
Reversing PD-1 Resistance in B16F10 Cells and Recovering Tumour Immunity Using a COX2 Inhibitor
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Background
2. Materials and Methods
2.1. Mice and Cells
2.2. Animal Experiments
2.3. Real-Time Quantitative Polymerase Chain Reaction (RT-qPCR)
2.4. Flow Cytometry
2.5. PGE2 Concentration Detection
2.6. Western Blotting
2.7. Statistical Analysis
3. Results
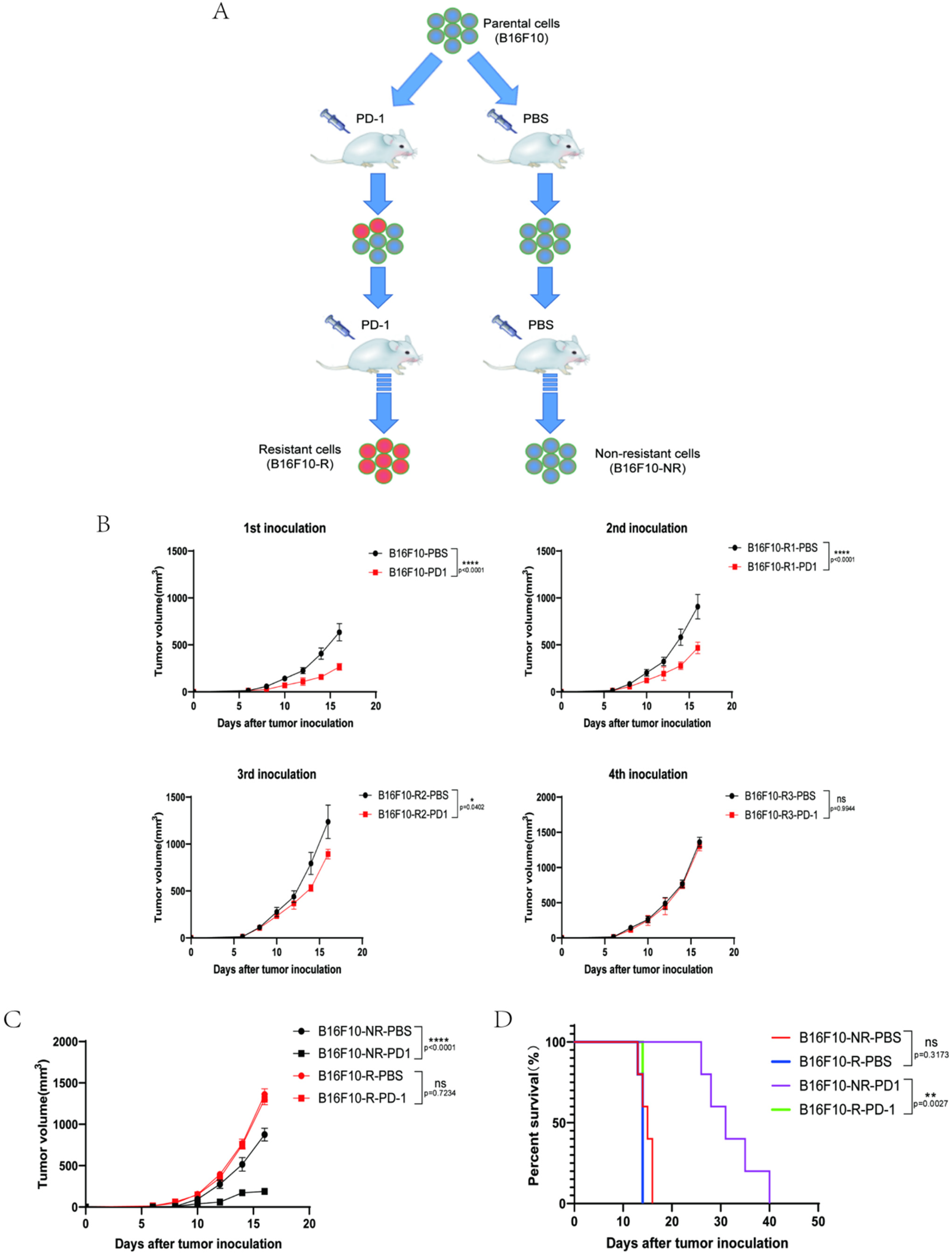
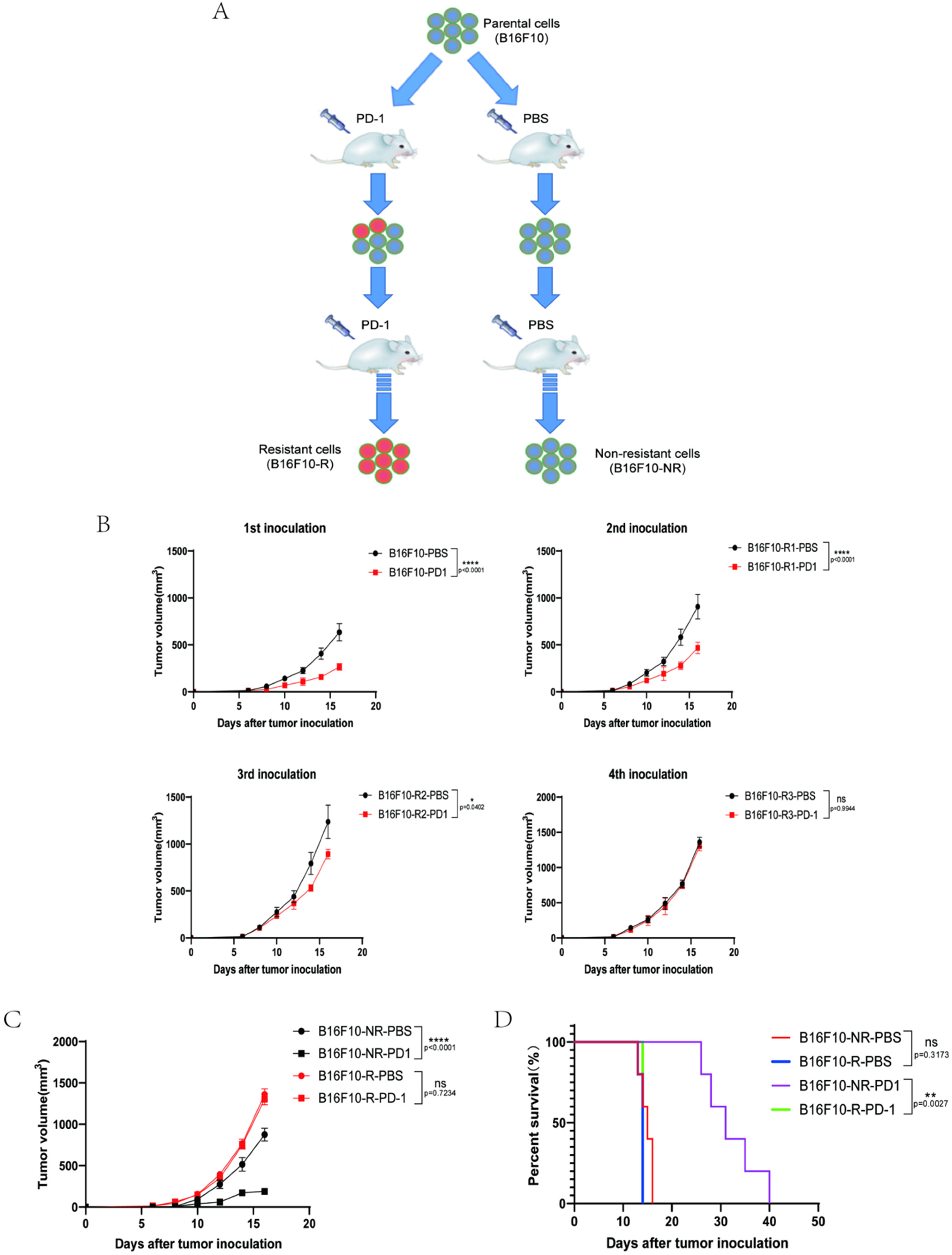
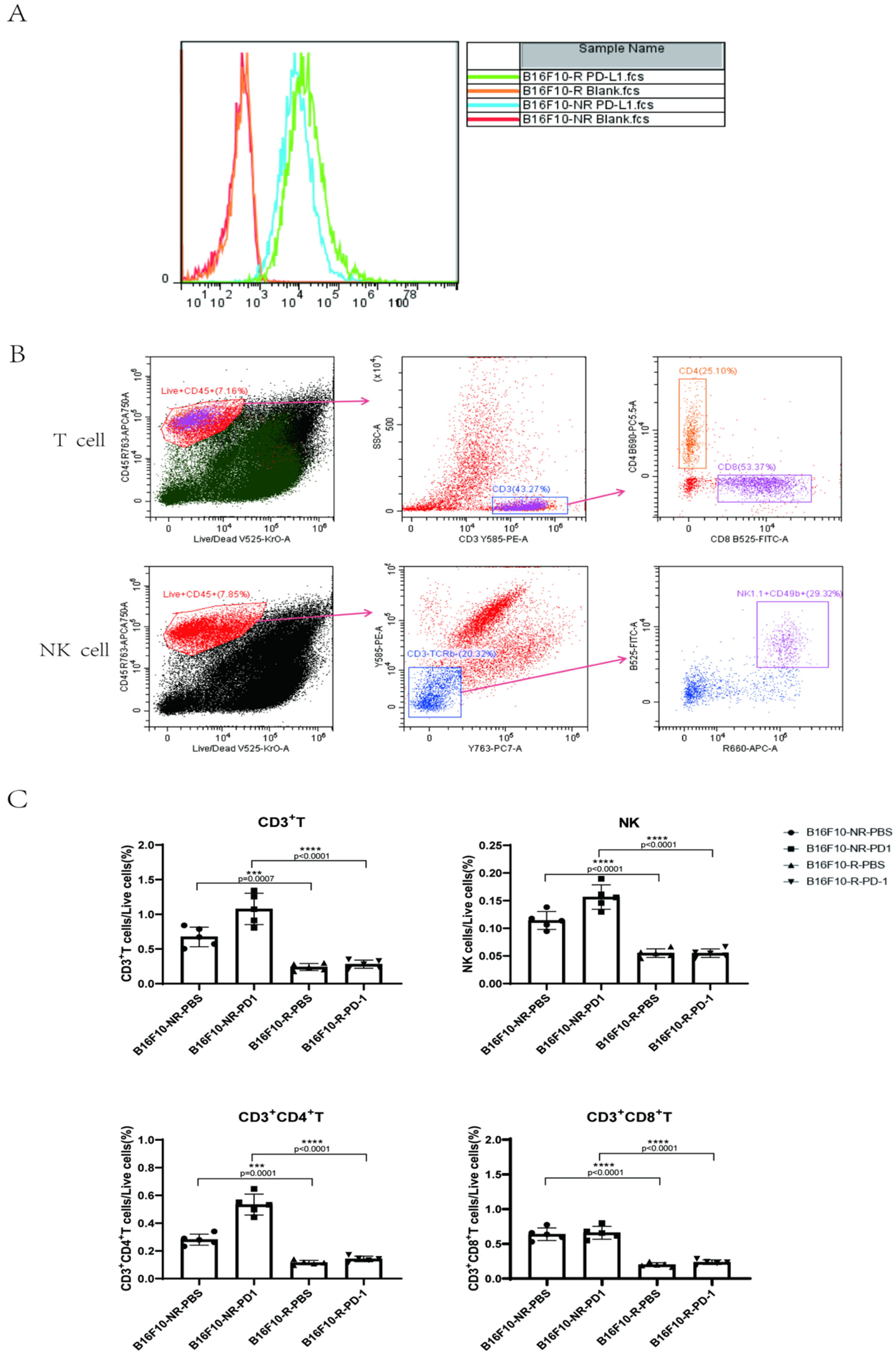
3.1. Development of a B16F10 Tumour Cell Line Resistant to Anti-PD-1 Therapy In Vivo
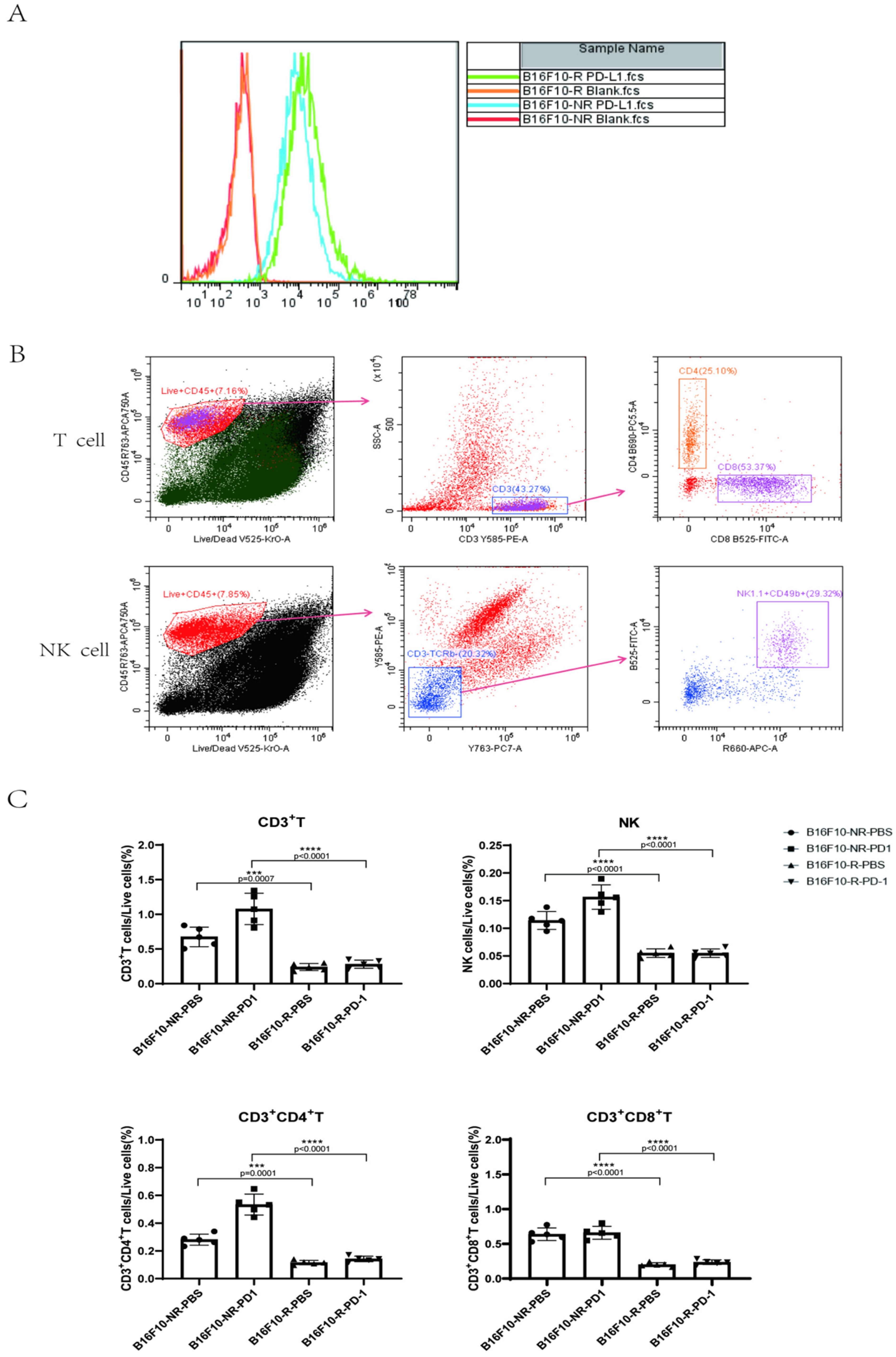
3.2. The Infiltrating Immune Cells Decreased Significantly in the TME of B16F10-R Tumours
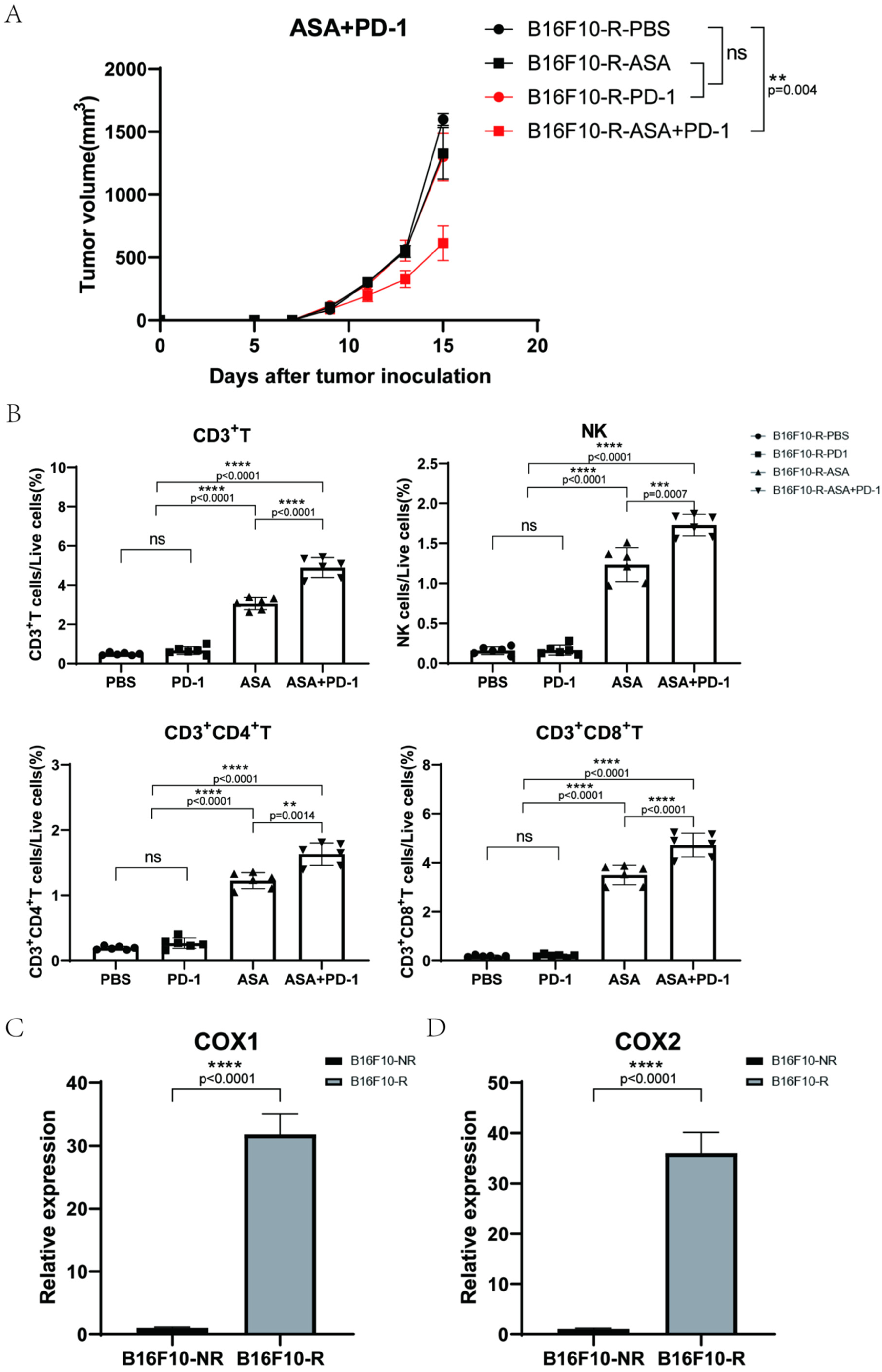
3.3. Aspirin Could Inhibit B16F10-R Tumour Growth
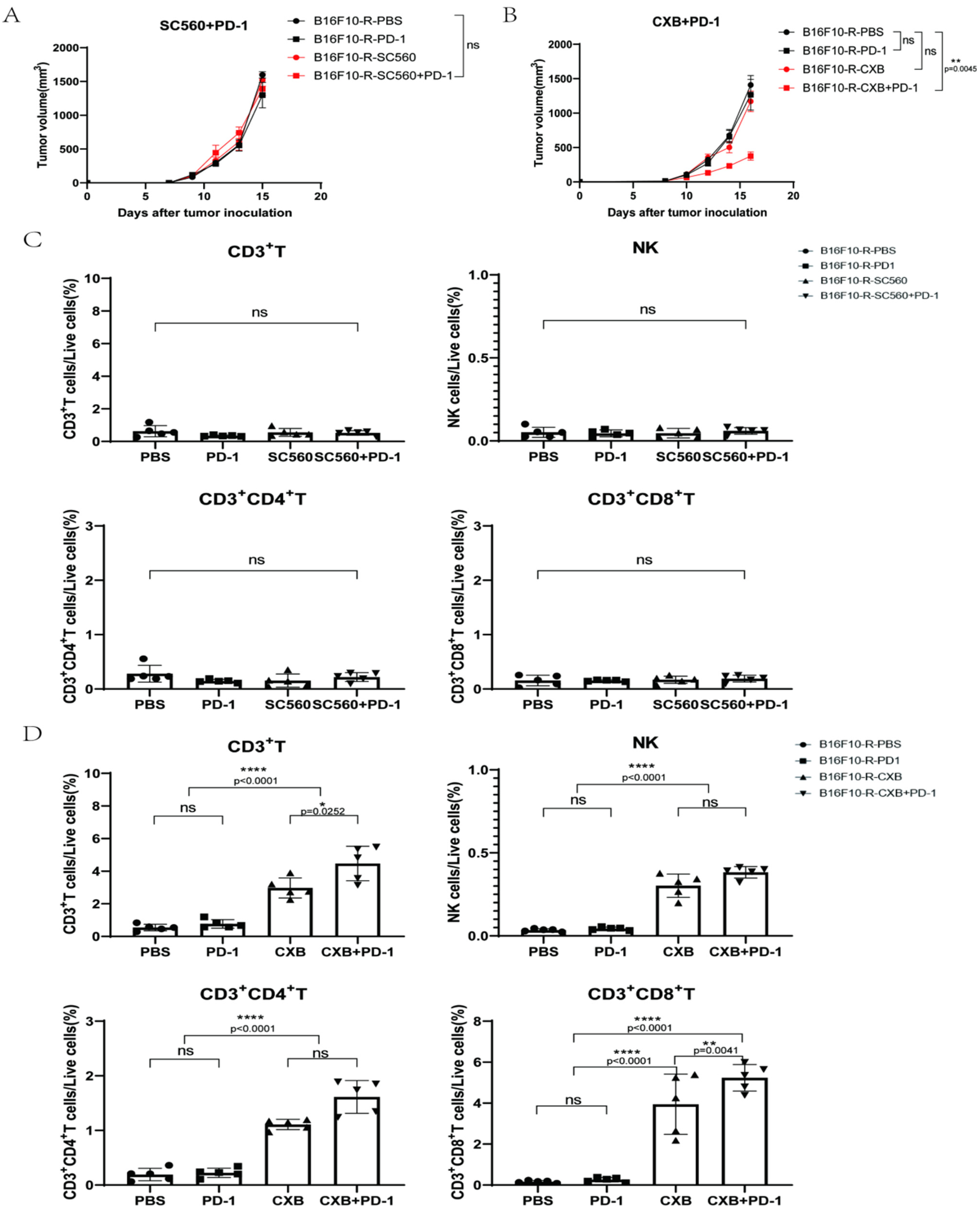
3.4. COX2 Inhibitor Can Inhibit B16F10-R Tumour Growth and Recover Immune Cell Infiltration
3.5. COX2 Knockout Abrogated the Acquired Resistance to Anti-PD-1 Treatment in B16F10-R Tumour Cells
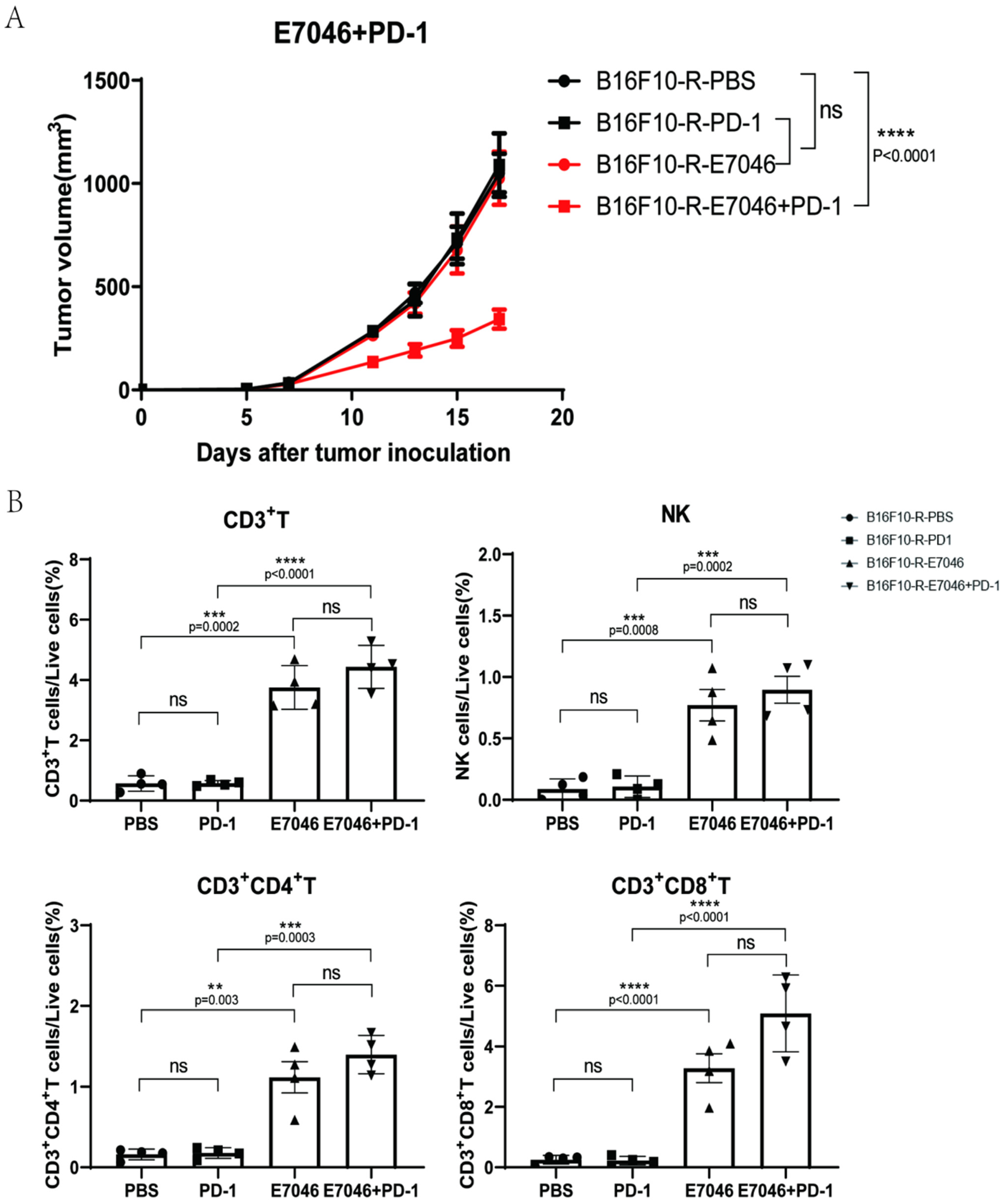
3.6. EP4 Inhibitor Could Inhibit B16F10-R Tumour Growth
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PD-1 | programmed cell death protein 1 |
| PD-L1 | programmed death-ligand 1 |
| COX2 | cyclooxygenase-2 |
