

Protein Phosphorylation and Redox Status: An as Yet Elusive Dyad in Chronic Lymphocytic Leukemia

 , ,
, ,  , and
, and {kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. A Quick Glance at Phosphorylation-Dependent Signaling in CLL
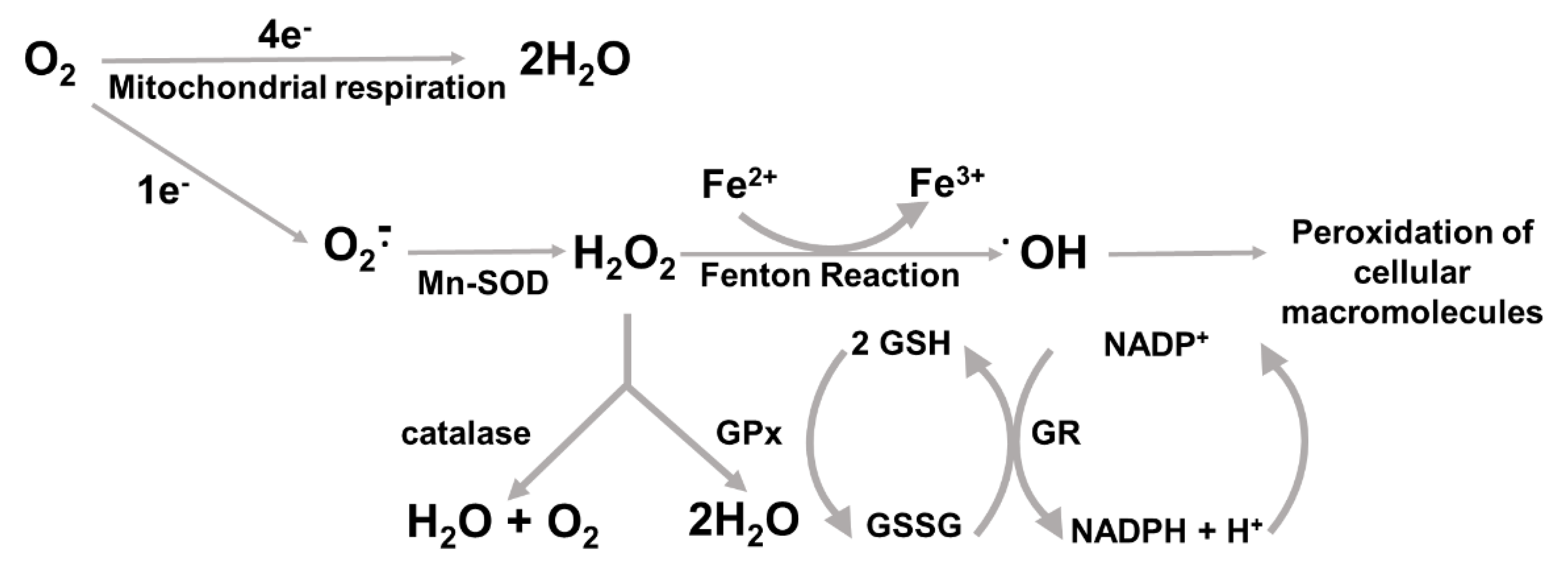
3. ROS and Antioxidant Response
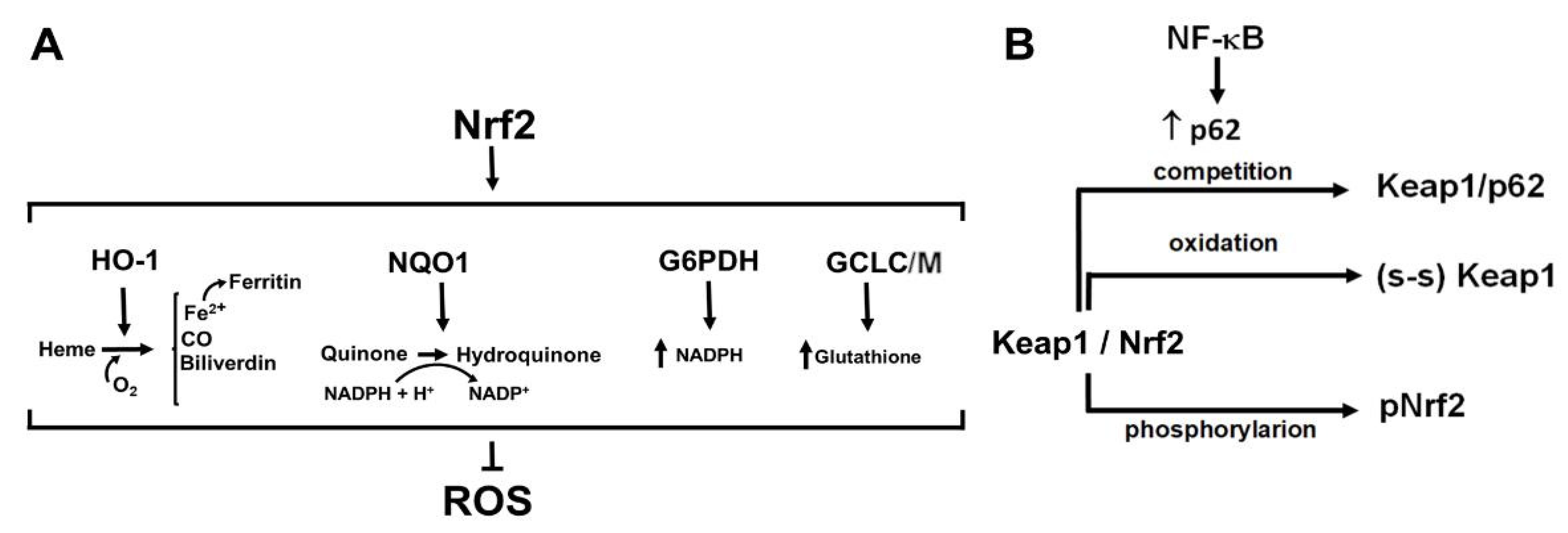
4. Nrf2, the Master Regulator of Antioxidant Responses: A Complex Regulation for Fine-Tuned Redox Homeostasis
5. NF-kB, Multifunctional Complexes for Pleiotropic Actions upon Oxidative Stress
6. The FOXO Family, the Paradox in the Struggle against Cellular Stress and Oxidation
7. ROS as Candidates for Therapeutic Intervention
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kipps, T.J.; Stevenson, F.K.; Wu, C.J.; Croce, C.M.; Packham, G.; Wierda, W.G.; O’Brien, S.; Gribben, J.; Rai, K. Chronic lymphocytic leukaemia. Nat. Rev. Dis. Prim. 2017, 3, 16096. [Google Scholar] [CrossRef] [Green Version]
- Fabbri, G.; Dalla-Favera, G.F.R. The molecular pathogenesis of chronic lymphocytic leukaemia. Nat. Cancer 2016, 16, 145–162. [Google Scholar] [CrossRef]
- Sharma, S.; Rai, K.R. Chronic lymphocytic leukemia (CLL) treatment: So many choices, such great options. Cancer 2019, 125, 1432–1440. [Google Scholar] [CrossRef]
- Soumerai, J.D.; Ni, A.; Darif, M.; Londhe, A.; Xing, G.; Mun, Y.; E Kay, N.; Shanafelt, T.D.; Rabe, K.G.; Byrd, J.C.; et al. Prognostic risk score for patients with relapsed or refractory chronic lymphocytic leukaemia treated with targeted therapies or chemoimmunotherapy: A retrospective, pooled cohort study with external validations. Lancet Haematol. 2019, 6, e366–e374. [Google Scholar] [CrossRef]
- Bond, D.A.; Huang, Y.; Fisher, J.L.; Ruppert, A.S.; Owen, D.H.; Bertino, E.M.; Rogers, K.A.; Bhat, S.A.; Grever, M.R.; Jaglowski, S.M.; et al. Second cancer incidence in CLL patients receiving BTK inhibitors. Leukemia 2020, 34, 3197–3205. [Google Scholar] [CrossRef] [PubMed]
- Burger, J.A. Treatment of Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2020, 383, 460–473. [Google Scholar] [CrossRef]
- Ghia, P.; Coutre, S.E.; Cheson, B.D.; Barrientos, J.C.; Hillmen, P.; Pettitt, A.R.; Zelenetz, A.D.; Shreay, S.; Hallek, M.; Furman, R.R. Impact of idelalisib on health-related quality of life in patients with relapsed chronic lymphocytic leukemia in a phase III randomized trial. Haematologica 2020, 105, e519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visentin, A.; Frezzato, F.; Severin, F.; Imbergamo, S.; Pravato, S.; Gargarella, L.R.; Manni, S.; Pizzo, S.; Ruggieri, E.; Facco, M.; et al. Lights and Shade of Next-Generation Pi3k Inhibitors in Chronic Lymphocytic Leukemia. OncoTargets Ther. 2020, 13, 9679–9688. [Google Scholar] [CrossRef]
- Perini, G.F.; Feres, C.C.P.; Teixeira, L.L.C.; Hamerschlak, N. BCL-2 Inhibition as Treatment for Chronic Lymphocytic Leukemia. Curr. Treat. Options Oncol. 2021, 22, 66–75. [Google Scholar] [CrossRef]
- Hacken, E.T.; Burger, J.A. Microenvironment dependency in Chronic Lymphocytic Leukemia: The basis for new targeted therapies. Pharmacol. Ther. 2014, 144, 338–348. [Google Scholar] [CrossRef]
- Vitale, C.; Griggio, V.; Riganti, C.; Todaro, M.; Kopecka, J.; Jones, R.; Salvetti, C.; Boccellato, E.; Perutelli, F.; Voena, C.; et al. Targeting HIF-1α Regulatory Pathways as a Strategy to Hamper Tumor-Microenvironment Interactions in CLL. Cancers 2021, 13, 2883. [Google Scholar] [CrossRef] [PubMed]
- Scielzo, C.; Ghia, P. Modeling the Leukemia Microenviroment In Vitro. Front. Oncol. 2020, 10, 607608–607616. [Google Scholar] [CrossRef]
- Von Bergwelt-Baildon, M.; Maecker, B.; Schultze, J.; Gribben, J.G. CD40 activation: Potential for specific immunotherapy in B-CLL. Ann. Oncol. 2004, 15, 853–857. [Google Scholar] [CrossRef]
- Poggi, A.; Prevosto, C.; Catellani, S.; Rocco, I.; Garuti, A.; Zocchi, M.R. Engagement of CD31 delivers an activating signal that contributes to the survival of chronic lymphocytic leukaemia cells. Br. J. Haematol. 2010, 151, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Kipps, T.J. The Pathogenesis of Chronic Lymphocytic Leukemia. Annu. Rev. Pathol. Mech. Dis. 2014, 9, 103–118. [Google Scholar] [CrossRef] [Green Version]
- Haiat, S.; Billard, C.; Quiney, C.; Ajchenbaum-Cymbalista, F.; Kolb, J.-P. Role of BAFF and APRIL in human B-cell chronic lymphocytic leukaemia. Immunology 2006, 118, 281–292. [Google Scholar] [CrossRef]
- Stevenson, F.K.; Krysov, S.; Davies, A.; Steele, A.J.; Packham, G. B-cell receptor signaling in chronic lymphocytic leukemia. Blood 2011, 118, 4313–4320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ten Hacken, E.; Gounari, M.; Ghia, P.; Burger, J.A. The importance of B cell receptor isotypes and stereotypes in chronic lymphocytic leukemia. Leukemia 2019, 33, 287–298. [Google Scholar] [CrossRef]
- Burger, J.A.; Wiestner, A. Targeting B cell receptor signalling in cancer: Preclinical and clinical advances. Nat. Cancer 2018, 18, 148–167. [Google Scholar] [CrossRef]
- Kipps, T.J.; Choi, M.Y. Targeted Therapy in Chronic Lymphocytic Leukemia. Cancer J. 2019, 25, 378–385. [Google Scholar] [CrossRef] [PubMed]
