
Molecular Targeting of the Most Functionally Complex Gene in Precision Oncology: p53

Abstract
:Simple Summary
Abstract
1. Introduction
2. Tumor Protein p53
3. p53 and Cancer
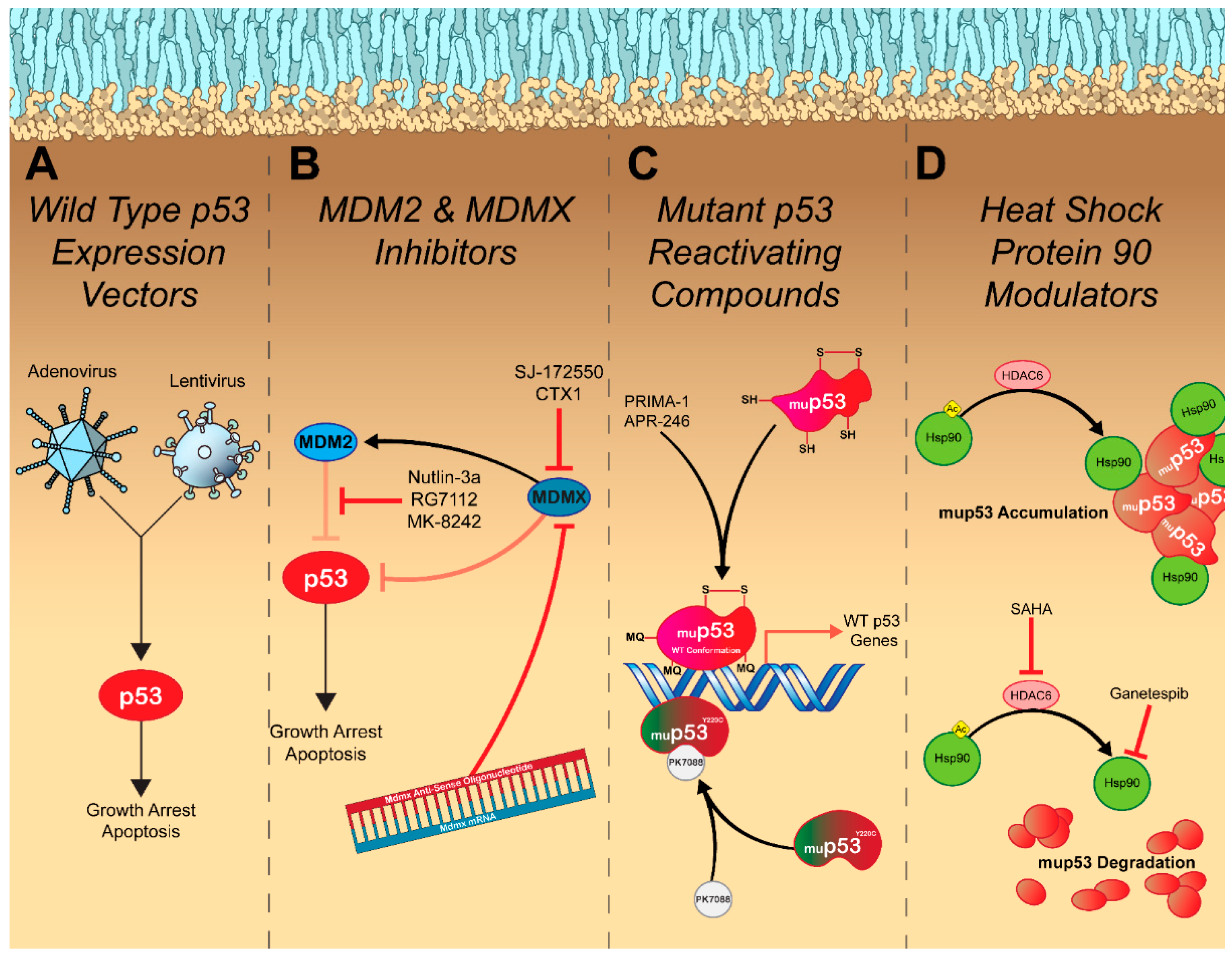
4. Restoring Endogenous p53 Signaling
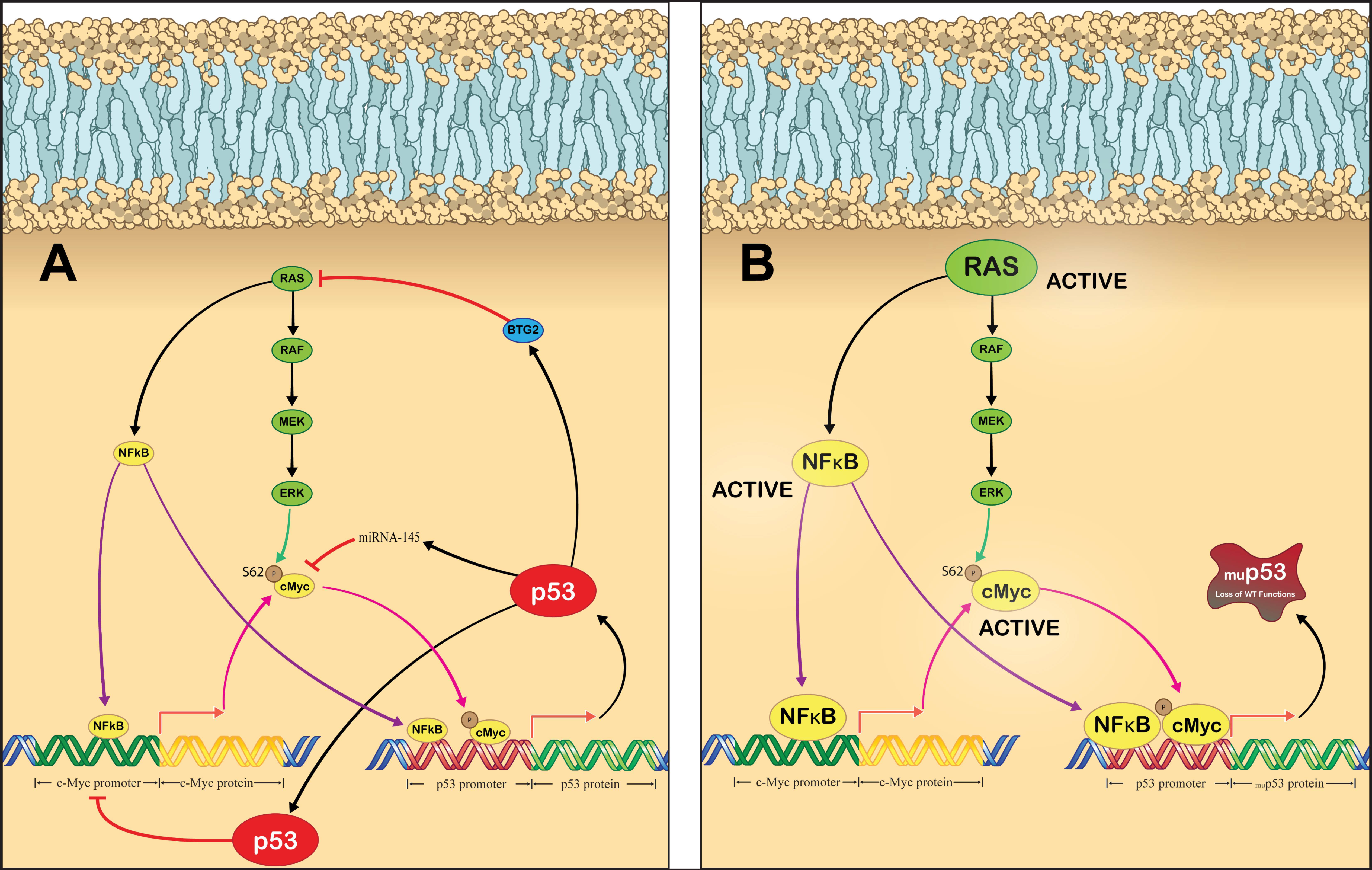
5. Inhibiting Oncogenic Gain of Function p53 Signaling
6. Exploiting Dysfunctional p53 Signaling
7. Future Directions
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Laversanne, M.; Weiderpass, E.; Soerjomataram, I. The Ever-Increasing Importance of Cancer as a Leading Cause of Premature Death Worldwide. Cancer 2021, 127, 3029–3030. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Knudson, A.G. Mutation and Cancer: Statistical Study of Retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, S.R.; Loeb, L.A.; Herr, A.J. Somatic Mutations in Aging, Cancer and Neurodegeneration. Mech. Ageing Dev. 2012, 133, 118–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoletov, K.; Beatty, P.H.; Lewis, J.D. Novel Therapeutic Targets for Cancer Metastasis. Expert Rev. Anticancer Ther. 2020, 20, 97–109. [Google Scholar] [CrossRef] [Green Version]
- Rudzinski, J.K.; Govindasamy, N.P.; Lewis, J.D.; Jurasz, P. The Role of the Androgen Receptor in Prostate Cancer-Induced Platelet Aggregation and Platelet-Induced Invasion. J. Thromb. Haemost. 2020, 18, 2976–2986. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Buckley, B.; Stoletov, K.; Jing, Y.; Ranson, M.; Lewis, J.D.; Kelso, M.; Fliegel, L. Roles of the Na+/H+ Exchanger Isoform 1 and Urokinase in Prostate Cancer Cell Migration and Invasion. Int. J. Mol. Sci. 2021, 22, 13263. [Google Scholar] [CrossRef]
