 180–196. [Google Scholar] [CrossRef]
- Ribatti, D.; Nico, B.; Vacca, A. Multiple Myeloma as a Model for the Role of Bone Marrow Niches in the Control of Angiogenesis. Int. Rev. Cell Mol. Biol. 2015, 314, 259–282. [Google Scholar] [CrossRef]
- Mondello, P.; Cuzzocrea, S.; Navarra, M.; Mian, M. Bone Marrow Micro-Environment Is a Crucial Player for Myelomagenesis and Disease Progression. Oncotarget 2017, 8, 20394–20409. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Rajkumar, S.V.; Mesa, R.A.; Fonseca, R.; Schroeder, G.; Plevak, M.F.; Dispenzieri, A.; Lacy, M.Q.; Lust, J.A.; Witzig, T.E.; Gertz, M.A.; et al. Bone Marrow Angiogenesis in 400 Patients with Monoclonal Gammopathy of Undetermined Significance, Multiple Myeloma, and Primary Amyloidosis. Clin. Cancer Res. 2002, 8, 2210–2216. [Google Scholar]
- Bhaskar, A.; Tiwary, B.N. Hypoxia Inducible Factor-1 Alpha and Multiple Myeloma. Int. J. Adv. Res. 2016, 4, 706–715. [Google Scholar]
- Angiogenic Switch during 5T2MM Murine Myeloma Tumorigenesis: Role of CD45 Heterogeneity | Blood | American Society of Hematology. Available online: https://ashpublications.org/blood/article/103/8/3131/18020/Angiogenic-switch-during-5T2MM-murine-myeloma (accessed on 24 May 2022).
- Ria, R.; Todoerti, K.; Berardi, S.; Coluccia, A.M.L.; De Luisi, A.; Mattioli, M.; Ronchetti, D.; Morabito, F.; Guarini, A.; Petrucci, M.T.; et al. Gene Expression Profiling of Bone Marrow Endothelial Cells in Patients with Multiple Myeloma. Clin. Cancer Res. 2009, 15, 5369–5378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solimando, A.G.; Summa, S.D.; Vacca, A.; Ribatti, D. Cancer-Associated Angiogenesis: The Endothelial Cell as a Checkpoint for Immunological Patrolling. Cancers 2020, 12, 3380. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Vacca, A.; Nico, B.; Roncali, L.; Dammacco, F. Postnatal Vasculogenesis. Mech. Dev. 2001, 100, 157–163. [Google Scholar] [CrossRef]
- Moschetta, M.; Mishima, Y.; Kawano, Y.; Manier, S.; Paiva, B.; Palomera, L.; Aljawai, Y.; Calcinotto, A.; Unitt, C.; Sahin, I.; et al. Targeting Vasculogenesis to Prevent Progression in Multiple Myeloma. Leukemia 2016, 30, 1103–1115. [Google Scholar] [CrossRef] [PubMed]
- Tenreiro, M.M.; Correia, M.L.; Brito, M.A. Endothelial Progenitor Cells in Multiple Myeloma Neovascularization: A Brick to the Wall. Angiogenesis 2017, 20, 443–462. [Google Scholar] [CrossRef]
- Reale, A.; Melaccio, A.; Lamanuzzi, A.; Saltarella, I.; Dammacco, F.; Vacca, A.; Ria, R. Functional and Biological Role of Endothelial Precursor Cells in Tumour Progression: A New Potential Therapeutic Target in Haematological Malignancies. Stem. Cells Int. 2015, 2016, e7954580. [Google Scholar] [CrossRef] [Green Version]
- Sweeney, M.; Foldes, G. It Takes Two: Endothelial-Perivascular Cell Cross-Talk in Vascular Development and Disease. Front. Cardiovasc. Med. 2018, 5, 154. [Google Scholar] [CrossRef]
- Dankbar, B.; Padró, T.; Leo, R.; Feldmann, B.; Kropff, M.; Mesters, R.M.; Serve, H.; Berdel, W.E.; Kienast, J. Vascular Endothelial Growth Factor and Interleukin-6 in Paracrine Tumor-Stromal Cell Interactions in Multiple Myeloma. Blood 2000, 95, 2630–2636. [Google Scholar] [CrossRef]
- Ria, R.; Melaccio, A.; Racanelli, V.; Vacca, A. Anti-VEGF Drugs in the Treatment of Multiple Myeloma Patients. J. Clin. Med. 2020, 9, 1765. [Google Scholar] [CrossRef]
- Somlo, G.; Lashkari, A.; Bellamy, W.; Zimmerman, T.M.; Tuscano, J.M.; O’Donnell, M.R.; Mohrbacher, A.F.; Forman, S.J.; Frankel, P.; Chen, H.X.; et al. Phase II Randomized Trial of Bevacizumab versus Bevacizumab and Thalidomide for Relapsed/Refractory Multiple Myeloma: A California Cancer Consortium Trial. Br. J. Haematol. 2011, 154, 533–535. [Google Scholar] [CrossRef] [Green Version]
- White, D.; Kassim, A.; Bhaskar, B.; Yi, J.; Wamstad, K.; Paton, V.E. Results from AMBER, a Randomized Phase 2 Study of Bevacizumab and Bortezomib versus Bortezomib in Relapsed or Refractory Multiple Myeloma. Cancer 2013, 119, 339–347. [Google Scholar] [CrossRef]
- Yordanova, A.; Hose, D.; Neben, K.; Witzens-Harig, M.; Gütgemann, I.; Raab, M.-S.; Moehler, T.; Goldschmidt, H.; Schmidt-Wolf, I.G. Sorafenib in Patients with Refractory or Recurrent Multiple Myeloma. Hematol. Oncol. 2013, 31, 197–200. [Google Scholar] [CrossRef]
- Srkalovic, G.; Hussein, M.A.; Hoering, A.; Zonder, J.A.; Popplewell, L.L.; Trivedi, H.; Mazzoni, S.; Sexton, R.; Orlowski, R.Z.; Barlogie, B. A Phase II Trial of BAY 43-9006 (Sorafenib) (NSC-724772) in Patients with Relapsing and Resistant Multiple Myeloma: SWOG S0434. Cancer Med. 2014, 3, 1275–1283. [Google Scholar] [CrossRef]
- Kumar, S.; Witzig, T.E.; Dispenzieri, A.; Lacy, M.Q.; Wellik, L.E.; Fonseca, R.; Lust, J.A.; Gertz, M.A.; Kyle, R.A.; Greipp, P.R.; et al. Effect of Thalidomide Therapy on Bone Marrow Angiogenesis in Multiple Myeloma. Leukemia 2004, 18, 624–627. [Google Scholar] [CrossRef] [Green Version]
- Terpos, E.; Katodritou, E.; Symeonidis, A.; Zagouri, F.; Gerofotis, A.; Christopoulou, G.; Gavriatopoulou, M.; Christoulas, D.; Ntanasis-Stathopoulos, I.; Kourakli, A.; et al. Effect of Induction Therapy with Lenalidomide, Doxorubicin and Dexamethasone on Bone Remodeling and Angiogenesis in Newly Diagnosed Multiple Myeloma. Int. J. Cancer 2019, 145, 559–568. [Google Scholar] [CrossRef]
- Deng, M.; Yuan, H.; Liu, S.; Hu, Z.; Xiao, H. Exosome-Transmitted LINC00461 Promotes Multiple Myeloma Cell Proliferation and Suppresses Apoptosis by Modulating MicroRNA/BCL-2 Expression. Cytotherapy 2019, 21, 96–106. [Google Scholar] [CrossRef]
- Scavelli, C.; Di Pietro, G.; Cirulli, T.; Coluccia, M.; Boccarelli, A.; Giannini, T.; Mangialardi, G.; Bertieri, R.; Coluccia, A.M.L.; Ribatti, D.; et al. Zoledronic Acid Affects Over-Angiogenic Phenotype of Endothelial Cells in Patients with Multiple Myeloma. Mol. Cancer Ther. 2007, 6, 3256–3262. [Google Scholar] [CrossRef] [Green Version]
