Simple Summary
Diclofenac is a widely used drug for its anti-inflammatory and pain alleviating properties. This review summarizes the current understanding about the drug diclofenac. The potential applications of diclofenac beyond its well-known anti-inflammatory properties for other diseases such as cancer are discussed, along with existing limitations.
Abstract
Diclofenac is a highly prescribed non-steroidal anti-inflammatory drug (NSAID) that relieves inflammation, pain, fever, and aches, used at different doses depending on clinical conditions. This drug inhibits cyclooxygenase-1 and cyclooxygenase-2 enzymes, which are responsible for the generation of prostaglandin synthesis. To improve current diclofenac-based therapies, we require new molecular systematic therapeutic approaches to reduce complex multifactorial effects. However, the critical challenge that appears with diclofenac and other drugs of the same class is their side effects, such as signs of stomach injuries, kidney problems, cardiovascular issues, hepatic issues, and diarrhea. In this article, we discuss why defining diclofenac-based mechanisms, pharmacological features, and its medicinal properties are needed to direct future drug development against neurodegeneration and imperfect ageing and to improve cancer therapy. In addition, we describe various advance molecular mechanisms and fundamental aspects linked with diclofenac which can strengthen and enable the better designing of new derivatives of diclofenac to overcome critical challenges and improve their applications.
1. Introduction
Non-steroidal anti-inflammatory drugs (NSAIDs) act by inhibiting eicosanoids resulting in anti-inflammatory, pain-relieving, and fever-reducing effects [1]. NSAIDs such as aspirin and indomethacin were synthesized before their therapeutic effects and mechanisms were clear. In 1971, John Vane suggested that the primary mechanism of the aspirin-like NSAIDs effect was cyclooxygenase (COX) enzymes inhibition [2]. Subsequently, improved understanding of the NSAID mechanism of action aided in the development of more NSAIDs, such as diclofenac, ibuprofen, and mefenamic acid. Different NSAIDs have distinct preferences for cyclooxygenase (COX) enzymes, some of which are non-selective to COX while others are specific COX-2 inhibitors [3]. COX enzymes, which are also called prostaglandin endoperoxide synthase, catalyzes the reaction of prostanoid formation (a subclass of eicosanoids), such as prostaglandins and thromboxane from arachidonic acid [4]. NSAIDs that interfere non-specifically with both cyclooxygenase-1 and cyclooxygenase-2 enzyme actions are grouped as non-selective COX inhibitors, whereas COX-2 specific NSAIDs are termed selective COX-2 inhibitors [5,6].
COX-2 inhibitors have an advantage over non-selective COX inhibitors due to a reduction in undesirable GI side effects, although they have been associated with an increased risk of thrombosis [7,8]. NSAIDs such as diclofenac, ibuprofen, and aspirin are common examples of non-specific COX inhibitors, whereas rofecoxib and celecoxib are categorized as selective inhibitors of cyclooxygenase-2 [5]. Progress in the knowledge of the NSAID mechanism of action has led to the identification of novel targets in addition to COX enzymes, associating them with their observed off-target effects [9,10]. However, few NSAID targets have also had potential therapeutic applications in diseases such as cancer and neurodegeneration [11,12,13,14].
Similar to other drugs, the discovery of diclofenac led to several studies elucidating its action mechanism, safety, off-target effects, and other pharmacological aspects [15,16]. Among NSAIDs, diclofenac is one of the most widely recommended [17]. Besides its use in conditions related to pain and inflammation, prior research have also found the role of diclofenac in the Wnt/β-catenin/T-cell factor pathway, Myc transport, and glucose metabolism, thereby suggesting its applications in cancer therapy [18,19]. The present review gives a comprehensive summary of the current understanding of the NSAID diclofenac. Additionally, findings that indicate the possibility of repurposing diclofenac therapy for diseases such as cancer and neurodegeneration are also discussed. Drug repurposing or repositioning is increasingly becoming an effective strategy in reinvestigating known old drugs for new therapeutic purposes, reducing the costs and time involved in de novo drug development [20,21,22]. Few recent examples include minoxidil, originally developed for hypertension, having effective outcomes in preventing hair loss [23]. Bromocriptine, developed for Parkinson’s disease, has been repositioned as a treatment for diabetes mellitus [24]. Similarly, thalidomide, initially developed to treat nausea in pregnant women, was granted approval as a multiple myeloma treatment with dexamethasone [25].
2. Diclofenac: Pharmacokinetics and Pharmacology
Diclofenac is a commonly recommended drug for pain, inflammation, and fever relief. The drug is used in conditions such as rheumatoid arthritis and pain related to surgery [26,27]. As the chemical name suggests, diclofenac is a phenyl acetic acid derivative available in the form of sodium, potassium, or sodium/misoprostol salt. Its molecular formula is C14H10Cl2NNaO2, with a molecular weight of 318.14 [28]. It was first synthesized by Alfred Sallmann and Rudolf Pfister in 1973. In contrast to other classical NSAIDs, diclofenac is known to inhibit the COX-2 enzyme with greater efficiency than the COX-1 enzyme [29]. Diclofenac is a weak acid and has limited solubility in both aqueous and hydrophobic media [29,30]. Diclofenac is known to be absorbed completely and is directly proportional to the dose applied [31,32]. The peak plasma concentration is observed within a range of 10 min to 2 h, depending on the dosage form, viz., enteric coated tablets, solution, etc., and individual-based parameters such as gastrointestinal pH [33,34,35]. Diclofenac sodium salt is a gradually releasing formulation that has high dissolution in the high pH environment present in the duodenum in comparison with the low pH environment in the stomach [36]. The potassium salt of diclofenac was developed to increase the rate of diclofenac absorption, which could be used in conditions where rapid pain relief is needed [33]. Nearly sixty percent of intact diclofenac reaches the circulation [32,37].
Diclofenac metabolism extensively occurs in the liver, where conjugation of diclofenac to glucuronic acid takes place [38]. The conjugation to uronic acid is aided by the enzyme UDP glucuronosyltransferase-2b7 (UGT2B7) [39]. The resultant metabolite, acyl glucuronide, reacts with the sulfhydryl groups of proteins. Acyl glucuronide can be metabolized into 4-hydroxy diclofenac acyl glucuronide by enzyme cytochrome P4502C8 (CYP2C8), which forms benzoquinone imine, resulting in the oxidative bioactivation of diclofenac [40,41]. For the most part, this phenyl acetic acid drug metabolizes into 4′ hydroxyl metabolite, along with other minor metabolites, viz., 3′ hydroxyl metabolite and 5′ hydroxy metabolite [42]. Cytochrome P450 enzyme catalyzes the 4′ and 3′ hydroxylation, and cytochrome P450 3A4 catalyzes the formation of the 5′ hydroxyl metabolite [43,44,45]. More than 60% of the administered dose of diclofenac is excreted through the urine, whereas the remainder is removed as bile conjugates or metabolites of diclofenac [29]. The concentration of major metabolite, 4′ hydroxyl derivative, has been observed at levels around 30% in urine and 20% in bile [46]. As diclofenac has a short half-life of 2 h, repeated doses are required to maintain its plasma concentration to manage certain serious conditions [47].
Following injury, membrane phospholipids are processed by phospholipase enzymes to form arachidonic acid [48]. Arachidonic acid is then further acted upon by COX and lipoxygenase enzymes to form prostaglandins and leukotrienes, respectively, that play key roles in the inflammatory and pain response [49]. Diclofenac has been observed to inhibit both the lipoxygenase- and COX-mediated inflammatory pathway [15]. Interestingly, diclofenac-mediated COX inhibition also has anti-proliferative characteristics [50]. In addition, diclofenac’s inhibitory effect on platelet aggregation has also been reported [51]. It has been observed that Mrp-2, a transport protein, plays a crucial part in the metabolism and transport of diclofenac. Rats deficient in Mrp-2 were much more resistant to the toxic intestinal effects of diclofenac [52,53]. Further studies investigating the transport and metabolism of diclofenac will help to design strategies that will improve efficacy and reduce toxicity. Figure 1 provides a schematic representation of diclofenac structure, currently available modes of its administration, its anti-inflammatory mechanism of action (upper right shaded region), and its hepatic metabolites (lower right shaded region).
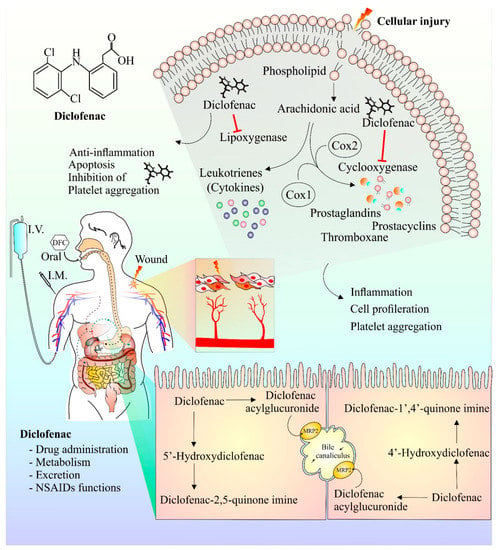
Figure 1.
Chemical structure, mechanism of action, mode of drug administration, and metabolism of diclofenac. Cell damage results in release of arachidonic acid, a constituent of plasma membrane. Arachidonic acid in presence of enzyme cyclooxygenase and lipoxygenase is metabolized into prostaglandins, prostacyclins, and cytokines. These components are responsible for the generation of anti-inflammatory responses at the site of injury, causing pain and inflammation. Inhibition of platelet aggregation and cell proliferation by diclofenac have also been reported. The anti-inflammatory and analgesic effect of diclofenac is ascribed to its ability to hinder cyclooxygenase and lipoxygenase enzyme actions. Diclofenac metabolism mainly occurs in the liver and the major metabolic component of diclofenac is 4′ hydroxyl diclofenac metabolite. Other minor metabolites such as 5′ hydroxy diclofenac are also formed. The activation/formation and inhibition are depicted with arrows and blunt heads, respectively.
The majority of diclofenac is removed through hepatic transformation and nearly 1% is eliminated unchanged through the renal pathway [43]. Various pathways, such as inhibition of the thromboxane-prostanoid receptor, nitric oxide cGMP activation, antinociceptive mechanism, inhibition of PPAR gamma, regulation of ion channels, specifically potassium channels, and NMDA receptor inhibition were found to be affected by diclofenac, in addition to previously established cyclooxygenase and lipoxygenase pathways [15]. The analgesic effects of diclofenac on muscle pain is reported to be mediated by peripheral NMDA pain receptors [54]. The non-specific interactions of diclofenac on COX enzymes have been linked to its gastrointestinal side effects [55]. In addition to gastrointestinal (GI) complexities, various other undesired effects have been observed from off-target interactions of diclofenac. The pathways affected by diclofenac have been studied for the treatment of other diseases. Although these studies are still at a preclinical level, there is a potential for diclofenac to be used in other therapies. The side effects and therapeutic potential of diclofenac are discussed in the next section of the review.