- Abou-Ouf, H.; Assem, H.; Ghosh, S.; Karnes, R.J.; Stoletov, K.; Palanisamy, N.; Lewis, J.D.; Bismar, T.A. High Serine-Arginine Protein Kinase 1 Expression with PTEN Loss Defines Aggressive Phenotype of Prostate Cancer Associated with Lethal Outcome and Decreased Overall Survival. Eur. Urol. Open Sci. 2021, 23, 1–8. [Google Scholar] [CrossRef]
- Yankaskas, C.L.; Bera, K.; Stoletov, K.; Serra, S.A.; Carrillo-Garcia, J.; Tuntithavornwat, S.; Mistriotis, P.; Lewis, J.D.; Valverde, M.A.; Konstantopoulos, K. The Fluid Shear Stress Sensor TRPM7 Regulates Tumor Cell Intravasation. Sci. Adv. 2021, 7, eabh3457. [Google Scholar] [CrossRef] [PubMed]
- Kanwar, N.; Carmine-Simmen, K.; Nair, R.; Wang, C.; Moghadas-Jafari, S.; Blaser, H.; Tran-Thanh, D.; Wang, D.; Wang, P.; Wang, J.; et al. Amplification of a Calcium Channel Subunit CACNG4 Increases Breast Cancer Metastasis. eBioMedicine 2020, 52, 102646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fortunato, A.; Boddy, A.; Mallo, D.; Aktipis, A.; Maley, C.C.; Pepper, J.W. Natural Selection in Cancer Biology: From Molecular Snowflakes to Trait Hallmarks. Cold Spring Harb. Perspect. Med. 2017, 7, a029652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, S.P.; Morin, R.D.; Khattra, J.; Prentice, L.; Pugh, T.; Burleigh, A.; Delaney, A.; Gelmon, K.; Guliany, R.; Senz, J.; et