| PGE2 | prostaglandin E2 |
| TME | tumour microenvironment |
| NK cells | natural killer cells |
References
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Zou, W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat. Rev. Cancer 2005, 5, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Salmaninejad, A.; Valilou, S.F.; Shabgah, A.G.; Aslani, S.; Alimardani, M.; Pasdar, A.; Sahebkar, A. PD-1/PD-L1 pathway: Basic biology and role in cancer immunotherapy. J. Cell. Physiol. 2019, 234, 16824–16837. [Google Scholar] [CrossRef]
- Frydenlund, N.; Mahalingam, M. PD-L1 and immune escape: Insights from melanoma and other lineage-unrelated malignancies. Hum. Pathol. 2017, 66, 13–33. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Chen, L. Inhibitory B7-family molecules in the tumour microenvironment. Nat. Rev. Immunol. 2008, 8, 467–477. [Google Scholar] [CrossRef]
- Tsushima, F.; Yao, S.; Shin, T.; Flies, A.; Flies, S.; Xu, H.; Tamada, K.; Pardoll, D.M.; Chen, L. Interaction between B7-H1 and PD-1 determines initiation and reversal of T-cell anergy. Blood 2007, 110, 180–185. [Google Scholar] [CrossRef]
- Daskivich, T.J.; Belldegrun, A. Words of wisdom. Re: Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. Eur. Urol. 2015, 67, 816–817. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.M.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef]