- D’Arena, G.; Seneca, E.; Migliaccio, I.; De Feo, V.; Giudice, A.; La Rocca, F.; Capunzo, M.; Calapai, G.; Festa, A.; Caraglia, M.; et al. Oxidative stress in chronic lymphocytic leukemia: Still a matter of debate. Leuk. Lymphoma 2019, 60, 867–875. [Google Scholar] [CrossRef]
- Tibaldi, E.; Federti, E.; Matte, A.; Iatcenko, I.; Wilson, A.B.; Riccardi, V.; Pagano, M.A.; De Franceschi, L. Oxidation Impacts the Intracellular Signaling Machinery in Hematological Disorders. Antioxidants 2020, 9, 353. [Google Scholar] [CrossRef] [PubMed]
- Yosifov, D.Y.; Idler, I.; Bhattacharya, N.; Reichenzeller, M.; Close, V.; Ezerina, D.; Scheffold, A.; Jebaraj, B.M.C.; Kugler, S.; Bloehdorn, J.; et al. Oxidative stress as candidate therapeutic target to overcome microenvironmental protection of CLL. Leukemia 2020, 34, 115–127. [Google Scholar] [CrossRef]
- Woolley, J.; Stanicka, J.; Cotter, T. Recent advances in reactive oxygen species measurement in biological systems. Trends Biochem. Sci. 2013, 38, 556–565. [Google Scholar] [CrossRef]
- Jitschin, R.; Hofmann, A.D.; Bruns, H.; Gießl, A.; Bricks, J.; Berger, J.; Saul, D.; Eckart, M.J.; Mackensen, A.; Mougiakakos, D. Mitochondrial metabolism contributes to oxidative stress and reveals therapeutic targets in chronic lymphocytic leukemia. Blood 2014, 123, 2663–2672. [Google Scholar] [CrossRef] [Green Version]
- Wu, R.P.; Hayashi, T.; Cottam, H.B.; Jin, G.; Yao, S.; Wu, C.C.N.; Rosenbach, M.D.; Corr, M.; Schwab, R.B.; Carson, D.A. Nrf2 responses and the therapeutic selectivity of electrophilic compounds in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2010, 107, 7479–7484. [Google Scholar] [CrossRef] [Green Version]
- Sajadimajd, S.; Khazaei, M. Oxidative Stress and Cancer: The Role of Nrf2. Curr. Cancer Drug Targets 2018, 18, 538–557. [Google Scholar] [CrossRef]
- Abdul-Aziz, A.; MacEwan, D.J.; Bowles, K.M.; Rushworth, S.A. Oxidative Stress Responses and NRF2 in Human Leukaemia. Oxidative Med. Cell. Longev. 2015, 2015, 454659. [Google Scholar] [CrossRef] [Green Version]
- Dubois, N.; Crompot, E.; Meuleman, N.; Bron, D.; Lagneaux, L.; Stamatopoulos, B. Importance of Crosstalk Between Chronic Lymphocytic Leukemia Cells and the Stromal Microenvironment: Direct Contact, Soluble Factors, and Extracellular Vesicles. Front. Oncol. 2020, 10, 1422. [Google Scholar] [CrossRef]
- Kurosaki, T.; Hikida, M. Tyrosine kinases and their substrates in B lymphocytes. Immunol. Rev. 2009, 228, 132–148. [Google Scholar] [CrossRef] [PubMed]
- Khan, W.N. Regulation of B Lymphocyte Development and Activation by Bruton’s Tyrosine Kinase. Immunol. Res. 2001, 23, 147–156. [Google Scholar] [CrossRef]
- Mohamed, A.J.; Yu, L.; Bäckesjö, C.-M.; Vargas, L.; Faryal, R.; Aints, A.; Christensson, B.; Berglöf, A.; Vihinen, M.; Nore, B.F.; et al. Bruton’s tyrosine kinase (Btk): Function, regulation, and transformation with special emphasis on the PH domain. Immunol. Rev. 2009, 228, 58–73. [Google Scholar] [CrossRef] [PubMed]
- Marshall, A.J.; Niiro, H.; Yun, T.J.; A Clark, E. Regulation of B-cell activation and differentiation by the phosphatidylinositol 3-kinase and phospholipase Cγ pathways. Immunol. Rev. 2000, 176, 30–46. [Google Scholar] [CrossRef] [PubMed]
- Gold, M.R.; Ingham, R.J.; McLeod, S.J.; Christian, S.L.; Scheid, M.P.; Duronio, V.; Santos, L.; Matsuuchi, L. Targets of B-cell antigen receptor signaling: The phosphatidylinositol 3-kinase/Akt/glycogen synthase kinase-3 signaling pathway and the Rap1 GTPase. Immunol. Rev. 2000, 176, 47–68. [Google Scholar] [CrossRef]
- Guo, B.; Su, T.T.; Rawlings, D.J. Protein kinase C family functions in B-cell activation. Curr. Opin. Immunol. 2004, 16, 367–373. [Google Scholar] [CrossRef]
- Xu, Y.; Harder, K.; Huntington, N.; Hibbs, M.; Tarlinton, D. Lyn Tyrosine KinaseAccentuating the Positive and the Negative. Immunity 2005, 22, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Trentin, L.; Frasson, M.; Donella-Deana, A.; Frezzato, F.; Pagano, M.A.; Tibaldi, E.; Gattazzo, C.; Zambello, R.; Semenzato, G.C.; Brunati, A.M. Geldanamycin-induced Lyn dissociation from aberrant Hsp90-stabilized cytosolic complex is an early event in apoptotic mechanisms in B-chronic lymphocytic leukemia. Blood 2008, 112, 4665–4674. [Google Scholar] [CrossRef]
- Zonta, F.; Pagano, M.A.; Trentin, L.; Tibaldi, E.; Frezzato, F.; Trimarco, V.; Facco, M.; Zagotto, G.; Pavan, V.; Ribaudo, G.; et al. Lyn sustains oncogenic signaling in chronic lymphocytic leukemia by strengthening SET-mediated inhibition of PP2A. Blood 2015, 125, 3747–3755. [Google Scholar] [CrossRef] [Green Version]
- Zonta, F.; Pagano, M.A.; Trentin, L.; Tibaldi, E.; Frezzato, F.; Gattazzo, C.; Martini, V.; Trimarco, V.; Mazzorana, M.; Bordin, L.; et al. Lyn-mediated procaspase 8 dimerization blocks apoptotic signaling in B-cell chronic lymphocytic leukemia. Blood 2014, 123, 875–883. [Google Scholar] [CrossRef] [Green Version]
- Frezzato, F.; Gattazzo, C.; Martini, V.; Trimarco, V.; Teramo, A.; Carraro, S.; Cabrelle, A.; Ave, E.; Facco, M.; Zambello, R.; et al. HS1, a Lyn Kinase Substrate, Is Abnormally Expressed in B-Chronic Lymphocytic Leukemia and Correlates with Response to Fludarabine-Based Regimen. PLoS ONE 2012, 7, e39902. [Google Scholar] [CrossRef]