al. Mutational Evolution in a Lobular Breast Tumour Profiled at Single Nucleotide Resolution. Nature 2009, 461, 809–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Friboulet, L.; Leshchiner, I.; Gainor, J.F.; Bergqvist, S.; Brooun, A.; Burke, B.J.; Deng, Y.-L.; Liu, W.; Dardaei, L.; et al. Resensitization to Crizotinib by the Lorlatinib ALK Resistance Mutation L1198F. N. Engl. J. Med. 2016, 374, 54–61. [Google Scholar] [CrossRef] [Green Version]
- Alfarouk, K.O.; Stock, C.-M.; Taylor, S.; Walsh, M.; Muddathir, A.K.; Verduzco, D.; Bashir, A.H.H.; Mohammed, O.Y.; Elhassan, G.O.; Harguindey, S.; et al. Resistance to Cancer Chemotherapy: Failure in Drug Response from ADME to P-Gp. Cancer Cell Int. 2015, 15, 71. [Google Scholar] [CrossRef] [Green Version]
- Croker, A.K.; Rodriguez-Torres, M.; Xia, Y.; Pardhan, S.; Leong, H.S.; Lewis, J.D.; Allan, A.L. Differential Functional Roles of ALDH1A1 and ALDH1A3 in Mediating Metastatic Behavior and Therapy Resistance of Human Breast Cancer Cells. Int. J. Mol. Sci. 2017, 18, 2039. [Google Scholar] [CrossRef]
- Zugazagoitia, J.; Guedes, C.; Ponce, S.; Ferrer, I.; Molina-Pinelo, S.; Paz-Ares, L. Current Challenges in Cancer Treatment. Clin. Ther. 2016, 38, 1551–1566. [Google Scholar] [CrossRef] [Green Version]
- Arnedos, M.; Soria, J.-C.; Andre, F.; Tursz, T. Personalized Treatments of Cancer Patients: A Reality in Daily Practice, a Costly Dream or a Shared Vision of the Future from the Oncology Community? Cancer Treat. Rev. 2014, 40, 1192–1198. [Google Scholar] [CrossRef]