- Zou, W.; Wolchok, J.D.; Chen, L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 2016, 8, 328rv4. [Google Scholar] [CrossRef]
- Balar, A.V.; Weber, J.S. PD-1 and PD-L1 antibodies in cancer: Current status and future directions. Cancer Immunol. Immunother. 2017, 66, 551–564. [Google Scholar] [CrossRef]
- Bu, X.; Mahoney, K.M.; Freeman, G.J. Learning from PD-1 resistance: New combination strategies. Trends Mol. Med. 2016, 22, 448–451. [Google Scholar] [CrossRef] [PubMed]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Curran, M.A.; Montalvo, W.; Yagita, H.; Allison, J.P. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc. Natl. Acad. Sci. USA 2010, 107, 4275–4280. [Google Scholar] [CrossRef] [PubMed]
- Dirks, J.; Egli, A.; Sester, U.; Sester, M.; Hirsch, H.H. Blockade of programmed death receptor-1 signaling restores expression of mostly proinflammatory cytokines in anergic cytomegalovirus-specific T cells. Transpl. Infect. Dis. 2013, 15, 79–89. [Google Scholar] [CrossRef]
- Kunzmann, A.T.; Murray, L.J.; Cardwell, C.R.; McShane, C.M.; McMenamin, U.C.; Cantwell, M.M. PTGS2 (cyclooxygenase-2) expression and survival among colorectal cancer patients: A systematic review. Cancer Epidemiol. Biomarkers Prev. 2013, 22, 1490–1497. [Google Scholar] [CrossRef]