3. Diclofenac: Side Effects, Adverse Drug Interactions, and Therapeutic Potential beyond Anti-Inflammation
3.1. Side Effects
The extensive understanding of the side effects of a particular drug is one of the most critical aspects of drug discovery. The undesired effects of a drug define the actual potential and limitations of a particular drug. As commonly known, NSAIDs that are non-specific inhibitors of COX enzymes, including diclofenac, are associated with the occurrence of GI side effects after long-term use. In the case of diclofenac, in addition to the GI complexities, other side effects observed include cardiological, hepatic, and renal toxicity [56,57,58]. Diclofenac has been found to exert prothrombotic effects by altering levels of PGI-2 and TxA 2 [59]. Additionally, diclofenac may also elevate the risks for heart attacks and myocardial infarctions [56,60]. A study comparing the cardiovascular risks of diclofenac with those of paracetamol, ibuprofen, and naproxen found that patients receiving diclofenac treatment had increased risk of cardiovascular events regardless of sex or age [61].
The use of topical diclofenac with DMSO on the knee of osteoarthritic patients has shown an occurrence of undesirable dermal reactions [62]. Some patients showed adverse GI effects, including diarrhea, nausea, abdominal pain, gastroesophageal reflux, and dyspepsia. Some patients also had undesired cardiovascular effects involving angina, hypertension, thrombosis, and myocardial infarction. Renal effects were also observed, as elevated creatinine levels were reported [62,63]. Further, in the case of a 35 year old patient, a severe life-threatening anaphylactic reaction to diclofenac was disclosed [64]. The side effects of diclofenac have been linked to its capacity to inhibit sodium and calcium channels in cardiac muscles [65] and change ROS levels [66,67]. Recently, diclofenac and other NSAIDs were found to interact with the Farnesoid X receptor that plays a crucial role in protecting the liver [10]. The toxic effects of diclofenac on the liver were also linked with lysosome dysfunction, mitochondrial injury, reactive oxygen species, modification in proteins due to drug metabolites, and immune-linked mechanisms [68,69]. Renal toxicity induced by diclofenac is attributed to mechanisms such as inhibition of renal prostaglandins [58] and destruction of proximal and distal tubules [70]. Recent findings have also shown that diclofenac can inhibit proteasomes, causing disturbance in proteostasis and mitochondrial dysfunction [71,72].
3.2. Adverse Drug Interactions
Adverse drug interactions have also been observed for diclofenac. Diclofenac, in combination with angiotensin-converting enzyme (ACE) inhibitors, have been found to cause increased systolic blood pressure, thereby reducing the effects of ACE inhibitors [73]. Cyclosporine A, used as an immunosuppressant in combination with diclofenac, has been found to cause increased risk of nephrotoxicity [74]. Aspirin reduces the uptake of diclofenac and a pharmacokinetic interaction exists between aspirin and diclofenac [75]. When used in combination with anti-coagulants, diclofenac has been found to increase bleeding complications [76]. The convulsive effects of quinolones, such as ciprofloxacin, has been observed to be elevated in the presence of diclofenac [77]. An incidence of renal failure followed by the death of a patient was documented when diclofenac was given with a dose of methotrexate [78]. Combinations of methotrexate and NSAIDs such as diclofenac, ketoprofen, and naproxen are reported to reduce excretion of methotrexate, leading to adverse drug–drug interactions [79]. Fixed-dose combinations of tramadol and diclofenac is not advised in patients suffering from serious renal impairment [80].
3.3. Therapeutic Potential beyond Anti-Inflammation
The off-target effects of diclofenac may also be utilized in the development of novel therapeutics. Diclofenac and meclofenamate sodium have been found to act as a novel voltage-gated potassium channel KCNQ2/KCNQ3 opener. The therapeutic application of this effect could be used for diseases associated with neuronal hyperexcitability, such as epilepsy [81,82]. In vivo work has shown encouraging results of diclofenac having an anticonvulsant effect. Diclofenac has been found to serve as a template for developing novel ion channel modulators [83]. Kv1.3 is also a target of diclofenac. This voltage-dependent potassium channel-mediated potassium-based current plays a key role in lymphocyte [84] and macrophage [85] activation. It has been observed that diclofenac reduces the immune response by affecting Kv1.3 channels [86], making diclofenac a good starting molecule for the development of autoimmune disorder therapeutics. The acid-sensing ion channel-1 is another diclofenac and NSAID target, which results in a reduction of the current induced by this acid-sensing channel [87]. This inhibition of current in sensory neurons has been thought to be another pathway of pain reduction, in addition to the classical prostaglandin inhibition by NSAIDs [87]. Diclofenac also inhibits phospholipase A2, which is considered to be the underlying mechanism through which diclofenac has therapeutic effects on acute pancreatitis [88]. However, indomethacin, another NSAID with a phospholipase A2 inhibitory effect, has been found to be the most potent agent in acute pancreatitis [89].
An interesting study reported that diclofenac could inhibit transthyretin amyloid fibril formation [90]. This property of diclofenac could be utilized in cases of senile systemic amyloidosis and familial amyloid polyneuropathy [91]. Inhibition of the AKR1C3 enzyme was found to be a therapeutic strategy in hormone-dependent cancers of the prostate and breast [92]. Diclofenac is one of the few known inhibitors of this enzyme. Further, it has been shown that diclofenac also has anti-bacterial and anti-mycotic effects. Diclofenac has been shown to inhibit the bacterial DNA replication of Escherichia coli and Listeria monocytogenes [93]. Altogether, in addition to the molecules involved in the COX-dependent anti-inflammatory pathway, the above studies provide a brief idea of other potential targets of diclofenac. These identified targets might have therapeutic value. Simultaneously, these targets must also be considered as causes of various side effects. Thus, a careful structural and metabolic study is required to overcome the existing limitations of diclofenac to properly utilize this drug in the development of therapeutics for other diseases.
Increased understanding of the pathways and mechanisms modified by diclofenac has provided crucial insights into diclofenac-mediated anti-nociceptive effects and its other possible applications. Diclofenac, like other NSAIDs, has been known to inhibit prostaglandin synthesis that results in its analgesic effects. Interestingly, diclofenac treatment is associated with the opening of potassium channels. The opening of potassium channels in afferent neurons is thought to produce anti-nociceptive effects [94]. Further, diclofenac-mediated inhibition of sodium currents in neurons has been found to play a crucial role in its anti-nociceptive outcomes [95]. Past studies have also shown evidence supporting diclofenac-mediated neuroprotective effects. The repressing effect of diclofenac on proton-induced currents in hippocampal interneurons may have a beneficial role in acidification-linked neuropathological conditions [96].
The potential of diclofenac as a neuroprotective agent and its involvement in the cell cycle and apoptosis is further discussed in the upcoming sections of this review. Some of the commonly known adverse drug–drug interactions and possible therapeutic applications of diclofenac are shown in Figure 2. These studies have provided important data with respect to the deleterious consequences of concurrent use of this drug. Thus, the adverse drug–drug interactions of diclofenac must be carefully assessed when designing combinatorial therapy with this drug. Further, as gender and individual differences may influence individual drug responses, methods such as utilizing individual donor characteristics possessing monocyte-derived hepatocyte-like cells may be beneficial in assessing these adverse drug–drug interactions on an individualistic basis [97]. Recently, using this method, enhanced toxicity while using diclofenac in combination with steroid hormones was observed in four out of nine patients [98]. However, aside from the adverse drug–drug interactions, the novel targets and applications identified so far (as depicted in Figure 2) make diclofenac an exciting molecule for repurposing this anti-inflammatory drug for other diseases. The following section provides a brief overview of our current understanding of the neuroprotective effects of diclofenac.
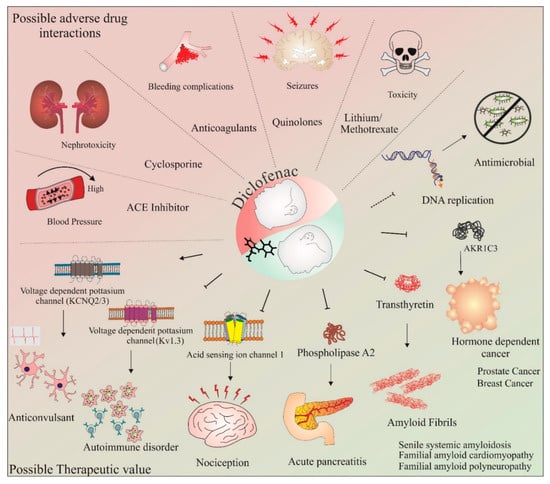
Figure 2.
Therapeutic value and adverse drug–drug interactions of diclofenac. The potential use of diclofenac in combination therapies targeting various conditions has been assessed in numerous studies. However, some combinations of drugs with diclofenac have shown adverse effects. These include diclofenac in combination with ACE inhibitors, cyclosporine, anticoagulants, quinolones, and lithium/methotrexate. These combinations have resulted in increased blood pressure, nephrotoxicity, bleeding complications, seizures, and toxicity, respectively. In addition, with regard to its anti-inflammatory effects, studies have shown the therapeutic value of diclofenac in various diseases and conditions, such as autoimmune disorders, nociception, pancreatitis, amyloid fibril formation, seizures, cancer, and as an antimicrobial agent. The activation/formation and inhibition are depicted with arrows and blunt heads, respectively. The dotted blunt end represents an inhibitory effect that requires further investigations on diclofenac’s mechanism of action.
4. Neuroprotective Abilities of Diclofenac: A Unique Possibility
Diclofenac has been found to have neuroprotective properties. A recent study suggested the neuroprotective role of diclofenac in chlorpromazine-induced catalepsy, having implications for Parkinson’s disease [99]. Previously, diclofenac had been shown to effectively impede Aβ 1-42 oligomerization and fibrillation, similar to another anti-inflammatory drug, celecoxib [100]. Misfolded transthyretin, a transport protein, can form aggregates in regions such as the heart and peripheral nerves and has been linked with amyloid diseases [101]. Diclofenac and its analogues have been found to inhibit aggregation of transthyretin amyloid [90]. Pancreatic amyloidosis includes islet amyloid peptide (IAPP) misfolding along with the occurrence of type 2 diabetes [102]. It was observed that diclofenac resulted in the inhibition of the oligomerization of the islet amyloid peptide (IAPP), causing a reduction in its cytotoxic effects [103]. Interestingly, the protective effect of diclofenac has been reported in protein misfolding diseases, such as Alzheimer’s [104]. Activation of glial cells by COX enzymes is known to be a common inflammatory mechanism in the brain, comprising cytokines, chemokines, and neurotoxins [105]. The impeding effect on COX enzymes by diclofenac is thought to aid in protecting brain cells from inflammation-induced toxicity [106]. Following an experimental focal penetrating traumatic brain injury in rats, the COX-2 inhibition-mediated protective effect of diclofenac appeared to be involved in reduced apoptosis and wound area [107]. Figure 3 provides a schema of various mechanisms involved in diclofenac-mediated protective effects.