- Bell, C.J.; Potts, K.G.; Hitt, M.M.; Pink, D.; Tuszynski, J.A.; Lewis, J.D. Novel Colchicine Derivative CR42-24 Demonstrates Potent Anti-Tumor Activity in Urothelial Carcinoma. Cancer Lett. 2022, 526, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Paproski, R.J.; Jovel, J.; Wong, G.K.S.; Lewis, J.D.; Zemp, R.J. Enhanced Detection of Cancer Biomarkers in Blood-Borne Extracellular Vesicles Using Nanodroplets and Focused Ultrasound. Cancer Res. 2017, 77, 3–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoletov, K.; Willetts, L.; Beatty, P.H.; Lewis, J.D. Discovery of Metastatic Regulators Using a Rapid and Quantitative Intravital Chick Chorioallantoic Membrane Model. J. Vis. Exp. 2021, 2021, e62077. [Google Scholar] [CrossRef]
- Wang, B.; Wu, H.; Chai, C.; Lewis, J.; Pichiorri, F.; Eisenstat, D.D.; Pomeroy, S.L.; Leng, R.P. MicroRNA-1301 Suppresses Tumor Cell Migration and Invasion by Targeting the p53/UBE4B Pathway in Multiple Human Cancer Cells. Cancer Lett. 2017, 401, 20–32. [Google Scholar] [CrossRef]
- Brown, D.W.; Wee, P.; Bhandari, P.; Vega, H.; Grin, L.; Sosnowski, D.; Hejazi, M.; Ablack, J.; Clancy, E.K.; Pink, D.; et al. Safe and Effective Delivery of Nucleic Acids Using Proteolipid Vehicles Formulated with Fusion-Associated Small Transmembrane Proteins. SSRN Preprint. 2022. Available online: https://ssrn.com/abstract=4241169 (accessed on 15 September 2022).