- Negi, R.R.; Rana, S.V.; Gupta, V.; Gupta, R.; Chadha, V.D.; Prasad, K.K.; Dhawan, D.K. Over-expression of cyclooxygenase-2 in Colorectal Cancer Patients. Asian Pac. J. Cancer Prev. 2019, 20, 1675–1681. [Google Scholar] [CrossRef]
- Xie, X.Q.; Luo, Y.; Ma, X.L.; Li, S.S.; Liu, L.; Zhang, H.; Li, P.; Wang, F. Clinical significance of circulating tumor cells and their expression of cyclooxygenase-2 in patients with nasopharyngeal carcinoma. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 6951–6961. [Google Scholar] [CrossRef]
- Nagaraju, G.P.; El-Rayes, B.F. Cyclooxygenase-2 in gastrointestinal malignancies. Cancer 2019, 125, 1221–1227. [Google Scholar] [CrossRef]
- Harris, R.E.; Casto, B.C.; Harris, Z.M. Cyclooxygenase-2 and the inflammogenesis of breast cancer. World J. Clin. Oncol. 2014, 5, 677–692. [Google Scholar] [CrossRef]
- Zelenay, S.; van der Veen, A.G.; Böttcher, J.P.; Snelgrove, K.J.; Rogers, N.; Acton, S.E.; Chakravarty, P.; Girotti, M.R.; Marais, R.; Quezada, S.A. Cyclooxygenase-dependent tumor growth through evasion of immunity. Cell 2015, 162, 1257–1270. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.H.; Perry, C.J.; Tsui, Y.C.; Staron, M.M.; Parish, I.A.; Dominguez, C.X.; Rosenberg, D.W.; Kaech, S.M. Prostaglandin E2 and programmed cell death 1 signaling coordinately impair CTL function and survival during chronic viral infection. Nat. Med. 2015, 21, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Kurtova, A.V.; Xiao, J.; Mo, Q.; Pazhanisamy, S.; Krasnow, R.; Lerner, S.P.; Chen, F.; Roh, T.T.; Lay, E.; Ho, P.L. Blocking PGE2-induced tumour repopulation abrogates bladder cancer chemoresistance. Nature 2015, 517, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Hangai, S.; Ao, T.; Kimura, Y.; Matsuki, K.; Kawamura, T.; Negishi, H.; Nishio, J.; Kodama, T.; Taniguchi, T.; Yanai, H. PGE2 induced in and released by dying cells functions as an inhibitory DAMP. Proc. Natl. Acad. Sci. USA 2016, 113, 3844–3849. [Google Scholar] [CrossRef] [PubMed]
- Larsson, K.; Kock, A.; Idborg, H.; Arsenian Henriksson, M.; Martinsson, T.; Johnsen, J.I.; Korotkova, M.; Kogner, P.; Jakobsson, P.-J. COX/mPGES-1/PGE2 pathway depicts an inflammatory-dependent high-risk neuroblastoma subset. Proc. Natl. Acad. Sci. USA 2015, 112, 8070–8075. [Google Scholar] [CrossRef]
- Xu, L.; Stevens, J.; Hilton, M.B.; Seaman, S.; Conrads, T.P.; Veenstra, T.D.; Logsdon, D.; Morris, H.; Swing, D.A.; Patel, N.L.; et al. COX-2 inhibition potentiates antiangiogenic cancer therapy and prevents metastasis in preclinical models. Sci. Transl. Med. 2014, 6, 242ra84. