


H3F3A K27M Mutation Promotes the Infiltrative Growth of High-Grade Glioma in Adults by Activating β-Catenin/USP1 Signaling


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Glioma Specimens
2.2. Antibodies and Reagents
2.3. Cell Culture
2.4. Western Blotting
2.5. RNA Isolation, cDNA Synthesis and RT-PCR
2.6. H3F3A Gene Sequencing
2.7. Lentivirus Packaging and Stable Cell Lines
2.8. Cell Counting Kit-8 (CCK-8) Assay
2.9. Colony Formation Assay
2.10. Wound Healing Assay
2.11. Transwell Invasion and Migration Assays
2.12. Orthotopic Mouse Model and In Vivo Imaging Analysis in Nude Mice
2.13. Hematoxylin and Eosin Staining
2.14. Statistical Analysis
3. Results
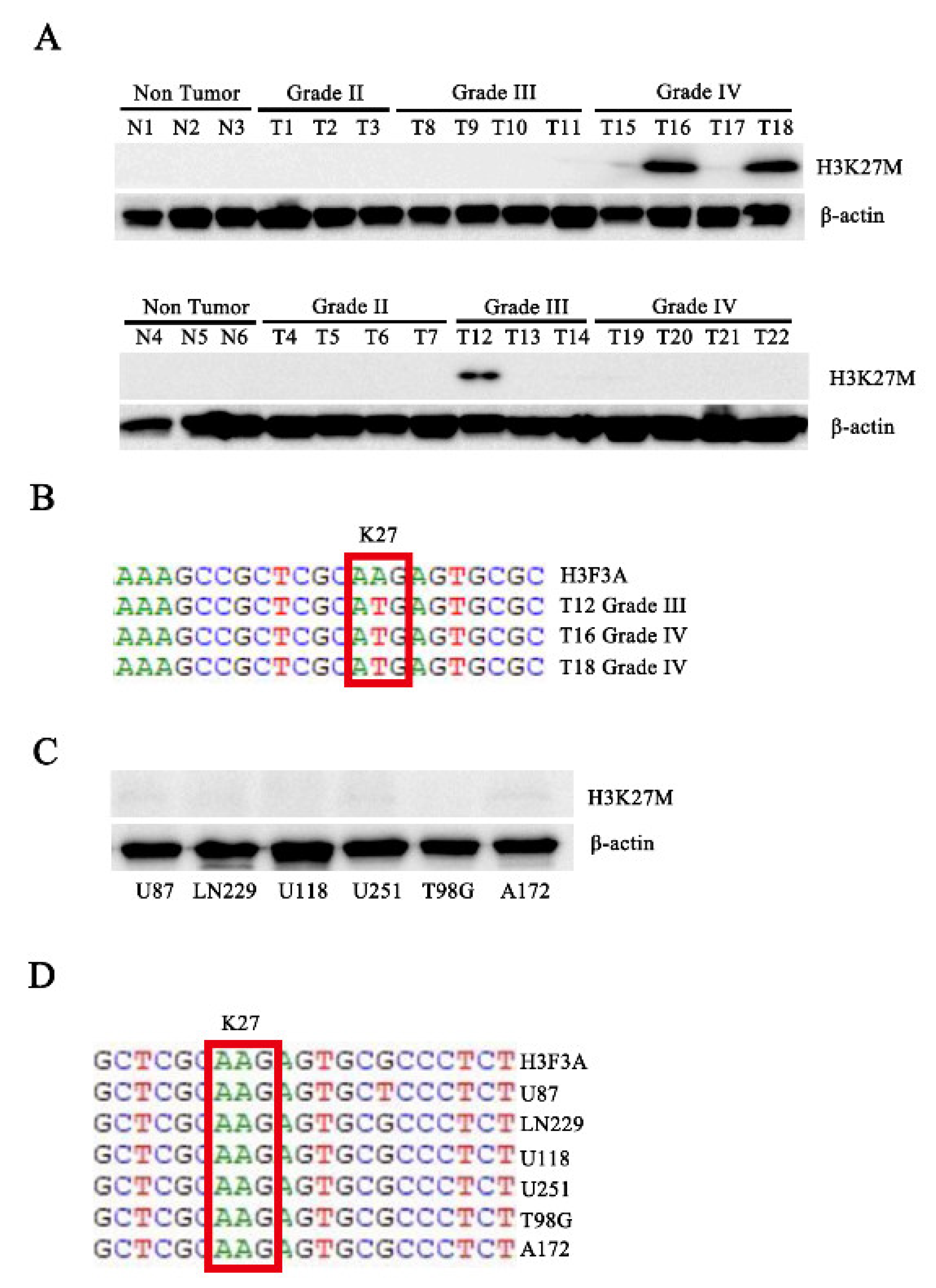
3.1. The H3.3K27M Mutation Exists in Human Patients with Glioma
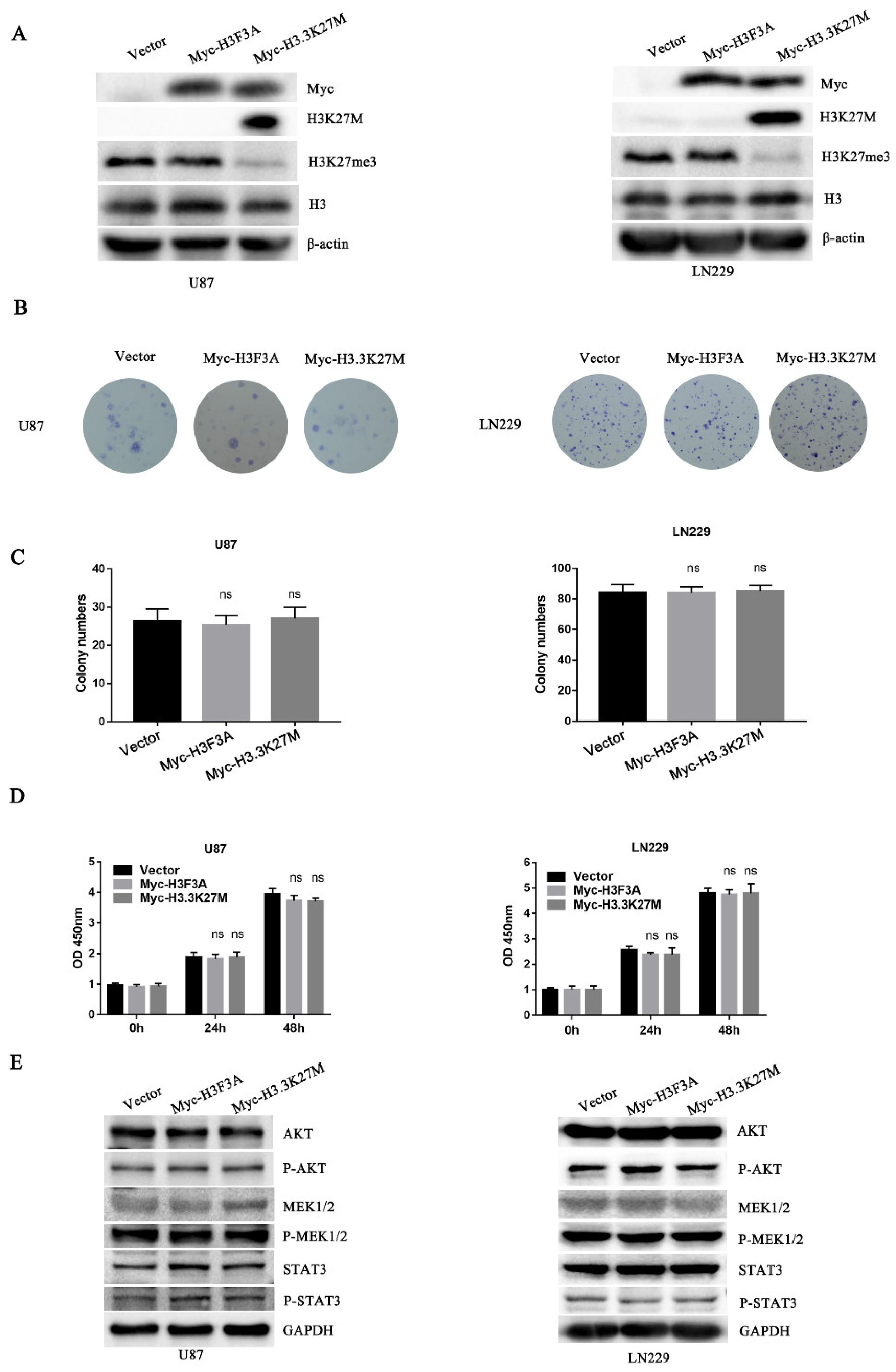
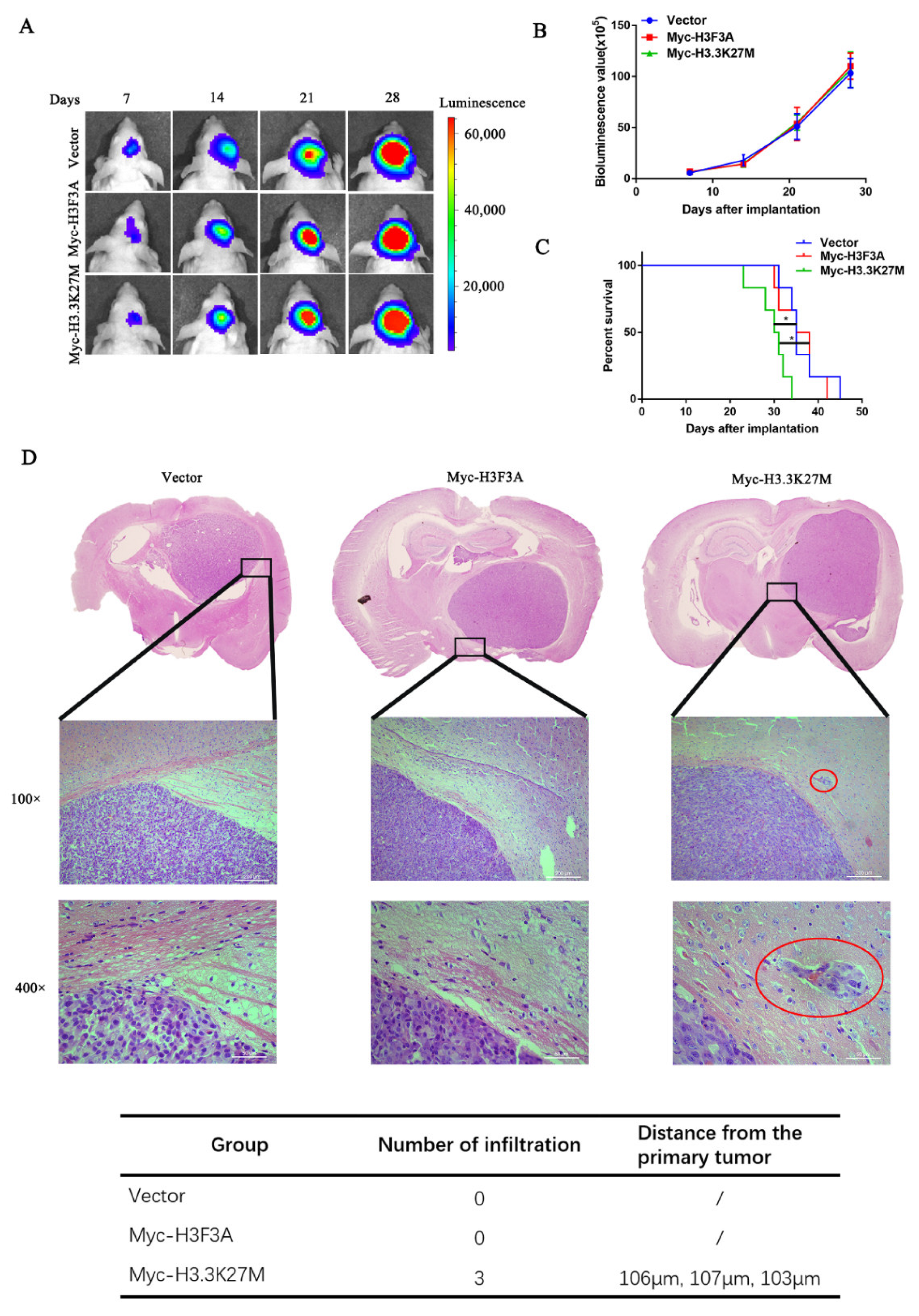
3.2. H3.3K27M has no Obvious Effect on Glioma Cell Proliferation but Promotes Glioma Cell Migration and Invasion In Vivo
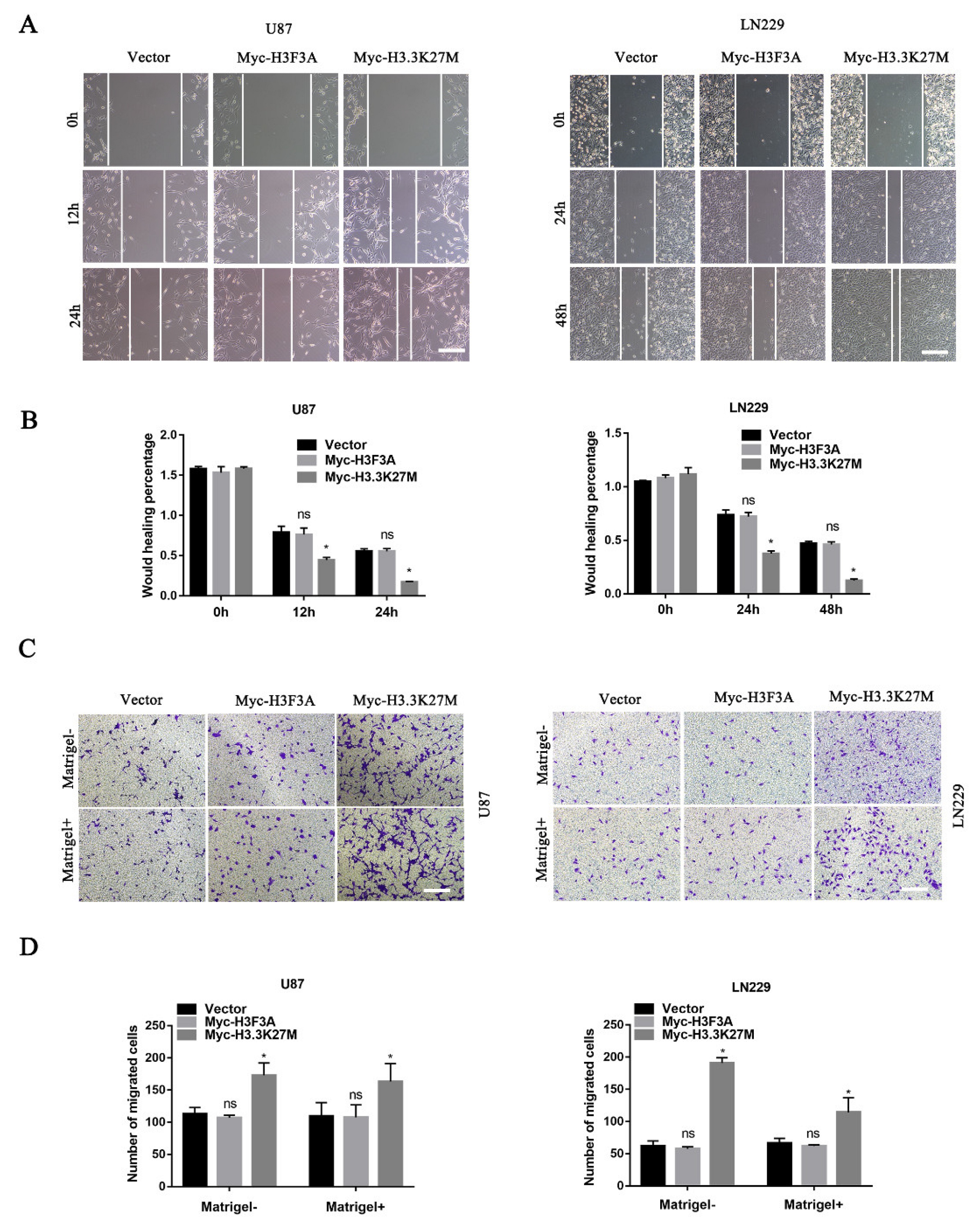
3.3. H3.3K27M Promotes Glioma Cell Migration and Invasion In Vitro
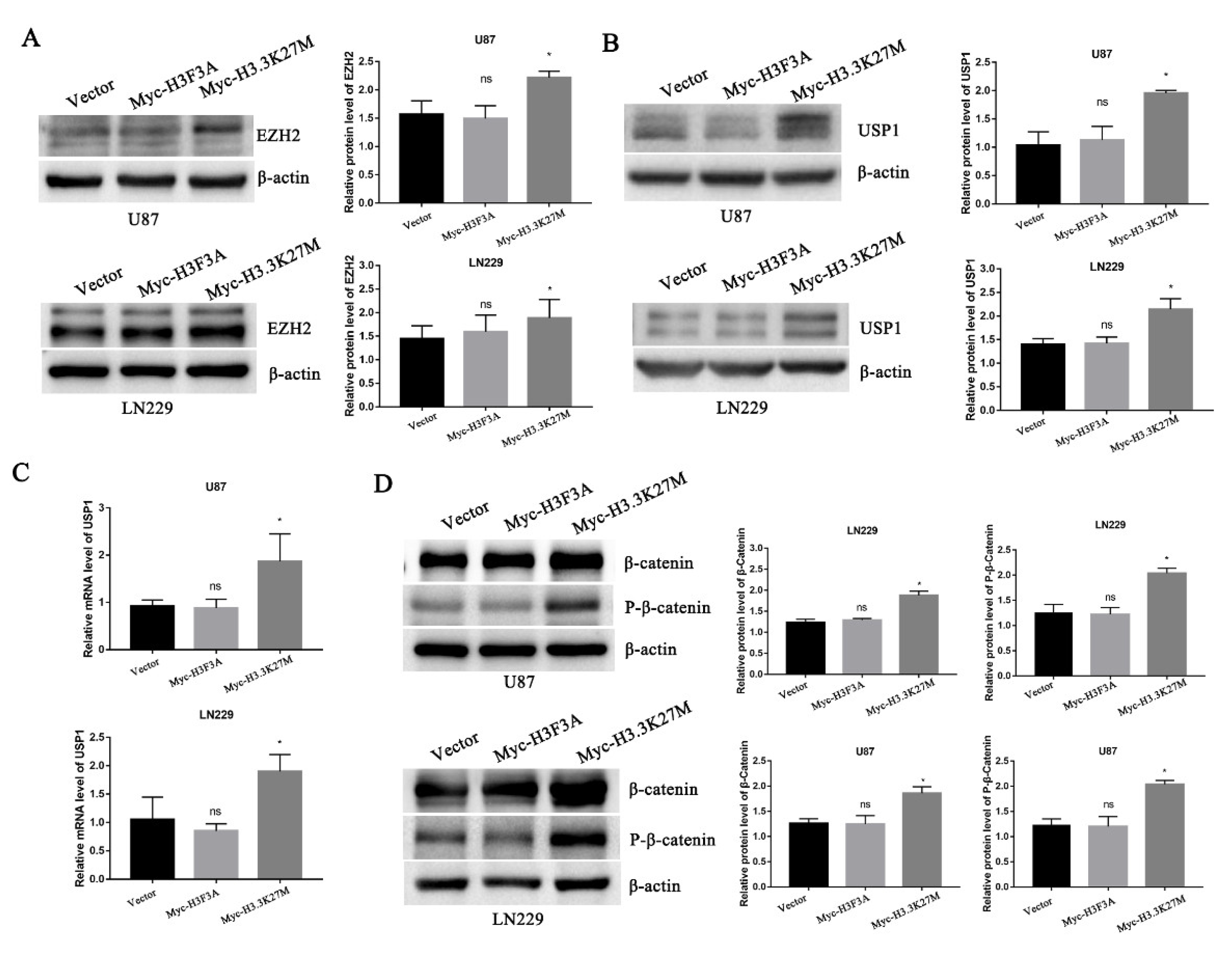
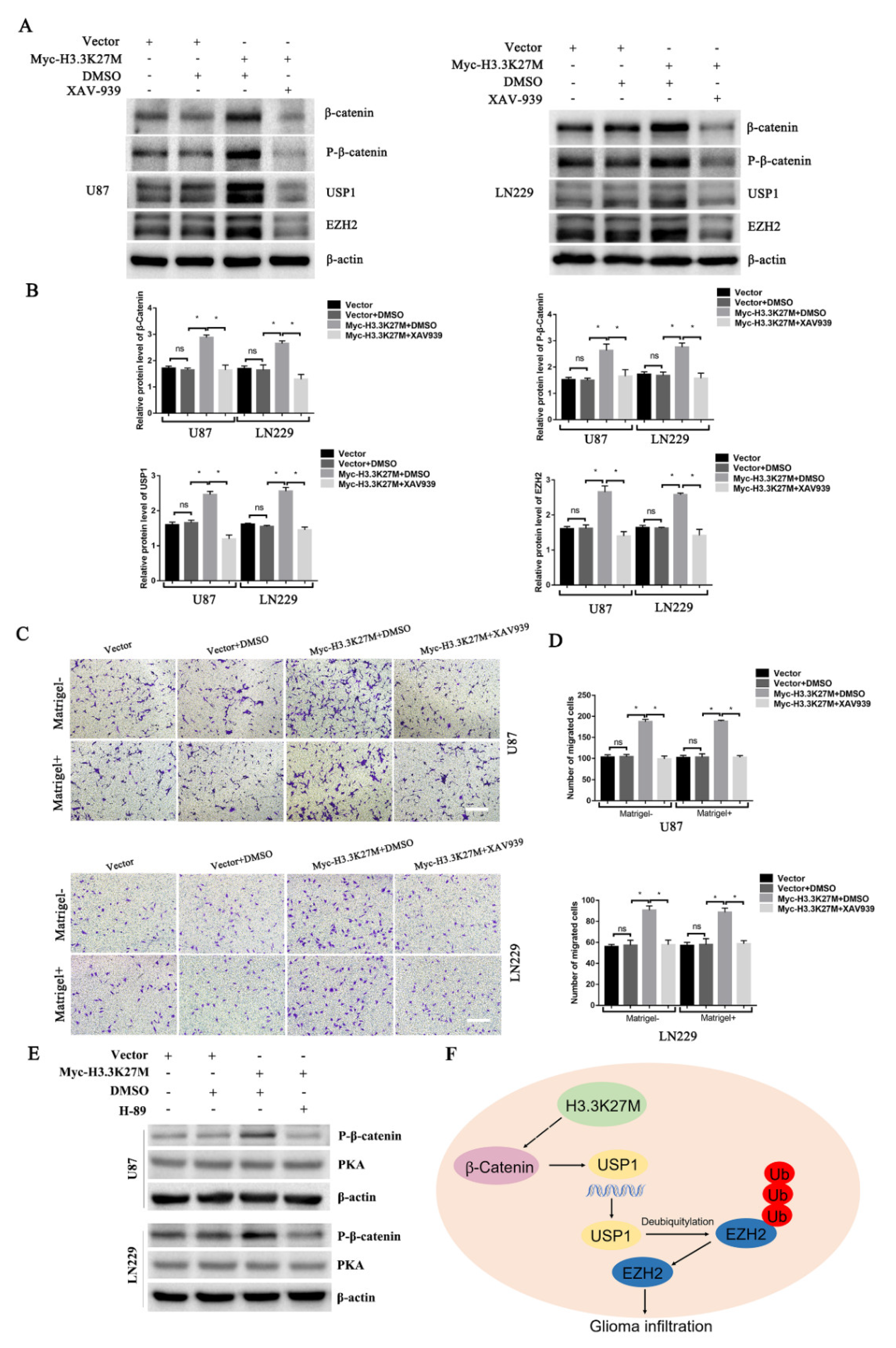
3.4. H3.3K27M Positively Regulates the β-Catenin/USP1 Signaling Pathway
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wesseling, P.; Capper, D. WHO 2016 Classification of gliomas. Neuropathol. Appl. Neurobiol. 2017, 44, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Gittleman, H.; Stetson, L.; Virk, S.M.; Barnholtz-Sloan, J.S. Epidemiology of Gliomas. In Current Understanding and Treatment of Gliomas; Springer: Cham, Switzerland, 2014; Volume 163, pp. 1–14. [Google Scholar] [CrossRef]