- Cooper, T.T.; Dieters-Castator, D.Z.; Liu, J.; Siegers, G.M.; Pink, D.; Lewis, J.D.; Fu, Y.; Steed, H.; Lajoie, G.A.; Postovit, L.-M. Plasma EV Biomarkers of High-Grade Serous Carcinoma Targeted Proteomics and Support Vector Classification Reveal Potential Biomarkers for the Early Detection of High-Grade Serous Ovarian Cancer. Ph.D. Thesis, Mayo Clinic College of Medicine and Science, Rochester, MN, USA, 2022. [Google Scholar] [CrossRef]
- Kedarisetti, P.; Bouvet, V.R.; Shi, W.; Bergman, C.N.; Dufour, J.; Kashani Ilkhechi, A.; Bell, K.L.; Paproski, R.J.; Lewis, J.D.; Wuest, F.R.; et al. Enrichment and Ratiometric Detection of Circulating Tumor Cells Using PSMA- and Folate Receptor-Targeted Magnetic and Surface-Enhanced Raman Scattering Nanoparticles. Biomed. Opt. Express 2020, 11, 6211. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.P. Cancer. p53, Guardian of the Genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 Network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef]
- Bieging, K.T.; Mello, S.S.; Attardi, L.D. Unravelling Mechanisms of p53-Mediated Tumour Suppression. Nat. Rev. Cancer 2014, 14, 359. [Google Scholar] [CrossRef] [Green Version]
- Freed-Pastor, W.A.; Prives, C. Mutant p53: One Name, Many Proteins. Genes Dev. 2012, 26, 1268–1286. [Google Scholar] [CrossRef]
- Stegh, A.H. Targeting the p53 Signaling Pathway in Cancer Therapy—The Promises, Challenges and Perils. Expert Opin. Ther. Targets 2012, 16, 67–83. [Google Scholar] [CrossRef]
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saldana-Meyer, R.; Recillas-Targa, F. Transcriptional and Epigenetic Regulation of the p53 Tumor Suppressor Gene. Epigenetics 2011, 6, 1068–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, S.L.; Levine, A.J. The p53 Pathway: Positive and Negative Feedback Loops. Oncogene 2005, 24, 2899–2908. [Google Scholar] [CrossRef] [Green Version]
- Oren, M. Decision Making by p53: Life, Death and Cancer. Cell Death Differ. 2003, 10, 431–442. [Google Scholar] [CrossRef]
- Lin, T.; Hou, P.F.; Meng, S.; Chen, F.; Jiang, T.; Li, M.L.; Shi, M.L.; Liu, J.J.; Zheng, J.N.; Bai, J. Emerging Roles of p53 Related LncRNAs in Cancer Progression: A Systematic Review. Int. J. Biol. Sci. 2019, 15, 1287–1298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 Promotes the Rapid Degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Kubbutat, M.H.; Jones, S.N.; Vousden, K.H. Regulation of p53 Stability by Mdm2. Nature 1997, 387, 299–303. [Google Scholar] [CrossRef]
- Shvarts, A.; Steegenga, W.T.; Riteco, N.; van Laar, T.; Dekker, P.; Bazuine, M.; van Ham, R.C.; van der Houven van Oordt, W.; Hateboer, G.; van der Eb, A.J.; et al. MDMX: A Novel p53-Binding Protein with Some Functional Properties of MDM2. EMBO J. 1996, 15, 5349–5357. [Google Scholar] [CrossRef]
- Jackson, M.W.; Berberich, S.J. MdmX Protects p53 from Mdm2-Mediated Degradation. Mol. Cell Biol. 2000, 20, 1001–1007. [Google Scholar] [CrossRef]
- Wade, M.; Wahl, G.M. Targeting Mdm2 and Mdmx in Cancer Therapy: Better Living through Medicinal Chemistry? Mol. Cancer Res. 2009, 7, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; El-Deiry, W.S. P73 or p53 Directly Regulates Human p53 Transcription to Maintain Cell Cycle Checkpoints. Cancer Res. 2006, 66, 6982–6989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bug, M.; Dobbelstein, M. Anthracyclines Induce the Accumulation of Mutant p53 through E2F1-Dependent and -Independent Mechanisms. Oncogene 2011, 30, 3612–3624. [Google Scholar] [CrossRef] [Green Version]