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Xie, K.; Li, B.; Xu, L.; Huang, L.; Feng, Y.; Pi, C.; Zhang, J.; Huang, T.; Jiang, M.; et al. Adaptive resistance in tumors to anti-PD-1 therapy through re-immunosuppression by upregulation of GPNMB expression. Int. Immunopharmacol. 2021, 101, 108199. [Google Scholar] [CrossRef]
- Böttcher, J.P.; Bonavita, E.; Chakravarty, P.; Blees, H.; Cabeza-Cabrerizo, M.; Sammicheli, S.; Rogers, N.C.; Sahai, E.; Zelenay, S.; e Sousa, C.R. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell 2018, 172, 1022–1037.e14. [Google Scholar] [CrossRef]
- Narumiya, S.; Sugimoto, Y.; Ushikubi, F. Prostanoid receptors: Structures, properties, and functions. Physiol. Rev. 1999, 79, 1193–1226. [Google Scholar] [CrossRef]
- Ma, X.; Holt, D.; Kundu, N.; Reader, J.; Goloubeva, O.; Take, Y.; Fulton, A.M. A prostaglandin E (PGE) receptor EP4 antagonist protects natural killer cells from PGE2-mediated immunosuppression and inhibits breast cancer metastasis. Oncoimmunology 2013, 2, e22647. [Google Scholar] [CrossRef]
- Huang, H.; Aladelokun, O.; Ideta, T.; Giardina, C.; Ellis, L.M.; Rosenberg, D.W. Inhibition of PGE 2/EP4 receptor signaling enhances oxaliplatin efficacy in resistant colon cancer cells through modulation of oxidative stress. Sci. Rep. 2019, 9, 4954. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Li, S.; Zhou, C.; Li, R.; Wang, H.; Luo, W.; Huang, Y.-S.; Chen, L.-K.; Cai, J.-L.; Wang, T.-X.; et al. Cisplatin induces chemoresistance through the PTGS2-mediated anti-apoptosis in gastric cancer. Int. J. Biochem. Cell B 2019, 116, 105610. [Google Scholar] [CrossRef] [PubMed]
- Bardhan, K.; Anagnostou, T.; Boussiotis, V.A. The PD1:PD-L1/2 pathway from discovery to clinical implementation. Front Immunol. 2016, 7, 550. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Ohashi, P.S. Clinical blockade of PD1 and LAG3--Potential mechanisms of action. Nat. Rev. Immunol. 2015, 15, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A. Adaptive immune resistance: How cancer protects from immune attack. Cancer Discov. 2015, 5, 915–919. [Google Scholar] [CrossRef]
- Kobayashi, K.; Kaira, K.; Kagamu, H. Recovery of the sensitivity to anti-PD-1 antibody by celecoxib in lung cancer. Anticancer Res. 2020, 40, 5309–5311. [Google Scholar] [CrossRef]
- Fujita, M.; Kohanbash, G.; Fellows-Mayle, W.; Hamilton, R.L.; Komohara, Y.; Decker, S.A.; Ohlfest, J.R.; Okada, H. COX-2 blockade suppresses gliomagenesis by inhibiting myeloid-derived suppressor cells. Cancer Res. 2011, 71, 2664–2674. [Google Scholar] [CrossRef]