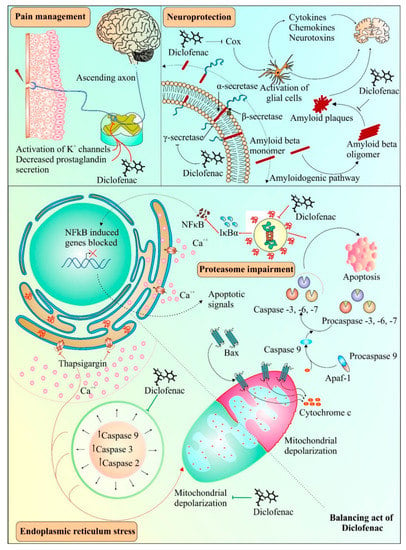
Figure 3.
Possible and known applications of diclofenac. Diclofenac can act as an analgesic agent by activating transient outward potassium channels in neurons and simultaneously reducing the production of prostaglandins. Diclofenac can also act as a potent inhibitor of oligomerization of β-amyloid fibrils and plaque formation, which can be used in the development of therapies for diseases involving amyloid aggregation. Alternatively, diclofenac also interferes with the activation of glial cells, which may further contribute to its neuroprotective properties. Diclofenac has both inhibitory and inducing effects on cell death under different conditions. Endoplasmic reticulum stress generated by thapsigargin leads to activation of caspases and causes mitochondrial depolarization. Diclofenac suppresses the intrinsic pathway of apoptosis by interfering with caspase activation and mitochondrial depolarization. On the other hand, diclofenac treatment can result in proteasomal dysfunction generating downstream apoptotic signals such as mitochondrial cytochrome c release, causing stimulation of caspases leading to apoptosis. The pathway shown under dotted dual faced arrow curve in the top right shows the role of diclofenac in the inhibition of β-amyloid fibrils and plaque formation. The activation/formation and inhibition are depicted with arrows and blunt heads, respectively. The green color coding on arrows and blunt ends depicts the cell survival, whereas, the red color indicates cell death promoting signals.
5. Diclofenac: Several Targets, Multiple Outcomes
Prior studies have suggested both pro- and anti-apoptotic effects of diclofenac [72,108,109]. As depicted in Figure 3, diclofenac alleviates apoptosis induced by thapsigargin-induced endoplasmic reticulum stress conditions [110]. Moreover, the suppression of caspase activity by diclofenac was due to the inhibitory effect of diclofenac on mitochondrial depolarization, and subsequently, on apoptosis [110]. Interestingly, various pathways of diclofenac inducing apoptosis and inhibiting proliferation of cancer cells have also been reported, as shown in Figure 3, indicating its dual role. These mechanisms include inhibition of proteasomes [72], enhancement of ROS production [111], increase in p73 activity [112], and inhibition of MYC expression and lactate transport [19]. The next section discusses the effect of diclofenac on apoptosis and the cell cycle. Prior studies found that deregulated or impaired cell cycles and apoptosis played a crucial role in cancer and metastasis development [113,114,115]. Thus, apoptosis and cell cycle mechanisms have long been considered central mechanisms for novel therapeutic development against tumor development and metastasis [116,117]. Therefore, drugs affecting the cell cycle or apoptosis may have cytostatic or cytotoxic effects leading to the control of cancer cell proliferation and metastasis [118,119,120].
To date, different naturally isolated and laboratory-synthesized molecules have been developed as anticancer agents [121,122,123]. Some of the synthetic molecule structures have been derived from its natural analogs, such as topotecan from camptothecin [124], whereas some anticancer drugs, such as doxorubicin and epirubicin, were developed from a natural molecule, daunorubicin, commonly found in Streptomyces bacteria. An important limitation in designing new anticancer drugs is their off-target effects and limited knowledge of their pharmacokinetics profile. An important strategy to overcome such problem is drug repurposing. Drug repurposing involves the application of a known drug for other diseases [22]. Examples include ropinirole, a Parkinson drug repurposed for restless leg syndrome; gabapentin, an anti-epileptic drug repurposed for neuropathic pain; and methotrexate, an anticancer drug repurposed for arthritis [125,126,127]. Diclofenac has also been investigated for its therapeutic role in several other diseases. As mentioned in the previous section and shown in Figure 2, various beneficial applications of diclofenac have been identified, such as its anticonvulsant and antimicrobial effects. In addition, the potential of diclofenac as an anticancer agent has also been studied and is discussed in the following section.
6. Diclofenac as an Anticancer Agent: Role of Cell Cycle Regulation and Apoptosis
The anticancer properties of diclofenac have been studied in different cancers such as neuroblastoma [128], osteoblast [129], glioma [130], colorectal cancer [131], fibrosarcoma [132], and pancreatic cancer [133]. Being two major pathways, cell cycle and apoptosis mechanisms have been deeply studied to understand the manipulative role of diclofenac in these pathways. Figure 4 depicts a few major apoptotic and cell cycle proteins and pathways that are regulated by diclofenac. The following subsections discuss findings that link underlying cell cycle regulatory and apoptotic mechanisms associated with the anticancer effects of diclofenac.
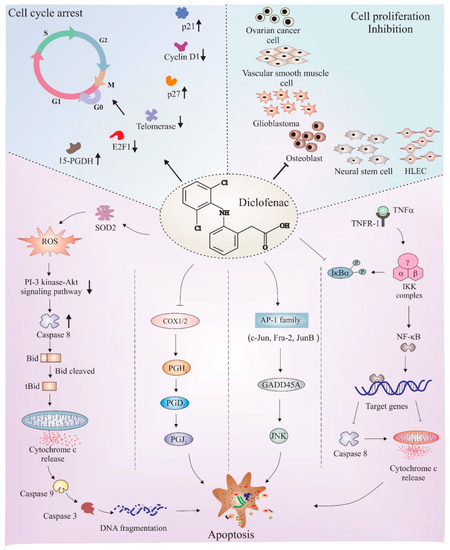
Figure 4.
Schematic depiction of mechanisms of diclofenac-mediated apoptosis and cell cycle arrest. Diclofenac is found to obstruct cell cycle in different cell lines, such as neural stem cells, human lymphatic endothelial cells (HLEC), osteoblasts, glioblastomas (GBM), ovarian cells, and VSMC. Studies found an upregulation of cell cycle inhibitory proteins, such as p27, p21, and 15-hydroxyprostaglandin dehydrogenase (15-PGDH), and downregulation of inducers of cell proliferation, such as E2F1 transcription factor and cyclin D1, all crucial factors responsible for causing cell cycle arrest after diclofenac treatment. Diclofenac has also been shown to have pro-apoptotic effects. The major downstream mechanisms that have been thought to be the reason for diclofenac-mediated apoptosis includes ROS-induced downregulation of the PI 3-kinase/Akt signaling pathway, inhibition of NF-kB activity, disturbance in proteasome activity, interference with COX enzyme activity, and induction of the JNK pathway via AP1 transcription factors. The activation/formation and inhibition are depicted with arrows and blunt heads, respectively. The upward and downward small arrows in bold depicts elevation and depletion in corresponding proteins or pathway. The bold arrows or blunt ends (on top left and right sections) points to downstream proteins or cancer types, respectively indicating the effect of diclofenac. The normal arrows or blunt ends in lower section represents individual pathways known to be involved in diclofenac mediated apoptosis.
6.1. Cell Cycle Regulation
Diclofenac treatment in glioma cells resulted in raised p21 expression, a cell cycle inhibitor, which is an effect associated with elevated levels of 15-PGDH (15 hydroxy prostaglandin dehydrogenase) [134]. Higher levels of p21 and p27 were also seen in vascular smooth muscle cells (VSMCs) treated with diclofenac. These outcomes were linked to the cell cycle inhibitory effect of diclofenac in the G1 phase of VSMCs [135]. Diclofenac also showed anti-carcinogenic effects in a colon cancer model. The anticarcinogenic effect of diclofenac was related with increased telomerase activity and depleted levels of CDK-4, CDK-2, cyclin E, and cyclin D1 [136]. E2f1, a transcription factor, is relevant for cancer progression and chemoresistance because it supports metastasis and invasion by inducing growth receptor signaling pathways when upregulated [137]. The anticarcinogenic outcome of diclofenac in ovarian cancer cells was suggested to be due to lowered expression of the E2f1 transcription factor [138]. Increased COX-2 levels were found to be the reason behind enhanced cellular proliferation and apoptosis resistance in various cancers [139,140]. Interestingly, diclofenac, a COX inhibitor, has been reported to have antitumor effects in rats with neuroblastoma [141].
6.2. Apoptosis
The influence of diclofenac on various apoptosis-inducing mechanisms has also been studied. The apoptotic effect of diclofenac in neuroblastoma cell lines was suggested to be linked with superoxide dismutase 2 (SOD2). SOD2 protects cell from ROS, and therefore, apoptosis or cellular damages [142,143]. Diclofenac impedes both expression levels and enzyme actions of SOD2 [128]. COX-2 has been found to inhibit the apoptosis mechanism activated through the Fas receptor; a receptor of the TNF family which induces apoptosis when it comes in contact with its agonists [144]. Further, a possible mechanism of the apoptotic effect of diclofenac is due to its COX inhibiting property, which causes inhibition of prostaglandins, such as prostaglandin H2 (PGH2), that have anti-apoptotic effects [15]. Another study by [145] proposed an activator protein-1-dependent mechanism of apoptosis induction by diclofenac in myeloid leukemia cells. The study proposed higher levels of activator protein-1 transcription factors, c-Jun, Jun-B, and Fra-2, which resulted in elevated levels of GADD45α, and therefore, JNK, leading to apoptosis. Another study found that the inhibition of TNF-α-mediated stimulation of NF-kB transcription factor activity by diclofenac sensitized hepatocytes towards apoptosis [146]. Taken together, various anticancer and pro-apoptotic mechanisms have been identified that supports the potential for diclofenac to be used as a therapeutic agent for different forms of cancer.
7. Discussion
Diclofenac is one of the oldest and most widely utilized NSAID. Several studies have elucidated its pharmacokinetic and pharmacological properties. Various formulations of diclofenac have been developed, such as ointment, injections, tablets, etc., which can be utilized as needed. Interestingly, peripheral N-methyl D-aspartate (NMDA) receptor antagonism was also identified as a possible analgesic mechanism of diclofenac in a recent study [54]. Such findings suggest that diclofenac, in addition to its commonly known targets, i.e., COX enzymes, may have other useful targets as well. Some of these off-target effects have been related to the undesirable effects of diclofenac, such as gastrointestinal bleeding, cardiotoxicity, hepatotoxicity, etc. Recently, a new study suggested diclofenac had undesirable effects on the cornea through a p53-mediated apoptosis mechanism [147]. In addition, adverse drug–drug interactions of diclofenac, when tested in combination with other drugs such as ACE inhibitors, antimicrobial agents, anticoagulants, and cyclosporine, have also been reported. A recently concluded study in mice found the concomitant use of cefepime and diclofenac led to multiple organ failure [148]. The potential of diclofenac as an antimicrobial agent, anticonvulsant, amyloid inhibitor, and anticancer agent has been proposed and tested. However, the anticancer effects of diclofenac have not been extensively studied among these.