- Guillerey, C.; Harjunpää, H.; Carrié, N.; Kassem, S.; Teo, T.; Miles, K.; Krumeich, S.; Weulersse, M.; Cuisinier, M.; Stannard, K.; et al. TIGIT Immune Checkpoint Blockade Restores CD8+ T-Cell Immunity against Multiple Myeloma. Blood 2018, 132, 1689–1694. [Google Scholar] [CrossRef] [Green Version]
- Noonan, K.A.; Huff, C.A.; Davis, J.; Lemas, M.V.; Fiorino, S.; Bitzan, J.; Ferguson, A.; Emerling, A.; Luznik, L.; Matsui, W.; et al. Adoptive Transfer of Activated Marrow-Infiltrating Lymphocytes Induces Measurable Antitumor Immunity in the Bone Marrow in Multiple Myeloma. Sci. Transl. Med. 2015, 7, 288ra78. [Google Scholar] [CrossRef] [Green Version]
- Teijeira, A.; Garasa, S.; Etxeberria, I.; Gato-Cañas, M.; Melero, I.; Delgoffe, G.M. Metabolic Consequences of T-Cell Costimulation in Anticancer Immunity. Cancer Immunol. Res. 2019, 7, 1564–1569. [Google Scholar] [CrossRef]
- Wherry, E.J. T Cell Exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef]
- Arai, Y.; Choi, U.; Corsino, C.I.; Koontz, S.M.; Tajima, M.; Sweeney, C.L.; Black, M.A.; Feldman, S.A.; Dinauer, M.C.; Malech, H.L. Enhanced Expression of CXCR4 Facilitates C-Kit-Targeted CAR-T Cell Trafficking to Bone Marrow and Enables Donor Stem Cell Engraftment. Biol. Blood Marrow Transplant. 2018, 24, S311. [Google Scholar] [CrossRef]
- Alsina, M.; Shah, N.; Raje, N.S.; Jagannath, S.; Madduri, D.; Kaufman, J.L.; Berdeja, J.G. Updated Results from the Phase I CRB-402 Study of Anti-Bcma CAR-T Cell Therapy Bb21217 in Patients with Relapsed and Refractory Multiple Myeloma: Correlation of Expansion and Duration of Response with T Cell Phenotypes. Blood 2020, 136, 25–26. [Google Scholar] [CrossRef]
- Le Calvez, B.; Moreau, P.; Touzeau, C. Immune Checkpoint Inhibitors for the Treatment of Myeloma: Novel Investigational Options. Expert Opin. Investig. Drugs 2021, 30, 965–973. [Google Scholar] [CrossRef]
- Ogawara, H.; Handa, H.; Yamazaki, T.; Toda, T.; Yoshida, K.; Nishimoto, N.; Al-ma’Quol, W.H.S.; Kaneko, Y.; Matsushima, T.; Tsukamoto, N.; et al. High Th1/Th2 Ratio in Patients with Multiple Myeloma. Leuk. Res. 2005, 29, 135–140. [Google Scholar] [CrossRef]
- Frassanito, M.A.; Cusmai, A.; Dammacco, F. Deregulated Cytokine Network and Defective Th1 Immune Response in Multiple Myeloma. Clin. Exp. Immunol. 2001, 125, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Verkleij, C.P.M.; Broekmans, M.E.C.; van Duin, M.; Frerichs, K.A.; Kuiper, R.; de Jonge, A.V.; Kaiser, M.; Morgan, G.; Axel, A.; Boominathan, R.; et al. Preclinical Activity and Determinants of Response of the GPRC5DxCD3 Bispecific Antibody Talquetamab in Multiple Myeloma. Blood Adv. 2021, 5, 2196–2215. [Google Scholar] [CrossRef] [PubMed]
- Green, D.J.; Pont, M.; Sather, B.D.; Cowan, A.J.; Turtle, C.J.; Till, B.G.; Nagengast, A.M.; Libby, E.N., III; Becker, P.S.; Coffey, D.G.; et al. Fully Human Bcma Targeted Chimeric Antigen Receptor T Cells Administered in a Defined Composition Demonstrate Potency at Low Doses in Advanced Stage High Risk Multiple Myeloma. Blood 2018, 132, 1011. [Google Scholar] [CrossRef]
- Garfall, A.L.; Dancy, E.K.; Cohen, A.D.; Hwang, W.-T.; Fraietta, J.A.; Davis, M.M.; Levine, B.L.; Siegel, D.L.; Stadtmauer, E.A.; Vogl, D.T.; et al. T-Cell Phenotypes Associated with Effective CAR T-Cell Therapy in Postinduction vs Relapsed Multiple Myeloma. Blood Adv. 2019, 3, 2812–2815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannopoulos, K.; Kaminska, W.; Hus, I.; Dmoszynska, A. The Frequency of T Regulatory Cells Modulates the Survival of Multiple Myeloma Patients: Detailed Characterisation of Immune Status in Multiple Myeloma. Br. J. Cancer 2012, 106, 546–552. [Google Scholar] [CrossRef]
- Alrasheed, N.; Lee, L.; Ghorani, E.; Henry, J.Y.; Conde, L.; Chin, M.; Galas-Filipowicz, D.; Furness, A.J.S.; Chavda, S.J.; Richards, H.; et al. Marrow-Infiltrating Regulatory T Cells Correlate with the Presence of Dysfunctional CD4+PD-1+ Cells and Inferior Survival in Patients with Newly Diagnosed Multiple Myeloma. Clin. Cancer Res. 2020, 26, 3443–3454. [Google Scholar] [CrossRef] [Green Version]
- Feyler, S.; Scott, G.B.; Parrish, C.; Jarmin, S.; Evans, P.; Short, M.; McKinley, K.; Selby, P.J.; Cook, G. Tumour Cell Generation of Inducible Regulatory T-Cells in Multiple Myeloma Is Contact-Dependent and Antigen-Presenting Cell-Independent. PLoS ONE 2012, 7, e35981. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, Y.; Nishikawa, H. Roles of Regulatory T Cells in Cancer Immunity. Int. Immunol. 2016, 28, 401–409. [Google Scholar] [CrossRef] [Green Version]
- Braga, W.M.T.; da Silva, B.R.; de Carvalho, A.C.; Maekawa, Y.H.; Bortoluzzo, A.B.; Rizzatti, E.G.; Atanackovic, D.; Colleoni, G.W.B. FOXP3 and CTLA4 Overexpression in Multiple Myeloma Bone Marrow as a Sign of Accumulation of CD4+ T Regulatory Cells. Cancer Immunol. Immunother. 2014, 63, 1189–1197. [Google Scholar] [CrossRef] [Green Version]
- Dahlhoff, J.; Manz, H.; Steinfatt, T.; Delgado-Tascon, J.; Seebacher, E.; Schneider, T.; Wilnit, A.; Mokhtari, Z.; Tabares, P.; Böckle, D.; et al. Transient Regulatory T-Cell Targeting Triggers Immune Control of Multiple Myeloma and Prevents Disease Progression. Leukemia 2022, 36, 790–800. [Google Scholar] [CrossRef]
- Zhou, L.; Ivanov, I.I.; Spolski, R.; Min, R.; Shenderov, K.; Egawa, T.; Levy, D.E.; Leonard, W.J.; Littman, D.R. IL-6 Programs TH-17 Cell Differentiation by Promoting Sequential Engagement of the IL-21 and IL-23 Pathways. Nat. Immunol. 2007, 8, 967–974. [Google Scholar] [CrossRef]
- Prabhala, R.H.; Pelluru, D.; Fulciniti, M.; Prabhala, H.K.; Nanjappa, P.; Song, W.; Pai, C.; Amin, S.; Tai, Y.-T.; Richardson, P.G.; et al. Elevated IL-17 Produced by TH17 Cells Promotes Myeloma Cell Growth and Inhibits Immune Function in Multiple Myeloma. Blood 2010, 115, 5385–5392. [Google Scholar] [CrossRef]
- Lei, L.; Sun, J.; Han, J.; Jiang, X.; Wang, Z.; Chen, L. Interleukin-17 Induces Pyroptosis in Osteoblasts through the NLRP3 Inflammasome Pathway in Vitro. Int. Immunopharmacol. 2021, 96, 107781. [Google Scholar] [CrossRef]