- Gattazzo, C.; Martini, V.; Frezzato, F.; Trimarco, V.; Tibaldi, E.; Castelli, M.; Facco, M.; Zonta, F.; Brunati, A.M.; Zambello, R.; et al. Cortactin, another player in the Lyn signaling pathway, is over-expressed and alternatively spliced in leukemic cells from patients with B-cell chronic lymphocytic leukemia. Haematologica 2014, 99, 1069–1077. [Google Scholar] [CrossRef] [Green Version]
- Motiwala, T.; Majumder, S.; Kutay, H.; Smith, D.S.; Neuberg, D.S.; Lucas, D.M.; Byrd, J.C.; Grever, M.; Jacob, S.T. Methylation and Silencing of Protein Tyrosine Phosphatase Receptor Type O in Chronic Lymphocytic Leukemia. Clin. Cancer Res. 2007, 13, 3174–3181. [Google Scholar] [CrossRef] [Green Version]
- O’Hayre, M.; Niederst, M.; Fecteau, J.F.; Nguyen, V.M.; Kipps, T.J.; Messmer, D.; Newton, A.C.; Handel, T.M. Mechanisms and consequences of the loss of PHLPP1 phosphatase in chronic lymphocytic leukemia (CLL). Leukemia 2012, 26, 1689–1692. [Google Scholar] [CrossRef]
- Pauls, S.; Marshall, A.J. Regulation of immune cell signaling by SHIP1: A phosphatase, scaffold protein, and potential therapeutic target. Eur. J. Immunol. 2017, 47, 932–945. [Google Scholar] [CrossRef] [Green Version]
- Cui, B.; Chen, L.; Zhang, S.; Mraz, M.; Fecteau, J.-F.; Yu, J.; Ghia, E.M.; Zhang, L.; Bao, L.; Rassenti, L.Z.; et al. MicroRNA-155 influences B-cell receptor signaling and associates with aggressive disease in chronic lymphocytic leukemia. Blood 2014, 124, 546–554. [Google Scholar] [CrossRef] [Green Version]
- Shehata, M.; Schnabl, S.; Demirtas, D.; Hilgarth, M.; Hubmann, R.; Ponath, E.; Badrnya, S.; Lehner, C.; Hoelbl, A.; Duechler, M.; et al. Reconstitution of PTEN activity by CK2 inhibitors and interference with the PI3-K/Akt cascade counteract the antiapoptotic effect of human stromal cells in chronic lymphocytic leukemia. Blood 2010, 116, 2513–2521. [Google Scholar] [CrossRef]
- Zou, Z.-J.; Fan, L.; Wang, L.; Xu, J.; Zhang, R.; Tian, T.; Li, J.-Y.; Xu, W. miR-26a and miR-214 down-regulate expression of the PTEN gene in chronic lymphocytic leukemia, but not PTEN mutation or promoter methylation. Oncotarget 2015, 6, 1276–1285. [Google Scholar] [CrossRef] [Green Version]
- Pagano, M.A.; Tibaldi, E.; Molino, P.; Frezzato, F.; Trimarco, V.; Facco, M.; Zagotto, G.; Ribaudo, G.; Leanza, L.; Peruzzo, R.; et al. Mitochondrial apoptosis is induced by Alkoxy phenyl-1-propanone derivatives through PP2A-mediated dephosphorylation of Bad and Foxo3A in CLL. Leukemia 2019, 33, 1148–1160. [Google Scholar] [CrossRef]
- Tibaldi, E.; Brunati, A.M.; Zonta, F.; Frezzato, F.; Gattazzo, C.; Zambello, R.; Gringeri, E.; Semenzato, G.C.; Pagano, M.A.; Trentin, L. Lyn-mediated SHP-1 recruitment to CD5 contributes to resistance to apoptosis of B-cell chronic lymphocytic leukemia cells. Leukemia 2011, 25, 1768–1781. [Google Scholar] [CrossRef] [Green Version]
- Tibaldi, E.; Pagano, M.A.; Frezzato, F.; Trimarco, V.; Facco, M.; Zagotto, G.; Ribaudo, G.; Pavan, V.; Bordin, L.; Visentin, A.; et al. Targeted activation of the SHP-1/PP2A signaling axis elicits apoptosis of chronic lymphocytic leukemia cells. Haematologica 2017, 102, 1401–1412. [Google Scholar] [CrossRef]
- Parvez, S.; Long, M.J.C.; Poganik, J.R.; Aye, Y. Redox Signaling by Reactive Electrophiles and Oxidants. Chem. Rev. 2018, 118, 8798–8888. [Google Scholar] [CrossRef]
- Russell, E.G.; Cotter, T.G. New Insight into the Role of Reactive Oxygen Species (ROS) in Cellular Signal-Transduction Processes. Int. Rev. Cell Mol. Biol. 2015, 319, 221–254. [Google Scholar] [CrossRef]
- Xiong, Y.; Tian, X.; Ai, H.-W. Molecular Tools to Generate Reactive Oxygen Species in Biological Systems. Bioconjug. Chem. 2019, 30, 1297–1303. [Google Scholar] [CrossRef]
- Ray, P.D.; Huang, B.-W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef]
- Surai, P.F.; Kochish, I.I.; Fisinin, V.I.; Kidd, M.T. Antioxidant Defence Systems and Oxidative Stress in Poultry Biology: An Update. Antioxidants 2019, 8, 235. [Google Scholar] [CrossRef] [Green Version]
- Ulrich, K.; Jakob, U. The role of thiols in antioxidant systems. Free Radic. Biol. Med. 2019, 140, 14–27. [Google Scholar] [CrossRef]
- Marengo, B.; Nitti, M.; Furfaro, A.L.; Colla, R.; De Ciucis, C.; Marinari, U.M.; Pronzato, M.A.; Traverso, N.; Domenicotti, C. Redox Homeostasis and Cellular Antioxidant Systems: Crucial Players in Cancer Growth and Therapy. Oxid. Med. Cell. Longev. 2016, 2016, 6235641. [Google Scholar] [CrossRef] [Green Version]
- Miller, A.-F. Superoxide dismutases: Ancient enzymes and new insights. FEBS Lett. 2012, 586, 585–595. [Google Scholar] [CrossRef]
- Wang, Y.; Branicky, R.; Noë, A.; Hekimi, S. Superoxide dismutases: Dual roles in controlling ROS damage and regulating ROS signaling. J. Cell Biol. 2018, 217, 1915–1928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glorieux, C.; Calderon, P.B. Catalase, a remarkable enzyme: Targeting the oldest antioxidant enzyme to find a new cancer treatment approach. Biol. Chem. 2017, 398, 1095–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glorieux, C.; Zamocky, M.; Sandoval, J.M.; Verrax, J.; Calderon, P.B. Regulation of catalase expression in healthy and cancerous cells. Free Radic. Biol. Med. 2015, 87, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohé, R. Glutathione peroxidases and redox-regulated transcription factors. Biol. Chem. 2006, 387, 1329–1335. [Google Scholar] [CrossRef] [PubMed]
