
Modeling Colorectal Cancer Progression Reveals Niche-Dependent Clonal Selection
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
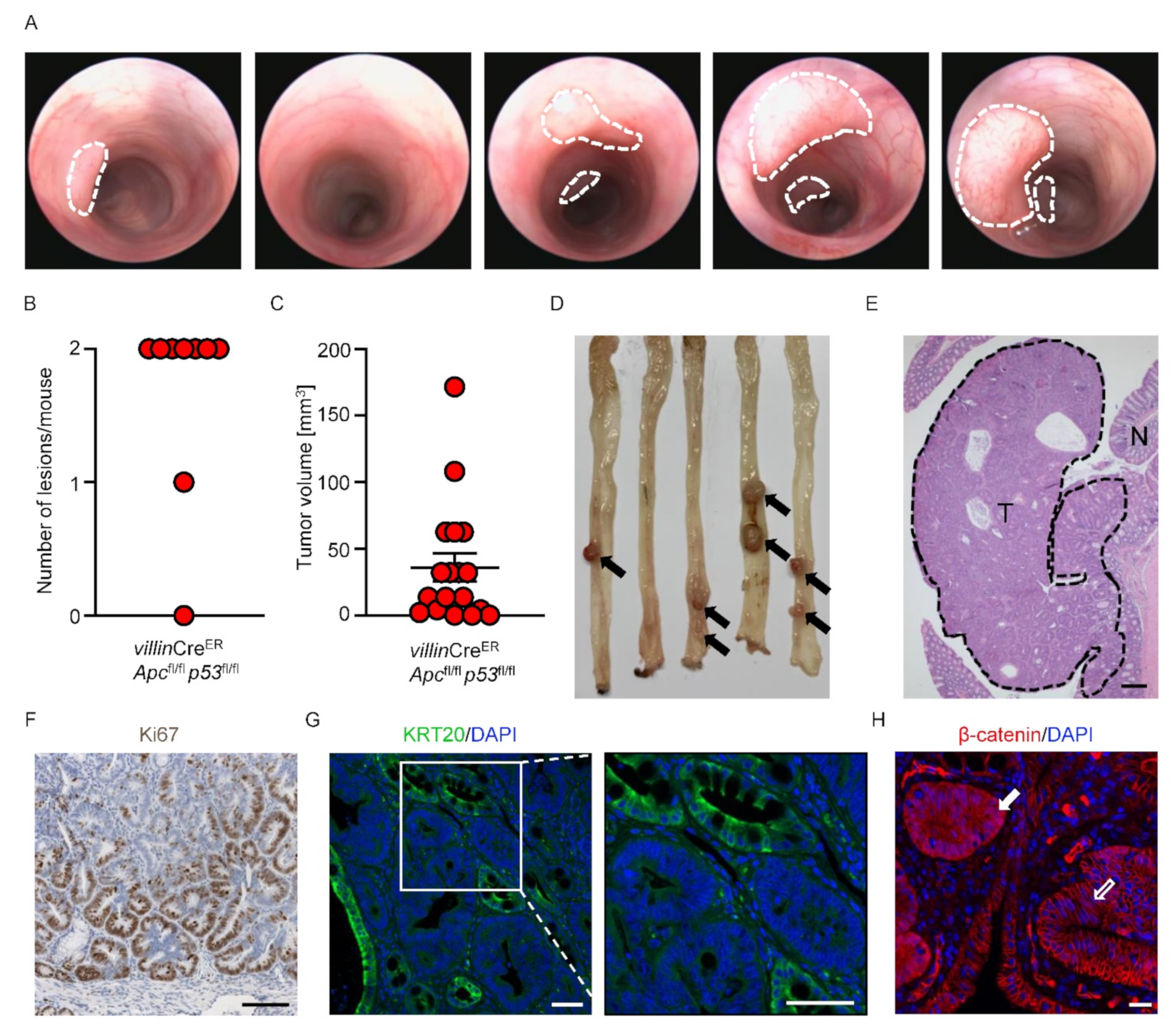
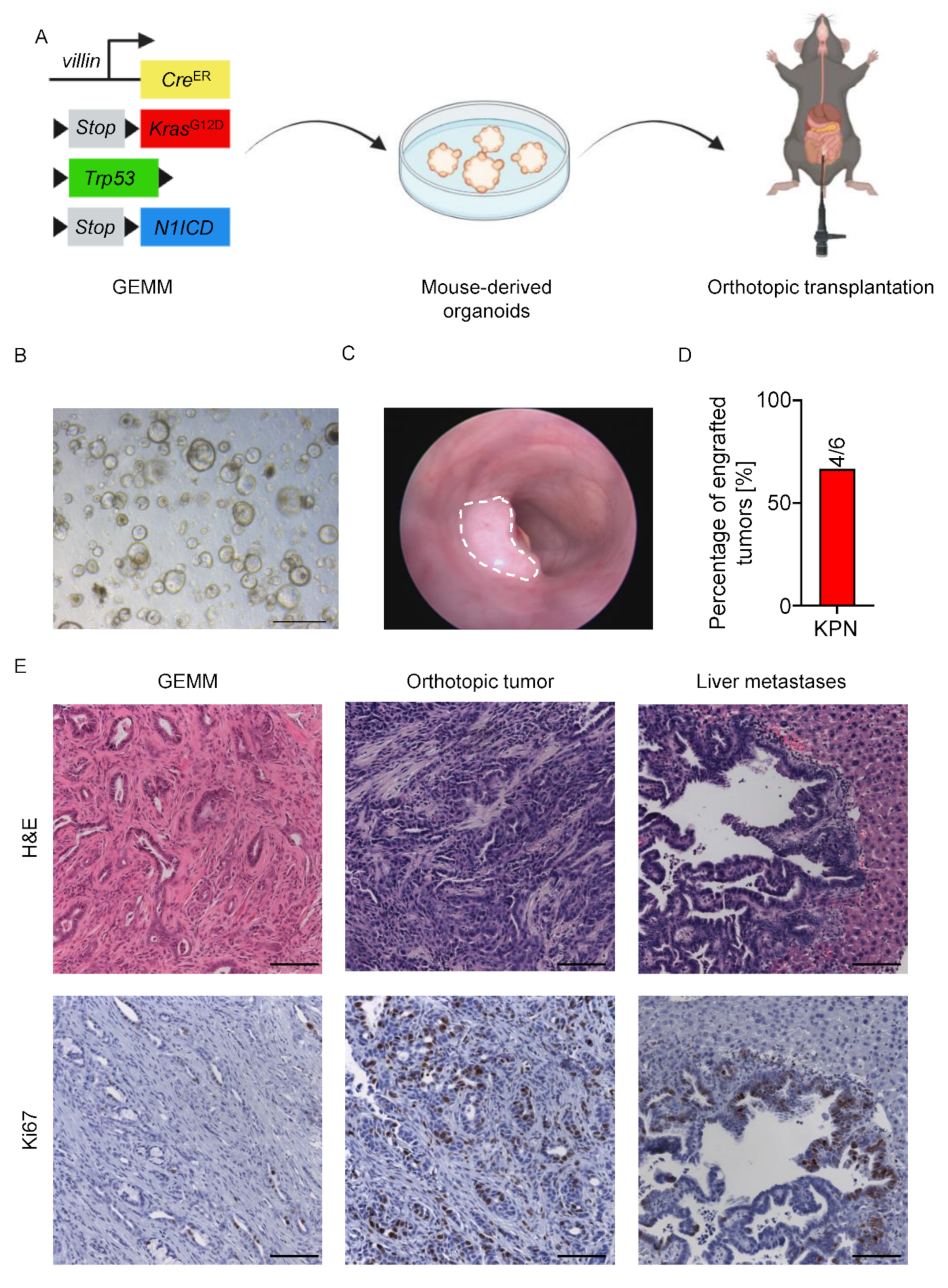
2.1. Animal Models
2.2. Production of Lentivirus
2.3. Culturing Organoids
2.4. Organoid Transduction and Selection
2.5. Flow Cytometry Analysis
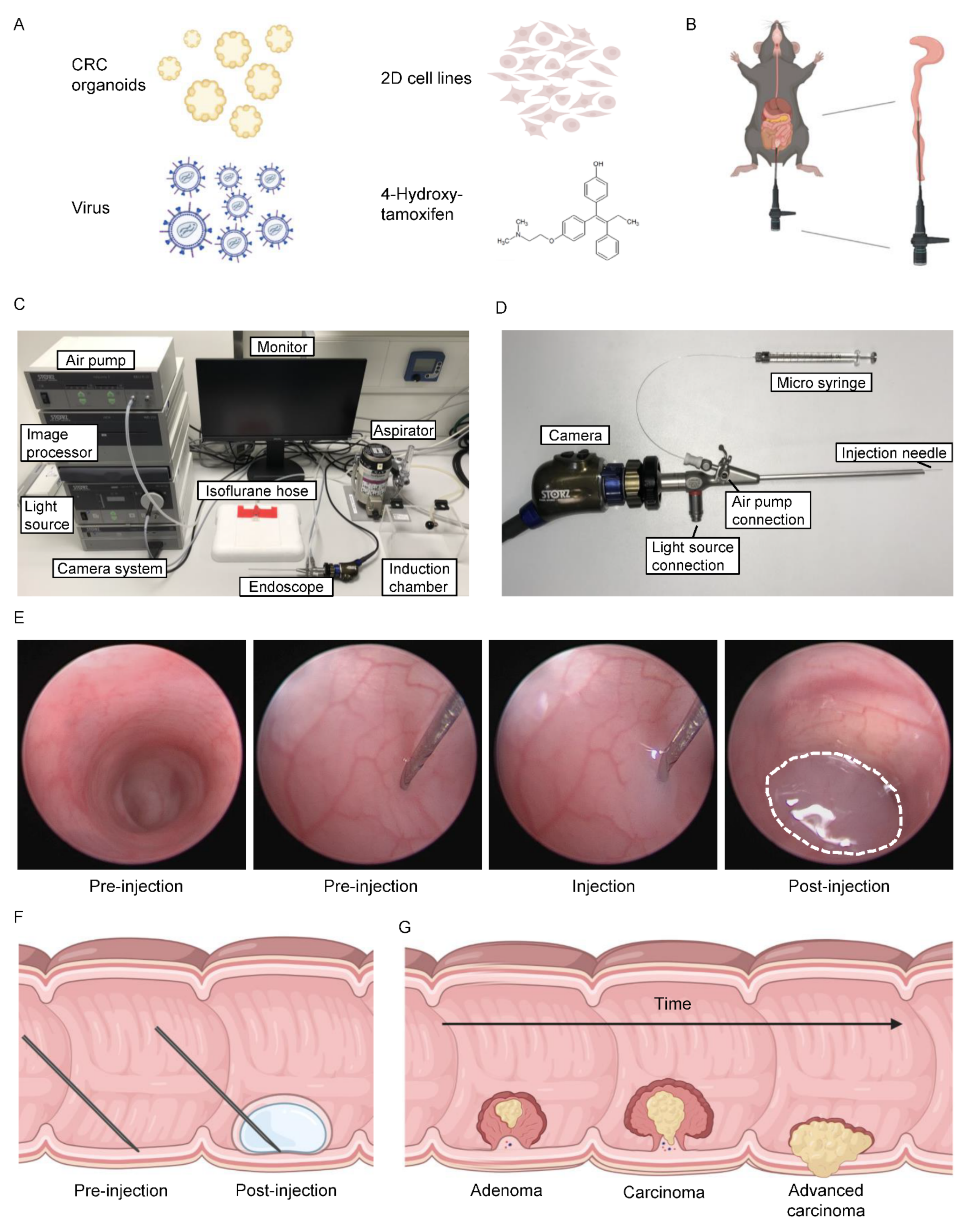
2.6. Murine Colonoscopy-Guided Mucosal Injection
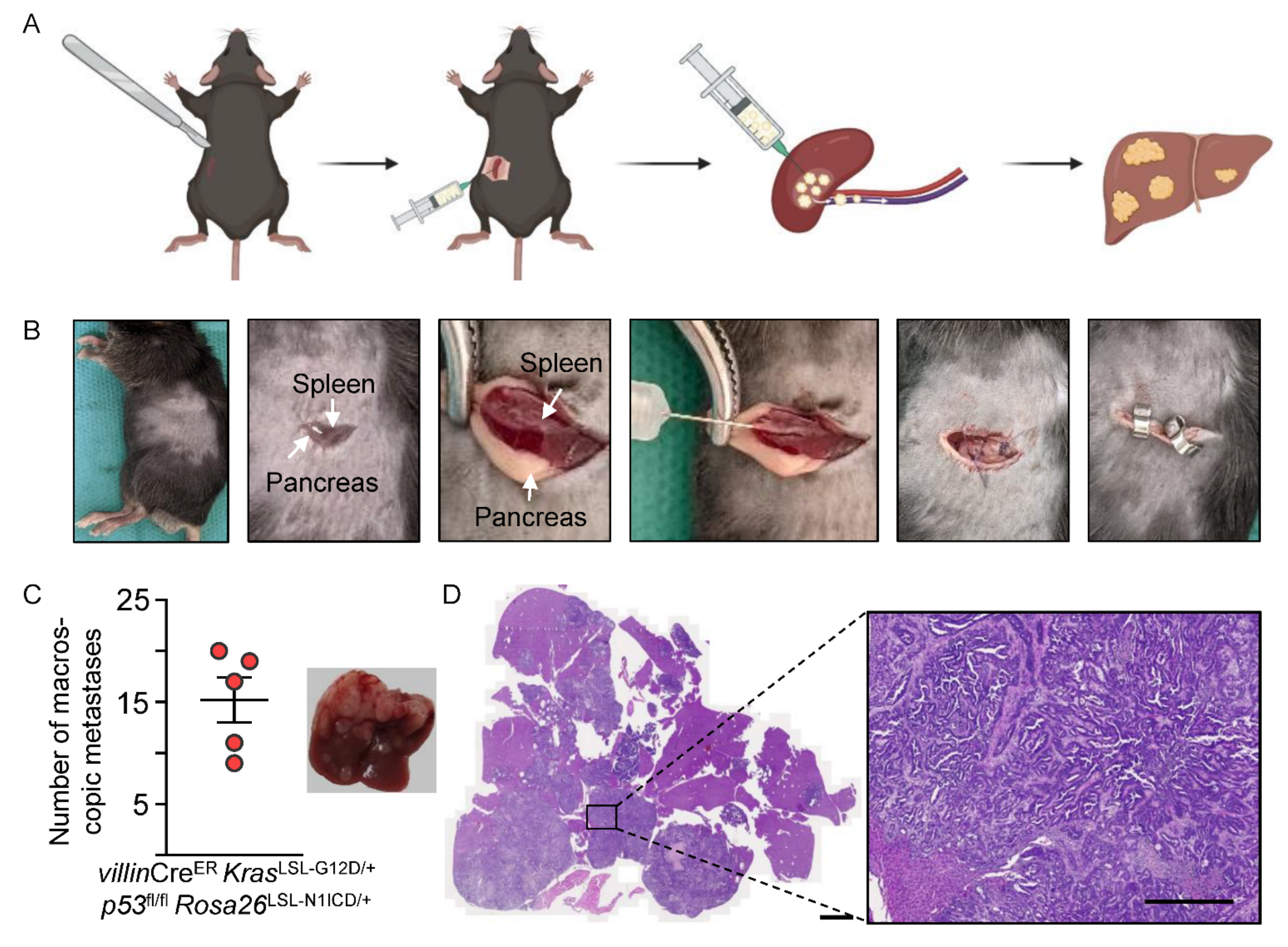
2.7. Intrasplenic Injection
2.8. Sub-Cutaneous Injection and Tumor Monitoring
2.9. Histology
2.10. Immunofluorescence
2.11. Immunohistochemistry
3. Results
3.1. Versatile Application of Orthotopic Colonoscopy-Guided Injections
3.2. Generation of Autochthonous Colonic Tumors
3.3. Orthotopic Mouse-Derived Organoid Transplantation
3.4. Modeling Liver-Specific Metastasis
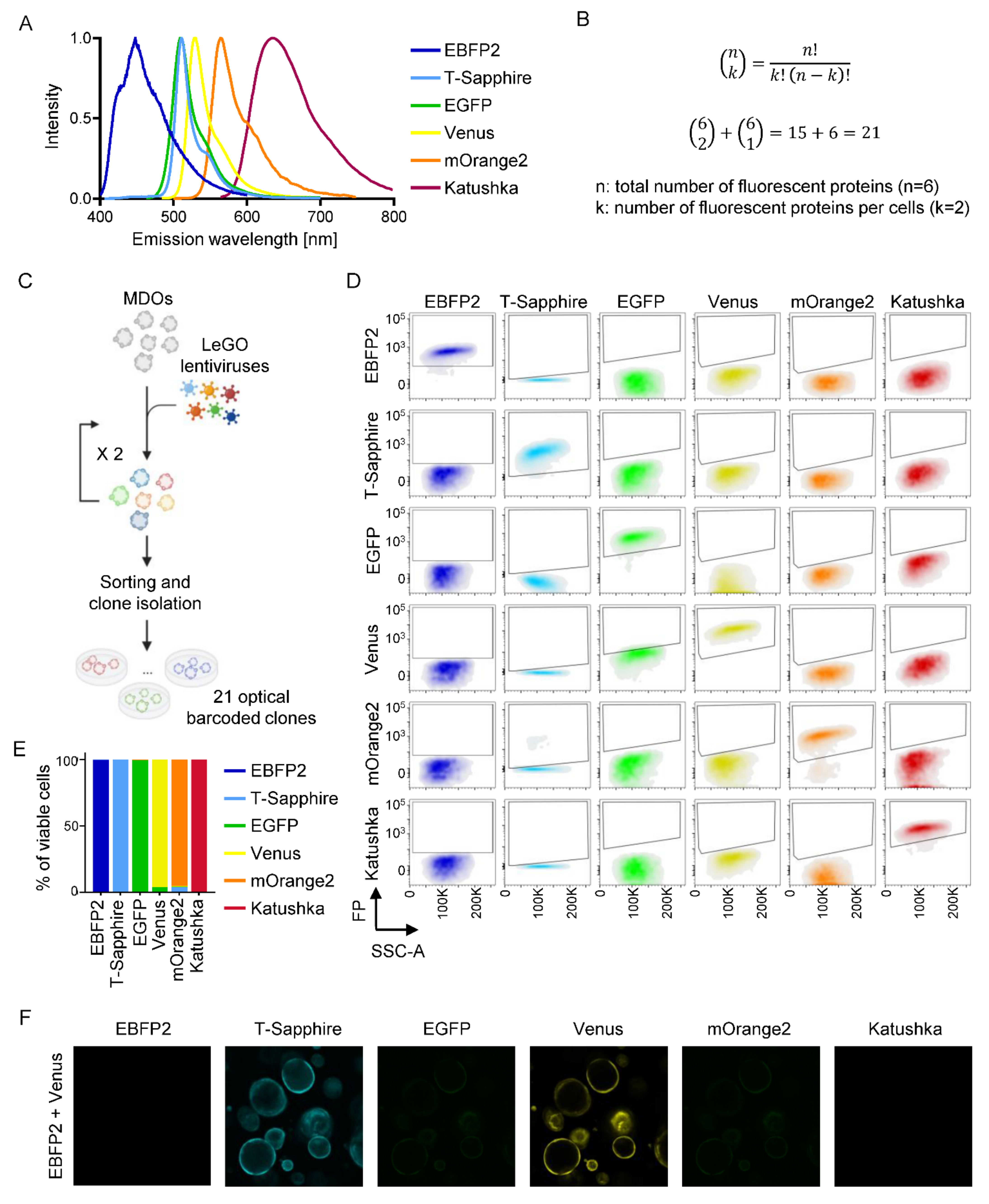
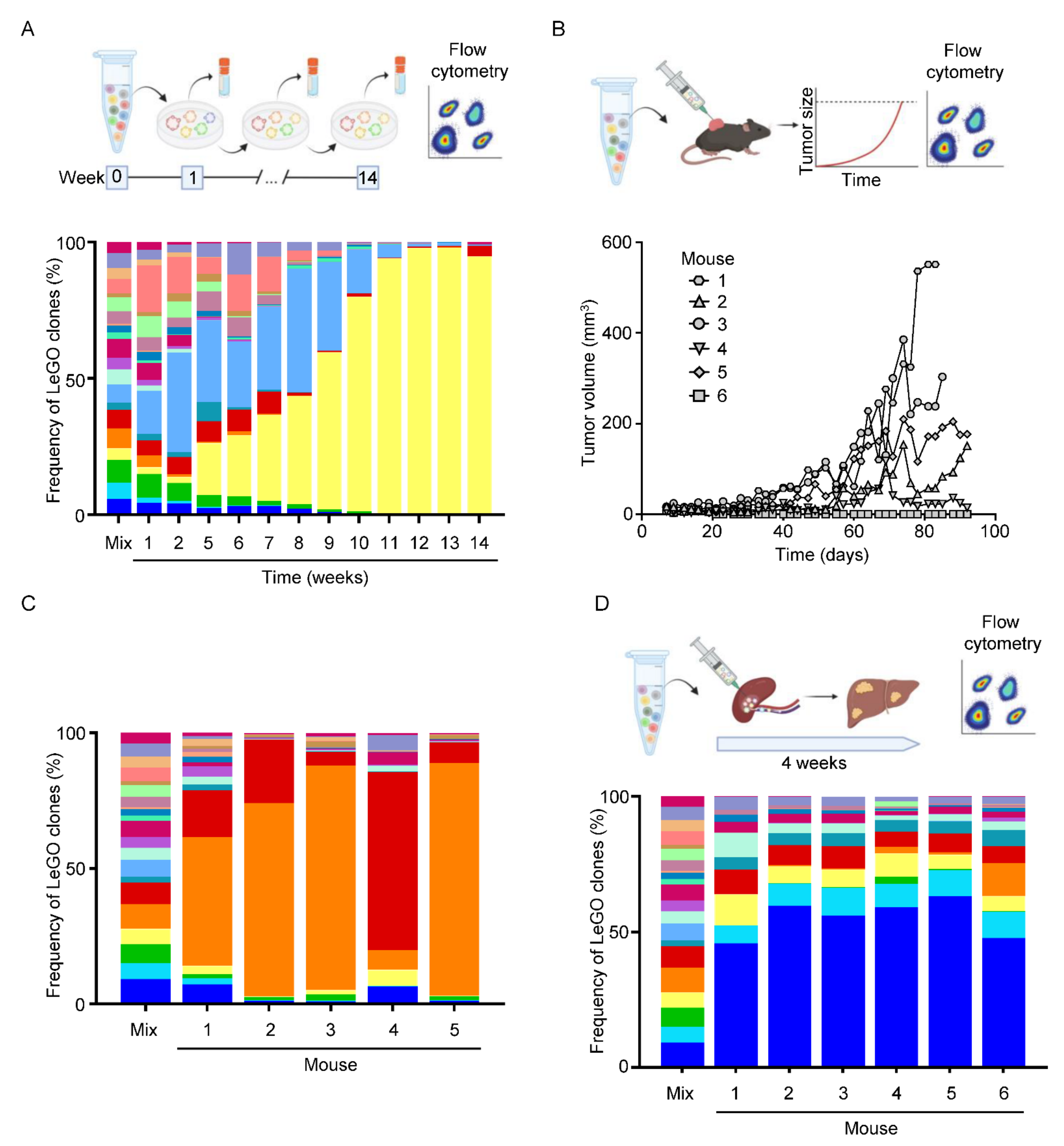
3.5. Optical Barcoding Reveals Niche-Dependent Clonal Selection
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Chen, C.; Holleczek, B.; Schöttker, B.; Hoffmeister, M.; Brenner, H. Strong Reduction of Colorectal Cancer Incidence and Mortality after Screening Colonoscopy: Prospective Cohort Study from Germany. Am. J. Gastroenterol. 2021, 116, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Goding Sauer, A.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef]
- Van den Berg, I.; van den Braak, R.R.J.C.; van Vugt, J.L.A.; Ijzermans, J.N.M.; Buettner, S. Actual survival after resection of primary colorectal cancer: Results from a prospective multicenter study. World J. Surg. Oncol. 2021, 19, 96. [Google Scholar] [CrossRef]
- Siravegna, G.; Mussolin, B.; Buscarino, M.; Corti, G.; Cassingena, A.; Crisafulli, G.; Ponzetti, A.; Cremolini, C.; Amatu, A.; Lauricella, C.; et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat. Med. 2015, 21, 795–801. [Google Scholar] [CrossRef]
- Angelova, M.; Mlecnik, B.; Vasaturo, A.; Bindea, G.; Fredriksen, T.; Lafontaine, L.; Buttard, B.; Morgand, E.; Bruni, D.; Jouret-Mourin, A.; et al. Evolution of Metastases in Space and Time under Immune Selection. Cell 2018, 175, 751–765.e16. [Google Scholar] [CrossRef]
- Jackstadt, R.; Sansom, O.J. Mouse models of intestinal cancer. J. Pathol. 2016, 238, 141–151. [Google Scholar] [CrossRef]