- Zhao, L.-J.; Gao, S.; Li, X. Effects of Thalidomide on the Ratio of Th17 to Treg Cells in Peripheral Blood and Expression of IL-17 and IL-35 in Patients with Multiple Myeloma. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2018, 26, 187–191. [Google Scholar] [CrossRef]
- Prabhala, R.H.; Fulciniti, M.; Pelluru, D.; Rashid, N.; Nigroiu, A.; Nanjappa, P.; Pai, C.; Lee, S.; Prabhala, N.S.; Bandi, R.L.; et al. Targeting IL-17A in Multiple Myeloma: A Potential Novel Therapeutic Approach in Myeloma. Leukemia 2016, 30, 379–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suen, H.; Brown, R.; Yang, S.; Weatherburn, C.; Ho, P.J.; Woodland, N.; Nassif, N.; Barbaro, P.; Bryant, C.; Hart, D.; et al. Multiple Myeloma Causes Clonal T-Cell Immunosenescence: Identification of Potential Novel Targets for Promoting Tumour Immunity and Implications for Checkpoint Blockade. Leukemia 2016, 30, 1716–1724. [Google Scholar] [CrossRef] [PubMed]
- Das, R.K.; Vernau, L.; Grupp, S.A.; Barrett, D.M. Naïve T-Cell Deficits at Diagnosis and after Chemotherapy Impair Cell Therapy Potential in Pediatric Cancers. Cancer Discov. 2019, 9, 492–499. [Google Scholar] [CrossRef] [Green Version]
- Arasanz, H.; Zuazo, M.; Bocanegra, A.; Gato, M.; Martínez-Aguillo, M.; Morilla, I.; Fernández, G.; Hernández, B.; López, P.; Alberdi, N.; et al. Early Detection of Hyperprogressive Disease in Non-Small Cell Lung Cancer by Monitoring of Systemic T Cell Dynamics. Cancers 2020, 12, 344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Battram, A.M.; Bachiller, M.; Lopez, V.; Fernández de Larrea, C.; Urbano-Ispizua, A.; Martín-Antonio, B. IL-15 Enhances the Persistence and Function of BCMA-Targeting CAR-T Cells Compared to IL-2 or IL-15/IL-7 by Limiting CAR-T Cell Dysfunction and Differentiation. Cancers 2021, 13, 3534. [Google Scholar] [CrossRef] [PubMed]
- Lanna, A.; Gomes, D.C.O.; Muller-Durovic, B.; McDonnell, T.; Escors, D.; Gilroy, D.W.; Lee, J.H.; Karin, M.; Akbar, A.N. A Sestrin-Dependent Erk-Jnk-P38 MAPK Activation Complex Inhibits Immunity during Aging. Nat. Immunol. 2017, 18, 354–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jinushi, M.; Vanneman, M.; Munshi, N.C.; Tai, Y.-T.; Prabhala, R.H.; Ritz, J.; Neuberg, D.; Anderson, K.C.; Carrasco, D.R.; Dranoff, G. MHC Class I Chain-Related Protein A Antibodies and Shedding Are Associated with the Progression of Multiple Myeloma. Proc. Natl. Acad. Sci. USA 2008, 105, 1285–1290. [Google Scholar] [CrossRef] [Green Version]
- Benson, D.M., Jr.; Bakan, C.E.; Mishra, A.; Hofmeister, C.C.; Efebera, Y.; Becknell, B.; Baiocchi, R.A.; Zhang, J.; Yu, J.; Smith, M.K.; et al. The PD-1/PD-L1 Axis Modulates the Natural Killer Cell versus Multiple Myeloma Effect: A Therapeutic Target for CT-011, a Novel Monoclonal Anti–PD-1 Antibody. Blood 2010, 116, 2286–2294. [Google Scholar] [CrossRef]
- Pazina, T.; MacFarlane, A.W.; Bernabei, L.; Dulaimi, E.; Kotcher, R.; Yam, C.; Bezman, N.A.; Robbins, M.D.; Ross, E.A.; Campbell, K.S.; et al. Alterations of NK Cell Phenotype in the Disease Course of Multiple Myeloma. Cancers 2021, 13, 226. [Google Scholar] [CrossRef]
- Cifaldi, L.; Prencipe, G.; Caiello, I.; Bracaglia, C.; Locatelli, F.; De Benedetti, F.; Strippoli, R. Inhibition of Natural Killer Cell Cytotoxicity by Interleukin-6: Implications for the Pathogenesis of Macrophage Activation Syndrome. Arthritis Rheumatol. 2015, 67, 3037–3046. [Google Scholar] [CrossRef]
- Tamura, H.; Ishibashi, M.; Yamashita, T.; Tanosaki, S.; Okuyama, N.; Kondo, A.; Hyodo, H.; Shinya, E.; Takahashi, H.; Dong, H.; et al. Marrow Stromal Cells Induce B7-H1 Expression on Myeloma Cells, Generating Aggressive Characteristics in Multiple Myeloma. Leukemia 2013, 27, 464–472. [Google Scholar] [CrossRef] [Green Version]
- Tamura, H.; Ishibashi, M.; Sunakawa-Kii, M.; Inokuchi, K. PD-L1-PD-1 Pathway in the Pathophysiology of Multiple Myeloma. Cancers 2020, 12, 924. [Google Scholar] [CrossRef]
- Elkabets, M.; Ribeiro, V.S.G.; Dinarello, C.A.; Ostrand-Rosenberg, S.; Di Santo, J.P.; Apte, R.N.; Vosshenrich, C.A.J. IL-1β Regulates a Novel Myeloid-Derived Suppressor Cell Subset That Impairs NK Cell Development and Function. Eur. J. Immunol. 2010, 40, 3347–3357. [Google Scholar] [CrossRef]
- Ponzetta, A.; Benigni, G.; Antonangeli, F.; Sciumè, G.; Sanseviero, E.; Zingoni, A.; Ricciardi, M.R.; Petrucci, M.T.; Santoni, A.; Bernardini, G. Multiple Myeloma Impairs Bone Marrow Localization of Effector Natural Killer Cells by Altering the Chemokine Microenvironment. Cancer Res. 2015, 75, 4766–4777. [Google Scholar] [CrossRef] [Green Version]
- Clara, J.A.; Childs, R.W. Harnessing Natural Killer Cells for the Treatment of Multiple Myeloma. Semin. Oncol. 2022, 49, 69–85. [Google Scholar] [CrossRef]
- Bachiller, M.; Battram, A.M.; Perez-Amill, L.; Martín-Antonio, B. Natural Killer Cells in Immunotherapy: Are We Nearly There? Cancers 2020, 12, 3139. [Google Scholar] [CrossRef]
- Flores-Borja, F.; Bosma, A.; Ng, D.; Reddy, V.; Ehrenstein, M.R.; Isenberg, D.A.; Mauri, C. CD19+CD24hiCD38hi B Cells Maintain Regulatory T Cells While Limiting TH1 and TH17 Differentiation. Sci. Transl. Med. 2013, 5, 173ra23. [Google Scholar] [CrossRef]
- Zhang, L.; Tai, Y.-T.; Ho, M.; Xing, L.; Chauhan, D.; Gang, A.; Qiu, L.; Anderson, K.C. Regulatory B Cell-Myeloma Cell Interaction Confers Immunosuppression and Promotes Their Survival in the Bone Marrow Milieu. Blood Cancer J. 2017, 7, e547. [Google Scholar] [CrossRef] [Green Version]
- Beider, K.; Bitner, H.; Leiba, M.; Gutwein, O.; Koren-Michowitz, M.; Ostrovsky, O.; Abraham, M.; Wald, H.; Galun, E.; Peled, A.; et al. Multiple Myeloma Cells Recruit Tumor-Supportive Macrophages through the CXCR4/CXCL12 Axis and Promote Their Polarization toward the M2 Phenotype. Oncotarget 2014, 5, 11283–11296. [Google Scholar] [CrossRef] [Green Version]