- Bush, N.A.O.; Chang, S.M.; Berger, M.S. Current and future strategies for treatment of glioma. Neurosurg. Rev. 2016, 40, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Montemurro, N.; Fanelli, G.N.; Scatena, C.; Ortenzi, V.; Pasqualetti, F.; Mazzanti, C.M.; Morganti, R.; Paiar, F.; Naccarato, A.G.; Perrini, P. Surgical outcome and molecular pattern characterization of recurrent glioblastoma multiforme: A single-center retrospective series. Clin. Neurol. Neurosurg. 2021, 207, 106735. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Nguyen, H.P.T.; Jones, J.J.; Stylli, S.S.; Whitehead, C.A.; Paradiso, L.; Luwor, R.B.; Areeb, Z.; Hanssen, E.; Cho, E.; et al. Extracellular Vesicles Secreted by Glioma Stem Cells Are Involved in Radiation Resistance and Glioma Progression. Int. J. Mol. Sci. 2022, 23, 2770. [Google Scholar] [CrossRef]
- Audia, J.E.; Campbell, R.M. Histone Modifications and Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019521. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [Green Version]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Xiang, Y.; Yan, K.; Zheng, Q.; Ke, H.; Cheng, J.; Xiong, W.; Shi, X.; Wei, L.; Zhao, M.; Yang, F.; et al. Histone Demethylase KDM4B Promotes DNA Damage by Activating Long Interspersed Nuclear Element-1. Cancer Res. 2019, 79, 86–98. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.; Wen, H.; Shi, X. The Histone Variant H3.3 in Transcriptional Regulation and Human Disease. J. Mol. Biol. 2016, 429, 1934–1945. [Google Scholar] [CrossRef]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.-Y.; Jones, D.T.W.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Khuong-Quang, D.-A.; Tönjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef]
- Gessi, M.; Capper, D.; Sahm, F.; Huang, K.; von Deimling, A.; Tippelt, S.; Fleischhack, G.; Scherbaum, D.; Alfer, J.; Juhnke, B.-O.; et al. Evidence of H3 K27M mutations in posterior fossa ependymomas. Acta Neuropathol. 2016, 132, 635–637. [Google Scholar] [CrossRef]
