
Potassium Ion Channels in Malignant Central Nervous System Cancers
Abstract
:Simple Summary
Abstract
1. Introduction
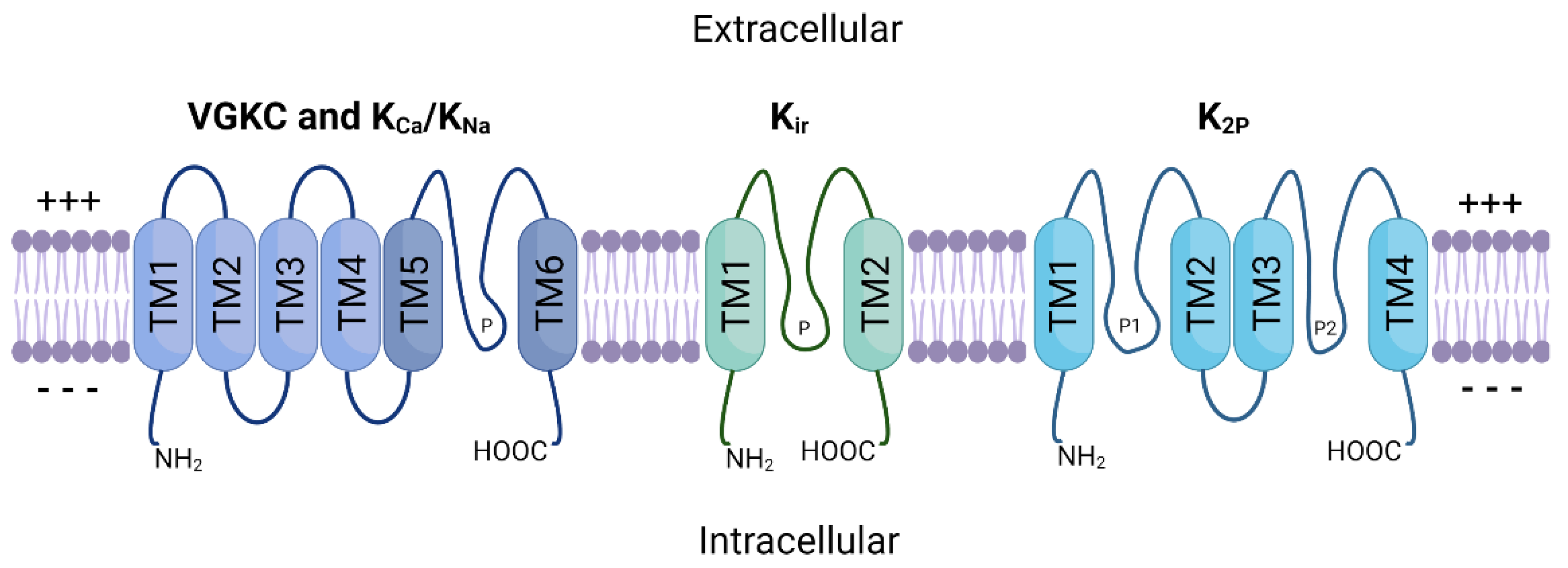
2. Potassium Ion Channels
Potassium Ion Channels in Non-CNS Cancer
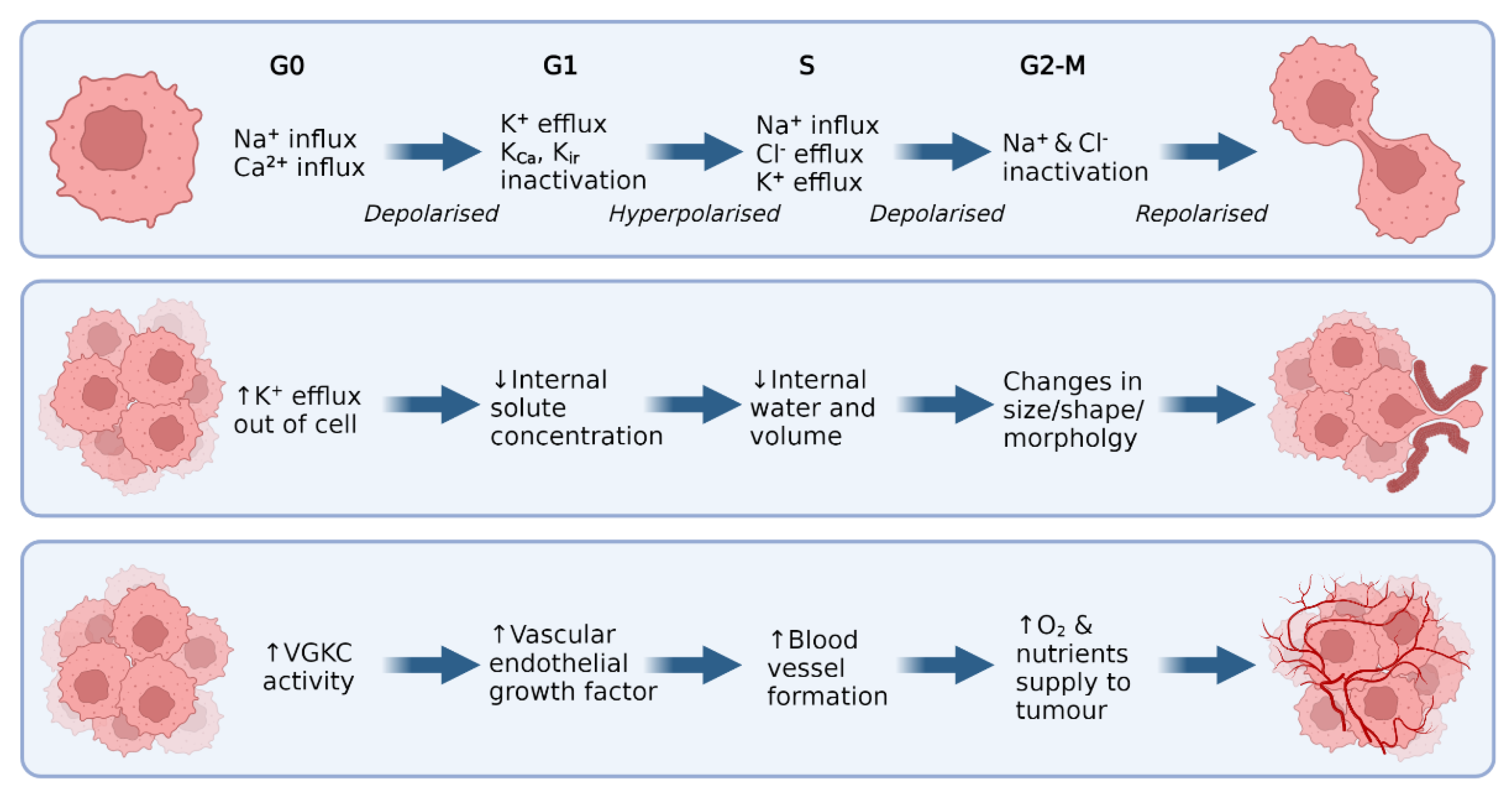
3. Malignant CNS Cancer
3.1. Targeted Therapies
3.2. Cellular Plasticity and Drug Resistance
4. Potassium Ion Channels in Malignant CNS Cancer
4.1. High-Grade Glioma
4.2. Low-Grade Glioma
4.3. Medulloblastoma
5. Potassium Ion Channels as Therapeutic Targets
6. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gabashvili, I.S.; Sokolowski, B.H.A.; Morton, C.C.; Giersch, A.B.S. Ion Channel Gene Expression in the Inner Ear. J. Assoc. Res. Otolaryngol. 2007, 8, 305–328. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Zhu, R.; Zhu, L.; Qiu, T.; Cao, Z.; Kang, T. Potassium Channels: Structures, Diseases, and Modulators. Chem. Biol. Drug Des. 2014, 83, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Ion Channels and the Electrical Properties of Membranes. In Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Abdul Kadir, L.; Stacey, M.; Barrett-Jolley, R. Emerging Roles of the Membrane Potential: Action Beyond the Action Potential. Front. Physiol. 2018, 9, 1661. [Google Scholar] [CrossRef] [PubMed]
- Chrysafides, S.M.; Bordes, S.; Sharma, S. Physiology, Resting Potential. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Deemyad, T.; Lüthi, J.; Spruston, N. Astrocytes Integrate and Drive Action Potential Firing in Inhibitory Subnetworks. Nat. Commun. 2018, 9, 4336. [Google Scholar] [CrossRef]
- Grant, A.O. Cardiac Ion Channels. Circ. Arrhythm. Electrophysiol. 2009, 2, 185–194. [Google Scholar] [CrossRef]
- Zavodnik, I.B.; Piasecka, A.; Szosland, K.; Bryszewska, M. Human Red Blood Cell Membrane Potential and Fluidity in Glucose Solutions. Scand. J. Clin. Lab. Investig. 1997, 57, 59–63. [Google Scholar] [CrossRef]
- Bordey, A.; Sontheimer, H. Electrophysiological Properties of Human Astrocytic Tumor Cells In Situ: Enigma of Spiking Glial Cells. J. Neurophysiol. 1998, 79, 2782–2793. [Google Scholar] [CrossRef]
- Blackiston, D.J.; McLaughlin, K.A.; Levin, M. Bioelectric Controls of Cell Proliferation. Cell Cycle 2009, 8, 3519–3528. [Google Scholar] [CrossRef]
- Cone, C.D.; Cone, C.M. Induction of Mitosis in Mature Neurons in Central Nervous System by Sustained Depolarization. Science 1976, 192, 155–158. [Google Scholar] [CrossRef]
- Griffin, M.; Khan, R.; Basu, S.; Smith, S. Ion Channels as Therapeutic Targets in High Grade Gliomas. Cancers 2020, 12, E3068. [Google Scholar] [CrossRef]
- Urrego, D.; Tomczak, A.P.; Zahed, F.; Stühmer, W.; Pardo, L.A. Potassium Channels in Cell Cycle and Cell Proliferation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130094. [Google Scholar] [CrossRef] [PubMed]
- Lastraioli, E.; Iorio, J.; Arcangeli, A. Ion Channel Expression as Promising Cancer Biomarker. Biochim. Biophys. Acta 2015, 1848, 2685–2702. [Google Scholar] [CrossRef]
- Kim, J.-B. Channelopathies. Korean J. Pediatr. 2014, 57, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Ion Channels and the Hallmarks of Cancer. Trends Mol. Med. 2010, 16, 107–121. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Jackson, W.F. Potassium Channels in Regulation of Vascular Smooth Muscle Contraction and Growth. Adv. Pharmacol. 2017, 78, 89–144. [Google Scholar] [PubMed]
- Neylon, C.B.; Lang, R.J.; Fu, Y.; Bobik, A.; Reinhart, P.H. Molecular Cloning and Characterization of the Intermediate-Conductance Ca2+-Activated K+ Channel in Vascular Smooth Muscle: Relationship between K(Ca) Channel Diversity and Smooth Muscle Cell Function. Circ. Res. 1999, 85, e33–e43. [Google Scholar] [CrossRef]
- Bi, D.; Toyama, K.; Lemaître, V.; Takai, J.; Fan, F.; Jenkins, D.P.; Wulff, H.; Gutterman, D.D.; Park, F.; Miura, H. The Intermediate Conductance Calcium-Activated Potassium Channel KCa3.1 Regulates Vascular Smooth Muscle Cell Proliferation via Controlling Calcium-Dependent Signaling. J. Biol. Chem. 2013, 288, 15843–15853. [Google Scholar] [CrossRef]
- Leanza, L.; Biasutto, L.; Manago, A.; Gulbins, E.; Zoratti, M.; Szabò, I. Intracellular Ion Channels and Cancer. Front. Physiol. 2013, 4, 227. [Google Scholar] [CrossRef]
- Rao, V.R.; Perez-Neut, M.; Kaja, S.; Gentile, S. Voltage-Gated Ion Channels in Cancer Cell Proliferation. Cancers 2015, 7, 849–875. [Google Scholar] [CrossRef]
- Pardo, L.A.; Stühmer, W. The Roles of K+ Channels in Cancer. Nat. Rev. Cancer 2014, 14, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Sontheimer, H. An Unexpected Role for Ion Channels in Brain Tumor Metastasis. Exp. Biol. Med. 2008, 233, 779–791. [Google Scholar] [CrossRef]
- Comes, N.; Serrano-Albarrás, A.; Capera, J.; Serrano-Novillo, C.; Condom, E.; Cajal, S.R.Y.; Ferreres, J.C.; Felipe, A. Involvement of Potassium Channels in the Progression of Cancer to a More Malignant Phenotype. Biochim. Biophys. Acta 2015, 1848, 2477–2492. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.B.; Zhao, S.G.; Liu, Y.H.; Hu, E.X.; Liu, B.X. Tetraethylammonium Inhibits Glioma Cells via Increasing Production of Intracellular Reactive Oxygen Species. Chemotherapy 2009, 55, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Chin, L.S.; Park, C.C.; Zitnay, K.M.; Sinha, M.; DiPatri, A.J.; Perillán, P.; Simard, J.M. 4-Aminopyridine Causes Apoptosis and Blocks an Outward Rectifier K+ Channel in Malignant Astrocytoma Cell Lines. J. Neurosci. Res. 1997, 48, 122–127. [Google Scholar] [CrossRef]
- Banderali, U.; Belke, D.; Singh, A.; Jayanthan, A.; Giles, W.R.; Narendran, A. Curcumin Blocks Kv11.1 (Erg) Potassium Current and Slows Proliferation in the Infant Acute Monocytic Leukemia Cell Line THP-1. Cell. Physiol. Biochem. 2011, 28, 1169–1180. [Google Scholar] [CrossRef]