- Andersen, M.N.; Andersen, N.F.; Rødgaard-Hansen, S.; Hokland, M.; Abildgaard, N.; Møller, H.J. The Novel Biomarker of Alternative Macrophage Activation, Soluble Mannose Receptor (SMR/SCD206): Implications in Multiple Myeloma. Leuk. Res. 2015, 39, 971–975. [Google Scholar] [CrossRef]
- Xu, R.; Li, Y.; Yan, H.; Zhang, E.; Huang, X.; Chen, Q.; Chen, J.; Qu, J.; Liu, Y.; He, J.; et al. CCL2 Promotes Macrophages-Associated Chemoresistance via MCPIP1 Dual Catalytic Activities in Multiple Myeloma. Cell Death Dis. 2019, 10, 781. [Google Scholar] [CrossRef]
- Russ, A.; Hua, A.B.; Montfort, W.R.; Rahman, B.; Riaz, I.B.; Khalid, M.U.; Carew, J.S.; Nawrocki, S.T.; Persky, D.; Anwer, F. Blocking “Don’t Eat Me” Signal of CD47-SIRPα in Hematological Malignancies, an in-Depth Review. Blood Rev. 2018, 32, 480–489. [Google Scholar] [CrossRef]
- Opperman, K.S.; Vandyke, K.; Clark, K.C.; Coulter, E.A.; Hewett, D.R.; Mrozik, K.M.; Schwarz, N.; Evdokiou, A.; Croucher, P.I.; Psaltis, P.J.; et al. Clodronate-Liposome Mediated Macrophage Depletion Abrogates Multiple Myeloma Tumor Establishment In Vivo. Neoplasia 2019, 21, 777–787. [Google Scholar] [CrossRef]
- Ries, C.H.; Cannarile, M.A.; Hoves, S.; Benz, J.; Wartha, K.; Runza, V.; Rey-Giraud, F.; Pradel, L.P.; Feuerhake, F.; Klaman, I.; et al. Targeting Tumor-Associated Macrophages with Anti-CSF-1R Antibody Reveals a Strategy for Cancer Therapy. Cancer Cell 2014, 25, 846–859. [Google Scholar] [CrossRef] [Green Version]
- Görgün, G.T.; Whitehill, G.; Anderson, J.L.; Hideshima, T.; Maguire, C.; Laubach, J.; Raje, N.; Munshi, N.C.; Richardson, P.G.; Anderson, K.C. Tumor-Promoting Immune-Suppressive Myeloid-Derived Suppressor Cells in the Multiple Myeloma Microenvironment in Humans. Blood 2013, 121, 2975–2987. [Google Scholar] [CrossRef] [Green Version]
- Jakubowiak, A.J.; Kumar, S.; Medhekar, R.; Pei, H.; Lefebvre, P.; Kaila, S.; He, J.; Lafeuille, M.-H.; Cortoos, A.; Londhe, A.; et al. Daratumumab Improves Depth of Response and Progression-Free Survival in Transplant-Ineligible, High-Risk, Newly Diagnosed Multiple Myeloma. Oncologist 2022, 27, e589–e596. [Google Scholar] [CrossRef] [PubMed]
- Htut, M.; Gasparetto, C.; Zonder, J.; Martin, T.G., III; Scott, E.C.; Chen, J.; Shemesh, S.; Brooks, C.L.; Chauhan, D.; Anderson, K.C.; et al. Results from Ongoing Phase 1/2 Trial of SL-401 in Combination with Pomalidomide and Dexamethasone in Relapsed or Refractory Multiple Myeloma. Blood 2016, 128, 5696. [Google Scholar] [CrossRef]
- Rossi, M.; Botta, C.; Correale, P.; Tassone, P.; Tagliaferri, P. Immunologic Microenvironment and Personalized Treatment in Multiple Myeloma. Expert Opin. Biol. Ther. 2013, 13 (Suppl. S1), S83–S93. [Google Scholar] [CrossRef]
- Yamamoto, L.; Amodio, N.; Gulla, A.; Anderson, K.C. Harnessing the Immune System Against Multiple Myeloma: Challenges and Opportunities. Front. Oncol. 2021, 10, 606368. [Google Scholar] [CrossRef] [PubMed]
- Moser-Katz, T.; Joseph, N.S.; Dhodapkar, M.V.; Lee, K.P.; Boise, L.H. Game of Bones: How Myeloma Manipulates Its Microenvironment. Front. Oncol. 2021, 10, 625199. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Smyth, M.J.; Martinet, L. Cancer Immunoediting and Immune Dysregulation in Multiple Myeloma. Blood 2020, 136, 2731–2740. [Google Scholar] [CrossRef]
- Whiteside, T.L. The Tumor Microenvironment and Its Role in Promoting Tumor Growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef] [Green Version]
- Kay, N.E.; Leong, T.L.; Bone, N.; Vesole, D.H.; Greipp, P.R.; Van Ness, B.; Oken, M.M.; Kyle, R.A. Blood Levels of Immune Cells Predict Survival in Myeloma Patients: Results of an Eastern Cooperative Oncology Group Phase 3 Trial for Newly Diagnosed Multiple Myeloma Patients. Blood 2001, 98, 23–28. [Google Scholar] [CrossRef]
- Raitakari, M.; Brown, R.D.; Gibson, J.; Joshua, D.E. T Cells in Myeloma. Hematol. Oncol. 2003, 21, 33–42. [Google Scholar] [CrossRef]
- Wei, F.; Cheng, X.-X.; Xue, J.Z.; Xue, S.-A. Emerging Strategies in TCR-Engineered T Cells. Front. Immunol. 2022, 13, 850358. [Google Scholar] [CrossRef]
- Kasakovski, D.; Xu, L.; Li, Y. T Cell Senescence and CAR-T Cell Exhaustion in Hematological Malignancies. J. Hematol. Oncol. 2018, 11, 91. [Google Scholar] [CrossRef]
- Ab Rahman, A.S.; Strother, R.M.; Paddison, J. New Zealand National Retrospective Cohort Study of Survival Outcomes of Patients with Metastatic Melanoma Receiving Immune-Checkpoint Inhibitors. Asia Pac. J. Clin. Oncol. 2022, 1–8. [Google Scholar] [CrossRef]
- Das, S.; Johnson, D.B. Immune-Related Adverse Events and Anti-Tumor Efficacy of Immune Checkpoint Inhibitors. J. Immunother. Cancer 2019, 7, 306. [Google Scholar] [CrossRef]
- Yamazaki, N.; Uhara, H.; Fukushima, S.; Uchi, H.; Shibagaki, N.; Kiyohara, Y.; Tsutsumida, A.; Namikawa, K.; Okuyama, R.; Otsuka, Y.; et al. Phase II Study of the Immune-Checkpoint Inhibitor Ipilimumab plus Dacarbazine in Japanese Patients with Previously Untreated, Unresectable or Metastatic Melanoma. Cancer Chemother. Pharm. 2015, 76, 969–975. [Google Scholar] [CrossRef] [Green Version]
- Miller, B.C.; Sen, D.R.; Al Abosy, R.; Bi, K.; Virkud, Y.V.; LaFleur, M.W.; Yates, K.B.; Lako, A.; Felt, K.; Naik, G.S.; et al. Subsets of Exhausted CD8+ T Cells Differentially Mediate Tumor Control and Respond to Checkpoint Blockade. Nat. Immunol. 2019, 20, 326–336. [Google Scholar] [CrossRef]
- Zelle-Rieser, C.; Thangavadivel, S.; Biedermann, R.; Brunner, A.; Stoitzner, P.; Willenbacher, E.; Greil, R.; Jöhrer, K. T Cells in Multiple Myeloma Display Features of Exhaustion and Senescence at the Tumor Site. J. Hematol. Oncol. 2016, 9, 116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visram, A.; Kourelis, T.V. Aging-Associated Immune System Changes in Multiple Myeloma: The Dark Side of the Moon. Cancer Treat. Res. Commun. 2021, 29, 100494. [Google Scholar] [CrossRef]