Diclofenac shows anticancer properties in neuroblastoma, colorectal cancer, and fibrosarcoma [149]. However, most of these studies are at the preclinical level. A simple search on www.clinicaltrials.gov using diclofenac as a key term returns more than five hundred results. Out of these, more than fifty studies are under active status. These trials involve either only diclofenac or diclofenac in combination with other drugs as a therapeutic option for various conditions, such as common bile duct disease, diabetic oculopathy, various forms of cancers, etc. A further refinement of the search shows that around five studies are underway for the application of diclofenac in cancer. The outcome of trials such as NCT04091022 and NCT02636569, which are involved in understanding the effect of diclofenac on preventing or reducing the incidence of non-melanoma skin cancer may provide crucial information about the potential of diclofenac use in cancer therapy. A recent outcome from a clinical trial using topical diclofenac on actinic keratosis (AK) patients, a skin condition having altered keratinocyte proliferation, showed reprogramming of metabolism and immune cell infiltration in AK lesions [150]. Another ongoing clinical study involves diclofenac as a constituent of a four-drug combination, TL-118, i.e., cimetidine, metronomic cyclophosphamide, diclofenac, and sulfasalazine [149,151]. Thus, diclofenac may serve as an important constituent in cancer therapy when used with various other drugs. Recently, diclofenac as a curcumin adjuvant has been shown to aid anti-Alzheimer effects of curcumin in mice [152].
A limitation of diclofenac use is its associated side effects. Thus, to use or develop diclofenac in new therapies, studies are needed to develop novel strategies for reducing the side effects of diclofenac. It has been observed that a combination of proton pump inhibitors and diclofenac significantly decreases the risk of peptic ulcers [153]. Additionally, thymoquinone, a natural compound, reduces renal toxicity caused by diclofenac [154]. An interesting study that used vitamin B12 in combination with diclofenac showed that a reduced diclofenac concentration was needed to generate an analgesic effect [155]. The use of omega-3 fatty acids has also shown protective effects against diclofenac-induced hepatotoxicity [68]. The combination of diclofenac and the curcuminoid complex showed better tolerability and functional capacities in knee osteoarthritis [156]. Recently, a study proposed the development of diclofenac analogs with reduced hepatotoxicity [157].
The use of novel techniques in drug delivery can also aid in increasing the availability of the drug at a respective site, leading to reduced dose requirements. A study using polymeric nanosuspensions as a drug carrier showed improved ocular availability of diclofenac in rabbit eyes [158]. Similarly, another study used nanocrystal suspensions of diclofenac for skin inflammation and found that the anti-inflammatory effects were superior in comparison with existing commercial counterparts. The increased drug availability or amount at the inflammation site was thought to be a possible reason behind this [159]. A study using nanofibers obtained from the polymer of poly (D, L-lactide-co-glycolide) to deliver diclofenac locally, showed increased survival rates in a mouse model of oral carcinoma [160].Taken together, such studies may be useful in designing novel therapies that would effectively reduce the adverse effects of diclofenac.
8. Conclusions
In conclusion, the current literature shows many possibilities of diclofenac to be used therapeutically beyond its well-known role in pain management and anti-inflammation. Although there has been progress in gaining knowledge on the mechanisms of action and potential applications of this drug, major work is still at the preclinical level. To get a better understanding of the potential of this drug, more clinical studies are required. Being a well-known drug, repurposing diclofenac in diseases such as cancer or neurodegeneration could be of huge value and may expedite research being done to develop novel drug combinations for these complex diseases.
Author Contributions
A.A. and A.U. performed the first draft, figures, and framework preparation and prepared the first draft of manuscript. R.D., S.S., A.K., D.K.A. and R.K.G. performed critical analysis, contributed to conceptualization, manuscript content alignment, and analysis, and helped in the finalization of the draft. A.M. finalized figures, analyzed the content of manuscript, and perform the final writing of the manuscript. All authors discussed the work of the manuscript and contributed to the finalization of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
AM received research grant EMR/2016/000716 from Science and Engineering Research Board (SERB), Department of Science & Technology, Government of India. DKA received Lung cancer concept awards (W81XWH-22-1-0038) and (W81XWH-22-1-0001) from the Department of Defense, USA.
Acknowledgments
The authors would like to thank Poonam and Bharat Pareek for their technical assistance and the entire lab management for their assistance during manuscript preparation.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| AKR1C3 | Aldo-keto-reductase family 1 member C3 |
| Mrp-2 | Multidrug resistance associated protein-2 |
| NF-kB | Nuclear factor kappa light chain enhancer of activated B cells |
| NMDA | N-methyl D-aspartate receptor |
| PGI 2 | Prostaglandin-I (2) |
| ROS | Reactive oxygen species |
| Tnf-α | Tumor necrosis factor alpha |
| TxA 2 | Thromboxane-A (2) |
References
- Harizi, H.; Corcuff, J.B.; Gualde, N. Arachidonic-acid-derived eicosanoids: Roles in biology and immunopathology. Trends Mol. Med. 2008, 14, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Vane, J.R.; Botting, R.M. The mechanism of action of aspirin. Thromb. Res. 2003, 110, 255–258. [Google Scholar] [CrossRef]
- Xue, D.; Zheng, Q.; Li, H.; Qian, S.; Zhang, B.; Pan, Z. Selective COX-2 inhibitor versus nonselective COX-1 and COX-2 inhibitor in the prevention of heterotopic ossification after total hip arthroplasty: A meta-analysis of randomised trials. Int. Orthop. 2011, 35, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.L.; Urade, Y.; Jakobsson, P.-J. Enzymes of the Cyclooxygenase Pathways of Prostanoid Biosynthesis. Chem. Rev. 2011, 111, 5821–5865. [Google Scholar] [CrossRef]
- Zarghi, A.; Arfaei, S. Selective COX-2 Inhibitors: A Review of Their Structure-Activity Relationships. Iran. J. Pharm. Res. IJPR 2011, 10, 655–683. [Google Scholar]
- Harris, R.E.; Beebe, J.; Alshafie, G.A. Reduction in cancer risk by selective and nonselective cyclooxygenase-2 (COX-2) inhibitors. J. Exp. Pharmacol. 2012, 4, 91–96. [Google Scholar] [CrossRef]
- Khan, M.; Fraser, A. Cox-2 inhibitors and the risk of cardiovascular thrombotic events. Ir. Med. J. 2012, 105, 119–121. [Google Scholar]
- Nussmeier, N.A.; Whelton, A.A.; Brown, M.T.; Langford, R.M.; Hoeft, A.; Parlow, J.L.; Boyce, S.W.; Verburg, K.M. Complications of the COX-2 inhibitors parecoxib and valdecoxib after cardiac surgery. N. Engl. J. Med. 2005, 352, 1081–1091. [Google Scholar] [CrossRef]
- Brueggemann, L.I.; Mani, B.K.; Mackie, A.R.; Cribbs, L.L.; Byron, K.L. Novel Actions of Nonsteroidal Anti-Inflammatory Drugs on Vascular Ion Channels: Accounting for Cardiovascular Side Effects and Identifying New Therapeutic Applications. Mol. Cell. Pharmacol. 2010, 2, 15–19. [Google Scholar]
- Lu, W.; Cheng, F.; Jiang, J.; Zhang, C.; Deng, X.; Xu, Z.; Zou, S.; Shen, X.; Tang, Y.; Huang, J. FXR antagonism of NSAIDs contributes to drug-induced liver injury identified by systems pharmacology approach. Sci. Rep. 2015, 5, 8114. [Google Scholar] [CrossRef]
- Weggen, S.; Eriksen, J.L.; Das, P.; Sagi, S.A.; Wang, R.; Pietrzik, C.U.; Findlay, K.A.; Smith, T.E.; Murphy, M.P.; Bulter, T.; et al. A subset of NSAIDs lower amyloidogenic Aβ42 independently of cyclooxygenase activity. Nature 2001, 414, 212. [Google Scholar] [CrossRef] [PubMed]
- Gurpinar, E.; Grizzle, W.E.; Piazza, G.A. NSAIDs inhibit tumorigenesis, but how? Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 1104–1113. [Google Scholar] [CrossRef] [PubMed]
- Asanuma, M.; Miyazaki, I.; Ogawa, N. Neuroprotective effects of nonsteroidal anti-inflammatory drugs on neurodegenerative diseases. Curr. Pharm. Des. 2004, 10, 695–700. [Google Scholar] [CrossRef] [PubMed]
- Dell’Omo, G.; Crescenti, D.; Vantaggiato, C.; Parravicini, C.; Borroni, A.P.; Rizzi, N.; Garofalo, M.; Pinto, A.; Recordati, C.; Scanziani, E.; et al. Inhibition of SIRT1 deacetylase and p53 activation uncouples the anti-inflammatory and chemopreventive actions of NSAIDs. Br. J. Cancer 2019, 120, 537–546. [Google Scholar] [CrossRef]
- Gan, T.J. Diclofenac: An update on its mechanism of action and safety profile. Curr. Med. Res. Opin. 2010, 26, 1715–1731. [Google Scholar] [CrossRef]