- Margis, R.; Dunand, C.; Teixeira, F.K.; Margis-Pinheiro, M. Glutathione peroxidase family—An evolutionary overview. FEBS J. 2008, 275, 3959–3970. [Google Scholar] [CrossRef]
- Dargel, R. Lipid peroxidation—A common pathogenetic mechanism? Exp. Toxicol. Pathol. 1992, 44, 169–181. [Google Scholar] [CrossRef]
- Poli, G.; Albano, E.; Dianzani, M.U. The role of lipid peroxidation in liver damage. Chem. Phys. Lipids 1987, 45, 117–142. [Google Scholar] [CrossRef]
- Tinkov, A.A.; Bjørklund, G.; Skalny, A.V.; Holmgren, A.; Skalnaya, M.G.; Chirumbolo, S.; Aaseth, J. The role of the thioredoxin/thioredoxin reductase system in the metabolic syndrome: Towards a possible prognostic marker? Cell Mol. Life Sci. 2018, 75, 1567–1586. [Google Scholar] [CrossRef]
- Fernandes, A.; Holmgren, A. Glutaredoxins: Glutathione-Dependent Redox Enzymes with Functions Far Beyond a Simple Thioredoxin Backup System. Antioxid. Redox Signal. 2004, 6, 63–74. [Google Scholar] [CrossRef]
- Couto, N.; Wood, J.; Barber, J. The role of glutathione reductase and related enzymes on cellular redox homoeostasis network. Free Radic. Biol. Med. 2016, 95, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Toroser, D.; Yarian, C.S.; Orr, W.C.; Sohal, R.S. Mechanisms of γ-glutamylcysteine ligase regulation. Biochim. Biophys. Acta 2006, 1760, 233–244. [Google Scholar] [CrossRef] [Green Version]
- Riganti, C.; Gazzano, E.; Polimeni, M.; Aldieri, E.; Ghigo, D. The pentose phosphate pathway: An antioxidant defense and a crossroad in tumor cell fate. Free Radic. Biol. Med. 2012, 53, 421–436. [Google Scholar] [CrossRef]
- Laborde, E. Glutathione transferases as mediators of signaling pathways involved in cell proliferation and cell death. Cell Death Differ. 2010, 17, 1373–1380. [Google Scholar] [CrossRef] [Green Version]
- Ross, D.; Siegel, D. The diverse functionality of NQO1 and its roles in redox control. Redox Biol. 2021, 41, 101950. [Google Scholar] [CrossRef] [PubMed]
- Drummond, G.S.; Baum, J.; Greenberg, M.; Lewis, D.; Abraham, N.G. HO-1 overexpression and underexpression: Clinical implications. Arch. Biochem. Biophys. 2019, 673, 108073. [Google Scholar] [CrossRef]
- Cheung, E.C.; Vousden, K.H. The role of ROS in tumour development and progression. Nat. Cancer 2022, 22, 280–297. [Google Scholar] [CrossRef]
- Helfinger, V.; Schröder, K. Redox control in cancer development and progression. Mol. Asp. Med. 2018, 63, 88–98. [Google Scholar] [CrossRef]
- Hegedűs, C.; Kovács, K.; Polgár, Z.; Regdon, Z.; Szabó, É.; Robaszkiewicz, A.; Forman, H.J.; Martner, A.; Virág, L. Redox control of cancer cell destruction. Redox Biol. 2018, 16, 59–74. [Google Scholar] [CrossRef]
- Kang, S.W.; Lee, S.; Lee, E.K. ROS and energy metabolism in cancer cells: Alliance for fast growth. Arch. Pharmacal. Res. 2015, 38, 338–345. [Google Scholar] [CrossRef]
- Barbato, A.; Scandura, G.; Puglisi, F.; Cambria, D.; La Spina, E.; Palumbo, G.A.; Lazzarino, G.; Tibullo, D.; Di Raimondo, F.; Giallongo, C.; et al. Mitochondrial Bioenergetics at the Onset of Drug Resistance in Hematological Malignancies: An Overview. Front. Oncol. 2020, 10, 604143–60414356. [Google Scholar] [CrossRef]
- Hull, T.D.; Boddu, R.; Guo, L.; Tisher, C.C.; Traylor, A.M.; Patel, B.; Joseph, R.; Prabhu, S.D.; Suliman, H.B.; Piantadosi, C.A.; et al. Heme oxygenase-1 regulates mitochondrial quality control in the heart. JCI Insight 2016, 1, e85817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sreekumar, P.; Ferrington, D.; Kannan, R. Glutathione Metabolism and the Novel Role of Mitochondrial GSH in Retinal Degeneration. Antioxidants 2021, 10, 661. [Google Scholar] [CrossRef]
- Zhou, Y.; Hileman, E.O.; Plunkett, W.; Keating, M.J.; Huang, P. Free radical stress in chronic lymphocytic leukemia cells and its role in cellular sensitivity to ROS-generating anticancer agents. Blood 2003, 101, 4098–4104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuo, X.L.; Chen, J.M.; Zhou, X.; Li, X.Z.; Mei, G.Y. Levels of Selenium, Zinc, Copper, and Antioxidant Enzyme Activity in Patients with Leukemia. Biol. Trace Element Res. 2006, 114, 41–54. [Google Scholar] [CrossRef]
- Sabry, S.A.; El-Senduny, F.F.; Abousamra, N.K.; El-Din, M.S.; Youssef, M.M. Oxidative stress in CLL patients leads to activation of Th9 cells: An experimental and comprehensive survey. Immunol. Med. 2020, 43, 36–46. [Google Scholar] [CrossRef] [Green Version]
- Maiti, G.P.; Sinha, S.; Mahmud, H.; Boysen, J.; Mendez, M.T.; Vesely, S.K.; Holter-Chakrabarty, J.; Kay, N.E.; Ghosh, A.K. SIRT3 overexpression and epigenetic silencing of catalase regulate ROS accumulation in CLL cells activating AXL signaling axis. Blood Cancer J. 2021, 11, 93–107. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, D.; Portales-Casamar, E.; Singh, A.; Srivastava, S.; Arenillas, D.; Happel, C.; Shyr, C.; Wakabayashi, N.; Kensler, T.W.; Wasserman, W.W.; et al. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res. 2010, 38, 5718–5734. [Google Scholar] [CrossRef]