- Avolio, M.; Trusolino, L. Rational Treatment of Metastatic Colorectal Cancer: A Reverse Tale of Men, Mice, and Culture Dishes. Cancer Discov. 2021, 11, 1644–1660. [Google Scholar] [CrossRef]
- Yaeger, R.; Chatila, W.K.; Lipsyc, M.D.; Hechtman, J.F.; Cercek, A.; Sanchez-Vega, F.; Jayakumaran, G.; Middha, S.; Zehir, A.; Donoghue, M.T.A.; et al. Clinical Sequencing Defines the Genomic Landscape of Metastatic Colorectal Cancer. Cancer Cell 2018, 33, 125–136.e3. [Google Scholar] [CrossRef] [Green Version]
- Leach, J.D.G.; Vlahov, N.; Tsantoulis, P.; Ridgway, R.A.; Flanagan, D.J.; Gilroy, K.; Sphyris, N.; Vázquez, E.G.; Vincent, D.F.; Faller, W.J.; et al. Oncogenic BRAF, unrestrained by TGFβ-receptor signalling, drives right-sided colonic tumorigenesis. Nat. Commun. 2021, 12, 3464. [Google Scholar] [CrossRef] [PubMed]
- Roper, J.; Tammela, T.; Cetinbas, N.M.; Akkad, A.; Roghanian, A.; Rickelt, S.; Almeqdadi, M.; Wu, K.; Oberli, M.A.; Sánchez-Rivera, F.J.; et al. In vivo genome editing and organoid transplantation models of colorectal cancer and metastasis. Nat. Biotechnol. 2017, 35, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Knight, J.R.P.; Alexandrou, C.; Skalka, G.L.; Vlahov, N.; Pennel, K.; Officer, L.; Teodosio, A.; Kanellos, G.; Gay, D.M.; May-Wilson, S.; et al. MNK inhibition sensitizes KRAS-mutant colorectal cancer to mTORC1 inhibition by reducing eIF4E phosphorylation and c-MYC expression. Cancer Discov. 2020, 11, 1228–1247. [Google Scholar] [CrossRef] [PubMed]
- Gay, D.M.; Ridgway, R.A.; Müller, M.; Hodder, M.C.; Hedley, A.; Clark, W.; Leach, J.D.; Jackstadt, R.; Nixon, C.; Huels, D.J.; et al. Loss of BCL9/9l suppresses Wnt driven tumourigenesis in models that recapitulate human cancer. Nat. Commun. 2019, 10, 723. [Google Scholar] [CrossRef]
- Ramesh, P.; Lannagan, T.R.M.; Jackstadt, R.; Taboada, L.A.; Lansu, N.; Wirapati, P.; van Hooff, S.R.; Dekker, D.; Pritchard, J.; Kirov, A.B.; et al. BCL-XL is crucial for progression through the adenoma-to-carcinoma sequence of colorectal cancer. Cell Death Differ. 2021, 28, 3282–3296. [Google Scholar] [CrossRef]