- Wehling, M. Non-steroidal anti-inflammatory drug use in chronic pain conditions with special emphasis on the elderly and patients with relevant comorbidities: Management and mitigation of risks and adverse effects. Eur. J. Clin. Pharmacol. 2014, 70, 1159–1172. [Google Scholar] [CrossRef]
- Alfaro, R.A.; Davis, D.D. Diclofenac. In StatPearls; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2022. [Google Scholar]
- Sareddy, G.R.; Kesanakurti, D.; Kirti, P.B.; Babu, P.P. Nonsteroidal anti-inflammatory drugs diclofenac and celecoxib attenuates Wnt/beta-catenin/Tcf signaling pathway in human glioblastoma cells. Neurochem. Res. 2013, 38, 2313–2322. [Google Scholar] [CrossRef]
- Gottfried, E.; Lang, S.A.; Renner, K.; Bosserhoff, A.; Gronwald, W.; Rehli, M.; Einhell, S.; Gedig, I.; Singer, K.; Seilbeck, A.; et al. New aspects of an old drug--diclofenac targets MYC and glucose metabolism in tumor cells. PLoS ONE 2013, 8, e66987. [Google Scholar] [CrossRef]
- Mehndiratta, M.M.; Wadhai, S.A.; Tyagi, B.K.; Gulati, N.S.; Sinha, M. Drug repositioning. Int. J. Epilepsy 2016, 3, 91–94. [Google Scholar] [CrossRef]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef]
- Corsello, S.M.; Bittker, J.A.; Liu, Z.; Gould, J.; McCarren, P.; Hirschman, J.E.; Johnston, S.E.; Vrcic, A.; Wong, B.; Khan, M.; et al. The Drug Repurposing Hub: A next-generation drug library and information resource. Nat. Med. 2017, 23, 405–408. [Google Scholar] [CrossRef] [PubMed]
- Massey, T.H.; Robertson, N.P. Repurposing drugs to treat neurological diseases. J. Neurol. 2018, 265, 446–448. [Google Scholar] [CrossRef] [PubMed]
- Shivaprasad, C.; Kalra, S. Bromocriptine in type 2 diabetes mellitus. Indian J. Endocrinol. Metab. 2011, 15, S17–S24. [Google Scholar] [CrossRef]
- Pushpakom, S. Chapter 1 Introduction and Historical Overview of Drug Repurposing Opportunities. In Drug Repurposing; The Royal Society of Chemistry: London, UK, 2022; pp. 1–13. [Google Scholar]
- Derry, P.; Derry, S.; Moore, R.A.; McQuay, H.J. Single dose oral diclofenac for acute postoperative pain in adults. Cochrane Database Syst. Rev. 2009, CD004768. [Google Scholar] [CrossRef]
- Atzeni, F.; Masala, I.F.; Sarzi-Puttini, P. A Review of Chronic Musculoskeletal Pain: Central and Peripheral Effects of Diclofenac. Pain Ther. 2018, 7, 163–177. [Google Scholar] [CrossRef]
- Rodrigues, E.B.; Farah, M.E.; Bottós, J.M.; Aggio, F.B. CHAPTER 29—Nonsteroidal anti-inflammatory drugs (NSAIDs) in the treatment of retinal diseases. In Retinal Pharmacotherapy; Nguyen, Q.D., Rodrigues, E.B., Farah, M.E., Mieler, W.F., Eds.; W.B. Saunders: Edinburgh, Scotland, 2010; pp. 196–200. [Google Scholar]
- Altman, R.; Bosch, B.; Brune, K.; Patrignani, P.; Young, C. Advances in NSAID Development: Evolution of Diclofenac Products Using Pharmaceutical Technology. Drugs 2015, 75, 859–877. [Google Scholar] [CrossRef]
- Fini, A.; Bassini, G.; Monastero, A.; Cavallari, C. Diclofenac Salts, VIII. Effect of the Counterions on the Permeation through Porcine Membrane from Aqueous Saturated Solutions. Pharmaceutics 2012, 4, 413–429. [Google Scholar] [CrossRef]
- Skoutakis, V.A.; Carter, C.A.; Mickle, T.R.; Smith, V.H.; Arkin, C.R.; Alissandratos, J.; Petty, D.E. Review of diclofenac and evaluation of its place in therapy as a nonsteroidal antiinflammatory agent. Drug Intell. Clin. Pharm. 1988, 22, 850–859. [Google Scholar] [CrossRef]
- Davies, N.M.; Anderson, K.E. Clinical pharmacokinetics of diclofenac. Therapeutic insights and pitfalls. Clin. Pharmacokinet. 1997, 33, 184–213. [Google Scholar] [CrossRef]
- Reiner, V.; Reiner, A.; Reiner, G.; Conti, M. Increased absorption rate of diclofenac from fast acting formulations containing its potassium salt. Arzneim. Forsch. 2001, 51, 885–890. [Google Scholar] [CrossRef]
- Macia, M.A.; Frias, J.; Carcas, A.J.; Guerra, P.; Valiente, R.; Lucero, M.L. Comparative bioavailability of a dispersible formulation of diclofenac and finding of double plasma peaks. Int. J. Clin. Pharmacol. Ther. 1995, 33, 333–339. [Google Scholar] [PubMed]
- Garbacz, G.; Weitschies, W. Investigation of dissolution behavior of diclofenac sodium extended release formulations under standard and biorelevant test conditions. Drug Dev. Ind. Pharm. 2010, 36, 518–530. [Google Scholar] [CrossRef] [PubMed]
- Birmingham, B.; Buvanendran, A. 40—Nonsteroidal Anti-inflammatory Drugs, Acetaminophen, and COX-2 Inhibitors. In Practical Management of Pain, 5th ed.; Benzon, H.T., Rathmell, J.P., Wu, C.L., Turk, D.C., Argoff, C.E., Hurley, R.W., Eds.; Mosby: Philadelphia, PA, USA, 2014; pp. 553–568.e555. [Google Scholar]
- John, V.A. The pharmacokinetics and metabolism of diclofenac sodium (Voltarol) in animals and man. Rheumatol. Rehabil. 1979, 2, 22–37. [Google Scholar] [PubMed]
- Lagas, J.S.; Sparidans, R.W.; Wagenaar, E.; Beijnen, J.H.; Schinkel, A.H. Hepatic clearance of reactive glucuronide metabolites of diclofenac in the mouse is dependent on multiple ATP-binding cassette efflux transporters. Mol. Pharmacol. 2010, 77, 687–694. [Google Scholar] [CrossRef]
- King, C.; Tang, W.; Ngui, J.; Tephly, T.; Braun, M. Characterization of Rat and Human UDP-Glucuronosyltransferases Responsible for the in Vitro Glucuronidation of Diclofenac. Toxicol. Sci. 2001, 61, 49–53. [Google Scholar] [CrossRef]
- Tang, W.; Stearns, R.A.; Bandiera, S.M.; Zhang, Y.; Raab, C.; Braun, M.P.; Dean, D.C.; Pang, J.; Leung, K.H.; Doss, G.A.; et al. Studies on cytochrome P-450-mediated bioactivation of diclofenac in rats and in human hepatocytes: Identification of glutathione conjugated metabolites. Drug Metab. Dispos. Biol. Fate Chem. 1999, 27, 365–372. [Google Scholar]
- Poon, G.K.; Chen, Q.; Teffera, Y.; Ngui, J.S.; Griffin, P.R.; Braun, M.P.; Doss, G.A.; Freeden, C.; Stearns, R.A.; Evans, D.C.; et al. Bioactivation of diclofenac via benzoquinone imine intermediates-identification of urinary mercapturic acid derivatives in rats and humans. Drug Metab. Dispos. Biol. Fate Chem. 2001, 29, 1608–1613. [Google Scholar]
- Marel, C.D.; Anderson, B.J.; Rømsing, J.; Jacqz-Aigrain, E.; Tibboel, D. Diclofenac and metabolite pharmacokinetics in children. Pediatric Anesth. 2004, 14, 443–451. [Google Scholar] [CrossRef]
- Kirchheiner, J.; Meineke, I.; Steinbach, N.; Meisel, C.; Roots, I.; Brockmöller, J. Pharmacokinetics of diclofenac and inhibition of cyclooxygenases 1 and 2: No relationship to the CYP2C9 genetic polymorphism in humans. Br. J. Clin. Pharmacol. 2003, 55, 51–61. [Google Scholar] [CrossRef]
- Dorado, P.; Berecz, R.; Cáceres, M.C.; Llerena, A. Analysis of diclofenac and its metabolites by high-performance liquid chromatography: Relevance of CYP2C9 genotypes in diclofenac urinary metabolic ratios. J. Chromatogr. B 2003, 789, 437–442. [Google Scholar] [CrossRef]
- Ngui, J.S.; Tang, W.; Stearns, R.A.; Shou, M.; Miller, R.R.; Zhang, Y.; Lin, J.H.; Baillie, T.A. Cytochrome P450 3A4-mediated interaction of diclofenac and quinidine. Drug Metab. Dispos. Biol. Fate Chem. 2000, 28, 1043–1050. [Google Scholar] [PubMed]
- Brogden, R.N.; Heel, R.C.; Pakes, G.E.; Speight, T.M.; Avery, G.S. Diclofenac Sodium: A Review of its Pharmacological Properties and Therapeutic Use in Rheumatic Diseases and Pain of Varying Origin. Drugs 1980, 20, 24–48. [Google Scholar] [CrossRef] [PubMed]
- Suhail, M.; Wu, P.-C.; Minhas, M.U. Development and characterization of pH-sensitive chondroitin sulfate-co-poly(acrylic acid) hydrogels for controlled release of diclofenac sodium. J. Saudi Chem. Soc. 2021, 25, 101212. [Google Scholar] [CrossRef]
- Lyons, J.L.; Tovar-y-Romo, L.B.; Thakur, K.T.; McArthur, J.C.; Haughey, N.J. Chapter 28—Pathobiology of CNS Human Immunodeficiency Virus Infection. In Neurobiology of Brain Disorders; Zigmond, M.J., Rowland, L.P., Coyle, J.T., Eds.; Academic Press: San Diego, CA, USA, 2015; pp. 444–466. [Google Scholar]
- Litalien, C.; Beaulieu, P. Chapter 117—Molecular Mechanisms of Drug Actions: From Receptors to Effectors. In Pediatric Critical Care, 4th ed.; Fuhrman, B.P., Zimmerman, J.J., Eds.; Mosby: Saint Louis, Missouri, 2011; pp. 1553–1568. [Google Scholar]
- Al-Nimer, M.S.M.; Hameed, H.G.; Mahmood, M.M. Antiproliferative effects of aspirin and diclofenac against the growth of cancer and fibroblast cells: In vitro comparative study. Saudi Pharm. J. SPJ Off. Publ. Saudi Pharm. Soc. 2015, 23, 483–486. [Google Scholar] [CrossRef]
- Power, I.; Chambers, W.A.; Greer, I.A.; Ramage, D.; Simon, E. Platelet function after intramuscular diclofenac. Anaesthesia 2007, 45, 916–919. [Google Scholar] [CrossRef]
- Niu, X.; de Graaf, I.A.; van de Vegte, D.; Langelaar-Makkinje, M.; Sekine, S.; Groothuis, G.M. Consequences of Mrp2 deficiency for diclofenac toxicity in the rat intestine ex vivo. Toxicol. Vitr. Int. J. Publ. Assoc. BIBRA 2015, 29, 168–175. [Google Scholar] [CrossRef]