- Joyon, N.; Tauziède-Espariat, A.; Alentorn, A.; Giry, M.; Castel, D.; Capelle, L.; Zanello, M.; Varlet, P.; Bielle, F. K27M mutation inH3F3Ain ganglioglioma grade I with spontaneous malignant transformation extends the histopathological spectrum of the histone H3 oncogenic pathway. Neuropathol. Appl. Neurobiol. 2017, 43, 271–276. [Google Scholar] [CrossRef]
- Karremann, M.; Gielen, G.H.; Hoffmann, M.; Wiese, M.; Colditz, N.; Warmuth-Metz, M.; Bison, B.; Claviez, A.; Van Vuurden, D.G.; Von Bueren, A.; et al. Diffuse high-grade gliomas with H3 K27M mutations carry a dismal prognosis independent of tumor location. Neuro-Oncology 2017, 20, 123–131. [Google Scholar] [CrossRef] [Green Version]
- Margueron, R.; Reinberg, D. The Polycomb complex PRC2 and its mark in life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef] [Green Version]
- Laugesen, A.; Højfeldt, J.W.; Helin, K. Molecular Mechanisms Directing PRC2 Recruitment and H3K27 Methylation. Mol. Cell 2019, 74, 8–18. [Google Scholar] [CrossRef] [Green Version]
- Mohammad, F.; Weissmann, S.; Leblanc, B.; Pandey, D.P.; Hojfeldt, J.; Comet, I.; Zheng, C.; Johansen, J.V.; Rapin, N.; Porse, N.R.B.T.; et al. EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat. Med. 2017, 23, 483–492. [Google Scholar] [CrossRef]
- Vinchure, O.S.; Sharma, V.; Tabasum, S.; Ghosh, S.; Singh, R.P.; Sarkar, C.; Kulshreshtha, R. Polycomb complex mediated epigenetic reprogramming alters TGF-β signaling via a novel EZH2/miR-490/TGIF2 axis thereby inducing migration and EMT potential in glioblastomas. Int. J. Cancer 2019, 145, 1254–1269. [Google Scholar] [CrossRef]