- Sikalidis, A.K.; Mazor, K.M.; Lee, J.-I.; Roman, H.B.; Hirschberger, L.L.; Stipanuk, M.H. Upregulation of capacity for glutathione synthesis in response to amino acid deprivation: Regulation of glutamate–cysteine ligase subunits. Amino Acids 2014, 46, 1285–1296. [Google Scholar] [CrossRef] [Green Version]
- Ryoo, I.-G.; Kwak, M.-K. Regulatory crosstalk between the oxidative stress-related transcription factor Nfe2l2/Nrf2 and mitochondria. Toxicol. Appl. Pharmacol. 2018, 359, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Padmavathi, G.; Ramkumar, K.M. MicroRNA mediated regulation of the major redox homeostasis switch, Nrf2, and its impact on oxidative stress-induced ischemic/reperfusion injury. Arch. Biochem. Biophys. 2021, 698, 108725. [Google Scholar] [CrossRef]
- Villeneuve, N.F.; Lau, A.; Zhang, D.D. Regulation of the Nrf2–Keap1 Antioxidant Response by the Ubiquitin Proteasome System: An Insight into Cullin-Ring Ubiquitin Ligases. Antioxid. Redox Signal. 2010, 13, 1699–1712. [Google Scholar] [CrossRef] [Green Version]
- Bellezza, I.; Giambanco, I.; Minelli, A.; Donato, R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 721–733. [Google Scholar] [CrossRef]
- Rotblat, B.; Melino, G.; Knight, R.A. NRF2 and p53: Januses in cancer? Oncotarget 2012, 3, 1272–1283. [Google Scholar] [CrossRef] [Green Version]
- Jain, A.; Lamark, T.; Sjøttem, E.; Larsen, K.B.; Awuh, J.A.; Øvervatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Lv, Y.-F.; Zhao, J.-L.; You, Q.-D.; Jiang, Z.-Y. Regulation of Nrf2 by phosphorylation: Consequences for biological function and therapeutic implications. Free Radic. Biol. Med. 2021, 168, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, S.F. Mechanisms for redox-regulation of protein kinase C. Front. Pharmacol. 2015, 6, 128. [Google Scholar] [CrossRef] [Green Version]
- Jalil, S.J.; Sacktor, T.C.; Shouval, H.Z. Atypical PKCs in memory maintenance: The roles of feedback and redundancy. Learn. Mem. 2015, 22, 344–353. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.-C.; Nguyen, T.; Pickett, C.B. Regulation of the antioxidant response element by protein kinase C-mediated phosphorylation of NF-E2-related factor 2. Proc. Natl. Acad. Sci. USA 2000, 97, 12475–12480. [Google Scholar] [CrossRef] [Green Version]
- Rada, P.; Rojo, A.I.; Chowdhry, S.; McMahon, M.; Hayes, J.D.; Cuadrado, A. SCF/β-TrCP Promotes Glycogen Synthase Kinase 3-Dependent Degradation of the Nrf2 Transcription Factor in a Keap1-Independent Manner. Mol. Cell. Biol. 2011, 31, 1121–1133. [Google Scholar] [CrossRef]
- Abhinav, K.; Jain, A.K.J. Show footnotes. GSK-3β Acts Upstream of Fyn Kinase in Regulation of Nuclear Export and Degradation of NF-E2 Related Factor 2. J. Biol. Chem. 2007, 282, 16502–16510. [Google Scholar] [CrossRef] [Green Version]
- Cross, D.A.; Alessi, D.R.; Cohen, P.; Andjelkovich, M.; Hemmings, B.A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 1995, 378, 785–789. [Google Scholar] [CrossRef]
- Xu, W.; Zhen, Y.; Zhou, S.-F.; Lu, N. Posttranslational regulation of phosphatase and tensin homolog (PTEN) and its functional impact on cancer behaviors. Drug Des. Dev. Ther. 2014, 8, 1745–1751. [Google Scholar] [CrossRef] [Green Version]
- Raman, D.; Pervaiz, S. Redox inhibition of protein phosphatase PP2A: Potential implications in oncogenesis and its progression. Redox Biol. 2019, 27, 101105. [Google Scholar] [CrossRef]
- Kazi, J.U.; Kabir, N.N.; Rönnstrand, L. Protein kinase C (PKC) as a drug target in chronic lymphocytic leukemia. Med. Oncol. 2013, 30, 757. [Google Scholar] [CrossRef] [Green Version]
- Martelli, A.M.; Paganelli, F.; Evangelisti, C.; Chiarini, F.; McCubrey, J.A. Pathobiology and Therapeutic Relevance of GSK-3 in Chronic Hematological Malignancies. Cells 2022, 11, 1812. [Google Scholar] [CrossRef]
- Strickland, I.; Ghosh, S. Use of cell permeable NBD peptides for suppression of inflammation. Ann. Rheum. Dis. 2006, 65, iii75–iii82. [Google Scholar] [CrossRef] [Green Version]
- López-Guerra, M.; Colomer, D. NF-κB as a therapeutic target in chronic lymphocytic leukemia. Expert Opin. Ther. Targets 2010, 14, 275–288. [Google Scholar] [CrossRef]
- Smale, S.T. Dimer-specific regulatory mechanisms within the NF-κB family of transcription factors. Immunol. Rev. 2012, 246, 193–204. [Google Scholar] [CrossRef]
- Christian, F.; Smith, E.L.; Carmody, R.J. The Regulation of NF-κB Subunits by Phosphorylation. Cells 2016, 5, 12. [Google Scholar] [CrossRef]
- Morgan, M.J.; Liu, Z.-G. Crosstalk of reactive oxygen species and NF-kappaκB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.-C. The non-canonical NF-κB pathway in immunity and inflammation. Nat. Rev. Immunol. 2017, 17, 545–558. [Google Scholar] [CrossRef]
- Dorrington, M.G.; Fraser, I.D.C.; Dorrington, M.G.; Fraser, I.D.C. NF-κB Signaling in Macrophages: Dynamics, Crosstalk, and Signal Integration. Front. Immunol. 2019, 10, 705. [Google Scholar] [CrossRef]
- Lingappan, K. NF-κB in oxidative stress. Curr. Opin. Toxicol. 2018, 7, 81–86. [Google Scholar] [CrossRef]
- Matthews, J.R.; Kaszubska, W.; Turcatti, G.; Wells, T.N.; Hay, R.T. Role of cysteine62in DNA recognition by the P50 subunit of NF-xB. Nucleic Acids Res. 1993, 21, 1727–1734. [Google Scholar] [CrossRef]