- Seitz, S.; Kretz-Rommel, A.; Oude Elferink, R.P.; Boelsterli, U.A. Selective protein adduct formation of diclofenac glucuronide is critically dependent on the rat canalicular conjugate export pump (Mrp2). Chem. Res. Toxicol. 1998, 11, 513–519. [Google Scholar] [CrossRef]
- Dong, X.D.; Svensson, P.; Cairns, B.E. The analgesic action of topical diclofenac may be mediated through peripheral NMDA receptor antagonism. Pain 2009, 147, 36–45. [Google Scholar] [CrossRef]
- Russell, R.I. Non-steroidal anti-inflammatory drugs and gastrointestinal damage—Problems and solutions. Postgrad. Med. J. 2001, 77, 82. [Google Scholar] [CrossRef]
- McGettigan, P.; Henry, D. Use of non-steroidal anti-inflammatory drugs that elevate cardiovascular risk: An examination of sales and essential medicines lists in low-, middle-, and high-income countries. PLoS Med. 2013, 10, e1001388. [Google Scholar] [CrossRef]
- Boelsterli, U.A. Diclofenac-induced liver injury: A paradigm of idiosyncratic drug toxicity. Toxicol. Appl. Pharmacol. 2003, 192, 307–322. [Google Scholar] [CrossRef]
- Dhanvijay, P.; Misra, A.K.; Varma, S.K. Diclofenac induced acute renal failure in a decompensated elderly patient. J. Pharmacol. Pharmacother. 2013, 4, 155–157. [Google Scholar] [CrossRef] [PubMed]
- Struthmann, L.; Hellwig, N.; Pircher, J.; Sohn, H.Y.; Buerkle, M.A.; Klauss, V.; Mannell, H.; Pohl, U.; Krotz, F. Prothrombotic effects of diclofenac on arteriolar platelet activation and thrombosis in vivo. J. Thromb. Haemost. JTH 2009, 7, 1727–1735. [Google Scholar] [CrossRef] [PubMed]
- Moore, N. Coronary Risks Associated with Diclofenac and Other NSAIDs: An Update. Drug Saf. 2020, 43, 301–318. [Google Scholar] [CrossRef]
- Schmidt, M.; Sørensen, H.T.; Pedersen, L. Diclofenac use and cardiovascular risks: Series of nationwide cohort studies. BMJ 2018, 362, k3426. [Google Scholar] [CrossRef]
- Shainhouse, J.Z.; Grierson, L.M.; Naseer, Z. A long-term, open-label study to confirm the safety of topical diclofenac solution containing dimethyl sulfoxide in the treatment of the osteoarthritic knee. Am. J. Ther. 2010, 17, 566–576. [Google Scholar] [CrossRef]
- Nair, B.; Taylor-Gjevre, R. A Review of Topical Diclofenac Use in Musculoskeletal Disease. Pharmaceuticals 2010, 3, 1892–1908. [Google Scholar] [CrossRef]
- Jha, A.A.; Bohra, V.; Behera, V. Severe anaphylactic reaction to diclofenac. Med. J. Armed Forces India 2015, 71, S279–S281. [Google Scholar] [CrossRef][Green Version]
- Yarishkin, O.V.; Hwang, E.M.; Kim, D.; Yoo, J.C.; Kang, S.S.; Kim, D.R.; Shin, J.-H.-J.; Chung, H.-J.; Jeong, H.-S.; Kang, D.; et al. Diclofenac, a Non-steroidal Anti-inflammatory Drug, Inhibits L-type Ca2+ Channels in Neonatal Rat Ventricular Cardiomyocytes. Korean J. Physiol. Pharmacol. Off. J. Korean Physiol. Soc. Korean Soc. Pharmacol. 2009, 13, 437–442. [Google Scholar] [CrossRef]
- Cantoni, L.; Valaperta, R.; Ponsoda, X.; Castell, J.V.; Barelli, D.; Rizzardini, M.; Mangolini, A.; Hauri, L.; Villa, P. Induction of hepatic heme oxygenase-1 by diclofenac in rodents: Role of oxidative stress and cytochrome P-450 activity. J. Hepatol. 2003, 38, 776–783. [Google Scholar] [CrossRef]
- Gomez-Lechon, M.J.; Ponsoda, X.; O’Connor, E.; Donato, T.; Castell, J.V.; Jover, R. Diclofenac induces apoptosis in hepatocytes by alteration of mitochondrial function and generation of ROS. Biochem. Pharmacol. 2003, 66, 2155–2167. [Google Scholar] [CrossRef] [PubMed]
- Adeyemi, W.J.; Olayaki, L.A. Diclofenac—Induced hepatotoxicity: Low dose of omega-3 fatty acids have more protective effects. Toxicol. Rep. 2018, 5, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.-H.; Lee, W.; Park, S.-H.; Lee, K.-Y.; Choi, Y.-J.; Choi, S.; Kang, D.; Kim, S.; Chang, T.-S.; Hong, S.-S.; et al. Diclofenac impairs autophagic flux via oxidative stress and lysosomal dysfunction: Implications for hepatotoxicity. Redox Biol. 2020, 37, 101751. [Google Scholar] [CrossRef] [PubMed]
- Yasmeen, T.; Qureshi, G.S.; Perveen, S. Adverse effects of diclofenac sodium on renal parenchyma of adult albino rats. J. Pak. Med. Assoc. JPMA 2007, 57, 349–351. [Google Scholar]
- Ghosh, R.; Goswami, S.K.; Feitoza, L.F.; Hammock, B.; Gomes, A.V. Diclofenac induces proteasome and mitochondrial dysfunction in murine cardiomyocytes and hearts. Int. J. Cardiol. 2016, 223, 923–935. [Google Scholar] [CrossRef]
- Amanullah, A.; Upadhyay, A.; Chhangani, D.; Joshi, V.; Mishra, R.; Yamanaka, K.; Mishra, A. Proteasomal Dysfunction Induced By Diclofenac Engenders Apoptosis Through Mitochondrial Pathway. J. Cell. Biochem. 2017, 118, 1014–1027. [Google Scholar] [CrossRef]
- Izhar, M.; Alausa, T.; Folker, A.; Hung, E.; Bakris, G.L. Effects of COX inhibition on blood pressure and kidney function in ACE inhibitor-treated blacks and hispanics. Hypertension 2004, 43, 573–577. [Google Scholar] [CrossRef]
- Colombo, M.D.; Perego, R.; Bellia, G. Cyclosporine-Associated Nephrotoxicity. Open J. Nephrol. 2013, 3, 13. [Google Scholar] [CrossRef]
- Willis, J.V.; Kendall, M.J.; Jack, D.B. A study of the effect of aspirin on the pharmacokinetics of oral and intravenous diclofenac sodium. Eur. J. Clin. Pharmacol. 1980, 18, 415–418. [Google Scholar] [CrossRef]
- Knijff-Dutmer, E.A.J.; Schut, G.A.; van de Laar, M.A.F.J. Concomitant Coumarin–NSAID Therapy and Risk for Bleeding. Ann. Pharmacother. 2003, 37, 12–16. [Google Scholar] [CrossRef]
- Shrivastava, M.P.; Makde, S.D.; Paranjpe, B.D. Interaction of ciprofloxacin with diclofenac and paracetamol in relation to it’s epileptogenic effect. Indian J. Physiol. Pharmacol. 1997, 41, 164–166. [Google Scholar] [PubMed]
- Grissinger, M. Severe Harm and Death Associated With Errors and Drug Interactions Involving Low-Dose Methotrexate. Pharm. Ther. 2018, 43, 191–248. [Google Scholar]
- Kawase, A.; Yamamoto, T.; Egashira, S.; Iwaki, M. Stereoselective Inhibition of Methotrexate Excretion by Glucuronides of Nonsteroidal Anti-inflammatory Drugs via Multidrug Resistance Proteins 2 and 4. J. Pharmacol. Exp. Ther. 2016, 356, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Shah, D.D.; Sorathia, Z.H. Tramadol/Diclofenac Fixed-Dose Combination: A Review of Its Use in Severe Acute Pain. Pain Ther. 2020, 9, 113–128. [Google Scholar] [CrossRef]
- Peretz, A.; Degani, N.; Nachman, R.; Uziyel, Y.; Gibor, G.; Shabat, D.; Attali, B. Meclofenamic Acid and Diclofenac, Novel Templates of KCNQ2/Q3 Potassium Channel Openers, Depress Cortical Neuron Activity and Exhibit Anticonvulsant Properties. Mol. Pharmacol. 2005, 67, 1053. [Google Scholar] [CrossRef]
- Ihara, Y.; Tomonoh, Y.; Deshimaru, M.; Zhang, B.; Uchida, T.; Ishii, A.; Hirose, S. Retigabine, a Kv7.2/Kv7.3-Channel Opener, Attenuates Drug-Induced Seizures in Knock-In Mice Harboring Kcnq2 Mutations. PLoS ONE 2016, 11, e0150095. [Google Scholar] [CrossRef]
- Gwanyanya, A.; Macianskiene, R.; Mubagwa, K. Insights into the effects of diclofenac and other non-steroidal anti-inflammatory agents on ion channels. J. Pharm. Pharmacol. 2012, 64, 1359–1375. [Google Scholar] [CrossRef]
- Lam, J.; Wulff, H. The Lymphocyte Potassium Channels Kv1.3 and KCa3.1 as Targets for Immunosuppression. Drug Dev. Res. 2011, 72, 573–584. [Google Scholar] [CrossRef]
- Martínez-Mármol, R.; Styrczewska, K.; Pérez-Verdaguer, M.; Vallejo-Gracia, A.; Comes, N.; Sorkin, A.; Felipe, A. Ubiquitination mediates Kv1.3 endocytosis as a mechanism for protein kinase C-dependent modulation. Sci. Rep. 2017, 7, 42395. [Google Scholar] [CrossRef]
- Villalonga, N.; David, M.; Bielanska, J.; Gonzalez, T.; Parra, D.; Soler, C.; Comes, N.; Valenzuela, C.; Felipe, A. Immunomodulatory effects of diclofenac in leukocytes through the targeting of Kv1.3 voltage-dependent potassium channels. Biochem. Pharmacol. 2010, 80, 858–866. [Google Scholar] [CrossRef]
- Voilley, N.; de Weille, J.; Mamet, J.; Lazdunski, M. Nonsteroid anti-inflammatory drugs inhibit both the activity and the inflammation-induced expression of acid-sensing ion channels in nociceptors. J. Neurosci. Off. J. Soc. Neurosci. 2001, 21, 8026–8033. [Google Scholar] [CrossRef]
- Murray, B.; Carter, R.; Imrie, C.; Evans, S.; O’Suilleabhain, C. Diclofenac reduces the incidence of acute pancreatitis after endoscopic retrograde cholangiopancreatography. Gastroenterology 2003, 124, 1786–1791. [Google Scholar] [CrossRef]
- Makela, A.; Kuusi, T.; Schroder, T. Inhibition of serum phospholipase-A2 in acute pancreatitis by pharmacological agents in vitro. Scand. J. Clin. Lab. Investig. 1997, 57, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Oza, V.B.; Smith, C.; Raman, P.; Koepf, E.K.; Lashuel, H.A.; Petrassi, H.M.; Chiang, K.P.; Powers, E.T.; Sachettinni, J.; Kelly, J.W. Synthesis, structure, and activity of diclofenac analogues as transthyretin amyloid fibril formation inhibitors. J. Med. Chem. 2002, 45, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Klabunde, T.; Petrassi, H.M.; Oza, V.B.; Raman, P.; Kelly, J.W.; Sacchettini, J.C. Rational design of potent human transthyretin amyloid disease inhibitors. Nat. Struct. Biol. 2000, 7, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Gobec, S.; Brozic, P.; Rizner, T.L. Nonsteroidal anti-inflammatory drugs and their analogues as inhibitors of aldo-keto reductase AKR1C3: New lead compounds for the development of anticancer agents. Bioorganic Med. Chem. Lett. 2005, 15, 5170–5175. [Google Scholar] [CrossRef]