- Mao, Y.; Sarhan, D.; Steven, A.; Seliger, B.; Kiessling, R.; Lundqvist, A. Inhibition of tumor-derived prostaglandin-E2 blocks the induction of myeloid-derived suppressor cells and recovers natural killer cell activity. Clin. Cancer Res. 2014, 20, 4096–4106. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pi, C.; Jing, P.; Li, B.; Feng, Y.; Xu, L.; Xie, K.; Huang, T.; Xu, X.; Gu, H.; Fang, J. Reversing PD-1 Resistance in B16F10 Cells and Recovering Tumour Immunity Using a COX2 Inhibitor. Cancers 2022, 14, 4134. https://doi.org/10.3390/cancers14174134
Pi C, Jing P, Li B, Feng Y, Xu L, Xie K, Huang T, Xu X, Gu H, Fang J. Reversing PD-1 Resistance in B16F10 Cells and Recovering Tumour Immunity Using a COX2 Inhibitor. Cancers. 2022; 14(17):4134. https://doi.org/10.3390/cancers14174134
Chicago/Turabian StylePi, Chenyu, Ping Jing, Bingyu Li, Yan Feng, Lijun Xu, Kun Xie, Tao Huang, Xiaoqing Xu, Hua Gu, and Jianmin Fang. 2022. "Reversing PD-1 Resistance in B16F10 Cells and Recovering Tumour Immunity Using a COX2 Inhibitor" Cancers 14, no. 17: 4134. https://doi.org/10.3390/cancers14174134
APA StylePi, C., Jing, P., Li, B., Feng, Y., Xu, L., Xie, K., Huang, T., Xu, X., Gu, H., & Fang, J. (2022). Reversing PD-1 Resistance in B16F10 Cells and Recovering Tumour Immunity Using a COX2 Inhibitor. Cancers, 14(17), 4134. https://doi.org/10.3390/cancers14174134