- Frassanito, M.A.; Cusmai, A.; Iodice, G.; Dammacco, F. Autocrine Interleukin-6 Production and Highly Malignant Multiple Myeloma: Relation with Resistance to Drug-Induced Apoptosis. Blood 2001, 97, 483–489. [Google Scholar] [CrossRef]
- Fontenot, J.D.; Gavin, M.A.; Rudensky, A.Y. Foxp3 Programs the Development and Function of CD4+CD25+ Regulatory T Cells. Nat. Immunol. 2003, 4, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Workman, C.J.; Szymczak-Workman, A.L.; Collison, L.W.; Pillai, M.R.; Vignali, D.A.A. The Development and Function of Regulatory T Cells. Cell Mol. Life Sci. 2009, 66, 2603–2622. [Google Scholar] [CrossRef] [Green Version]
- Curiel, T.J. Regulatory T Cells and Treatment of Cancer. Curr. Opin. Immunol. 2008, 20, 241–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadjiaggelidou, C.; Katodritou, E. Regulatory T-Cells and Multiple Myeloma: Implications in Tumor Immune Biology and Treatment. J. Clin. Med. 2021, 10, 4588. [Google Scholar] [CrossRef] [PubMed]
- Raja, K.R.M.; Rihova, L.; Zahradova, L.; Klincova, M.; Penka, M.; Hajek, R. Increased T Regulatory Cells Are Associated with Adverse Clinical Features and Predict Progression in Multiple Myeloma. PLoS ONE 2012, 7, e47077. [Google Scholar] [CrossRef]
- Prabhala, R.H.; Neri, P.; Bae, J.E.; Tassone, P.; Shammas, M.A.; Allam, C.K.; Daley, J.F.; Chauhan, D.; Blanchard, E.; Thatte, H.S.; et al. Dysfunctional T Regulatory Cells in Multiple Myeloma. Blood 2006, 107, 301–304. [Google Scholar] [CrossRef] [Green Version]
- Noonan, K.; Marchionni, L.; Anderson, J.; Pardoll, D.; Roodman, G.D.; Borrello, I. A Novel Role of IL-17–Producing Lymphocytes in Mediating Lytic Bone Disease in Multiple Myeloma. Blood 2010, 116, 3554–3563. [Google Scholar] [CrossRef] [Green Version]
- Foglietta, M.; Castella, B.; Mariani, S.; Coscia, M.; Godio, L.; Ferracini, R.; Ruggeri, M.; Muccio, V.; Omedé, P.; Palumbo, A.; et al. The Bone Marrow of Myeloma Patients Is Steadily Inhabited by a Normal-Sized Pool of Functional Regulatory T Cells Irrespectiveof the Disease Status. Haematologica 2014, 99, 1605–1610. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Fu, J.; Zhou, Y. Metabolism Controls the Balance of Th17/T-Regulatory Cells. Front. Immunol. 2017, 8, 1632. [Google Scholar] [CrossRef] [Green Version]
- Rossi, M.; Altomare, E.; Botta, C.; Gallo Cantafio, M.E.; Sarvide, S.; Caracciolo, D.; Riillo, C.; Gaspari, M.; Taverna, D.; Conforti, F.; et al. MiR-21 Antagonism Abrogates Th17 Tumor Promoting Functions in Multiple Myeloma. Leukemia 2021, 35, 823–834. [Google Scholar] [CrossRef]
- Ma, T.; Zhang, Y.; Zhou, X.; Xie, P.; Li, J. A Unique Role of T Helper 17 Cells in Different Treatment Stages of Multiple Myeloma. Clin. Lymphoma Myeloma Leuk. 2020, 20, 190–197. [Google Scholar] [CrossRef] [Green Version]
- Dhodapkar, K.M.; Barbuto, S.; Matthews, P.; Kukreja, A.; Mazumder, A.; Vesole, D.; Jagannath, S.; Dhodapkar, M.V. Dendritic Cells Mediate the Induction of Polyfunctional Human IL17-Producing Cells (Th17-1 Cells) Enriched in the Bone Marrow of Patients with Myeloma. Blood 2008, 112, 2878–2885. [Google Scholar] [CrossRef]
- Kale, A.; Sharma, A.; Stolzing, A.; Desprez, P.-Y.; Campisi, J. Role of Immune Cells in the Removal of Deleterious Senescent Cells. Immun. Ageing 2020, 17, 16. [Google Scholar] [CrossRef]
- Iannello, A.; Thompson, T.W.; Ardolino, M.; Lowe, S.W.; Raulet, D.H. P53-Dependent Chemokine Production by Senescent Tumor Cells Supports NKG2D-Dependent Tumor Elimination by Natural Killer Cells. J. Exp. Med. 2013, 210, 2057–2069. [Google Scholar] [CrossRef]
- Liu, Q.; Sun, Z.; Chen, L. Memory T Cells: Strategies for Optimizing Tumor Immunotherapy. Protein Cell 2020, 11, 549–564. [Google Scholar] [CrossRef] [Green Version]
- Thomas, R.; Wang, W.; Su, D.-M. Contributions of Age-Related Thymic Involution to Immunosenescence and Inflammaging. Immun. Ageing 2020, 17, 2. [Google Scholar] [CrossRef] [Green Version]
- Coder, B.D.; Wang, H.; Ruan, L.; Su, D.-M. Thymic Involution Perturbs Negative Selection Leading to Autoreactive T Cells That Induce Chronic Inflammation. J. Immunol. 2015, 194, 5825–5837. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, L.P.; Teixeira, V.R.; Alencar-Silva, T.; Simonassi-Paiva, B.; Pereira, R.W.; Pogue, R.; Carvalho, J.L. Hallmarks of Aging and Immunosenescence: Connecting the Dots. Cytokine Growth Factor Rev. 2021, 59, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, I.J.; Lalinde Ruiz, N.; Llano León, M.; Martínez Enríquez, L.; del Montilla Velásquez, M.P.; Ortiz Aguirre, J.P.; Rodríguez Bohórquez, O.M.; Velandia Vargas, E.A.; Hernández, E.D.; Parra López, C.A. Immunosenescence Study of T Cells: A Systematic Review. Front. Immunol. 2021, 11, 3460. [Google Scholar] [CrossRef] [PubMed]
- Fahy, G.M.; Brooke, R.T.; Watson, J.P.; Good, Z.; Vasanawala, S.S.; Maecker, H.; Leipold, M.D.; Lin, D.T.S.; Kobor, M.S.; Horvath, S. Reversal of Epigenetic Aging and Immunosenescent Trends in Humans. Aging Cell 2019, 18, e13028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Zhao, L.; Wan, Y.Y.; Zhu, B. Mechanism of Action of IL-7 and Its Potential Applications and Limitations in Cancer Immunotherapy. Int. J. Mol. Sci. 2015, 16, 10267–10280. [Google Scholar] [CrossRef] [Green Version]
- Di Mitri, D.; Azevedo, R.I.; Henson, S.M.; Libri, V.; Riddell, N.E.; Macaulay, R.; Kipling, D.; Soares, M.V.D.; Battistini, L.; Akbar, A.N. Reversible Senescence in Human CD4+CD45RA+CD27- Memory T Cells. J. Immunol. 2011, 187, 2093–2100. [Google Scholar] [CrossRef] [Green Version]
- Aiello, A.; Farzaneh, F.; Candore, G.; Caruso, C.; Davinelli, S.; Gambino, C.M.; Ligotti, M.E.; Zareian, N.; Accardi, G. Immunosenescence and Its Hallmarks: How to Oppose Aging Strategically? A Review of Potential Options for Therapeutic Intervention. Front. Immunol. 2019, 10, 2247. [Google Scholar] [CrossRef] [Green Version]