- Pineda-Molina, E.; Klatt, P.; Vázquez, J.; Marina, A.; de Lacoba, M.G.; Pérez-Sala, D.; Lamas, S. Glutathionylation of the p50 Subunit of NF-κB: A Mechanism for Redox-Induced Inhibition of DNA Binding. Biochemistry 2001, 40, 14134–14142. [Google Scholar] [CrossRef]
- Jamaluddin, M.; Wang, S.; Boldogh, I.; Tian, B.; Brasier, A.R. TNF-α-induced NF-κB/RelA Ser276 phosphorylation and enhanceosome formation is mediated by an ROS-dependent PKAc pathway. Cell. Signal. 2007, 19, 1419–1433. [Google Scholar] [CrossRef]
- Byun, M.-S.; Choi, J.; Jue, D.-M. Cysteine-179 of IκB kinase β plays a critical role in enzyme activation by promoting phosphorylation of activation loop serines. Exp. Mol. Med. 2006, 38, 546–552. [Google Scholar] [CrossRef]
- Herscovitch, M.; Comb, W.; Ennis, T.; Coleman, K.; Yong, S.; Armstead, B.; Kalaitzidis, D.; Alimchandani, S.; Gilmore, T.D. Intermolecular disulfide bond formation in the NEMO dimer requires Cys54 and Cys347. Biochem. Biophys. Res. Commun. 2008, 367, 103–108. [Google Scholar] [CrossRef] [Green Version]
- Imbert, V.; A Rupec, R.; Livolsi, A.; Pahl, H.L.; Traenckner, E.-M.; Mueller-Dieckmann, C.; Farahifar, D.; Rossi, B.; Auberger, P.; A Baeuerle, P.; et al. Tyrosine Phosphorylation of IκB-α Activates NF-κB without Proteolytic Degradation of IκB-α. Cell 1996, 86, 787–798. [Google Scholar] [CrossRef]
- Fan, C.; Li, Q.; Ross, D.; Engelhardt, J.F. Tyrosine Phosphorylation of IκBα Activates NFκB through a Redox-regulated and c-Src-dependent Mechanism Following Hypoxia/Reoxygenation. J. Biol. Chem. 2003, 278, 2072–2080. [Google Scholar] [CrossRef] [Green Version]
- Bubici, C.; Papa, S.; Dean, K.; Franzoso, G. Mutual cross-talk between reactive oxygen species and nuclear factor-kappa B: Molecular basis and biological significance. Oncogene 2006, 25, 6731–6748. [Google Scholar] [CrossRef] [Green Version]
- Béraud, C.; Henzel, W.J.; Baeuerle, P.A. Involvement of regulatory and catalytic subunits of phosphoinositide 3-kinase in NF-κB activation. Proc. Natl. Acad. Sci. USA 1999, 96, 429–434. [Google Scholar] [CrossRef] [Green Version]
- Abu-Amer, Y.; Ross, F.P.; McHugh, K.P.; Livolsi, A.; Peyron, J.-F.; Teitelbaum, S.L. Tumor Necrosis Factor-α Activation of Nuclear Transcription Factor-κB in Marrow Macrophages Is Mediated by c-Src Tyrosine Phosphorylation of IκBα. J. Biol. Chem. 1998, 273, 29417–29423. [Google Scholar] [CrossRef] [Green Version]
- Kil, I.S.; Kim, S.Y.; Park, J.-W. Glutathionylation regulates IκB. Biochem. Biophys. Res. Commun. 2008, 373, 169–173. [Google Scholar] [CrossRef]
- Wu, M.; Bian, Q.; Liu, Y.; Fernandes, A.F.; Taylor, A.; Pereira, P.; Shang, F. Sustained oxidative stress inhibits NF-κB activation partially via inactivating the proteasome. Free Radic. Biol. Med. 2009, 46, 62–69. [Google Scholar] [CrossRef] [Green Version]
- Djavaheri-Mergny, M.; Javelaud, D.; Wietzerbin, J.; Besançon, F. NF-κB activation prevents apoptotic oxidative stress via an increase of both thioredoxin and MnSOD levels in TNFα-treated Ewing sarcoma cells. FEBS Lett. 2004, 578, 111–115. [Google Scholar] [CrossRef] [Green Version]
- Kairisalo, M.; Korhonen, L.; Blomgren, K.; Lindholm, D. X-linked inhibitor of apoptosis protein increases mitochondrial antioxidants through NF-κB activation. Biochem. Biophys. Res. Commun. 2007, 364, 138–144. [Google Scholar] [CrossRef]
- Tanaka, T.; Nakamura, H.; Nishiyama, A.; Hosoi, F.; Masutani, H.; Wada, H.; Yodoi, J. Redox regulation by thioredoxin superfamily; protection against oxidative stress and aging. Free Radic. Res. 2000, 33, 851–855. [Google Scholar] [CrossRef]
- Lavrovsky, Y.; Schwartzman, M.L.; Levere, R.D.; Kappas, A.; Abraham, N.G. Identification of binding sites for transcription factors NF-kappa B and AP-2 in the promoter region of the human heme oxygenase 1 gene. Proc. Natl. Acad. Sci. USA 1994, 91, 5987–5991. [Google Scholar] [CrossRef]
- Mansouri, L.; Papakonstantinou, N.; Ntoufa, S.; Stamatopoulos, K.; Rosenquist, R. NF-κB activation in chronic lymphocytic leukemia: A point of convergence of external triggers and intrinsic lesions. Semin. Cancer Biol. 2016, 39, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Muzio, M.; Fonte, E.; Caligaris-Cappio, F. Toll-like Receptors in Chronic Lymphocytic Leukemia. Mediterr. J. Hematol. Infect. Dis. 2012, 4, e2012055. [Google Scholar] [CrossRef]
- Pontoriero, M.; Fiume, G.; Vecchio, E.; de Laurentiis, A.; Albano, F.; Iaccino, E.; Mimmi, S.; Pisano, A.; Agosti, V.; Giovannone, E.; et al. Activation of NF-κB in B cell receptor signaling through Bruton’s tyrosine kinase-dependent phosphorylation of IκB-α. Klin. Wochenschr. 2019, 97, 675–690. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, L.; Yu, J.; Ghia, E.M.; Choi, M.Y.; Zhang, L.; Zhang, S.; Sanchez-Lopez, E.; Widhopf, G.F., 2nd; Messer, K.; et al. Cirmtuzumab blocks Wnt5a/ROR1 stimulation of NF-κB to repress autocrine STAT3 activation in chronic lymphocytic leukemia. Blood 2019, 134, 1084–1094. [Google Scholar] [CrossRef]