- Kogan-Sakin, I.; Tabach, Y.; Buganim, Y.; Molchadsky, A.; Solomon, H.; Madar, S.; Kamer, I.; Stambolsky, P.; Shelly, A.; Goldfinger, N.; et al. Mutant p53(R175H) Upregulates Twist1 Expression and Promotes Epithelial-Mesenchymal Transition in Immortalized Prostate Cells. Cell Death Differ. 2011, 18, 271–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fei, P.; El-Deiry, W.S. p53 and Radiation Responses. Oncogene 2003, 22, 5774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirley, S.H.; Rundhaug, J.E.; Tian, J.; Cullinan-Ammann, N.; Lambertz, I.; Conti, C.J.; Fuchs-Young, R. Transcriptional Regulation of Estrogen Receptor-α by p53 in Human Breast Cancer Cells. Cancer Res. 2009, 69, 3405–3414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartek, J.; Lukas, J. Chk1 and Chk2 Kinases in Checkpoint Control and Cancer. Cancer Cell 2003, 3, 421–429. [Google Scholar] [CrossRef] [Green Version]
- Maya, R.; Balass, M.; Kim, S.T.; Shkedy, D.; Martinez Leal, J.F.; Shifman, O.; Moas, M.; Buschmann, T.; Ronai, Z.; Shiloh, Y.; et al. ATM-Dependent Phosphorylation of Mdm2 on Serine 395: Role in p53 Activation by DNA Damage. Genes Dev. 2001, 15, 1067–1077. [Google Scholar] [CrossRef] [Green Version]
- Shieh, S.Y.; Ikeda, M.; Taya, Y.; Prives, C. DNA Damage-Induced Phosphorylation of p53 Alleviates Inhibition by MDM2. Cell 1997, 91, 325–334. [Google Scholar] [CrossRef] [Green Version]
- Chehab, N.H.; Malikzay, A.; Stavridi, E.S.; Halazonetis, T.D. Phosphorylation of Ser-20 Mediates Stabilization of Human p53 in Response to DNA Damage. Proc. Natl. Acad. Sci. USA 1999, 96, 13777–13782. [Google Scholar] [CrossRef]
- Tibbetts, R.S.; Brumbaugh, K.M.; Williams, J.M.; Sarkaria, J.N.; Cliby, W.A.; Shieh, S.Y.; Taya, Y.; Prives, C.; Abraham, R.T. A Role for ATR in the DNA Damage-Induced Phosphorylation of p53. Genes Dev. 1999, 13, 152–157. [Google Scholar] [CrossRef] [Green Version]
- Pereg, Y.; Shkedy, D.; de Graaf, P.; Meulmeester, E.; Edelson-Averbukh, M.; Salek, M.; Biton, S.; Teunisse, A.F.A.S.; Lehmann, W.D.; Jochemsen, A.G.; et al. Phosphorylation of Hdmx Mediates Its Hdm2- and ATM-Dependent Degradation in Response to DNA Damage. Proc. Natl. Acad. Sci. USA 2005, 102, 5056–5061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai, H.; Wiederschain, D.; Kitao, H.; Stuart, J.; Tsai, K.K.C.; Yuan, Z.-M. DNA Damage-Induced MDMX Degradation Is Mediated by MDM2*. J. Biol. Chem. 2003, 278, 45946–45953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Y.; Dai, M.-S.; Lu, S.Z.; Xu, Y.; Luo, Z.; Zhao, Y.; Lu, H. 14-3-3γ Binds to MDMX That Is Phosphorylated by UV-Activated Chk1, Resulting in p53 Activation. EMBO J. 2006, 25, 1207–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barak, Y.; Juven, T.; Haffner, R.; Oren, M. Mdm2 Expression Is Induced by Wild Type p53 Activity. EMBO J. 1993, 12, 461–468. [Google Scholar] [CrossRef]
- Lu, X.; Ma, O.; Nguyen, T.A.; Jones, S.N.; Oren, M.; Donehower, L.A. The Wip1 Phosphatase Acts as a Gatekeeper in the p53-Mdm2 Autoregulatory Loop. Cancer Cell 2007, 12, 342–354. [Google Scholar] [CrossRef] [Green Version]
- Komori, H.; Enomoto, M.; Nakamura, M.; Iwanaga, R.; Ohtani, K. Distinct E2F-Mediated Transcriptional Program Regulates P14ARF Gene Expression. EMBO J. 2005, 24, 3724–3736. [Google Scholar] [CrossRef] [Green Version]