- Jackstadt, R.; van Hooff, S.R.; Leach, J.D.; Cortes-Lavaud, X.; Lohuis, J.O.; Ridgway, R.A.; Wouters, V.M.; Roper, J.; Kendall, T.J.; Roxburgh, C.S.; et al. Epithelial NOTCH Signaling Rewires the Tumor Microenvironment of Colorectal Cancer to Drive Poor-Prognosis Subtypes and Metastasis. Cancer Cell 2019, 36, 319–336.e7. [Google Scholar] [CrossRef]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]
- Dunne, P.D.; Alderdice, M.; O’Reilly, P.G.; Roddy, A.C.; McCorry, A.M.B.; Richman, S.; Maughan, T.; McDade, S.S.; Johnston, P.G.; Longley, D.B.; et al. Cancer-cell intrinsic gene expression signatures overcome intratumoural heterogeneity bias in colorectal cancer patient classification. Nat. Commun. 2017, 8, 15657. [Google Scholar] [CrossRef]
- Isella, C.; Brundu, F.; Bellomo, S.E.; Galimi, F.; Zanella, E.; Porporato, R.; Petti, C.; Fiori, A.; Orzan, F.; Senetta, R.; et al. Selective analysis of cancer-cell intrinsic transcriptional traits defines novel clinically relevant subtypes of colorectal cancer. Nat. Commun. 2017, 8, 15107. [Google Scholar] [CrossRef]
- Lannagan, T.R.; Jackstadt, R.; Leedham, S.J.; Sansom, O.J. Advances in colon cancer research: In vitro and animal models. Curr. Opin. Genet. Dev. 2021, 66, 50–56. [Google Scholar] [CrossRef]
- Drost, J.; Clevers, H. Organoids in cancer research. Nat. Rev. Cancer 2018, 18, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Fujii, M.; Shimokawa, M.; Date, S.; Takano, A.; Matano, M.; Nanki, K.; Ohta, Y.; Toshimitsu, K.; Nakazato, Y.; Kawasaki, K.; et al. A Colorectal Tumor Organoid Library Demonstrates Progressive Loss of Niche Factor Requirements during Tumorigenesis. Cell Stem Cell 2016, 18, 827–838. [Google Scholar] [CrossRef] [PubMed]
- Morral, C.; Stanisavljevic, J.; Hernando-Momblona, X.; Mereu, E.; Álvarez-Varela, A.; Cortina, C.; Stork, D.; Slebe, F.; Turon, G.; Whissell, G.; et al. Zonation of Ribosomal DNA Transcription Defines a Stem Cell Hierarchy in Colorectal Cancer. Cell Stem Cell 2020, 26, 845–861.e12. [Google Scholar] [CrossRef] [PubMed]
- Shimokawa, M.; Ohta, Y.; Nishikori, S.; Matano, M.; Takano, A.; Fujii, M.; Date, S.; Sugimoto, S.; Kanai, T.; Sato, T. Visualization and targeting of LGR5+ human colon cancer stem cells. Nature 2017, 545, 187–192. [Google Scholar] [CrossRef]
- De Sousa E Melo, F.; Kurtova, A.V.; Harnoss, J.M.; Kljavin, N.; Hoeck, J.D.; Hung, J.; Anderson, J.E.; Storm, E.E.; Modrusan, Z.; Koeppen, H.; et al. A distinct role for Lgr5+ stem cells in primary and metastatic colon cancer. Nature 2017, 543, 676–680. [Google Scholar] [CrossRef]