- Mannick, J.B.; Morris, M.; Hockey, H.-U.P.; Roma, G.; Beibel, M.; Kulmatycki, K.; Watkins, M.; Shavlakadze, T.; Zhou, W.; Quinn, D.; et al. TORC1 Inhibition Enhances Immune Function and Reduces Infections in the Elderly. Sci. Transl. Med. 2018, 10, eaaq1564. [Google Scholar] [CrossRef] [Green Version]
- Weichhart, T.; Haidinger, M.; Katholnig, K.; Kopecky, C.; Poglitsch, M.; Lassnig, C.; Rosner, M.; Zlabinger, G.J.; Hengstschläger, M.; Müller, M.; et al. Inhibition of MTOR Blocks the Anti-Inflammatory Effects of Glucocorticoids in Myeloid Immune Cells. Blood 2011, 117, 4273–4283. [Google Scholar] [CrossRef] [Green Version]
- El-Sherbiny, Y.M.; Meade, J.L.; Holmes, T.D.; McGonagle, D.; Mackie, S.L.; Morgan, A.W.; Cook, G.; Feyler, S.; Richards, S.J.; Davies, F.E.; et al. The Requirement for DNAM-1, NKG2D, and NKp46 in the Natural Killer Cell-Mediated Killing of Myeloma Cells. Cancer Res. 2007, 67, 8444–8449. [Google Scholar] [CrossRef] [Green Version]
- Blom, B.; van Hoeven, V.; Hazenberg, M.D. ILCs in Hematologic Malignancies: Tumor Cell Killers and Tissue Healers. Semin. Immunol. 2019, 41, 101279. [Google Scholar] [CrossRef]
- Martín-Antonio, B.; Suñe, G.; Perez-Amill, L.; Castella, M.; Urbano-Ispizua, A. Natural Killer Cells: Angels and Devils for Immunotherapy. Int. J. Mol. Sci. 2017, 18, 1868. [Google Scholar] [CrossRef] [Green Version]
- Martín-Antonio, B.; Suñe, G.; Najjar, A.; Perez-Amill, L.; Antoñana-Vildosola, A.; Castella, M.; León, S.; Velasco-de Andrés, M.; Lozano, F.; Lozano, E.; et al. Extracellular NK Histones Promote Immune Cell Anti-Tumor Activity by Inducing Cell Clusters through Binding to CD138 Receptor. J. Immunother. Cancer 2019, 7, 259. [Google Scholar] [CrossRef]
- Carbone, E.; Neri, P.; Mesuraca, M.; Fulciniti, M.T.; Otsuki, T.; Pende, D.; Groh, V.; Spies, T.; Pollio, G.; Cosman, D.; et al. HLA Class I, NKG2D, and Natural Cytotoxicity Receptors Regulate Multiple Myeloma Cell Recognition by Natural Killer Cells. Blood 2005, 105, 251–258. [Google Scholar] [CrossRef] [Green Version]
- Nersesian, S.; Schwartz, S.L.; Grantham, S.R.; MacLean, L.K.; Lee, S.N.; Pugh-Toole, M.; Boudreau, J.E. NK Cell Infiltration Is Associated with Improved Overall Survival in Solid Cancers: A Systematic Review and Meta-Analysis. Transl. Oncol. 2021, 14, 100930. [Google Scholar] [CrossRef]
- Konjević, G.; Vuletić, A.; Mirjačić Martinović, K.; Colović, N.; Čolović, M.; Jurišić, V. Decreased CD161 Activating and Increased CD158a Inhibitory Receptor Expression on NK Cells Underlies Impaired NK Cell Cytotoxicity in Patients with Multiple Myeloma. J. Clin. Pathol. 2016, 69, 1009–1016. [Google Scholar] [CrossRef]
- Hanna, J.; Goldman-Wohl, D.; Hamani, Y.; Avraham, I.; Greenfield, C.; Natanson-Yaron, S.; Prus, D.; Cohen-Daniel, L.; Arnon, T.I.; Manaster, I.; et al. Decidual NK Cells Regulate Key Developmental Processes at the Human Fetal-Maternal Interface. Nat. Med. 2006, 12, 1065–1074. [Google Scholar] [CrossRef]
- Jabrane-Ferrat, N. Features of Human Decidual NK Cells in Healthy Pregnancy and During Viral Infection. Front. Immunol. 2019, 10, 1397. [Google Scholar] [CrossRef]
- Fu, B.; Zhou, Y.; Ni, X.; Tong, X.; Xu, X.; Dong, Z.; Sun, R.; Tian, Z.; Wei, H. Natural Killer Cells Promote Fetal Development through the Secretion of Growth-Promoting Factors. Immunity 2017, 47, 1100–1113.e6. [Google Scholar] [CrossRef] [Green Version]
- Sojka, D.K.; Yang, L.; Yokoyama, W.M. Uterine Natural Killer Cells. Front. Immunol. 2019, 10, 960. [Google Scholar] [CrossRef]
- Huhn, O.; Zhao, X.; Esposito, L.; Moffett, A.; Colucci, F.; Sharkey, A.M. How Do Uterine Natural Killer and Innate Lymphoid Cells Contribute to Successful Pregnancy? Front. Immunol. 2021, 12, 607669. [Google Scholar] [CrossRef]
- Shah, N.; Martin-Antonio, B.; Yang, H.; Ku, S.; Lee, D.A.; Cooper, L.J.N.; Decker, W.K.; Li, S.; Robinson, S.N.; Sekine, T.; et al. Antigen Presenting Cell-Mediated Expansion of Human Umbilical Cord Blood Yields Log-Scale Expansion of Natural Killer Cells with Anti-Myeloma Activity. PLoS ONE 2013, 8, e76781. [Google Scholar] [CrossRef]
- Martin-Antonio, B.; Najjar, A.; Robinson, S.N.; Chew, C.; Li, S.; Yvon, E.; Thomas, M.W.; Mc Niece, I.; Orlowski, R.; Muñoz-Pinedo, C.; et al. Transmissible Cytotoxicity of Multiple Myeloma Cells by Cord Blood-Derived NK Cells Is Mediated by Vesicle Trafficking. Cell Death Differ. 2015, 22, 96–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, X.; Zhang, L.; Wei, W. Regulatory B Cells in Inflammatory Diseases and Tumor. Int. Immunopharmacol. 2019, 67, 281–286. [Google Scholar] [CrossRef]
- Zhou, M.; Wen, Z.; Cheng, F.; Ma, J.; Li, W.; Ren, H.; Sheng, Y.; Dong, H.; Lu, L.; Hu, H.-M.; et al. Tumor-Released Autophagosomes Induce IL-10-Producing B Cells with Suppressive Activity on T Lymphocytes via TLR2-MyD88-NF-ΚB Signal Pathway. Oncoimmunology 2016, 5, e1180485. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.; Guo, T.; Cui, J.; Zhang, L.; Pan, L. Onset of Regulatory B Cells Occurs at Initial Stage of B Cell Dysfunction in Multiple Myeloma. Blood 2019, 134, 1780. [Google Scholar] [CrossRef]
- Zheng, Y.; Cai, Z.; Wang, S.; Zhang, X.; Qian, J.; Hong, S.; Li, H.; Wang, M.; Yang, J.; Yi, Q. Macrophages Are an Abundant Component of Myeloma Microenvironment and Protect Myeloma Cells from Chemotherapy Drug-Induced Apoptosis. Blood 2009, 114, 3625–3628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Yang, J.; Qian, J.; Qiu, P.; Hanabuchi, S.; Lu, Y.; Wang, Z.; Liu, Z.; Li, H.; He, J.; et al. PSGL-1/Selectin and ICAM-1/CD18 Interactions Are Involved in Macrophage-Induced Drug Resistance in Myeloma. Leukemia 2013, 27, 702–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Hu, W.; Xia, Z.; Liang, Y.; Lu, Y.; Lin, S.; Tang, H. High Numbers of CD163+ Tumor-Associated Macrophages Correlate with Poor Prognosis in Multiple Myeloma Patients Receiving Bortezomib-Based Regimens. J. Cancer 2019, 10, 3239–3245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, P.J. Macrophage Polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef]