- Ma, L.; Lin, K.; Chang, G.; Chen, Y.; Yue, C.; Guo, Q.; Zhang, S.; Jia, Z.; Huang, T.T.; Zhou, A.; et al. Aberrant Activation of β-Catenin Signaling Drives Glioma Tumorigenesis via USP1-Mediated Stabilization of EZH2. Cancer Res. 2019, 79, 72–85. [Google Scholar] [CrossRef] [Green Version]
- Fanelli, G.; Grassini, D.; Ortenzi, V.; Pasqualetti, F.; Montemurro, N.; Perrini, P.; Naccarato, A.; Scatena, C. Decipher the Glioblastoma Microenvironment: The First Milestone for New Groundbreaking Therapeutic Strategies. Genes 2021, 12, 445. [Google Scholar] [CrossRef]
- Maimaiti, B.; Mijiti, S.; Jiang, T.; Xie, Y.; Zhao, W.; Cheng, Y.; Meng, H. Case Report: H3K27M-Mutant Glioblastoma Simultaneously Present in the Brain and Long-Segment Spinal Cord Accompanied by Acute Pulmonary Embolism. Front. Oncol. 2022, 11, 763854. [Google Scholar] [CrossRef]
- Niu, X.; Wang, C.; Zhou, X.; Yang, Y.; Liu, Y.; Zhang, Y.; Mao, Q. Pineal Region Glioblastomas: Clinical Characteristics, Treatment, and Survival Outcome. World Neurosurg. 2020, 146, e799–e810. [Google Scholar] [CrossRef]
- Sturm, D.; Witt, H.; Hovestadt, V.; Khuong-Quang, D.-A.; Jones, D.T.; Konermann, C.; Pfaff, E.; Tönjes, M.; Sill, M.; Bender, S.; et al. Hotspot Mutations in H3F3A and IDH1 Define Distinct Epigenetic and Biological Subgroups of Glioblastoma. Cancer Cell 2012, 22, 425–437. [Google Scholar] [CrossRef] [Green Version]
- Manjunath, N.; Jha, P.; Singh, J.; Raheja, A.; Kaur, K.; Suri, A.; Garg, A.; Sharma, M.C.; Sarkar, C.; Mohan, M.; et al. Clinico-pathological and molecular characterization of diffuse midline gliomas: Is there a prognostic significance? Neurol. Sci. 2020, 42, 925–934. [Google Scholar] [CrossRef]
- Feng, J.; Hao, S.; Pan, C.; Wang, Y.; Wu, Z.; Zhang, J.; Yan, H.; Zhang, L.; Wan, H. The H3.3 K27M mutation results in a poorer prognosis in brainstem gliomas than thalamic gliomas in adults. Hum. Pathol. 2015, 46, 1626–1632. [Google Scholar] [CrossRef]