- El Marjou, F.; Janssen, K.-P.; Chang, B.H.-J.; Li, M.; Hindie, V.; Chan, L.; Louvard, D.; Chambon, P.; Metzger, D.; Robine, S. Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis 2004, 39, 186–193. [Google Scholar] [CrossRef]
- Kuraguchi, M.; Wang, X.-P.; Bronson, R.T.; Rothenberg, R.; Ohene-Baah, N.Y.; Lund, J.J.; Kucherlapati, M.; Maas, R.L.; Kucherlapati, R. Adenomatous polyposis coli (APC) is required for normal development of skin and thymus. PLoS Genet. 2006, 2, e146. [Google Scholar] [CrossRef]
- Marino, S.; Vooijs, M.; van Der Gulden, H.; Jonkers, J.; Berns, A. Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev. 2000, 14, 994–1004. [Google Scholar] [CrossRef]
- Chen, C.; Neumann, J.; Kühn, F.; Lee, S.M.L.; Drefs, M.; Andrassy, J.; Werner, J.; Bazhin, A.V.; Schiergens, T.S. Establishment of an Endoscopy-Guided Minimally Invasive Orthotopic Mouse Model of Colorectal Cancer. Cancers 2020, 12, 3007. [Google Scholar] [CrossRef]
- Zigmond, E.; Halpern, Z.; Elinav, E.; Brazowski, E.; Jung, S.; Varol, C. Utilization of murine colonoscopy for orthotopic implantation of colorectal cancer. PLoS ONE 2011, 6, e28858. [Google Scholar] [CrossRef] [Green Version]
- Roper, J.; Tammela, T.; Akkad, A.; Almeqdadi, M.; Santos, S.B.; Jacks, T.; Yilmaz, Ö.H. Colonoscopy-based colorectal cancer modeling in mice with CRISPR-Cas9 genome editing and organoid transplantation. Nat. Protoc. 2018, 13, 217–234. [Google Scholar] [CrossRef] [PubMed]
- Najumudeen, A.K.; Ceteci, F.; Fey, S.K.; Hamm, G.; Steven, R.T.; Hall, H.; Nikula, C.J.; Dexter, A.; Murta, T.; Race, A.M.; et al. The amino acid transporter SLC7A5 is required for efficient growth of KRAS-mutant colorectal cancer. Nat. Genet. 2021, 53, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Reiter, J.G.; Hung, W.-T.; Lee, I.-H.; Nagpal, S.; Giunta, P.; Degner, S.; Liu, G.; Wassenaar, E.C.E.; Jeck, W.R.; Taylor, M.S.; et al. Lymph node metastases develop through a wider evolutionary bottleneck than distant metastases. Nat. Genet. 2020, 52, 692–700. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Ding, J.; Ma, Z.; Sun, R.; Seoane, J.A.; Shaffer, J.S.; Suarez, C.J.; Berghoff, A.S.; Cremolini, C.; Falcone, A.; et al. Quantitative evidence for early metastatic seeding in colorectal cancer. Nat. Genet. 2019, 51, 1113–1122. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Li, Z.; Ma, Z.; Curtis, C. Multi-cancer analysis of clonality and the timing of systemic spread in paired primary tumors and metastases. Nat. Genet. 2020, 52, 701–708. [Google Scholar] [CrossRef]