- Zinatizadeh, M.R.; Schock, B.; Chalbatani, G.M.; Zarandi, P.K.; Jalali, S.A.; Miri, S.R. The Nuclear Factor Kappa B (NF-kB) signaling in cancer development and immune diseases. Gene Funct. Dis. 2020, 8, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Lopez, E.; Ghia, E.M.; Antonucci, L.; Sharma, N.; Rassenti, L.Z.; Xu, J.; Sun, B.; Kipps, T.J.; Karin, M. NF-κB-p62-NRF2 survival signaling is associated with high ROR1 expression in chronic lymphocytic leukemia. Cell Death Differ. 2020, 27, 2206–2216. [Google Scholar] [CrossRef] [PubMed]
- Moscat, J.; Diaz-Meco, M.T. p62 at the Crossroads of Autophagy, Apoptosis, and Cancer. Cell 2009, 137, 1001–1004. [Google Scholar] [CrossRef] [Green Version]
- Cui, B.; Ghia, E.M.; Chen, L.; Rassenti, L.Z.; DeBoever, C.; Widhopf, G.F.; Yu, J.; Neuberg, D.S.; Wierda, W.G.; Rai, K.R.; et al. High-level ROR1 associates with accelerated disease progression in chronic lymphocytic leukemia. Blood 2016, 128, 2931–2940. [Google Scholar] [CrossRef] [Green Version]
- Gui, T.; Burgering, B.M.T. FOXOs: Masters of the equilibrium. FEBS J. 2021. [Google Scholar] [CrossRef] [PubMed]
- Ticchioni, M.; Essafi, M.; Jeandel, P.Y.; Davi, F.; Cassuto, J.P.; Deckert, M.; Bernard, A. Homeostatic chemokines increase survival of B-chronic lymphocytic leukemia cells through inactivation of transcription factor FOXO3a. Oncogene 2007, 26, 7081–7091. [Google Scholar] [CrossRef] [PubMed]
- Jiramongkol, Y.; Lam, E.W.-F. FOXO transcription factor family in cancer and metastasis. Cancer Metastasis Rev. 2020, 39, 681–709. [Google Scholar] [CrossRef]
- Lam, E.W.-F.; Brosens, J.; Gomes, A.R.; Koo, C.Y. Forkhead box proteins: Tuning forks for transcriptional harmony. Nat. Cancer 2013, 13, 482–495. [Google Scholar] [CrossRef] [PubMed]
- Cosimo, E.; Tarafdar, A.; Moles, M.W.; Holroyd, A.K.; Malik, N.; Catherwood, M.A.; Hay, J.; Dunn, K.M.; Macdonald, A.M.; Guichard, S.M.; et al. AKT/mTORC2 Inhibition Activates FOXO1 Function in CLL Cells Reducing B-Cell Receptor-Mediated Survival. Clin. Cancer Res. 2019, 25, 1574–1587. [Google Scholar] [CrossRef] [Green Version]
- Klotz, L.-O.; Sánchez-Ramos, C.; Prieto-Arroyo, I.; Urbánek, P.; Steinbrenner, H.; Monsalve, M. Redox regulation of FoxO transcription factors. Redox Biol. 2015, 6, 51–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olmos, Y.; Sanchez-Gomez, F.J.; Wild, B.; Garcia-Quintans, N.; Cabezudo, S.; Lamas, S.; Monsalve, M. SirT1 Regulation of Antioxidant Genes Is Dependent on the Formation of a FoxO3a/PGC-1α Complex. Antioxid. Redox Signal. 2013, 19, 1507–1521. [Google Scholar] [CrossRef]
- Vogt, P.K.; Jiang, H.; Aoki, M. Triple Layer Control: Phosphorylation, Acetylation and Ubiquitination of FOXO Proteins. Cell Cycle 2005, 4, 908–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tzivion, G.; Dobson, M.; Ramakrishnan, G. FoxO transcription factors; Regulation by AKT and 14-3-3 proteins. Biochim. Biophys. Acta 2011, 1813, 1938–1945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farhan, M.; Wang, H.; Gaur, U.; Little, P.; Xu, J.; Zheng, W. FOXO Signaling Pathways as Therapeutic Targets in Cancer. Int. J. Biol. Sci. 2017, 13, 815–827. [Google Scholar] [CrossRef]
- Yang, J.-Y.; Hung, M.-C. A New Fork for Clinical Application: Targeting Forkhead Transcription Factors in Cancer. Clin. Cancer Res. 2009, 15, 752–757. [Google Scholar] [CrossRef] [Green Version]
- Daitoku, H.; Sakamaki, J.-I.; Fukamizu, A. Regulation of FoxO transcription factors by acetylation and protein–protein interactions. Biochim. Biophys. Acta 2011, 1813, 1954–1960. [Google Scholar] [CrossRef] [PubMed]
- Dansen, T.B.; Smits, L.M.M.; van Triest, M.H.; de Keizer, P.L.J.; van Leenen, D.; Koerkamp, M.G.; Szypowska, A.; Meppelink, A.; Brenkman, A.B.; Yodoi, J.; et al. Redox-sensitive cysteines bridge p300/CBP-mediated acetylation and FoxO4 activity. Nat. Chem. Biol. 2009, 5, 664–672. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, H.; Daitoku, H.; Hatta, M.; Aoyama, H.; Yoshimochi, K.; Fukamizu, A. Acetylation of Foxo1 alters its DNA-binding ability and sensitivity to phosphorylation. Proc. Natl. Acad. Sci. USA 2005, 102, 11278–11283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Wang, Y.; Zhu, W.-G. Applications of post-translational modifications of FoxO family proteins in biological functions. J. Mol. Cell Biol. 2011, 3, 276–282. [Google Scholar] [CrossRef] [Green Version]
- Alpha-Tocopherol, Beta Carotene Cancer Prevention Study Group. The Effect of Vitamin E and Beta Carotene on the Incidence of Lung Cancer and Other Cancers in Male Smokers. N. Engl. J. Med. 1994, 330, 1029–1035. [Google Scholar] [CrossRef]
- Sayin, V.I.; Ibrahim, M.X.; Larsson, E.; Nilsson, J.A.; Lindahl, P.; Bergo, M.O. Antioxidants Accelerate Lung Cancer Progression in Mice. Sci. Transl. Med. 2014, 6, 221ra15. [Google Scholar] [CrossRef]
- Satia, J.A.; Littman, A.J.; Slatore, C.; Galanko, J.A.; White, E. Long-term Use of -Carotene, Retinol, Lycopene, and Lutein Supplements and Lung Cancer Risk: Results from the VITamins and Lifestyle (VITAL) Study. Am. J. Epidemiol. 2009, 169, 815–828. [Google Scholar] [CrossRef]