- Yeo, M.S.; Subhash, V.V.; Suda, K.; Balcıoğlu, H.E.; Zhou, S.; Thuya, W.L.; Loh, X.Y.; Jammula, S.; Peethala, P.C.; Tan, S.H.; et al. FBXW5 Promotes Tumorigenesis and Metastasis in Gastric Cancer via Activation of the FAK-Src Signaling Pathway. Cancers 2019, 11, 836. [Google Scholar] [CrossRef] [Green Version]
- Lewis, P.W.; Müller, M.M.; Koletsky, M.S.; Cordero, F.; Lin, S.; Banaszynski, L.A.; Garcia, B.A.; Muir, T.W.; Becher, O.J.; Allis, C.D. Inhibition of PRC2 Activity by a Gain-of-Function H3 Mutation Found in Pediatric Glioblastoma. Science 2013, 340, 857–861. [Google Scholar] [CrossRef] [Green Version]
- Chan, K.-M.; Fang, D.; Gan, H.; Hashizume, R.; Yu, C.; Schroeder, M.; Gupta, N.; Mueller, S.; James, C.D.; Jenkins, R.; et al. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev. 2013, 27, 985–990. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Liu, J.; Xue, H.; Li, C.; Liu, Q.; Zhou, Y.; Wang, T.; Wang, H.; Qian, H.; Wen, T. A TGF-β-MTA1-SOX4-EZH2 signaling axis drives epithelial–mesenchymal transition in tumor metastasis. Oncogene 2019, 39, 2125–2139. [Google Scholar] [CrossRef]
- Chen, C.-W.; Fu, M.; Du, Z.-H.; Zhao, F.; Yang, W.-W.; Xu, L.-H.; Li, S.-L.; Ge, X.-Y. Long Noncoding RNA MRPL23-AS1 Promotes Adenoid Cystic Carcinoma Lung Metastasis. Cancer Res. 2020, 80, 2273–2285. [Google Scholar] [CrossRef]
- Ma, J.; Zhang, J.; Weng, Y.-C.; Wang, J.-C. EZH2-Mediated microRNA-139-5p Regulates Epithelial-Mesenchymal Transition and Lymph Node Metastasis of Pancreatic Cancer. Mol. Cells 2018, 41, 868–880. [Google Scholar] [CrossRef]