- Mohme, M.; Maire, C.L.; Riecken, K.; Zapf, S.; Aranyossy, T.; Westphal, M.; Lamszus, K.; Fehse, B. Optical Barcoding for Single-Clone Tracking to Study Tumor Heterogeneity. Mol. Ther. 2017, 25, 621–633. [Google Scholar] [CrossRef]
- De Sousa E Melo, F.; Harnoss, J.M.; Kljavin, N.; Scott, R.; Sohn, C.; Leong, K.G.; de Sauvage, F.J. Modeling Colorectal Cancer Progression through Orthotopic Implantation of Organoids. Methods Mol. Biol. 2020, 2171, 331–346. [Google Scholar]
- Enquist, I.B.; Good, Z.; Jubb, A.M.; Fuh, G.; Wang, X.; Junttila, M.R.; Jackson, E.L.; Leong, K.G. Lymph node-independent liver metastasis in a model of metastatic colorectal cancer. Nat. Commun. 2014, 5, 3530. [Google Scholar] [CrossRef]
- Kasashima, H.; Duran, A.; Cid-Diaz, T.; Muta, Y.; Kinoshita, H.; Batlle, E.; Diaz-Meco, M.T.; Moscat, J. Mouse model of colorectal cancer: Orthotopic co-implantation of tumor and stroma cells in cecum and rectum. STAR Protoc. 2021, 2, 100297. [Google Scholar] [CrossRef]
- Fumagalli, A.; Drost, J.; Suijkerbuijk, S.J.E.; van Boxtel, R.; de Ligt, J.; Offerhaus, G.J.; Begthel, H.; Beerling, E.; Tan, E.H.; Sansom, O.J.; et al. Genetic dissection of colorectal cancer progression by orthotopic transplantation of engineered cancer organoids. Proc. Natl. Acad. Sci. USA 2017, 114, E2357–E2364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cullin, N.; Antunes, C.A.; Straussman, R.; Stein-Thoeringer, C.K.; Elinav, E. Microbiome and cancer. Cancer Cell 2021, 39, 1317–1341. [Google Scholar] [CrossRef] [PubMed]
- Betzler, A.M.; Nanduri, L.K.; Hissa, B.; Blickensdörfer, L.; Muders, M.H.; Roy, J.; Jesinghaus, M.; Steiger, K.; Weichert, W.; Kloor, M.; et al. Differential Effects of Trp53 Alterations in Murine Colorectal Cancer. Cancers 2021, 13, 808. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, M.; Mizutani, T.; Mochizuki, W.; Matsumoto, T.; Nozaki, K.; Sakamaki, Y.; Ichinose, S.; Okada, Y.; Tanaka, T.; Watanabe, M.; et al. Small intestinal stem cell identity is maintained with functional Paneth cells in heterotopically grafted epithelium onto the colon. Genes Dev. 2014, 28, 1752–1757. [Google Scholar] [CrossRef]