- Venturini, E.; Leanza, L.; Azzolini, M.; Kadow, S.; Mattarei, A.; Weller, M.; Tabatabai, G.; Edwards, M.J.; Zoratti, M.; Paradisi, C.; et al. Targeting the Potassium Channel Kv1.3 Kills Glioblastoma Cells. Neurosignals 2017, 25, 26–38. [Google Scholar] [CrossRef]
- Kuang, Q.; Purhonen, P.; Hebert, H. Structure of Potassium Channels. Cell. Mol. Life Sci. 2015, 72, 3677–3693. [Google Scholar] [CrossRef]
- Sun, X.; Zaydman, M.A.; Cui, J. Regulation of Voltage-Activated K+ Channel Gating by Transmembrane β Subunits. Front. Pharmacol. 2012, 3, 63. [Google Scholar] [CrossRef]
- Jiang, Y.; Lee, A.; Chen, J.; Ruta, V.; Cadene, M.; Chait, B.T.; MacKinnon, R. X-ray Structure of a Voltage-Dependent K+ Channel. Nature 2003, 423, 33–41. [Google Scholar] [CrossRef]
- Doyle, D.A.; Cabral, J.M.; Pfuetzner, R.A.; Kuo, A.; Gulbis, J.M.; Cohen, S.L.; Chait, B.T.; MacKinnon, R. The Structure of the Potassium Channel: Molecular Basis of K+ Conduction and Selectivity. Science 1998, 280, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Rasmusson, R.L.; Morales, M.J.; Wang, S.; Liu, S.; Campbell, D.L.; Brahmajothi, M.V.; Strauss, H.C. Inactivation of Voltage-Gated Cardiac K+ Channels. Circ. Res. 1998, 82, 739–750. [Google Scholar] [CrossRef] [PubMed]
- Sigworth, F.J. Voltage Gating of Ion Channels. Q. Rev. Biophys. 1994, 27, 1–40. [Google Scholar] [CrossRef] [PubMed]
- Sabater, V.G.; Rigby, M.; Burrone, J. Voltage-Gated Potassium Channels Ensure Action Potential Shape Fidelity in Distal Axons. J. Neurosci. 2021, 41, 5372–5385. [Google Scholar] [CrossRef]
- He, K.; McCord, M.C.; Hartnett, K.A.; Aizenman, E. Regulation of Pro-Apoptotic Phosphorylation of Kv2.1 K+ Channels. PLoS ONE 2015, 10, e0129498. [Google Scholar] [CrossRef]
- Wang, Q.; Curran, M.E.; Splawski, I.; Burn, T.C.; Millholland, J.M.; VanRaay, T.J.; Shen, J.; Timothy, K.W.; Vincent, G.M.; de Jager, T.; et al. Positional Cloning of a Novel Potassium Channel Gene: KVLQT1 Mutations Cause Cardiac Arrhythmias. Nat. Genet. 1996, 12, 17–23. [Google Scholar] [CrossRef]
- Isacoff, E.Y.; Jan, L.Y.; Minor, D.L. Conduits of Life’s Spark: A Perspective on Ion Channel Research since the Birth of Neuron. Neuron 2013, 80, 658–674. [Google Scholar] [CrossRef]
- Kaczmarek, L.K. Slack, Slick and Sodium-Activated Potassium Channels. ISRN Neurosci. 2013, 2013, 354262. [Google Scholar] [CrossRef]
- Huang, X.; Jan, L.Y. Targeting Potassium Channels in Cancer. J. Cell Biol. 2014, 206, 151–162. [Google Scholar] [CrossRef]
- Kshatri, A.S.; Gonzalez-Hernandez, A.; Giraldez, T. Physiological Roles and Therapeutic Potential of Ca2+ Activated Potassium Channels in the Nervous System. Front. Mol. Neurosci. 2018, 11, 258. [Google Scholar] [CrossRef] [Green Version]
- Sweet, T.-B.; Cox, D.H. Measuring the Influence of the BKCa β1 Subunit on Ca2+ Binding to the BKCa Channel. J. Gen. Physiol. 2009, 133, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Lee, U.S.; Cui, J. BK Channel Activation: Structural and Functional Insights. Trends Neurosci. 2010, 33, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Contet, C.; Goulding, S.P.; Kuljis, D.A.; Barth, A.L. BK Channels in the Central Nervous System. Int. Rev. Neurobiol. 2016, 128, 281–342. [Google Scholar] [PubMed]
- Howarth, C. The Contribution of Astrocytes to the Regulation of Cerebral Blood Flow. Front. Neurosci. 2014, 8, 103. [Google Scholar] [CrossRef] [PubMed]
- Filosa, J.A.; Bonev, A.D.; Straub, S.V.; Meredith, A.L.; Wilkerson, M.K.; Aldrich, R.W.; Nelson, M.T. Local Potassium Signaling Couples Neuronal Activity to Vasodilation in the Brain. Nat. Neurosci. 2006, 9, 1397–1403. [Google Scholar] [CrossRef] [PubMed]
- Yuan, A.; Santi, C.M.; Wei, A.; Wang, Z.W.; Pollak, K.; Nonet, M.; Kaczmarek, L.; Crowder, C.M.; Salkoff, L. The Sodium-Activated Potassium Channel Is Encoded by a Member of the Slo Gene Family. Neuron 2003, 37, 765–773. [Google Scholar] [CrossRef]
- Budelli, G.; Hage, T.A.; Wei, A.; Rojas, P.; Jong, Y.-J.I.; O’Malley, K.; Salkoff, L. Na+-Activated K+ Channels Express a Large Delayed Outward Current in Neurons during Normal Physiology. Nat. Neurosci. 2009, 12, 745–750. [Google Scholar] [CrossRef]
- Bausch, A.E.; Dieter, R.; Nann, Y.; Hausmann, M.; Meyerdierks, N.; Kaczmarek, L.K.; Ruth, P.; Lukowski, R. The Sodium-Activated Potassium Channel Slack Is Required for Optimal Cognitive Flexibility in Mice. Learn. Mem. 2015, 22, 323–335. [Google Scholar] [CrossRef]
- Barcia, G.; Fleming, M.R.; Deligniere, A.; Gazula, V.-R.; Brown, M.R.; Langouet, M.; Chen, H.; Kronengold, J.; Abhyankar, A.; Cilio, R.; et al. De Novo Gain-of-Function KCNT1 Channel Mutations Cause Malignant Migrating Partial Seizures of Infancy. Nat. Genet. 2012, 44, 1255–1259. [Google Scholar] [CrossRef]
- Hager, N.A.; McAtee, C.K.; Lesko, M.A.; O’Donnell, A.F. Inwardly Rectifying Potassium Channel Kir2.1 and Its “Kir-Ious” Regulation by Protein Trafficking and Roles in Development and Disease. Front. Cell Dev. Biol. 2021, 9, 796136. [Google Scholar] [CrossRef]
- Belus, M.T.; Rogers, M.A.; Elzubeir, A.; Josey, M.; Rose, S.; Andreeva, V.; Yelick, P.C.; Bates, E.A. Kir2.1 Is Important for Efficient BMP Signaling in Mammalian Face Development. Dev. Biol. 2018, 444 (Suppl. 1), S297–S307. [Google Scholar] [CrossRef]
- Sakmann, B.; Noma, A.; Trautwein, W. Acetylcholine Activation of Single Muscarinic K+ Channels in Isolated Pacemaker Cells of the Mammalian Heart. Nature 1983, 303, 250–253. [Google Scholar] [CrossRef] [PubMed]
- Nishida, M.; MacKinnon, R. Structural Basis of Inward Rectification: Cytoplasmic Pore of the G Protein-Gated Inward Rectifier GIRK1 at 1.8 Å Resolution. Cell 2002, 111, 957–965. [Google Scholar] [CrossRef]