- Mazumdar, K.; Dastidar, S.G.; Park, J.H.; Dutta, N.K. The anti-inflammatory non-antibiotic helper compound diclofenac: An antibacterial drug target. Eur. J. Clin. Microbiol. Infect. Dis. Off. Publ. Eur. Soc. Clin. Microbiol. 2009, 28, 881–891. [Google Scholar] [CrossRef]
- Ortiz, M.I.; Torres-Lopez, J.E.; Castaneda-Hernandez, G.; Rosas, R.; Vidal-Cantu, G.C.; Granados-Soto, V. Pharmacological evidence for the activation of K(+) channels by diclofenac. Eur. J. Pharmacol. 2002, 438, 85–91. [Google Scholar] [CrossRef]
- Lee, H.M.; Kim, H.I.; Shin, Y.K.; Lee, C.S.; Park, M.; Song, J.H. Diclofenac inhibition of sodium currents in rat dorsal root ganglion neurons. Brain Res. 2003, 992, 120–127. [Google Scholar] [CrossRef]
- Dorofeeva, N.A.; Barygin, O.I.; Staruschenko, A.; Bolshakov, K.V.; Magazanik, L.G. Mechanisms of non-steroid anti-inflammatory drugs action on ASICs expressed in hippocampal interneurons. J. Neurochem. 2008, 106, 429–441. [Google Scholar] [CrossRef]
- Benesic, A.; Rahm, N.L.; Ernst, S.; Gerbes, A.L. Human monocyte-derived cells with individual hepatocyte characteristics: A novel tool for personalized in vitro studies. Lab. Investig. 2012, 92, 926–936. [Google Scholar] [CrossRef]
- Benesic, A.; Jalal, K.; Gerbes, A.L. Drug-Drug Combinations Can Enhance Toxicity as Shown by Monocyte-Derived Hepatocyte-like Cells From Patients With Idiosyncratic Drug-Induced Liver Injury. Toxicol. Sci. 2019, 171, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Naeem, S.; Najam, R.; Khan, S.S.; Mirza, T.; Sikandar, B. Neuroprotective effect of diclofenac on chlorpromazine induced catalepsy in rats. Metab. Brain Dis. 2019, 34, 1191–1199. [Google Scholar] [CrossRef]
- Parmar, H.S.; Assaiya, A.; Agrawal, R.; Tiwari, S.; Mufti, I.; Jain, N.; Manivannan, E.; Banerjee, T.; Kumar, A. Inhibition of Aβ(1-42)Oligomerization, Fibrillization and Acetylcholinesterase Activity by Some Anti-Inflammatory Drugs: An in vitro Study. Anti-Inflamm. Anti-Allergy Agents Med. Chem. 2017, 15, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Galant, N.J.; Bugyei-Twum, A.; Rakhit, R.; Walsh, P.; Sharpe, S.; Arslan, P.E.; Westermark, P.; Higaki, J.N.; Torres, R.; Tapia, J.; et al. Substoichiometric inhibition of transthyretin misfolding by immune-targeting sparsely populated misfolding intermediates: A potential diagnostic and therapeutic for TTR amyloidoses. Sci. Rep. 2016, 6, 25080. [Google Scholar] [CrossRef] [PubMed]
- Tomita, T. Amyloidosis of pancreatic islets in primary amyloidosis (AL type). Pathol. Int. 2005, 55, 223–227. [Google Scholar] [CrossRef]
- Fortin, J.S.; Benoit-Biancamano, M.O. Inhibition of islet amyloid polypeptide aggregation and associated cytotoxicity by nonsteroidal anti-inflammatory drugs. Can. J. Physiol. Pharmacol. 2016, 94, 35–48. [Google Scholar] [CrossRef]
- Scharf, S.; Mander, A.; Ugoni, A.; Vajda, F.; Christophidis, N. A double-blind, placebo-controlled trial of diclofenac/misoprostol in Alzheimer’s disease. Neurology 1999, 53, 197–201. [Google Scholar] [CrossRef]
- Tzeng, S.F.; Hsiao, H.Y.; Mak, O.T. Prostaglandins and cyclooxygenases in glial cells during brain inflammation. Curr. Drug Targets Inflamm. Allergy 2005, 4, 335–340. [Google Scholar] [CrossRef]
- Ajmone-Cat, M.A.; Bernardo, A.; Greco, A.; Minghetti, L. Non-Steroidal Anti-Inflammatory Drugs and Brain Inflammation: Effects on Microglial Functions. Pharmaceuticals 2010, 3, 1949–1964. [Google Scholar] [CrossRef]
- Dehlaghi Jadid, K.; Davidsson, J.; Lidin, E.; Hånell, A.; Angéria, M.; Mathiesen, T.; Risling, M.; Günther, M. COX-2 Inhibition by Diclofenac Is Associated with Decreased Apoptosis and Lesion Area After Experimental Focal Penetrating Traumatic Brain Injury in Rats. Front. Neurol. 2019, 10, 811. [Google Scholar] [CrossRef]
- Hassan, H.; Varney, M.; Dave, B.J.; Singh, R.K. Diclofenac Induces Apoptosis and Suppresses Diffuse Large B-Cell Lymphoma Proliferation Independent of P53 Status. Blood 2014, 124, 5485. [Google Scholar] [CrossRef]
- Ashton, M.; Hanson, P.J. Disparate effects of non-steroidal anti-inflammatory drugs on apoptosis in guinea-pig gastric mucous cells: Inhibition of basal apoptosis by diclofenac. Br. J. Pharmacol. 2002, 135, 407–416. [Google Scholar] [CrossRef]
- Yamazaki, T.; Muramoto, M.; Oe, T.; Morikawa, N.; Okitsu, O.; Nagashima, T.; Nishimura, S.; Katayama, Y.; Kita, Y. Diclofenac, a non-steroidal anti-inflammatory drug, suppresses apoptosis induced by endoplasmic reticulum stresses by inhibiting caspase signaling. Neuropharmacology 2006, 50, 558–567. [Google Scholar] [CrossRef] [PubMed]
- Inoue, A.; Muranaka, S.; Fujita, H.; Kanno, T.; Tamai, H.; Utsumi, K. Molecular mechanism of diclofenac-induced apoptosis of promyelocytic leukemia: Dependency on reactive oxygen species, Akt, Bid, cytochrome and caspase pathway. Free. Radic. Biol. Med. 2004, 37, 1290–1299. [Google Scholar] [CrossRef]
- Hassan, H.M.; Varney, M.L.; Chaturvedi, N.K.; Joshi, S.S.; Weisenburger, D.D.; Singh, R.K.; Dave, B.J. Modulation of p73 isoforms expression induces anti-proliferative and pro-apoptotic activity in mantle cell lymphoma independent of p53 status. Leuk. Lymphoma 2016, 57, 2874–2889. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.; Watari, H.; AbuAlmaaty, A.; Ohba, Y.; Sakuragi, N. Apoptosis and Molecular Targeting Therapy in Cancer. BioMed Res. Int. 2014, 2014, 150845. [Google Scholar] [CrossRef]
- Sandhu, C.; Slingerland, J. Deregulation of the cell cycle in cancer. Cancer Detect. Prev. 2000, 24, 107–118. [Google Scholar]
- Kohrman, A.Q.; Matus, D.Q. Divide or Conquer: Cell Cycle Regulation of Invasive Behavior. Trends Cell Biol. 2017, 27, 12–25. [Google Scholar] [CrossRef]
- Lapenna, S.; Giordano, A. Cell cycle kinases as therapeutic targets for cancer. Nat. Rev. Drug Discov. 2009, 8, 547–566. [Google Scholar] [CrossRef]
- Senderowicz, A.M. The Cell Cycle as a Target for Cancer Therapy: Basic and Clinical Findings with the Small Molecule Inhibitors Flavopiridol and UCN-01. Oncologist 2002, 7, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Pegram, M.D.; Konecny, G.E.; O’Callaghan, C.; Beryt, M.; Pietras, R.; Slamon, D.J. Rational Combinations of Trastuzumab With Chemotherapeutic Drugs Used in the Treatment of Breast Cancer. JNCI J. Natl. Cancer Inst. 2004, 96, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Ciardiello, F.; Caputo, R.; Bianco, R.; Damiano, V.; Pomatico, G.; De Placido, S.; Bianco, A.R.; Tortora, G. Antitumor Effect and Potentiation of Cytotoxic Drugs Activity in Human Cancer Cells by ZD-1839 (Iressa), an Epidermal Growth Factor Receptor-selective Tyrosine Kinase Inhibitor. Clin. Cancer Res. 2000, 6, 2053. [Google Scholar] [PubMed]
- Sparreboom, A.; de Jonge, M.J.A.; Verweij, J. The use of oral cytotoxic and cytostatic drugs in cancer treatment. Eur. J. Cancer 2002, 38, 18–22. [Google Scholar] [CrossRef]
- Amanullah, A.; Upadhyay, A.; Joshi, V.; Mishra, R.; Jana, N.R.; Mishra, A. Progressing neurobiological strategies against proteostasis failure: Challenges in neurodegeneration. Prog. Neurobiol. 2017, 159, 1–38. [Google Scholar] [CrossRef]
- Gordaliza, M. Natural products as leads to anticancer drugs. Clin. Transl. Oncol. Off. Publ. Fed. Span. Oncol. Soc. Natl. Cancer Inst. Mex. 2007, 9, 767–776. [Google Scholar] [CrossRef]
- Towle, M.J.; Salvato, K.A.; Budrow, J.; Wels, B.F.; Kuznetsov, G.; Aalfs, K.K.; Welsh, S.; Zheng, W.; Seletsky, B.M.; Palme, M.H.; et al. In Vitro and In Vivo Anticancer Activities of Synthetic Macrocyclic Ketone Analogues of Halichondrin B. Cancer Res. 2001, 61, 1013. [Google Scholar]
- Pommier, Y.; Leo, E.; Zhang, H.; Marchand, C. DNA Topoisomerases and Their Poisoning by Anticancer and Antibacterial Drugs. Chem. Biol. 2010, 17, 421–433. [Google Scholar] [CrossRef]
- Papapetropoulos, A.; Szabo, C. Inventing new therapies without reinventing the wheel: The power of drug repurposing. Br. J. Pharmacol. 2018, 175, 165–167. [Google Scholar] [CrossRef]
- Fox, A.; Gentry, C.; Patel, S.; Kesingland, A.; Bevan, S. Comparative activity of the anti-convulsants oxcarbazepine, carbamazepine, lamotrigine and gabapentin in a model of neuropathic pain in the rat and guinea-pig. Pain 2003, 105, 355–362. [Google Scholar] [CrossRef]
- Friedman, B.; Cronstein, B. Methotrexate mechanism in treatment of rheumatoid arthritis. Jt. Bone Spine 2018, 86, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Cecere, F.; Iuliano, A.; Albano, F.; Zappelli, C.; Castellano, I.; Grimaldi, P.; Masullo, M.; De Vendittis, E.; Ruocco, M.R. Diclofenac-Induced Apoptosis in the Neuroblastoma Cell Line SH-SY5Y: Possible Involvement of the Mitochondrial Superoxide Dismutase. J. Biomed. Biotechnol. 2010, 2010, 11. [Google Scholar] [CrossRef] [PubMed]