- Sugimoto, S.; Ohta, Y.; Fujii, M.; Matano, M.; Shimokawa, M.; Nanki, K.; Date, S.; Nishikori, S.; Nakazato, Y.; Nakamura, T.; et al. Reconstruction of the Human Colon Epithelium In Vivo. Cell Stem Cell 2018, 22, 171–176.e5. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, K.; Wu, C.; O’Rourke, K.P.; Szeglin, B.C.; Zheng, Y.; Sauvé, C.-E.G.; Adileh, M.; Wasserman, I.; Marco, M.R.; Kim, A.S.; et al. A rectal cancer organoid platform to study individual responses to chemoradiation. Nat. Med. 2019, 25, 1607–1614. [Google Scholar] [CrossRef]
- Yui, S.; Azzolin, L.; Maimets, M.; Pedersen, M.T.; Fordham, R.P.; Hansen, S.L.; Larsen, H.L.; Guiu, J.; Alves, M.R.P.; Rundsten, C.F.; et al. YAP/TAZ-Dependent Reprogramming of Colonic Epithelium Links ECM Remodeling to Tissue Regeneration. Cell Stem Cell 2018, 22, 35–49.e7. [Google Scholar] [CrossRef]
- Yui, S.; Nakamura, T.; Sato, T.; Nemoto, Y.; Mizutani, T.; Zheng, X.; Ichinose, S.; Nagaishi, T.; Okamoto, R.; Tsuchiya, K.; et al. Functional engraftment of colon epithelium expanded in vitro from a single adult Lgr5+ stem cell. Nat. Med. 2012, 18, 618–623. [Google Scholar] [CrossRef]
- O’Rourke, K.P.; Loizou, E.; Livshits, G.; Schatoff, E.M.; Baslan, T.; Manchado, E.; Simon, J.; Romesser, P.B.; Leach, B.; Han, T.; et al. Transplantation of engineered organoids enables rapid generation of metastatic mouse models of colorectal cancer. Nat. Biotechnol. 2017, 35, 577–582. [Google Scholar] [CrossRef]
- Jackstadt, R.; Röh, S.; Neumann, J.; Jung, P.; Hoffmann, R.; Horst, D.; Berens, C.; Bornkamm, G.W.; Kirchner, T.; Menssen, A.; et al. AP4 is a mediator of epithelial-mesenchymal transition and metastasis in colorectal cancer. J. Exp. Med. 2013, 210, 1331–1350. [Google Scholar] [CrossRef]
- Tomlinson, J.S.; Jarnagin, W.R.; DeMatteo, R.P.; Fong, Y.; Kornprat, P.; Gonen, M.; Kemeny, N.; Brennan, M.F.; Blumgart, L.H.; D’Angelica, M. Actual 10-year survival after resection of colorectal liver metastases defines cure. J. Clin. Oncol. 2007, 25, 4575–4580. [Google Scholar] [CrossRef] [PubMed]