- Anumonwo, J.M.; Lopatin, A.N. Cardiac Strong Inward Rectifier Potassium Channels. J. Mol. Cell. Cardiol. 2010, 48, 45. [Google Scholar] [CrossRef] [PubMed]
- Feliciangeli, S.; Chatelain, F.C.; Bichet, D.; Lesage, F. The Family of K2P Channels: Salient Structural and Functional Properties. J. Physiol. 2015, 593, 2587–2603. [Google Scholar] [CrossRef]
- Zúñiga, L.; Zúñiga, R. Understanding the Cap Structure in K2P Channels. Front. Physiol. 2016, 7, 228. [Google Scholar] [CrossRef]
- Miller, A.N.; Long, S.B. Crystal Structure of the Human Two-Pore Domain Potassium Channel K2P1. Science 2012, 335, 432–436. [Google Scholar] [CrossRef]
- Brohawn, S.G.; del Mármol, J.; MacKinnon, R. Crystal Structure of the Human K2P TRAAK, a Lipid- and Mechano-Sensitive K+ Ion Channel. Science 2012, 335, 436–441. [Google Scholar] [CrossRef]
- Gada, K.; Plant, L.D. Two-Pore Domain Potassium Channels: Emerging Targets for Novel Analgesic Drugs: IUPHAR Review 26. Br. J. Pharmacol. 2019, 176, 256–266. [Google Scholar] [CrossRef]
- Hemmerlein, B.; Weseloh, R.M.; de Queiroz, F.M.; Knötgen, H.; Sánchez, A.; Rubio, M.E.; Martin, S.; Schliephacke, T.; Jenke, M.; Stühmer, W.; et al. Overexpression of Eag1 Potassium Channels in Clinical Tumours. Mol. Cancer 2006, 5, 41. [Google Scholar] [CrossRef] [Green Version]
- Luis, E.; Anaya-Hernández, A.; León-Sánchez, P.; Durán-Pastén, M.L. The Kv10.1 Channel: A Promising Target in Cancer. Int. J. Mol. Sci. 2022, 23, 8458. [Google Scholar] [CrossRef] [PubMed]
- Bijlenga, P.; Occhiodoro, T.; Liu, J.H.; Bader, C.R.; Bernheim, L.; Fischer-Lougheed, J. An Ether -à-Go-Go K+ Current, Ih-Eag, Contributes to the Hyperpolarization of Human Fusion-Competent Myoblasts. J. Physiol. 1998, 512, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Brüggemann, A.; Pardo, L.A.; Stühmer, W.; Pongs, O. Ether-à-Go-Go Encodes a Voltage-Gated Channel Permeable to K+ and Ca2+ and Modulated by CAMP. Nature 1993, 365, 445–448. [Google Scholar] [CrossRef] [PubMed]
- Pardo, L.A.; Brüggemann, A.; Camacho, J.; Stühmer, W. Cell Cycle-Related Changes in the Conducting Properties of r-Eag K+ Channels. J. Cell Biol. 1998, 143, 767–775. [Google Scholar] [CrossRef]
- Pardo, L.A.; del Camino, D.; Sánchez, A.; Alves, F.; Brüggemann, A.; Beckh, S.; Stühmer, W. Oncogenic Potential of EAG K+ Channels. EMBO J. 1999, 18, 5540–5547. [Google Scholar] [CrossRef]
- Aissaoui, D.; Mlayah-Bellalouna, S.; Jebali, J.; Abdelkafi-Koubaa, Z.; Souid, S.; Moslah, W.; Othman, H.; Luis, J.; ElAyeb, M.; Marrakchi, N.; et al. Functional Role of Kv1.1 and Kv1.3 Channels in the Neoplastic Progression Steps of Three Cancer Cell Lines, Elucidated by Scorpion Peptides. Int. J. Biol. Macromol. 2018, 111, 1146–1155. [Google Scholar] [CrossRef] [PubMed]
- Abdul, M.; Hoosein, N. Expression and Activity of Potassium Ion Channels in Human Prostate Cancer. Cancer Lett. 2002, 186, 99–105. [Google Scholar] [CrossRef]
- Abdul, M.; Hoosein, N. Reduced Kv1.3 Potassium Channel Expression in Human Prostate Cancer. J. Membr. Biol. 2006, 214, 99–102. [Google Scholar] [CrossRef]
- Jang, S.-H.; Kang, K.-S.; Ryu, P.-D.; Lee, S.-Y. Kv1.3 Voltage-Gated K+ Channel Subunit as a Potential Diagnostic Marker and Therapeutic Target for Breast Cancer. BMB Rep. 2009, 42, 535–539. [Google Scholar] [CrossRef]
- Brevet, M.; Haren, N.; Sevestre, H.; Merviel, P.; Ouadid-Ahidouch, H. DNA Methylation of K(v)1.3 Potassium Channel Gene Promoter Is Associated with Poorly Differentiated Breast Adenocarcinoma. Cell. Physiol. Biochem. 2009, 24, 25–32. [Google Scholar] [CrossRef]
- Wu, J.; Zhong, D.; Wu, X.; Sha, M.; Kang, L.; Ding, Z. Voltage-Gated Potassium Channel Kv1.3 Is Highly Expressed in Human Osteosarcoma and Promotes Osteosarcoma Growth. Int. J. Mol. Sci. 2013, 14, 19245–19256. [Google Scholar] [CrossRef] [PubMed]
- Szabo, I.; Trentin, L.; Trimarco, V.; Semenzato, G.; Leanza, L. Biophysical Characterization and Expression Analysis of Kv1.3 Potassium Channel in Primary Human Leukemic B Cells. Cell. Physiol. Biochem. 2015, 37, 965–978. [Google Scholar] [CrossRef] [PubMed]
- Felipe, A.; Bielanska, J.; Comes, N.; Vallejo, A.; Roig, S.; Cajal, S.R.Y.; Condom, E.; Hernández-Losa, J.; Ferreres, J.C. Targeting the Voltage-Dependent K+ Channels Kv1.3 and Kv1.5 as Tumor Biomarkers for Cancer Detection and Prevention. Curr. Med. Chem. 2012, 19, 661–674. [Google Scholar] [CrossRef] [PubMed]
- Bielanska, J.; Hernández-Losa, J.; Moline, T.; Somoza, R.; Cajal, S.R.Y.; Condom, E.; Ferreres, J.C.; Felipe, A. Increased Voltage-Dependent K+ Channel Kv1.3 and Kv1.5 Expression Correlates with Leiomyosarcoma Aggressiveness. Oncol. Lett. 2012, 4, 227–230. [Google Scholar] [CrossRef]
- Vallejo-Gracia, A.; Bielanska, J.; Hernández-Losa, J.; Castellví, J.; Ruiz-Marcellan, M.C.; Cajal, S.R.Y.; Condom, E.; Manils, J.; Soler, C.; Comes, N.; et al. Emerging Role for the Voltage-Dependent K+ Channel Kv1.5 in B-Lymphocyte Physiology: Expression Associated with Human Lymphoma Malignancy. J. Leukoc. Biol. 2013, 94, 779–789. [Google Scholar] [CrossRef]
- Lan, M.; Shi, Y.; Han, Z.; Hao, Z.; Pan, Y.; Liu, N.; Guo, C.; Hong, L.; Wang, J.; Qiao, T.; et al. Expression of Delayed Rectifier Potassium Channels and Their Possible Roles in Proliferation of Human Gastric Cancer Cells. Cancer Biol. Ther. 2005, 4, 1342–1347. [Google Scholar] [CrossRef]
- Wu, J.; Chen, Z.; Liu, Q.; Zeng, W.; Wu, X.; Lin, B. Silencing of Kv1.5 Gene Inhibits Proliferation and Induces Apoptosis of Osteosarcoma Cells. Int. J. Mol. Sci. 2015, 16, 26914–26926. [Google Scholar] [CrossRef]
- Suzuki, T.; Takimoto, K. Selective Expression of HERG and Kv2 Channels Influences Proliferation of Uterine Cancer Cells. Int. J. Oncol. 2004, 25, 153–159. [Google Scholar] [CrossRef]