- Zhao, M.; Hu, X.; Xu, Y.; Wu, C.; Chen, J.; Ren, Y.; Kong, L.; Sun, S.; Zhang, L.; Jin, R.; et al. Targeting of EZH2 inhibits epithelial-mesenchymal transition in head and neck squamous cell carcinoma via regulating the STAT3/VEGFR2 axis. Int. J. Oncol. 2019, 55, 1165–1175. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Liao, W.; Wu, Q.; Huang, X.; Pan, Z.; Chen, W.; Gu, S.; Huang, Z.; Wang, Y.; Tang, X.; et al. LINC00152 upregulates ZEB1 expression and enhances epithelial-mesenchymal transition and oxaliplatin resistance in esophageal cancer by interacting with EZH2. Cancer Cell Int. 2020, 20, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Stazi, G.; Taglieri, L.; Nicolai, A.; Romanelli, A.; Fioravanti, R.; Morrone, S.; Sabatino, M.; Ragno, R.; Taurone, S.; Nebbioso, M.; et al. Dissecting the role of novel EZH2 inhibitors in primary glioblastoma cell cultures: Effects on proliferation, epithelial-mesenchymal transition, migration, and on the pro-inflammatory phenotype. Clin. Epigenetics 2019, 11, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, J.; An, W.; Chen, C.; Wang, W.; Zhu, C.; Chen, F.; Chen, H.; Zheng, W.; Gong, J. MiR-32 Inhibits Proliferation and Metastasis by Targeting EZH2 in Glioma. Technol. Cancer Res. Treat. 2019, 18. [Google Scholar] [CrossRef]
- Chen, Y.; Hou, S.; Jiang, R.; Sun, J.; Cheng, C.; Qian, Z. EZH2 is a potential prognostic predictor of glioma. J. Cell. Mol. Med. 2020, 25, 925–936. [Google Scholar] [CrossRef]
- Delaney, K.; Strobino, M.; Wenda, J.M.; Pankowski, A.; Steiner, F.A. H3.3K27M-induced chromatin changes drive ectopic replication through misregulation of the JNK pathway in C. elegans. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.-Y.; Bush, K.; Klein, R.H.; Cervantes, V.; Lewis, N.; Naqvi, A.; Carcaboso, A.M.; Lechpammer, M.; Knoepfler, P.S. Reciprocal H3.3 gene editing identifies K27M and G34R mechanisms in pediatric glioma including NOTCH signaling. Commun. Biol. 2020, 3, 1–15. [Google Scholar] [CrossRef]