- Fridman, W.H.; Miller, I.; Sautès-Fridman, C.; Byrne, A.T. Therapeutic Targeting of the Colorectal Tumor Stroma. Gastroenterology 2020, 158, 303–321. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Wang, X.; Lu, J.; Salfenmoser, M.; Wirsik, N.M.; Schleußner, N.; Imle, A.; Valls, A.F.; Radhakrishnan, P.; Liang, J.; et al. Reduction of Liver Metastasis Stiffness Improves Response to Bevacizumab in Metastatic Colorectal Cancer. Cancer Cell 2020, 37, 800–817.e7. [Google Scholar] [CrossRef] [PubMed]
- Gambaro, K.; Marques, M.; McNamara, S.; du Tertre, M.C.; Diaz, Z.; Hoffert, C.; Srivastava, A.; Hébert, S.; Samson, B.; Lespérance, B.; et al. Copy number and transcriptome alterations associated with metastatic lesion response to treatment in colorectal cancer. Clin. Transl. Med. 2021, 11, e401. [Google Scholar] [CrossRef]
- Mamlouk, S.; Childs, L.H.; Aust, D.; Heim, D.; Melching, F.; Oliveira, C.; Wolf, T.; Durek, P.; Schumacher, D.; Bläker, H.; et al. DNA copy number changes define spatial patterns of heterogeneity in colorectal cancer. Nat. Commun. 2017, 8, 14093. [Google Scholar] [CrossRef] [PubMed]
- Berthelet, J.; Wimmer, V.C.; Whitfield, H.J.; Serrano, A.; Boudier, T.; Mangiola, S.; Merdas, M.; El-Saafin, F.; Baloyan, D.; Wilcox, J.; et al. The site of breast cancer metastases dictates their clonal composition and reversible transcriptomic profile. Sci. Adv. 2021, 7, eabf4408. [Google Scholar] [CrossRef]
- Reeves, M.Q.; Kandyba, E.; Harris, S.; Del Rosario, R.; Balmain, A. Multicolour lineage tracing reveals clonal dynamics of squamous carcinoma evolution from initiation to metastasis. Nat. Cell Biol. 2018, 20, 699–709. [Google Scholar] [CrossRef] [PubMed]
- Maddipati, R.; Stanger, B.Z. Pancreatic Cancer Metastases Harbor Evidence of Polyclonality. Cancer Discov 2015, 5, 1086–1097. [Google Scholar] [CrossRef]
- Westcott, P.M.K.; Sacks, N.J.; Schenkel, J.M.; Ely, Z.A.; Smith, O.; Hauck, H.; Jaeger, A.M.; Zhang, D.; Backlund, C.M.; Beytagh, M.C.; et al. Low neoantigen expression and poor T-cell priming underlie early immune escape in colorectal cancer. Nat. Cancer 2021, 2, 1071–1085. [Google Scholar] [CrossRef]
- Grzelak, C.A.; Goddard, E.T.; Lederer, E.E.; Rajaram, K.; Dai, J.; Shor, R.E.; Lim, A.R.; Kim, J.; Beronja, S.; Funnell, A.P.W.; et al. Elimination of fluorescent protein immunogenicity permits modeling of metastasis in immune-competent settings. Cancer Cell 2022, 40, 1–2. [Google Scholar] [CrossRef]