- Song, M.S.; Park, S.M.; Park, J.S.; Byun, J.H.; Jin, H.J.; Seo, S.H.; Ryu, P.D.; Lee, S.Y. Kv3.1 and Kv3.4, Are Involved in Cancer Cell Migration and Invasion. Int. J. Mol. Sci. 2018, 19, E1061. [Google Scholar] [CrossRef]
- Menéndez, S.T.; Rodrigo, J.P.; Allonca, E.; García-Carracedo, D.; Alvarez-Alija, G.; Casado-Zapico, S.; Fresno, M.F.; Rodríguez, C.; Suárez, C.; García-Pedrero, J.M. Expression and Clinical Significance of the Kv3.4 Potassium Channel Subunit in the Development and Progression of Head and Neck Squamous Cell Carcinomas. J. Pathol. 2010, 221, 402–410. [Google Scholar] [CrossRef]
- Jang, S.H.; Choi, C.; Hong, S.-G.; Yarishkin, O.V.; Bae, Y.M.; Kim, J.G.; O’Grady, S.M.; Yoon, K.-A.; Kang, K.-S.; Ryu, P.D.; et al. Silencing of Kv4.1 Potassium Channels Inhibits Cell Proliferation of Tumorigenic Human Mammary Epithelial Cells. Biochem. Biophys. Res. Commun. 2009, 384, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Fujii, T.; Takahashi, Y.; Takahashi, Y.; Suzuki, T.; Ukai, M.; Tauchi, K.; Horikawa, N.; Tsukada, K.; Sakai, H. Up-Regulation of Kv7.1 Channels in Thromboxane A2-Induced Colonic Cancer Cell Proliferation. Pflug. Arch. 2014, 466, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-J.; Jang, S.H.; Jeong, Y.A.; Ryu, P.D.; Kim, D.-Y.; Lee, S.Y. Involvement of Kv4.1 K+ Channels in Gastric Cancer Cell Proliferation. Biol. Pharm. Bull. 2010, 33, 1754–1757. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.-W.; Luo, H.; Jin, X.; Yan, J.; Ai, Y. Aberrant Expression of Eag1 Potassium Channels in Gastric Cancer Patients and Cell Lines. Med. Oncol. 2007, 24, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhong, D.; Lin, B.; Zhai, W.; Ding, Z.; Wu, J. P38 MAPK Regulates the Expression of Ether à Go-Go Potassium Channel in Human Osteosarcoma Cells. Radiol. Oncol. 2013, 47, 42–49. [Google Scholar] [CrossRef]
- Wu, J.; Zhong, D.; Fu, X.; Liu, Q.; Kang, L.; Ding, Z. Silencing of Ether à Go-Go 1 by ShRNA Inhibits Osteosarcoma Growth and Cell Cycle Progression. Int. J. Mol. Sci. 2014, 15, 5570–5581. [Google Scholar] [CrossRef]
- Cheng, Y.Y.; Wright, C.M.; Kirschner, M.B.; Williams, M.; Sarun, K.H.; Sytnyk, V.; Leshchynska, I.; Edelman, J.J.; Vallely, M.P.; McCaughan, B.C.; et al. KCa1.1, a Calcium-Activated Potassium Channel Subunit Alpha 1, Is Targeted by MiR-17-5p and Modulates Cell Migration in Malignant Pleural Mesothelioma. Mol. Cancer 2016, 15, 44. [Google Scholar] [CrossRef]
- Ohya, S.; Kajikuri, J.; Endo, K.; Kito, H.; Elboray, E.E.; Suzuki, T. Ca2+-Activated K+ Channel KCa 1.1 as a Therapeutic Target to Overcome Chemoresistance in Three-Dimensional Sarcoma Spheroid Models. Cancer Sci. 2021, 112, 3769–3783. [Google Scholar] [CrossRef]
- Guéguinou, M.; Harnois, T.; Crottes, D.; Uguen, A.; Deliot, N.; Gambade, A.; Chantôme, A.; Haelters, J.P.; Jaffrès, P.A.; Jourdan, M.L.; et al. SK3/TRPC1/Orai1 Complex Regulates SOCE-Dependent Colon Cancer Cell Migration: A Novel Opportunity to Modulate Anti-EGFR MAb Action by the Alkyl-Lipid Ohmline. Oncotarget 2016, 7, 36168–36184. [Google Scholar] [CrossRef]
- Potier, M.; Joulin, V.; Roger, S.; Besson, P.; Jourdan, M.-L.; Leguennec, J.-Y.; Bougnoux, P.; Vandier, C. Identification of SK3 Channel as a New Mediator of Breast Cancer Cell Migration. Mol. Cancer Ther. 2006, 5, 2946–2953. [Google Scholar] [CrossRef] [Green Version]
- Györffy, B.; Lanczky, A.; Eklund, A.C.; Denkert, C.; Budczies, J.; Li, Q.; Szallasi, Z. An Online Survival Analysis Tool to Rapidly Assess the Effect of 22,277 Genes on Breast Cancer Prognosis Using Microarray Data of 1809 Patients. Breast Cancer Res. Treat. 2010, 123, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Bulk, E.; Ay, A.-S.; Hammadi, M.; Ouadid-Ahidouch, H.; Schelhaas, S.; Hascher, A.; Rohde, C.; Thoennissen, N.H.; Wiewrodt, R.; Schmidt, E.; et al. Epigenetic Dysregulation of KCa 3.1 Channels Induces Poor Prognosis in Lung Cancer. Int. J. Cancer 2015, 137, 1306–1317. [Google Scholar] [CrossRef] [PubMed]
- Rabjerg, M.; Oliván-Viguera, A.; Hansen, L.K.; Jensen, L.; Sevelsted-Møller, L.; Walter, S.; Jensen, B.L.; Marcussen, N.; Köhler, R. High Expression of KCa3.1 in Patients with Clear Cell Renal Carcinoma Predicts High Metastatic Risk and Poor Survival. PLoS ONE 2015, 10, e0122992. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.H.; Shen, B.; Yao, H.L.; Jia, Y.C.; Ren, J.; Feng, Y.J.; Wang, Y.Z. Blockage of Intermediate-Conductance-Ca2+-Activated K+ Channels Inhibits Progression of Human Endometrial Cancer. Oncogene 2007, 26, 5107–5114. [Google Scholar] [CrossRef]
- Jäger, H.; Dreker, T.; Buck, A.; Giehl, K.; Gress, T.; Grissmer, S. Blockage of Intermediate-Conductance Ca2+-Activated K+ Channels Inhibit Human Pancreatic Cancer Cell Growth in Vitro. Mol. Pharmacol. 2004, 65, 630–638. [Google Scholar] [CrossRef]
- Lee, I.; Park, C.; Kang, W.K. Knockdown of Inwardly Rectifying Potassium Channel Kir2.2 Suppresses Tumorigenesis by Inducing Reactive Oxygen Species-Mediated Cellular Senescence. Mol. Cancer Ther. 2010, 9, 2951–2959. [Google Scholar] [CrossRef]
- Adam, M.A.; Untch, B.R.; Olson, J.A. Parathyroid Carcinoma: Current Understanding and New Insights into Gene Expression and Intraoperative Parathyroid Hormone Kinetics. Oncologist 2010, 15, 61–72. [Google Scholar] [CrossRef]
- Liu, G.; Wu, K.; Sheng, Y. Elucidation of the Molecular Mechanisms of Anaplastic Thyroid Carcinoma by Integrated MiRNA and MRNA Analysis. Oncol. Rep. 2016, 36, 3005–3013. [Google Scholar] [CrossRef]
- Jiang, S.; Zhu, L.; Yang, J.; Hu, L.; Gu, J.; Xing, X.; Sun, Y.; Zhang, Z. Integrated Expression Profiling of Potassium Channels Identifys KCNN4 as a Prognostic Biomarker of Pancreatic Cancer. Biochem. Biophys. Res. Commun. 2017, 494, 113–119. [Google Scholar] [CrossRef]