- Khalife, J.; Ghose, J.; Martella, M.; Viola, D.; Rocci, A.; Troadec, E.; Terrazas, C.; Satoskar, A.R.; Gunes, E.G.; Dona, A.; et al. MiR-16 Regulates Crosstalk in NF-ΚB Tolerogenic Inflammatory Signaling between Myeloma Cells and Bone Marrow Macrophages. JCI Insight 2019, 4, 129348. [Google Scholar] [CrossRef]
- Wang, Q.; Lu, Y.; Li, R.; Jiang, Y.; Zheng, Y.; Qian, J.; Bi, E.; Zheng, C.; Hou, J.; Wang, S.; et al. Therapeutic Effects of CSF1R-Blocking Antibodies in Multiple Myeloma. Leukemia 2018, 32, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Park, C.; Guenthner, N.; Gurley, S.; Zhang, L.; Lubben, B.; Adebayo, O.; Bash, H.; Chen, Y.; Maksimos, M.; et al. Tumor-Associated Macrophages in Multiple Myeloma: Advances in Biology and Therapy. J Immunother. Cancer 2022, 10, e003975. [Google Scholar] [CrossRef] [PubMed]
- Favaloro, J.; Liyadipitiya, T.; Brown, R.; Yang, S.; Suen, H.; Woodland, N.; Nassif, N.; Hart, D.; Fromm, P.; Weatherburn, C.; et al. Myeloid Derived Suppressor Cells Are Numerically, Functionally and Phenotypically Different in Patients with Multiple Myeloma. Leuk. Lymphoma 2014, 55, 2893–2900. [Google Scholar] [CrossRef] [PubMed]
- De Veirman, K.; Van Ginderachter, J.A.; Lub, S.; De Beule, N.; Thielemans, K.; Bautmans, I.; Oyajobi, B.O.; De Bruyne, E.; Menu, E.; Lemaire, M.; et al. Multiple Myeloma Induces Mcl-1 Expression and Survival of Myeloid-Derived Suppressor Cells. Oncotarget 2015, 6, 10532–10547. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.-E.; Lim, J.-Y.; Ryu, D.-B.; Kim, T.W.; Yoon, J.-H.; Cho, B.-S.; Eom, K.-S.; Kim, Y.-J.; Kim, H.-J.; Lee, S.; et al. Circulating Immune Cell Phenotype Can Predict the Outcome of Lenalidomide plus Low-Dose Dexamethasone Treatment in Patients with Refractory/Relapsed Multiple Myeloma. Cancer Immunol. Immunother. 2016, 65, 983–994. [Google Scholar] [CrossRef]
- Wang, J.; Veirman, K.D.; Beule, N.D.; Maes, K.; Bruyne, E.D.; Valckenborgh, E.V.; Vanderkerken, K.; Menu, E. The Bone Marrow Microenvironment Enhances Multiple Myeloma Progression by Exosome-Mediated Activation of Myeloid-Derived Suppressor Cells. Oncotarget 2015, 6, 43992–44004. [Google Scholar] [CrossRef] [Green Version]
- Uckun, F.M. Dual Targeting of Multiple Myeloma Stem Cells and Myeloid-Derived Suppressor Cells for Treatment of Chemotherapy-Resistant Multiple Myeloma. Front. Oncol. 2021, 11, 760382. [Google Scholar] [CrossRef]
- Li, K.; Shi, H.; Zhang, B.; Ou, X.; Ma, Q.; Chen, Y.; Shu, P.; Li, D.; Wang, Y. Myeloid-Derived Suppressor Cells as Immunosuppressive Regulators and Therapeutic Targets in Cancer. Sig. Transduct. Target. Ther. 2021, 6, 362. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Cellular Compartment or Process | Molecules and/or Cell Population Involved | Impact on MM Disease | Therapeutic Strategy Proposed |
|---|---|---|---|
| ECM | 1. CXCR4/CXCL12. 2. CD138 and VLA-4 (MM)/Fibronectin (ECM). | 1. MM homing into the BM [30]. 2. NFkB activation, tumor survival, drug resistance [32]. | 1–2: AMD3100 (CXCR4 inhibitor), and Bortezomib (VLA-4 downregulation) [34]. |
| BM-MSCs | 1. VLA-4 (MM)/VCAM-1 (BM-MSCs). 2. LFA-1 (MM)/ICAM-1 (BM-MSCs). 3. IL6 secretion by BM-MSCs induced by MM cells. 4. Notch pathways and DKK1. 5. LINC00461 in BM-MSCs exosomes 6. Ligation of BAFF. 7. APRIL secretion (BM-MSCs)/BCMA and TACI (MM) [51] 8. TNFα. | 1. NFkB activation, MM survival [39]. 2. Disease progression [40]. 3. Enhanced secretion of VEGF and bFGF by MM that re-stimulates IL6 production [43]. 4. IL6, VEGF, and IGF-1 secretion in BM-MSCs [46,47]. 5. MM cell proliferation and drug resistance [98]. 6–7. MM proliferation [50]. 8. Enhanced LFA-1, ICAM-1, VCAM-1, and VLA-4 (MM) and ICAM-1 (BM-MSCs), increased binding of MM to BM-MSCs and further IL6 secretion [54]. | 1. Natalizumab: anti-α4 integrin (NCT00675428). 2. LFA878: LFA-1 inhibitor (preclinical studies) [41]. 3. Tocilizumab: anti-IL6R [57]. 4. BHQ880: anti-DKK1 [58]. 5. LINC00461 knockdown (preclinical studies) [98]. 6. Tabalumab: anti-BAFF [59]. 7. APRIL-based CARs target BCMA or TACI [52]. 8. Anti- TNFα. However, these drugs in other inflammatory conditions increase the risk of MM [56]. |
| Osteoclast/osteoblast imbalance | 1. MIP1α and MIP1β (MM). 2. RANKL (osteocytes)/RANK (osteoclasts). 3. MM induce RANKL and IL6 secretion by BM-MSCs. 4. VLA-4 (MM)/VCAM-1 (osteoblasts and BM-MSCs). | 1. Osteoclast activation [63,64], IL6 secretion [65], CHSY1 up-regulation, Notch signaling, MM survival, recruitment of osteoclast precursors [66]. 2–3. Osteoclast activity [67,68]. 4. RUNX2 decreased activity, decreased osteoblast differentiation [72], decreased OPG secretion, osteoclast formation and bone degradation [73]. | 1-2-3. Amino-bisphosphonates that inhibit osteoclast activity [69]. 2–3. Denosumab: anti-RANKL [70] 4. Natalizumab: anti-α4 integrin (NCT00675428). 4. BHQ880: anti-DKK1 [75]. |
| Angiogenesis in the vascular niche | 1. VEGF production (MM). 2. EGFR-2, Tie2/Tek, β3-integrin and endoglin in MM endothelial cells. 3. MM cells induce HGF, VEGF and IL8 secretion in BM-MSCs. 4. IGF1 and IL6 secretion by MM endothelial cells. | 1. Angiogenesis [82]. 2. Enhanced MM cell interaction with new blood vessels and further dissemination [83]. 3. Neovascularization [89]. 4. MM growth, enhanced MM production of VEGF, PDGF, Ang-1, HGF, and IL1. Enhanced angiogenesis [90]. | 1. Amino-bisphosphonates are anti-angiogenic [69,99]. 1–3. Bevacizumab: anti-VEGF [92,93]. 2. Derivatives of quinolone and quinazoline inhibit VEGFRs, EGFR, and PDGFR [94,95]. 4. Immunomodulators [96,97]. |
| Effector CD8 T cells | 1. CXCR4 (MILs)/CXCL12 (BM-MSCs). 2. CM phenotype of MILs. 3. PD-1, CTLA-4, LAG-3, or TIGIT (T cells) with PD-L1, CD80/CD86, MHC-II, and CD155 (MM). 4. TIGIT expression on T cells in MM [100]. | 1. Trafficking of MILs to the BM [101]. 2. Enhanced CR in patients [101]. 3. Inhibition of T cell activity [102,103] 4. Dysfunctional T cells with decreased proliferation and cytokine production [100]. | 1. Administer MILs with enhanced CXCR4 expression that has shown efficacy in CAR-T cells [104]. 2. Addition of PI3K inhibitors during the production of MILs [105]. 3. ICI treatments targeting others than PD-1/PD-L1 due to their toxicity in MM [106]. 4. TIGIT inhibition [100]. |