- Van der Heijden, M.; Miedema, D.M.; Waclaw, B.; Veenstra, V.L.; Lecca, M.C.; Nijman, L.E.; van Dijk, E.; van Neerven, S.M.; Lodestijn, S.C.; Lenos, K.J.; et al. Spatiotemporal regulation of clonogenicity in colorectal cancer xenografts. Proc. Natl. Acad. Sci. USA 2019, 116, 6140–6145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamprecht, S.; Schmidt, E.M.; Blaj, C.; Hermeking, H.; Jung, A.; Kirchner, T.; Horst, D. Multicolor lineage tracing reveals clonal architecture and dynamics in colon cancer. Nat. Commun. 2017, 8, 1406. [Google Scholar] [CrossRef] [PubMed]
- Ramadan, R.; van Driel, M.S.; Vermeulen, L.; van Neerven, S.M. Intestinal stem cell dynamics in homeostasis and cancer. Trends Cancer 2022, 8, 416–425. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fluorescent Protein | Laser (nm) | Dicroic Mirror (nm LP) | Bandpass Filter (nm) |
|---|---|---|---|
| EBFP2 | 405 | - | 450/50 |
| T-Sapphire | 405 | 505 | 530/30 |
| EGFP | 488 | 505 | 510/20 |
| Venus | 488 | 550 | 542/27 |
| mOrange2/tdTomato | 561 | 570 | 582/15 |
| dKatushka | 561 | 735 | 660/20 |
| Fluorescent Protein | Laser (nm) | Dicroic Mirror (nm LP) | Bandpass Filter (nm) |
|---|---|---|---|
| EBFP2 | 405 | - | 450/50 |
| T-Sapphire | 405 | 505 | 525/50 |
| EGFP | 488 | 505 | 515/20 |
| Venus | 488 | 550 | 560/40 |
| mOrange2/tdTomato | 561 | 570 | 586/10 |
| dKatushka | 561 | 735 | 780/60 |
| * ZombieNIR | 640 | 750 | 780/60 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vaquero-Siguero, N.; Schleussner, N.; Volk, J.; Mastel, M.; Meier, J.; Jackstadt, R. Modeling Colorectal Cancer Progression Reveals Niche-Dependent Clonal Selection. Cancers 2022, 14, 4260. https://doi.org/10.3390/cancers14174260
Vaquero-Siguero N, Schleussner N, Volk J, Mastel M, Meier J, Jackstadt R. Modeling Colorectal Cancer Progression Reveals Niche-Dependent Clonal Selection. Cancers. 2022; 14(17):4260. https://doi.org/10.3390/cancers14174260
Chicago/Turabian StyleVaquero-Siguero, Nuria, Nikolai Schleussner, Julia Volk, Manuel Mastel, Jasmin Meier, and Rene Jackstadt. 2022. "Modeling Colorectal Cancer Progression Reveals Niche-Dependent Clonal Selection" Cancers 14, no. 17: 4260. https://doi.org/10.3390/cancers14174260