- Voloshyna, I.; Besana, A.; Castillo, M.; Matos, T.; Weinstein, I.B.; Mansukhani, M.; Robinson, R.B.; Cordon-Cardo, C.; Feinmark, S.J. TREK-1 Is a Novel Molecular Target in Prostate Cancer. Cancer Res. 2008, 68, 1197–1203. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.J.; Cho, Y.G.; Jeong, S.W.; Kim, Y.S.; Kim, S.Y.; Nam, S.W.; Lee, S.H.; Yoo, N.J.; Lee, J.Y.; Park, W.S. Altered Expression of KCNK9 in Colorectal Cancers. Apmis 2004, 112, 588–594. [Google Scholar] [CrossRef] [PubMed]
- Mu, D.; Chen, L.; Zhang, X.; See, L.-H.; Koch, C.M.; Yen, C.; Tong, J.J.; Spiegel, L.; Nguyen, K.C.Q.; Servoss, A.; et al. Genomic Amplification and Oncogenic Properties of the KCNK9 Potassium Channel Gene. Cancer Cell 2003, 3, 297–302. [Google Scholar] [CrossRef]
- Serrano-Novillo, C.; Capera, J.; Colomer-Molera, M.; Condom, E.; Ferreres, J.C.; Felipe, A. Implication of Voltage-Gated Potassium Channels in Neoplastic Cell Proliferation. Cancers 2019, 11, 287. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Dubuc, A.M.; Hashizume, R.; Berg, J.; He, Y.; Wang, J.; Chiang, C.; Cooper, M.K.; Northcott, P.A.; Taylor, M.D.; et al. Voltage-Gated Potassium Channel EAG2 Controls Mitotic Entry and Tumor Growth in Medulloblastoma via Regulating Cell Volume Dynamics. Genes Dev. 2012, 26, 1780–1796. [Google Scholar] [CrossRef]
- deHart, G.W.; Jin, T.; McCloskey, D.E.; Pegg, A.E.; Sheppard, D. The Alpha9beta1 Integrin Enhances Cell Migration by Polyamine-Mediated Modulation of an Inward-Rectifier Potassium Channel. Proc. Natl. Acad. Sci. USA 2008, 105, 7188–7193. [Google Scholar] [CrossRef]
- Masi, A.; Becchetti, A.; Restano-Cassulini, R.; Polvani, S.; Hofmann, G.; Buccoliero, A.M.; Paglierani, M.; Pollo, B.; Taddei, G.L.; Gallina, P.; et al. HERG1 Channels Are Overexpressed in Glioblastoma Multiforme and Modulate VEGF Secretion in Glioblastoma Cell Lines. Br. J. Cancer 2005, 93, 781–792. [Google Scholar] [CrossRef]
- Crociani, O.; Zanieri, F.; Pillozzi, S.; Lastraioli, E.; Stefanini, M.; Fiore, A.; Fortunato, A.; D’Amico, M.; Masselli, M.; De Lorenzo, E.; et al. HERG1 Channels Modulate Integrin Signaling to Trigger Angiogenesis and Tumor Progression in Colorectal Cancer. Sci. Rep. 2013, 3, 3308. [Google Scholar] [CrossRef]
- Umaru, B.; Pyriochou, A.; Kotsikoris, V.; Papapetropoulos, A.; Topouzis, S. ATP-Sensitive Potassium Channel Activation Induces Angiogenesis In Vitro and In Vivo. J. Pharmacol. Exp. Ther. 2015, 354, 79–87. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Weller, M.; Wick, W.; Aldape, K.; Brada, M.; Berger, M.; Pfister, S.M.; Nishikawa, R.; Rosenthal, M.; Wen, P.Y.; Stupp, R.; et al. Glioma. Nat. Rev. Dis. Prim. 2015, 1, 15017. [Google Scholar] [CrossRef]
- Miller, K.D.; Ostrom, Q.T.; Kruchko, C.; Patil, N.; Tihan, T.; Cioffi, G.; Fuchs, H.E.; Waite, K.A.; Jemal, A.; Siegel, R.L.; et al. Brain and Other Central Nervous System Tumor Statistics, 2021. CA Cancer J. Clin. 2021, 71, 381–406. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tyler, E.; Lustick, M.; Klein, D.; Walter, K.A. Healthcare Costs for High-Grade Glioma. Anticancer Res. 2019, 39, 1375–1381. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Brada, M.; van den Bent, M.J.; Tonn, J.-C.; Pentheroudakis, G.; ESMO Guidelines Working Group. High-Grade Glioma: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2014, 25 (Suppl. 3), iii93–iii101. [Google Scholar] [CrossRef] [PubMed]
- Yao, M.; Li, S.; Wu, X.; Diao, S.; Zhang, G.; He, H.; Bian, L.; Lu, Y. Cellular Origin of Glioblastoma and Its Implication in Precision Therapy. Cell. Mol. Immunol. 2018, 15, 737–739. [Google Scholar] [CrossRef]
- Wang, Y.; Jiang, T. Understanding High Grade Glioma: Molecular Mechanism, Therapy and Comprehensive Management. Cancer Lett. 2013, 331, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Claus, E.B.; Walsh, K.M.; Wiencke, J.K.; Molinaro, A.M.; Wiemels, J.L.; Schildkraut, J.M.; Bondy, M.L.; Berger, M.; Jenkins, R.; Wrensch, M. Survival and Low-Grade Glioma: The Emergence of Genetic Information. Neurosurg. Focus 2015, 38, E6. [Google Scholar] [CrossRef]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO Classification of Tumours of the Central Nervous System. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef]
- Forst, D.A.; Nahed, B.V.; Loeffler, J.S.; Batchelor, T.T. Low-Grade Gliomas. Oncologist 2014, 19, 403–413. [Google Scholar] [CrossRef]
- Millard, N.E.; De Braganca, K.C. Medulloblastoma. J. Child Neurol. 2016, 31, 1341–1353. [Google Scholar] [CrossRef]
- Shergalis, A.; Bankhead, A.; Luesakul, U.; Muangsin, N.; Neamati, N. Current Challenges and Opportunities in Treating Glioblastoma. Pharmacol. Rev. 2018, 70, 412–445. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Cohen, A.L.; Colman, H. Targeted Therapeutics in Patients with High-Grade Gliomas: Past, Present, and Future. Curr. Treat. Options Oncol. 2016, 17, 42. [Google Scholar] [CrossRef] [PubMed]
- An, Z.; Aksoy, O.; Zheng, T.; Fan, Q.-W.; Weiss, W.A. Epidermal Growth Factor Receptor and EGFRvIII in Glioblastoma: Signaling Pathways and Targeted Therapies. Oncogene 2018, 37, 1561–1575. [Google Scholar] [CrossRef] [PubMed]
- Swartz, A.M.; Li, Q.-J.; Sampson, J.H. Rindopepimut: A Promising Immunotherapeutic for the Treatment of Glioblastoma Multiforme. Immunotherapy 2014, 6, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Inman, S. Rindopepimut Misses OS Endpoint in Phase III Glioblastoma Trial. Available online: https://www.onclive.com/view/rindopepimut-misses-os-endpoint-in-phase-iii-glioblastoma-trial (accessed on 11 June 2022).