- Krischak, G.D.; Augat, P.; Blakytny, R.; Claes, L.; Kinzl, L.; Beck, A. The non-steroidal anti-inflammatory drug diclofenac reduces appearance of osteoblasts in bone defect healing in rats. Arch. Orthop. Trauma Surg. 2007, 127, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Leidgens, V.; Seliger, C.; Jachnik, B.; Welz, T.; Leukel, P.; Vollmann-Zwerenz, A.; Bogdahn, U.; Kreutz, M.; Grauer, O.M.; Hau, P. Ibuprofen and Diclofenac Restrict Migration and Proliferation of Human Glioma Cells by Distinct Molecular Mechanisms. PLoS ONE 2015, 10, e0140613. [Google Scholar] [CrossRef]
- Hamoya, T.; Fujii, G.; Miyamoto, S.; Takahashi, M.; Totsuka, Y.; Wakabayashi, K.; Toshima, J.; Mutoh, M. Effects of NSAIDs on the risk factors of colorectal cancer: A mini review. Genes Environ. Off. J. Jpn. Environ. Mutagen Soc. 2016, 38, 6. [Google Scholar] [CrossRef]
- Hofer, M.; Hoferova, Z.; Fedorocko, P.; Mackova, N.O. Hematopoiesis-stimulating and anti-tumor effects of repeated administration of diclofenac in mice with transplanted fibrosarcoma cells. Physiol. Res. 2002, 51, 629–632. [Google Scholar]
- Mayorek, N.; Naftali-Shani, N.; Grunewald, M. Diclofenac inhibits tumor growth in a murine model of pancreatic cancer by modulation of VEGF levels and arginase activity. PLoS ONE 2010, 5, e12715. [Google Scholar] [CrossRef]
- Wakimoto, N.; Wolf, I.; Yin, D.; Kelly, J.; Akagi, T.; Abramovitz, L.; Black, K.L.; Tai, H.-H.; Koeffler, H.P. Nonsteroidal Anti-inflammatory Drugs Suppress Glioma via 15-Hydroxyprostaglandin Dehydrogenase. Cancer Res. 2008, 68, 6978. [Google Scholar] [CrossRef]
- Brooks, G.; Yu, X.M.; Wang, Y.; Crabbe, M.J.; Shattock, M.J.; Harper, J.V. Non-steroidal anti-inflammatory drugs (NSAIDs) inhibit vascular smooth muscle cell proliferation via differential effects on the cell cycle. J. Pharm. Pharmacol. 2003, 55, 519–526. [Google Scholar] [CrossRef]
- Rana, C.; Piplani, H.; Vaish, V.; Nehru, B.; Sanyal, S.N. Downregulation of telomerase activity by diclofenac and curcumin is associated with cell cycle arrest and induction of apoptosis in colon cancer. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2015, 36, 5999–6010. [Google Scholar] [CrossRef]
- Engelmann, D.; Pützer, B.M. The Dark Side of E2F1: In Transit beyond Apoptosis. Cancer Res. 2012, 72, 571. [Google Scholar] [CrossRef] [PubMed]
- Valle, B.L.; D’Souza, T.; Becker, K.G.; Wood, W.H.; Zhang, Y.; Wersto, R.P.; Morin, P.J. Non-Steroidal Anti-inflammatory Drugs Decrease E2F1 Expression and Inhibit Cell Growth in Ovarian Cancer Cells. PLoS ONE 2013, 8, e61836. [Google Scholar] [CrossRef] [PubMed]
- Sobolewski, C.; Cerella, C.; Dicato, M.; Ghibelli, L.; Diederich, M. The Role of Cyclooxygenase-2 in Cell Proliferation and Cell Death in Human Malignancies. Int. J. Cell Biol. 2010, 2010, 215158. [Google Scholar] [CrossRef]
- Liu, B.; Qu, L.; Yan, S. Cyclooxygenase-2 promotes tumor growth and suppresses tumor immunity. Cancer Cell Int. 2015, 15, 106. [Google Scholar] [CrossRef] [PubMed]
- Johnsen, J.I.; Lindskog, M.; Ponthan, F.; Pettersen, I.; Elfman, L.; Orrego, A.; Sveinbjornsson, B.; Kogner, P. Cyclooxygenase-2 is expressed in neuroblastoma, and nonsteroidal anti-inflammatory drugs induce apoptosis and inhibit tumor growth in vivo. Cancer Res. 2004, 64, 7210–7215. [Google Scholar] [CrossRef] [PubMed]
- Hosoki, A.; Yonekura, S.; Zhao, Q.L.; Wei, Z.L.; Takasaki, I.; Tabuchi, Y.; Wang, L.L.; Hasuike, S.; Nomura, T.; Tachibana, A.; et al. Mitochondria-targeted superoxide dismutase (SOD2) regulates radiation resistance and radiation stress response in HeLa cells. J. Radiat. Res. 2012, 53, 58–71. [Google Scholar] [CrossRef]
- Petrache, I.; Medler, T.R.; Richter, A.T.; Kamocki, K.; Chukwueke, U.; Zhen, L.; Gu, Y.; Adamowicz, J.; Schweitzer, K.S.; Hubbard, W.C.; et al. Superoxide dismutase protects against apoptosis and alveolar enlargement induced by ceramide. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 295, L44–L53. [Google Scholar] [CrossRef]
- Nzeako, U.C.; Guicciardi, M.E.; Yoon, J.-H.; Bronk, S.F.; Gores, G.J. COX-2 inhibits Fas-mediated apoptosis in cholangiocarcinoma cells. Hepatology 2003, 35, 552–559. [Google Scholar] [CrossRef]
- Singh, R.; Cadeddu, R.P.; Frobel, J.; Wilk, C.M.; Bruns, I.; Zerbini, L.F.; Prenzel, T.; Hartwig, S.; Brunnert, D.; Schroeder, T.; et al. The non-steroidal anti-inflammatory drugs Sulindac sulfide and Diclofenac induce apoptosis and differentiation in human acute myeloid leukemia cells through an AP-1 dependent pathway. Apoptosis Int. J. Program. Cell Death 2011, 16, 889–901. [Google Scholar] [CrossRef]
- Fredriksson, L.; Herpers, B.; Benedetti, G.; Matadin, Q.; Puigvert, J.C.; de Bont, H.; Dragovic, S.; Vermeulen, N.P.; Commandeur, J.N.; Danen, E.; et al. Diclofenac inhibits tumor necrosis factor-alpha-induced nuclear factor-kappaB activation causing synergistic hepatocyte apoptosis. Hepatology 2011, 53, 2027–2041. [Google Scholar] [CrossRef]
- Li, H.; Fan, T.-J.; Zou, P.; Xu, B. Diclofenac Sodium Triggers p53-Dependent Apoptosis in Human Corneal Epithelial Cells via ROS-Mediated Crosstalk. Chem. Res. Toxicol. 2021, 34, 70–79. [Google Scholar] [CrossRef]
- Aboubakr, M.; Abdelkader, A.; Habotta, O.A.; Adel, N.; Emam, M.A.; Abdelhiee, E.Y.; Shanab, O.; Shoghy, K.; Elnoury, H.; Soliman, M.M.; et al. Cefepime and diclofenac sodium combined treatment-potentiated multiple organ injury: Role of oxidative damage and disrupted lipid metabolism. J. Biochem. Mol. Toxicol. 2021, 35, e22929. [Google Scholar] [CrossRef]
- Pantziarka, P.; Sukhatme, V.; Bouche, G.; Meheus, L.; Sukhatme, V.P. Repurposing Drugs in Oncology (ReDO)—Diclofenac as an anti-cancer agent. Ecancermedicalscience 2016, 10, 610. [Google Scholar] [CrossRef] [PubMed]
- Singer, K.; Dettmer, K.; Unger, P.; Schönhammer, G.; Renner, K.; Peter, K.; Siska, P.J.; Berneburg, M.; Herr, W.; Oefner, P.J.; et al. Topical Diclofenac Reprograms Metabolism and Immune Cell Infiltration in Actinic Keratosis. Front. Oncol. 2019, 9, 605. [Google Scholar] [CrossRef] [PubMed]
- Breuer, S.; Maimon, O.; Appelbaum, L.; Peretz, T.; Hubert, A. TL-118-anti-angiogenic treatment in pancreatic cancer: A case report. Med. Oncol. 2013, 30, 585. [Google Scholar] [CrossRef] [PubMed]
- Pande, S.; Patel, C.; Sarkar, D.; Acharya, S. Lactobacillus Rhamnosus UBLR-58 and Diclofenac Potentiate the Anti- Alzheimer Activity of Curcumin in Mice. Curr. Enzym. Inhib. 2021, 17, 49–56. [Google Scholar] [CrossRef]
- Hoer, A.; Gothe, H.; Schiffhorst, G.; Sterzel, A.; Grass, U.; Haussler, B. Comparison of the effects of diclofenac or other non-steroidal anti-inflammatory drugs (NSAIDs) and diclofenac or other NSAIDs in combination with proton pump inhibitors (PPI) on hospitalisation due to peptic ulcer disease. Pharmacoepidemiol. Drug Saf. 2007, 16, 854–858. [Google Scholar] [CrossRef]
- Aycan, İ.Ö.; Elpek, Ö.; Akkaya, B.; Kıraç, E.; Tuzcu, H.; Kaya, S.; Coşkunfırat, N.; Aslan, M. Diclofenac induced gastrointestinal and renal toxicity is alleviated by thymoquinone treatment. Food Chem. Toxicol. 2018, 118, 795–804. [Google Scholar] [CrossRef]
- Tamaddonfard, E.; Samadi, F.; Egdami, K. The effects of vitamin B(12) and diclofenac and their combination on cold and mechanical allodynia in a neuropathic pain model in rats. Vet. Res. Forum Int. Q. J. 2013, 4, 19–24. [Google Scholar]
- Shep, D.; Khanwelkar, C.; Gade, P.; Karad, S. Efficacy and safety of combination of curcuminoid complex and diclofenac versus diclofenac in knee osteoarthritis: A randomized trial. Medicine 2020, 99, e19723. [Google Scholar] [CrossRef]
- Tateishi, Y.; Ohe, T.; Ogawa, M.; Takahashi, K.; Nakamura, S.; Mashino, T. Development of Novel Diclofenac Analogs Designed to Avoid Metabolic Activation and Hepatocyte Toxicity. ACS Omega 2020, 5, 32608–32616. [Google Scholar] [CrossRef] [PubMed]
- Agnihotri, S.M.; Vavia, P.R. Diclofenac-loaded biopolymeric nanosuspensions for ophthalmic application. Nanomed. Nanotechnol. Biol. Med. 2009, 5, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Pireddu, R.; Caddeo, C.; Valenti, D.; Marongiu, F.; Scano, A.; Ennas, G.; Lai, F.; Fadda, A.M.; Sinico, C. Diclofenac acid nanocrystals as an effective strategy to reduce in vivo skin inflammation by improving dermal drug bioavailability. Colloids Surf. B Biointerfaces 2016, 143, 64–70. [Google Scholar] [CrossRef]
- Will, O.M.; Purcz, N.; Chalaris, A.; Heneweer, C.; Boretius, S.; Purcz, L.; Nikkola, L.; Ashammakhi, N.; Kalthoff, H.; Gluer, C.C.; et al. Increased survival rate by local release of diclofenac in a murine model of recurrent oral carcinoma. Int. J. Nanomed. 2016, 11, 5311–5321. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).