| CD4 conventional T cells | 1. Reduced CD4/CD8 ratio, lower number of CD4 T and Th2 cells in MM [107]. 2. IL6 secretion inhibits polarization of naïve T cells into Th1 cells [108]. 3. GPRC5D (MM)/CD4 T cells [109]. | 1–2. Tumor escape to immune surveillance [108]. 3. Inhibition of CD4 T-cell anti-MM activity. | 1. Optimization of CD4/CD8 ratio in cellular immunotherapy products [110,111]. 2. Tocilizumab (anti-IL6R). 3. Bispecific antibody against GPRC5D. (talquetamab) enhances anti-MM activity of CD4 T cells [109]. |
| T-reg cells | 1. Increased T-regs in the BM of MM [112,113]. 2. IL10 and TGFβ secretion by T-regs. 3. CTLA-4 and ICOS expression in T-regs. 4. ICOS (T-reg)/ICOSL (MM) [114]. 5. GPRC5D (MM)/T-regs [109]. | 1. Shorter time to progression [112,113]. 2. Interruption of CD4 T cell-mediated generation of CD8 T cell responses [115] 3. T-reg suppressive activity [116]. 4. Generation of functional T-regs [114]. 5. Inhibition of CD4 conventional and T-reg activity. | 1–2. Optimized MIL product with lower number of T-regs induces CR [101]. 2. Transient T-reg depletion [117]. 3, 4. Inhibition of T-regs with anti-ICOSL MoAb [114]. 5. Talquetamab enhances anti-MM activity of T-reg cells by themselves [109]. |
| Th17 cells | 1. IL6 induces IL21 that with TGFβ induces Th17 differentiation [118]. | 1. MM growth [119], osteoblast cell death [120], osteoclasts activation, tumor growth and MBD [119]. | Thalidomide normalizes the ratio of Th17 and T-reg cells in PB [121]. Anti-IL17 Ab show anti-MM activity [122]. |
| Age in T cells | High number of immunosenescent T cells (CD57, KLRG1, CD160, CD28−, PD1low, and CTLA4low) [123]. | Enhanced by chemotherapy [124] and ICI treatments [125]. | Addition of PI3K inhibitors [105], IL15 [126] and sestrins inhibition [127] during the production of the immunotherapy product. |
| NK cells | 1. MM cells downregulate NKG2D and NKp80 on NK cells [128]. 2. PDL1 (MM)/PD1 (NK cells) [129]. 3. BM-MSCs-derived IL6. 4. Tumor-derived IL1β in MDSCs. 5. Increased CXCL9 and CXCL10, decreased CXCL12, down-regulation of CXCR3 on NK cells. 6. CD56bright NK cells highly activated in BM and PB [130]. | 1. Inhibition of NK activity [128]. 2. Inhibition of NK activity [129]. 3. NK inhibition [131], PD-L1 on MM cells, impacting the NK and T cell activity [132,133]. 4. NK inhibition [134]. 5. Driving of NK cells outside the BM [135]. 6. Additional markers to characterize a possible angiogenic activity of CD56bright NK cells. | 1-2-3-4-5: Combination of IMiDs and MoAb enhance endogenous NK cell activity and ADCC of NK cells. BiKEs/TRiKEs redirect endogenous NK cells to tumor cells. Ab recruiting molecules bind tumor-associated antigens with endogenous IgG inducing NK-mediated ADCC. ALT-803: IL-15 superagonist that stimulates NK cells and T cells. CAR-NKs targeting SLAMF7, CD138 or NKG2D ligands on MM [136,137]. 6. Previous selection of in vitro expanded CD56dim NK cells. |
| Regulatory B cells | MM cells promote B-reg cell survival and their accumulation in the BM. | 1. IL10 secretion of B-reg cells inhibits CD4 T cell differentiation into Th1 and Th17 cells, and favors polarization into T-regs [138]. 2. B-regs avoid NK-ADCC in MM [139]. | Strategies to target B-reg cells have not been described yet. Novel research to decipher cellular interactions with B-regs and how B-regs exert their suppressive activity is required. |
| TAMs | 1. CXCL12 (MM and BM-MSCs)/CXCR4 (monocytes). 2. M2 macrophage immunosuppresion through IL6, IL10, IL8, TNFα, CD206, CD163, CCL2. 3. CD47 (MM)/SIRPα (macrophages). | 1. Monocytes recruitment and M2 polarization in BM [140]. 2. MM proliferation and progression [141,142]. 3. Immune checkpoint resulting in a “don’t eat me” signal in M2 macrophages and immune evasion [143]. | 1. AMD-3100: CXCR4 inhibitor (preclinical studies) [140]. 2. Clodronate liposome to deplete resident M2 macrophages in BM (preclinical studies) [144]. 2. Anti-CSF1R to reprogram TAMs to promote M1 phenotype (preclinical studies) [145]. 3. Antibodies anti-CD47 (SRF231: NCT03512340 and AO-176: NCT04445701). 3. SIRPα-IgG1 Fc fusion proteins (TTI-621: NCT02663518 and TTI-622: NCT03530683). |
| MDSCs | 1. IL10, CCL5, MIP-1 or IL6 from MM cells generate MDSCs 2. ARG1, ROS, COX2, iNOS, IL6, IL10 and IL18 (MDSCs) | 1–2. Inhibit immune responses, induce T-regs, promote angiogenesis and differentiate into osteoclasts [146]. | 1. Daratumumab: anti-CD38 (dual targeting of MM cells and MDSCs) [147] 2. Tagraxofusb: CD123-targeted agent [148] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hervás-Salcedo, R.; Martín-Antonio, B. A Journey through the Inter-Cellular Interactions in the Bone Marrow in Multiple Myeloma: Implications for the Next Generation of Treatments. Cancers 2022, 14, 3796. https://doi.org/10.3390/cancers14153796
Hervás-Salcedo R, Martín-Antonio B. A Journey through the Inter-Cellular Interactions in the Bone Marrow in Multiple Myeloma: Implications for the Next Generation of Treatments. Cancers. 2022; 14(15):3796. https://doi.org/10.3390/cancers14153796
Chicago/Turabian StyleHervás-Salcedo, Rosario, and Beatriz Martín-Antonio. 2022. "A Journey through the Inter-Cellular Interactions in the Bone Marrow in Multiple Myeloma: Implications for the Next Generation of Treatments" Cancers 14, no. 15: 3796. https://doi.org/10.3390/cancers14153796
APA StyleHervás-Salcedo, R., & Martín-Antonio, B. (2022). A Journey through the Inter-Cellular Interactions in the Bone Marrow in Multiple Myeloma: Implications for the Next Generation of Treatments. Cancers, 14(15), 3796. https://doi.org/10.3390/cancers14153796