- Batchelor, T.T.; Mulholland, P.; Neyns, B.; Nabors, L.B.; Campone, M.; Wick, A.; Mason, W.; Mikkelsen, T.; Phuphanich, S.; Ashby, L.S.; et al. Phase III Randomized Trial Comparing the Efficacy of Cediranib as Monotherapy, and in Combination with Lomustine, versus Lomustine Alone in Patients with Recurrent Glioblastoma. J. Clin. Oncol. 2013, 31, 3212–3218. [Google Scholar] [CrossRef]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A Randomized Trial of Bevacizumab for Newly Diagnosed Glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef]
- Chinot, O.L.; Wick, W.; Mason, W.; Henriksson, R.; Saran, F.; Nishikawa, R.; Carpentier, A.F.; Hoang-Xuan, K.; Kavan, P.; Cernea, D.; et al. Bevacizumab plus Radiotherapy-Temozolomide for Newly Diagnosed Glioblastoma. N. Engl. J. Med. 2014, 370, 709–722. [Google Scholar] [CrossRef]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849. [Google Scholar] [CrossRef]
- Debanne, D.; Daoudal, G.; Sourdet, V.; Russier, M. Brain Plasticity and Ion Channels. J. Physiol. Paris 2003, 97, 403–414. [Google Scholar] [CrossRef]
- Liebau, S.; Kleger, A.; Levin, M.; Yu, S.P. Stem Cells and Ion Channels. Stem Cells Int. 2013, 2013, 238635. [Google Scholar] [CrossRef]
- Bouffet, E.; Hansford, J.; Garré, M.L.; Hara, J.; Plant-Fox, A.; Aerts, I.; Locatelli, F.; Van der Lugt, J.; Papusha, L.; Sahm, F.; et al. Primary Analysis of a Phase II Trial of Dabrafenib plus Trametinib (Dab + Tram) in BRAF V600–Mutant Pediatric Low-Grade Glioma (PLGG). J. Clin. Oncol. 2022, 40, LBA2002. [Google Scholar] [CrossRef]
- Sharpe, H.J.; Pau, G.; Dijkgraaf, G.J.; Basset-Seguin, N.; Modrusan, Z.; Januario, T.; Tsui, V.; Durham, A.B.; Dlugosz, A.A.; Haverty, P.M.; et al. Genomic Analysis of Smoothened Inhibitor Resistance in Basal Cell Carcinoma. Cancer Cell 2015, 27, 327–341. [Google Scholar] [CrossRef] [PubMed]
- Robinson, G.W.; Kaste, S.C.; Chemaitilly, W.; Bowers, D.C.; Laughton, S.; Smith, A.; Gottardo, N.G.; Partap, S.; Bendel, A.; Wright, K.D.; et al. Irreversible Growth Plate Fusions in Children with Medulloblastoma Treated with a Targeted Hedgehog Pathway Inhibitor. Oncotarget 2017, 8, 69295–69302. [Google Scholar] [CrossRef] [PubMed]
- Frappaz, D.; Barritault, M.; Montané, L.; Laigle-Donadey, F.; Chinot, O.; Le Rhun, E.; Bonneville-Levard, A.; Hottinger, A.F.; Meyronnet, D.; Bidaux, A.-S.; et al. MEVITEM-a Phase I/II Trial of Vismodegib + Temozolomide vs. Temozolomide in Patients with Recurrent/Refractory Medulloblastoma with Sonic Hedgehog Pathway Activation. Neuro Oncol. 2021, 23, 1949–1960. [Google Scholar] [CrossRef] [PubMed]
- Dimitrova, V.; Arcaro, A. Targeting the PI3K/AKT/MTOR Signaling Pathway in Medulloblastoma. Curr Mol Med. 2015, 15, 82–93. [Google Scholar] [CrossRef]
- Holzhauser, S.; Lukoseviciute, M.; Andonova, T.; Ursu, R.G.; Dalianis, T.; Wickström, M.; Kostopoulou, O.N. Targeting Fibroblast Growth Factor Receptor (FGFR) and Phosphoinositide 3-Kinase (PI3K) Signaling Pathways in Medulloblastoma Cell Lines. Anticancer Res. 2020, 40, 53–66. [Google Scholar] [CrossRef]
- Holzhauser, S.; Lukoseviciute, M.; Papachristofi, C.; Vasilopoulou, C.; Herold, N.; Wickström, M.; Kostopoulou, O.N.; Dalianis, T. Effects of PI3K and FGFR Inhibitors Alone and in Combination, and with/without Cytostatics in Childhood Neuroblastoma Cell Lines. Int. J. Oncol. 2021, 58, 211–225. [Google Scholar] [CrossRef]
- Ghasemi, D.R.; Fleischhack, G.; Milde, T.; Pajtler, K.W. The Current Landscape of Targeted Clinical Trials in Non-WNT/Non-SHH Medulloblastoma. Cancers 2022, 14, 679. [Google Scholar] [CrossRef]
- Sturm, D.; Witt, H.; Hovestadt, V.; Khuong-Quang, D.-A.; Jones, D.T.W.; Konermann, C.; Pfaff, E.; Tönjes, M.; Sill, M.; Bender, S.; et al. Hotspot Mutations in H3F3A and IDH1 Define Distinct Epigenetic and Biological Subgroups of Glioblastoma. Cancer Cell 2012, 22, 425–437. [Google Scholar] [CrossRef]
- Greenall, S.A.; Donoghue, J.F.; Van Sinderen, M.; Dubljevic, V.; Budiman, S.; Devlin, M.; Street, I.; Adams, T.E.; Johns, T.G. EGFRvIII-Mediated Transactivation of Receptor Tyrosine Kinases in Glioma: Mechanism and Therapeutic Implications. Oncogene 2015, 34, 5277–5287. [Google Scholar] [CrossRef]
- Pillay, V.; Allaf, L.; Wilding, A.L.; Donoghue, J.F.; Court, N.W.; Greenall, S.A.; Scott, A.M.; Johns, T.G. The Plasticity of Oncogene Addiction: Implications for Targeted Therapies Directed to Receptor Tyrosine Kinases. Neoplasia 2009, 11, 448–458. [Google Scholar] [CrossRef] [Green Version]
- Friedmann-Morvinski, D. Glioblastoma Heterogeneity and Cancer Cell Plasticity. Crit. Rev. Oncog. 2014, 19, 327–336. [Google Scholar] [CrossRef]
- Osuka, S.; Meir, E.G.V. Overcoming Therapeutic Resistance in Glioblastoma: The Way Forward. J. Clin. Investig. 2017, 127, 415–426. [Google Scholar] [CrossRef]
- Bourkoula, E.; Mangoni, D.; Ius, T.; Pucer, A.; Isola, M.; Musiello, D.; Marzinotto, S.; Toffoletto, B.; Sorrentino, M.; Palma, A.; et al. Glioma-Associated Stem Cells: A Novel Class of Tumor-Supporting Cells Able to Predict Prognosis of Human Low-Grade Gliomas. Stem Cells 2014, 32, 1239–1253. [Google Scholar] [CrossRef]
- Sampetrean, O.; Saya, H. Characteristics of Glioma Stem Cells. Brain Tumor Pathol. 2013, 30, 209–214. [Google Scholar] [CrossRef]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.L.; Rich, J.N. Cancer Stem Cells in Glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef]
- Hadjipanayis, C.G.; Van Meir, E.G. Tumor Initiating Cells in Malignant Gliomas: Biology and Implications for Therapy. J. Mol. Med. 2009, 87, 363–374. [Google Scholar] [CrossRef]
- Singh, N.; Miner, A.; Hennis, L.; Mittal, S. Mechanisms of Temozolomide Resistance in Glioblastoma—A Comprehensive Review. Cancer Drug Resist. 2021, 4, 17–43. [Google Scholar] [CrossRef]
- Kim, H.; Zheng, S.; Amini, S.S.; Virk, S.M.; Mikkelsen, T.; Brat, D.J.; Grimsby, J.; Sougnez, C.; Muller, F.; Hu, J.; et al. Whole-Genome and Multisector Exome Sequencing of Primary and Post-Treatment Glioblastoma Reveals Patterns of Tumor Evolution. Genome Res. 2015, 25, 316–327. [Google Scholar] [CrossRef]
- Orzan, F.; De Bacco, F.; Crisafulli, G.; Pellegatta, S.; Mussolin, B.; Siravegna, G.; D’Ambrosio, A.; Comoglio, P.M.; Finocchiaro, G.; Boccaccio, C. Genetic Evolution of Glioblastoma Stem-Like Cells from Primary to Recurrent Tumor. Stem Cells 2017, 35, 2218–2228. [Google Scholar] [CrossRef]
- Zhang, Y.; Dube, C.; Gibert, M.; Cruickshanks, N.; Wang, B.; Coughlan, M.; Yang, Y.; Setiady, I.; Deveau, C.; Saoud, K.; et al. The P53 Pathway in Glioblastoma. Cancers 2018, 10, E297. [Google Scholar] [CrossRef] [Green Version]
- Cordner, R.; Black, K.L.; Wheeler, C.J. Exploitation of Adaptive Evolution in Glioma Treatment. CNS Oncol. 2013, 2, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Ruggieri, P.; Mangino, G.; Fioretti, B.; Catacuzzeno, L.; Puca, R.; Ponti, D.; Miscusi, M.; Franciolini, F.; Ragona, G.; Calogero, A. The Inhibition of KCa3.1 Channels Activity Reduces Cell Motility in Glioblastoma Derived Cancer Stem Cells. PLoS ONE 2012, 7, e47825. [Google Scholar] [CrossRef] [PubMed]