- Pajovic, S.; Siddaway, R.; Bridge, T.; Sheth, J.; Rakopoulos, P.; Kim, B.; Ryall, S.; Agnihotri, S.; Phillips, L.; Yu, M.; et al. Epigenetic activation of a RAS/MYC axis in H3.3K27M-driven cancer. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef]
- Ehteda, A.; Simon, S.; Franshaw, L.; Giorgi, F.M.; Liu, J.; Joshi, S.; Rouaen, J.R.; Pang, C.N.I.; Pandher, R.; Mayoh, C.; et al. Dual targeting of the epigenome via FACT complex and histone deacetylase is a potent treatment strategy for DIPG. Cell Rep. 2021, 35, 108994. [Google Scholar] [CrossRef]
- Pan, X.; Ma, L.; Wang, J. The clinicopathological significance and prognostic value of β-catenin Ser45-phosphorylation expression in esophageal squamous cell carcinoma. Int. J. Clin. Exp. Pathol. 2019, 12, 3507–3513. [Google Scholar]
- Daoud, E.V.; Rajaram, V.; Cai, C.; Oberle, R.J.; Martin, G.R.; Raisanen, J.M.; White, C.; Foong, C.; Mickey, B.; Pan, E.; et al. Adult Brainstem Gliomas With H3K27M Mutation: Radiology, Pathology, and Prognosis. J. Neuropathol. Exp. Neurol. 2018, 77, 302–311. [Google Scholar] [CrossRef] [Green Version]
- Cordero, F.J.; Huang, Z.; Grenier, C.; He, X.; Hu, G.; McLendon, R.E.; Murphy, S.K.; Hashizume, R.; Becher, O.J. Histone H3.3K27M Represses p16 to Accelerate Gliomagenesis in a Murine Model of DIPG. Mol. Cancer Res. 2017, 15, 1243–1254. [Google Scholar] [CrossRef] [Green Version]
- Taurin, S.; Sandbo, N.; Qin, Y.; Browning, D.; Dulin, N.O. Phosphorylation of β-Catenin by Cyclic AMP-dependent Protein Kinase. J. Biol. Chem. 2006, 281, 9971–9976. [Google Scholar] [CrossRef] [Green Version]
- Hino, S.-I.; Tanji, C.; Nakayama, K.I.; Kikuchi, A. Phosphorylation of β-Catenin by Cyclic AMP-Dependent Protein Kinase Stabilizes β-Catenin through Inhibition of Its Ubiquitination. Mol. Cell. Biol. 2005, 25, 9063–9072. [Google Scholar] [CrossRef] [Green Version]