- Martínez, M.E.; Jacobs, E.T.; Baron, J.A.; Marshall, J.R.; Byers, T. Dietary Supplements and Cancer Prevention: Balancing Potential Benefits Against Proven Harms. JNCI J. Natl. Cancer Inst. 2012, 104, 732–739. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.H.; Stocker, R.; Vollbracht, C.; Paulsen, G.; Riley, D.; Daiber, A.; Cuadrado, A. Antioxidants in Translational Medicine. Antioxid. Redox Signal. 2015, 23, 1130–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjelakovic, G.; Nikolova, D.; Gluud, L.L.; Simonetti, R.G.; Gluud, C. Mortality in Randomized Trials of Antioxidant Supplements for Primary and Secondary Prevention: Systematic review and meta-analysis. JAMA 2007, 297, 842–857. [Google Scholar] [CrossRef]
- Gori, T.; Münzel, T. Oxidative stress and endothelial dysfunction: Therapeutic implications. Ann. Med. 2011, 43, 259–272. [Google Scholar] [CrossRef]
- Seifried, H.E.; Anderson, D.E.; Fisher, E.I.; Milner, J.A. A review of the interaction among dietary antioxidants and reactive oxygen species. J. Nutr. Biochem. 2007, 18, 567–579. [Google Scholar] [CrossRef]
- Ren, D.; Villeneuve, N.F.; Jiang, T.; Wu, T.; Lau, A.; Toppin, H.A.; Zhang, D.D. Brusatol enhances the efficacy of chemotherapy by inhibiting the Nrf2-mediated defense mechanism. Proc. Natl. Acad. Sci. USA 2011, 108, 1433–1438. [Google Scholar] [CrossRef] [Green Version]
- Tsuchida, K.; Tsujita, T.; Hayashi, M.; Ojima, A.; Keleku-Lukwete, N.; Katsuoka, F.; Otsuki, A.; Kikuchi, H.; Oshima, Y.; Suzuki, M.; et al. Halofuginone enhances the chemo-sensitivity of cancer cells by suppressing NRF2 accumulation. Free Radic. Biol. Med. 2017, 103, 236–247. [Google Scholar] [CrossRef]
- Yasuda, D.; Ohe, T.; Takahashi, K.; Imamura, R.; Kojima, H.; Okabe, T.; Ichimura, Y.; Komatsu, M.; Yamamoto, M.; Nagano, T.; et al. Inhibitors of the protein–protein interaction between phosphorylated p62 and Keap1 attenuate chemoresistance in a human hepatocellular carcinoma cell line. Free Radic. Res. 2020, 54, 859–871. [Google Scholar] [CrossRef]
- Kitamura, H.; Motohashi, H. NRF2 addiction in cancer cells. Cancer Sci. 2018, 109, 900–911. [Google Scholar] [CrossRef] [Green Version]
- Baird, L.; Yamamoto, M. NRF2-Dependent Bioactivation of Mitomycin C as a Novel Strategy to Target KEAP1-NRF2 Pathway Activation in Human Cancer. Mol. Cell. Biol. 2021, 41, e00473-20. [Google Scholar] [CrossRef]
- Kim, S.J.; Kim, H.S.; Seo, Y.R. Understanding of ROS-Inducing Strategy in Anticancer Therapy. Oxid. Med. Cell. Longev. 2019, 2019, 5381692. [Google Scholar] [CrossRef]
- Salimi, A.; Roudkenar, M.H.; Sadeghi, L.; Mohseni, A.; Seydi, E.; Pirahmadi, N.; Pourahmad, J. Ellagic acid, a polyphenolic compound, selectively induces ROS-mediated apoptosis in cancerous B-lymphocytes of CLL patients by directly targeting mitochondria. Redox Biol. 2015, 6, 461–471. [Google Scholar] [CrossRef] [Green Version]
- Salimi, A.; Roudkenar, M.H.; Sadeghi, L.; Mohseni, A.R.; Seydi, E.; Pirahmadi, N.; Pourahmad, J. Selective Anticancer Activity of Acacetin Against Chronic Lymphocytic Leukemia Using Both In Vivo and In Vitro Methods: Key Role of Oxidative Stress and Cancerous Mitochondria. Nutr. Cancer 2016, 68, 1404–1416. [Google Scholar] [CrossRef]
- Liu, J.; Chen, G.; Pelicano, H.; Liao, J.; Huang, J.; Feng, L.; Keating, M.J.; Huang, P. Targeting p53-deficient chronic lymphocytic leukemia cells in vitro and in vivo by ROS-mediated mechanism. Oncotarget 2016, 7, 71378–71389. [Google Scholar] [CrossRef]
- Mato, A.R.; Roeker, L.; Eyre, T.A.; Nabhan, C.; Lamanna, N.; Hill, B.T.; Brander, D.M.; Barr, P.M.; Lansigan, F.; Cheson, B.D.; et al. A retrospective comparison of venetoclax alone or in combination with an anti-CD20 monoclonal antibody in R/R CLL. Blood Adv. 2019, 3, 1568–1573. [Google Scholar] [CrossRef] [Green Version]
- Camp, N.; Garrett, M.; Gopal, A.K.; James, R. Ibrutinib Selects for Cells with Elevated Reactive Oxygen Species and Downregulated Phosphatases. Blood 2019, 134, 3795. [Google Scholar] [CrossRef]
- Nguyen, L.X.T.; Troadec, E.; Kalvala, A.; Kumar, B.; Hoang, D.H.; Viola, D.; Zhang, B.; Nguyen, D.Q.; Aldoss, I.; Ghoda, L.; et al. The Bcl-2 inhibitor venetoclax inhibits Nrf2 antioxidant pathway activation induced by hypomethylating agents in AML. J. Cell. Physiol. 2019, 234, 14040–14049. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pagano, M.A.; Frezzato, F.; Visentin, A.; Trentin, L.; Brunati, A.M. Protein Phosphorylation and Redox Status: An as Yet Elusive Dyad in Chronic Lymphocytic Leukemia. Cancers 2022, 14, 4881. https://doi.org/10.3390/cancers14194881
Pagano MA, Frezzato F, Visentin A, Trentin L, Brunati AM. Protein Phosphorylation and Redox Status: An as Yet Elusive Dyad in Chronic Lymphocytic Leukemia. Cancers. 2022; 14(19):4881. https://doi.org/10.3390/cancers14194881
Chicago/Turabian StylePagano, Mario Angelo, Federica Frezzato, Andrea Visentin, Livio Trentin, and Anna Maria Brunati. 2022. "Protein Phosphorylation and Redox Status: An as Yet Elusive Dyad in Chronic Lymphocytic Leukemia" Cancers 14, no. 19: 4881. https://doi.org/10.3390/cancers14194881
APA StylePagano, M. A., Frezzato, F., Visentin, A., Trentin, L., & Brunati, A. M. (2022). Protein Phosphorylation and Redox Status: An as Yet Elusive Dyad in Chronic Lymphocytic Leukemia. Cancers, 14(19), 4881. https://doi.org/10.3390/cancers14194881