- Rosa, P.; Sforna, L.; Carlomagno, S.; Mangino, G.; Miscusi, M.; Pessia, M.; Franciolini, F.; Calogero, A.; Catacuzzeno, L. Overexpression of Large-Conductance Calcium-Activated Potassium Channels in Human Glioblastoma Stem-Like Cells and Their Role in Cell Migration. J. Cell. Physiol. 2017, 232, 2478–2488. [Google Scholar] [CrossRef] [PubMed]
- Pchelintseva, E.; Djamgoz, M.B.A. Mesenchymal Stem Cell Differentiation: Control by Calcium-Activated Potassium Channels. J. Cell. Physiol. 2018, 233, 3755–3768. [Google Scholar] [CrossRef]
- Ocasio, J.K.; Babcock, B.; Malawsky, D.; Weir, S.J.; Loo, L.; Simon, J.M.; Zylka, M.J.; Hwang, D.; Dismuke, T.; Sokolsky, M.; et al. ScRNA-Seq in Medulloblastoma Shows Cellular Heterogeneity and Lineage Expansion Support Resistance to SHH Inhibitor Therapy. Nat. Commun. 2019, 10, 5829. [Google Scholar] [CrossRef]
- Vanner, R.J.; Remke, M.; Gallo, M.; Selvadurai, H.J.; Coutinho, F.; Lee, L.; Kushida, M.; Head, R.; Morrissy, S.; Zhu, X.; et al. Quiescent Sox2+ Cells Drive Hierarchical Growth and Relapse in Sonic Hedgehog Subgroup Medulloblastoma. Cancer Cell 2014, 26, 33–47. [Google Scholar] [CrossRef]
- Mansouri, S.; Nejad, R.; Karabork, M.; Ekinci, C.; Solaroglu, I.; Aldape, K.D.; Zadeh, G. Sox2: Regulation of Expression and Contribution to Brain Tumors. CNS Oncol. 2016, 5, 159–173. [Google Scholar] [CrossRef]
- Metz, E.P.; Wuebben, E.L.; Wilder, P.J.; Cox, J.L.; Datta, K.; Coulter, D.; Rizzino, A. Tumor Quiescence: Elevating SOX2 in Diverse Tumor Cell Types Downregulates a Broad Spectrum of the Cell Cycle Machinery and Inhibits Tumor Growth. BMC Cancer 2020, 20, 941. [Google Scholar] [CrossRef]
- Swiderska-Syn, M.; Mir-Pedrol, J.; Oles, A.; Schleuger, O.; Salvador, A.D.; Greiner, S.M.; Seward, C.; Yang, F.; Babcock, B.R.; Shen, C.; et al. Noncanonical Activation of GLI Signaling in SOX2+ Cells Drives Medulloblastoma Relapse. Sci. Adv. 2022, 8, eabj9138. [Google Scholar] [CrossRef]
- Ransom, C.B.; Sontheimer, H. BK Channels in Human Glioma Cells. J. Neurophysiol. 2001, 85, 790–803. [Google Scholar] [CrossRef] [Green Version]
- Wondergem, R.; Bartley, J.W. Menthol Increases Human Glioblastoma Intracellular Ca2+, BK Channel Activity and Cell Migration. J. Biomed. Sci. 2009, 16, 90. [Google Scholar] [CrossRef] [PubMed]
- Abdullaev, I.F.; Rudkouskaya, A.; Mongin, A.A.; Kuo, Y.-H. Calcium-Activated Potassium Channels BK and IK1 Are Functionally Expressed in Human Gliomas but Do Not Regulate Cell Proliferation. PLoS ONE 2010, 5, e12304. [Google Scholar] [CrossRef] [PubMed]
- Weaver, A.K.; Liu, X.; Sontheimer, H. Role for Calcium-Activated Potassium Channels (BK) in Growth Control of Human Malignant Glioma Cells. J. Neurosci. Res. 2004, 78, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Kraft, R.; Krause, P.; Jung, S.; Basrai, D.; Liebmann, L.; Bolz, J.; Patt, S. BK Channel Openers Inhibit Migration of Human Glioma Cells. Pflug. Arch. 2003, 446, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Newman, E.A. Inward-Rectifying Potassium Channels in Retinal Glial (Müller) Cells. J. Neurosci. 1993, 13, 3333–3345. [Google Scholar] [CrossRef] [PubMed]
- Olsen, M.L.; Sontheimer, H. Mislocalization of Kir Channels in Malignant Glia. Glia 2004, 46, 63–73. [Google Scholar] [CrossRef]
- Traynelis, S.F.; Dingledine, R. Potassium-Induced Spontaneous Electrographic Seizures in the Rat Hippocampal Slice. J. Neurophysiol. 1988, 59, 259–276. [Google Scholar] [CrossRef]
- Brandalise, F.; Ratto, D.; Leone, R.; Olivero, F.; Roda, E.; Locatelli, C.A.; Grazia Bottone, M.; Rossi, P. Deeper and Deeper on the Role of BK and Kir4.1 Channels in Glioblastoma Invasiveness: A Novel Summative Mechanism? Front. Neurosci. 2020, 14, 595664. [Google Scholar] [CrossRef]
- Grimaldi, A.; D’Alessandro, G.; Di Castro, M.A.; Lauro, C.; Singh, V.; Pagani, F.; Sforna, L.; Grassi, F.; Di Angelantonio, S.; Catacuzzeno, L.; et al. Kv1.3 Activity Perturbs the Homeostatic Properties of Astrocytes in Glioma. Sci. Rep. 2018, 8, 7654. [Google Scholar] [CrossRef]
- Preussat, K.; Beetz, C.; Schrey, M.; Kraft, R.; Wölfl, S.; Kalff, R.; Patt, S. Expression of Voltage-Gated Potassium Channels Kv1.3 and Kv1.5 in Human Gliomas. Neurosci. Lett. 2003, 346, 33–36. [Google Scholar] [CrossRef]
- Martínez, R.; Stühmer, W.; Martin, S.; Schell, J.; Reichmann, A.; Rohde, V.; Pardo, L. Analysis of the Expression of Kv10.1 Potassium Channel in Patients with Brain Metastases and Glioblastoma Multiforme: Impact on Survival. BMC Cancer 2015, 15, 839. [Google Scholar] [CrossRef] [PubMed]
- Sales, T.T.; Resende, F.F.B.; Chaves, N.L.; Titze-De-Almeida, S.S.; Báo, S.N.; Brettas, M.L.; Titze-De-Almeida, R. Suppression of the Eag1 Potassium Channel Sensitizes Glioblastoma Cells to Injury Caused by Temozolomide. Oncol Lett. 2016, 12, 2581–2589. [Google Scholar] [CrossRef] [PubMed]
- Lastraioli, E.; Perrone, G.; Sette, A.; Fiore, A.; Crociani, O.; Manoli, S.; D’Amico, M.; Masselli, M.; Iorio, J.; Callea, M.; et al. HERG1 Channels Drive Tumour Malignancy and May Serve as Prognostic Factor in Pancreatic Ductal Adenocarcinoma. Br. J. Cancer 2015, 112, 1076–1087. [Google Scholar] [CrossRef]
- Staudacher, I.; Jehle, J.; Staudacher, K.; Pledl, H.-W.; Lemke, D.; Schweizer, P.A.; Becker, R.; Katus, H.A.; Thomas, D. HERG K+ Channel-Dependent Apoptosis and Cell Cycle Arrest in Human Glioblastoma Cells. PLoS ONE 2014, 9, e88164. [Google Scholar] [CrossRef]
- Patt, S.; Preussat, K.; Beetz, C.; Kraft, R.; Schrey, M.; Kalff, R.; Schönherr, K.; Heinemann, S.H. Expression of Ether à Go-Go Potassium Channels in Human Gliomas. Neurosci. Lett. 2004, 368, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-Y.; Li, J.-Y.; Liu, X.; Yan, X.-Y.; Wang, W.; Wu, F.; Liang, T.-Y.; Yang, F.; Hu, H.-M.; Mao, H.-X.; et al. A Three Ion Channel Genes-Based Signature Predicts Prognosis of Primary Glioblastoma Patients and Reveals a Chemotherapy Sensitive Subtype. Oncotarget 2016, 7, 74895–74903. [Google Scholar] [CrossRef]
- Venkatesh, H.S.; Morishita, W.; Geraghty, A.C.; Silverbush, D.; Gillespie, S.M.; Arzt, M.; Tam, L.T.; Espenel, C.; Ponnuswami, A.; Ni, L.; et al. Electrical and Synaptic Integration of Glioma into Neural Circuits. Nature 2019, 573, 539–545. [Google Scholar] [CrossRef]
- Venkatesh, H.S.; Johung, T.B.; Caretti, V.; Noll, A.; Tang, Y.; Nagaraja, S.; Gibson, E.M.; Mount, C.W.; Polepalli, J.; Mitra, S.S.; et al. Neuronal Activity Promotes Glioma Growth through Neuroligin-3 Secretion. Cell 2015, 161, 803–816. [Google Scholar] [CrossRef]
- Augustus, M.; Pineau, D.; Aimond, F.; Azar, S.; Lecca, D.; Scamps, F.; Muxel, S.; Darlix, A.; Ritchie, W.; Gozé, C.; et al. Identification of CRYAB+ KCNN3+ SOX9+ Astrocyte-Like and EGFR+ PDGFRA+ OLIG1+ Oligodendrocyte-Like Tumoral Cells in Diffuse IDH1-Mutant Gliomas and Implication of NOTCH1 Signalling in Their Genesis. Cancers 2021, 13, 2107. [Google Scholar] [CrossRef] [PubMed]