- Yost, C.; Torres, M.; Miller, J.R.; Huang, E.; Kimelman, D.; Moon, R.T. The axis-inducing activity, stability, and subcellular distribution of beta-catenin is regulated in Xenopus embryos by glycogen synthase kinase 3. Genes Dev. 1996, 10, 1443–1454. [Google Scholar] [CrossRef] [Green Version]
- Dimitrova, Y.; Li, J.; Lee, Y.-T.; Rios-Esteves, J.; Friedman, D.B.; Choi, H.-J.; Weis, W.; Wang, C.-Y.; Chazin, W.J. Direct Ubiquitination of β-Catenin by Siah-1 and Regulation by the Exchange Factor TBL1. J. Biol. Chem. 2010, 285, 13507–13516. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, Z.; Zhu, Y.; Feng, X.; Liu, X.; Zhou, K.; Wang, Q.; Zhang, H.; Shi, H. H3F3A K27M Mutation Promotes the Infiltrative Growth of High-Grade Glioma in Adults by Activating β-Catenin/USP1 Signaling. Cancers 2022, 14, 4836. https://doi.org/10.3390/cancers14194836
Sun Z, Zhu Y, Feng X, Liu X, Zhou K, Wang Q, Zhang H, Shi H. H3F3A K27M Mutation Promotes the Infiltrative Growth of High-Grade Glioma in Adults by Activating β-Catenin/USP1 Signaling. Cancers. 2022; 14(19):4836. https://doi.org/10.3390/cancers14194836
Chicago/Turabian StyleSun, Zhiyuan, Yufu Zhu, Xia Feng, Xiaoyun Liu, Kunlin Zhou, Qing Wang, Hengzhu Zhang, and Hengliang Shi. 2022. "H3F3A K27M Mutation Promotes the Infiltrative Growth of High-Grade Glioma in Adults by Activating β-Catenin/USP1 Signaling" Cancers 14, no. 19: 4836. https://doi.org/10.3390/cancers14194836
APA StyleSun, Z., Zhu, Y., Feng, X., Liu, X., Zhou, K., Wang, Q., Zhang, H., & Shi, H. (2022). H3F3A K27M Mutation Promotes the Infiltrative Growth of High-Grade Glioma in Adults by Activating β-Catenin/USP1 Signaling. Cancers, 14(19), 4836. https://doi.org/10.3390/cancers14194836