- Arvind, S.; Arivazhagan, A.; Santosh, V.; Chandramouli, B.A. Differential Expression of a Novel Voltage Gated Potassium Channel--Kv 1.5 in Astrocytomas and Its Impact on Prognosis in Glioblastoma. Br. J. Neurosurg. 2012, 26, 16–20. [Google Scholar] [CrossRef]
- Comes, N.; Bielanska, J.; Vallejo-Gracia, A.; Serrano-Albarrás, A.; Marruecos, L.; Gómez, D.; Soler, C.; Condom, E.; Ramón y Cajal, S.; Hernández-Losa, J.; et al. The Voltage-Dependent K+ Channels Kv1.3 and Kv1.5 in Human Cancer. Front. Physiol. 2013, 4, 283. [Google Scholar] [CrossRef]
- Liu, X.; Chang, Y.; Reinhart, P.H.; Sontheimer, H.; Chang, Y. Cloning and Characterization of Glioma BK, a Novel BK Channel Isoform Highly Expressed in Human Glioma Cells. J. Neurosci. 2002, 22, 1840–1849. [Google Scholar] [CrossRef]
- Huang, X.; He, Y.; Dubuc, A.M.; Hashizume, R.; Zhang, W.; Reimand, J.; Yang, H.; Wang, T.A.; Stehbens, S.J.; Younger, S.; et al. EAG2 Potassium Channel with Evolutionarily Conserved Function as a Brain Tumor Target. Nat. Neurosci. 2015, 18, 1236–1246. [Google Scholar] [CrossRef]
- Francisco, M.A.; Wanggou, S.; Fan, J.J.; Dong, W.; Chen, X.; Momin, A.; Abeysundara, N.; Min, H.-K.; Chan, J.; McAdam, R.; et al. Chloride Intracellular Channel 1 Cooperates with Potassium Channel EAG2 to Promote Medulloblastoma Growth. J. Exp. Med. 2020, 217, e20190971. [Google Scholar] [CrossRef]
- Fan, J.; Pusong, R.; Morrissy, S.; Farooq, H.; Taylor, M.; Huang, X. MEDU-28. Eliminating the Root of Medulloblastoma by Targeting a Voltage-Gated Potassium Channel. Neuro Oncol. 2019, 21, ii109. [Google Scholar] [CrossRef]
- Huang, G.; Xu, Q.; Cui, Y.; Li, N.; Bian, X.; Lv, S. Medulloblastoma Stem Cells: Promising Targets in Medulloblastoma Therapy. Cancer Sci. 2016, 107, 583–589. [Google Scholar] [CrossRef]
- Santos, R.; Ursu, O.; Gaulton, A.; Bento, A.P.; Donadi, R.S.; Bologa, C.G.; Karlsson, A.; Al-Lazikani, B.; Hersey, A.; Oprea, T.I.; et al. A Comprehensive Map of Molecular Drug Targets. Nat. Rev. Drug Discov. 2017, 16, 19–34. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A Major Update to the DrugBank Database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Vandenberg, J.I.; Perry, M.D.; Perrin, M.J.; Mann, S.A.; Ke, Y.; Hill, A.P. HERG K+ Channels: Structure, Function, and Clinical Significance. Physiol. Rev. 2012, 92, 1393–1478. [Google Scholar] [CrossRef]
- Brendorp, B.; Pedersen, O.; Torp-Pedersen, C.; Sahebzadah, N.; Køber, L. A Benefit-Risk Assessment of Class III Antiarrhythmic Agents. Drug Saf. 2002, 25, 847–865. [Google Scholar] [CrossRef]
- Guardado-Mendoza, R.; Prioletta, A.; Jiménez-Ceja, L.M.; Sosale, A.; Folli, F. The Role of Nateglinide and Repaglinide, Derivatives of Meglitinide, in the Treatment of Type 2 Diabetes Mellitus. Arch. Med. Sci. 2013, 9, 936–943. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Channel | Cancer Type | Highlights | References |
|---|---|---|---|
| Voltage-gated potassium channels | |||
| Kv1.1 | Breast | ↑1 Expression relates to ↑ metastasis and tumourigenesis | [68] |
| Prostate | ↓ Expression in higher grade tumours, although variable between patients | [69,70] | |
| Kv1.3 | Breast | ↑ Expression regulates migration but not apoptosis or proliferation | [68] |
| Breast | Inhibition reduced malignant cell proliferation | [71] | |
| ↓ Expression in grade III tumours | [72] | ||
| ↑ Expression in patient samples and cell lines | [73] | ||
| Colon | Kv1.3 is a regulator of migration but not apoptosis or proliferation | [68] | |
| Leukemia | No observed relationship with malignancy, acts as a tumour suppressor | [74,75] | |
| Leiomyosarcoma | ↑ Expression in more aggressive tumours | [76] | |
| Smooth muscle | ↑ Expression in more severe phenotypes | [73] | |
| Kv1.5 | Lymphoma | Expression reduces with increased malignancy | [77] |
| Stomach | Involved with malignant cell proliferation via Ca2+ regulation | [78] | |
| Osteosarcoma | Inhibition halts proliferation via cell cycle arrest at G0/G1 | [79] | |
| Cervical | Regulates cell cycle of malignant cells (works with Kv9.3) | [80] | |
| Leiomyosarcoma | ↑ Expression in more aggressive tumours | [76] | |
| Kv2.1 | Stomach | Involved in malignant cell proliferation via Ca2+ regulation | [78] |
| Lung | ↑ Expression and regulates migration in more aggressive malignancies | [81] | |
| Kv3.4 | Oral | Regulates invasion and tumourigenesis | [82] |
| Breast | Inhibition results in ↓ cell proliferation | [83] | |
| Kv4.1 | Colon | ↑ Expression and role in cell proliferation | [84] |
| Breast | ↑ Expression in more severe phenotypes, knockdown inhibits proliferation | [83] | |
| Gastric | ↑ Expression in human gastric cancer cell lines | [85] | |
| Kv7.1 | Breast | Expression induces oncogenesis and growth | [67] |
| Kv10.1 | Stomach | Atypical expression and regulates proliferation | [86] |
| Osteosarcoma | Inhibition results in ↓ cell proliferation via arrest at G1 | [87,88] | |
| Calcium-activated potassium channels | |||
| KCa1.1 | Mesothelial | ↑ Expression in more malignant phenotype, knockdown inhibits migration | [89] |
| Sarcoma | Inhibition sensitised cells to paclitaxel, doxorubicin, and cisplatin | [90] | |
| KCa2.3 | Colorectal | Forms a lipid raft ion channel complex with TRPC1/Orai1 to enhance migration, knockdown significantly reduced migration | [91] |
| Breast | ↑ Expression in highly metastasizing cell lines, knockdown greatly reduced migration | [92] | |
| KCa3.1 | Breast | ↑ Expression linked to lower overall survival | [93] |
| Lung | Inhibition reduced tumour growth in vivo | [94] | |
| Renal | ↑ Expression linked to lower overall survival and increased metastasis | [95] | |
| Endometrial | Inhibition reduced malignant cell growth in vitro and in vivo | [96] | |
| Pancreatic | Inhibition reduced malignant cell growth in vitro | [97] | |
| Inward-rectifying potassium channels | |||
| Kir2.2 | Prostate, stomach, breast | Knockdown increased reactive oxygen species leading to cell cycle arrest | [98] |
| Kir5.1 | Parathyroid | ↑ Expression in parathyroid carcinoma | [99] |
| Thyroid | ↓ Expression in anaplastic thyroid carcinoma | [100] | |
| Pancreas | ↓ Expression in pancreatic ductal adenocarcinoma (data set) | [101] | |
| Two-pore domain potassium channels | |||
| TREK-1 | Prostate | ↑ Expression in cancer vs. healthy prostate cancer tissue | [102] |
| TASK-3 | Colorectal | ↑ Expression in colorectal cancer samples | [103] |
| Breast | Significant overexpression in 44% of breast tumours | [104] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boyle, Y.; Johns, T.G.; Fletcher, E.V. Potassium Ion Channels in Malignant Central Nervous System Cancers. Cancers 2022, 14, 4767. https://doi.org/10.3390/cancers14194767
Boyle Y, Johns TG, Fletcher EV. Potassium Ion Channels in Malignant Central Nervous System Cancers. Cancers. 2022; 14(19):4767. https://doi.org/10.3390/cancers14194767
Chicago/Turabian StyleBoyle, Yasmin, Terrance G. Johns, and Emily V. Fletcher. 2022. "Potassium Ion Channels in Malignant Central Nervous System Cancers" Cancers 14, no. 19: 4767. https://doi.org/10.3390/cancers14194767