- Pomerantz, J.; Schreiber-Agus, N.; Liégeois, N.J.; Silverman, A.; Alland, L.; Chin, L.; Potes, J.; Chen, K.; Orlow, I.; Lee, H.-W.; et al. The Ink4a Tumor Suppressor Gene Product, P19ARF, Interacts with MDM2 and Neutralizes MDM2′s Inhibition of p53. Cell 1998, 92, 713–723. [Google Scholar] [CrossRef] [Green Version]
- Fogal, V.; Hsieh, J.-K.; Royer, C.; Zhong, S.; Lu, X. Cell Cycle-Dependent Nuclear Retention of p53 by E2F1 Requires Phosphorylation of p53 at Ser315. EMBO J. 2005, 24, 2768–2782. [Google Scholar] [CrossRef] [Green Version]
- Gartel, A.L.; Radhakrishnan, S.K. Lost in Transcription: P21 Repression, Mechanisms, and Consequences. Cancer Res. 2005, 65, 3980–3985. [Google Scholar] [CrossRef]
- Gencel-Augusto, J.; Lozano, G. REVIEW p53 Tetramerization: At the Center of the Dominant-Negative Effect of Mutant p53. Genes Dev. 2020, 34, 1128–1146. [Google Scholar] [CrossRef]
- Fischer, M. Census and Evaluation of p53 Target Genes. Oncogene 2017, 36, 3943–3956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kracikova, M.; Akiri, G.; George, A.; Sachidanandam, R.; Aaronson, S.A. A Threshold Mechanism Mediates p53 Cell Fate Decision between Growth Arrest and Apoptosis. Cell Death Differ. 2013, 20, 576–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.-P.; Liu, F.; Wang, W. Two-Phase Dynamics of p53 in the DNA Damage Response. Proc. Natl. Acad. Sci. USA 2011, 108, 8990–8995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, A.B.; Schumacher, B. p53 in the DNA-Damage-Repair Process. Cold Spring Harb. Perspect. Med. 2016, 6, a026070. [Google Scholar] [CrossRef] [Green Version]
- Mijit, M.; Caracciolo, V.; Melillo, A.; Amicarelli, F.; Giordano, A. Role of p53 in the Regulation of Cellular Senescence. Biomolecules 2020, 10, 420. [Google Scholar] [CrossRef] [Green Version]
- Yosef, R.; Pilpel, N.; Papismadov, N.; Gal, H.; Ovadya, Y.; Vadai, E.; Miller, S.; Porat, Z.; Ben-Dor, S.; Krizhanovsky, V. P21 Maintains Senescent Cell Viability under Persistent DNA Damage Response by Restraining JNK and Caspase Signaling. EMBO J. 2017, 36, 2280–2295. [Google Scholar] [CrossRef]
- Gupta, S.; Silveira, D.A.; Mombach, J.C.M. Towards DNA-Damage Induced Autophagy: A Boolean Model of p53-Induced Cell Fate Mechanisms. DNA Repair 2020, 96, 102971. [Google Scholar] [CrossRef]
- Yun, C.W.; Lee, S.H. The Roles of Autophagy in Cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef] [Green Version]
- Mrakovcic, M.; Fröhlich, L.F. p53-Mediated Molecular Control of Autophagy in Tumor Cells. Biomolecules 2018, 8, 14. [Google Scholar] [CrossRef]
- Crighton, D.; Wilkinson, S.; O’Prey, J.; Syed, N.; Smith, P.; Harrison, P.R.; Gasco, M.; Garrone, O.; Crook, T.; Ryan, K.M. DRAM, a p53-Induced Modulator of Autophagy, Is Critical for Apoptosis. Cell 2006, 126, 121–134. [Google Scholar] [CrossRef] [Green Version]
- Kenzelmann Broz, D.; Mello, S.S.; Bieging, K.T.; Jiang, D.; Dusek, R.L.; Brady, C.A.; Sidow, A.; Attardi, L.D. Global Genomic Profiling Reveals an Extensive p53-Regulated Autophagy Program Contributing to Key p53 Responses. Genes Dev. 2013, 27, 1016–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, E. Autophagy and p53. Cold Spring Harb. Perspect. Med. 2016, 6, a026120. [Google Scholar] [CrossRef] [PubMed]
- Mercer, W.E.; Shields, M.T.; Amin, M.; Sauve, G.J.; Appella, E.; Romano, J.W.; Ullrich, S.J. Negative Growth Regulation in a Glioblastoma Tumor Cell Line That Conditionally Expresses Human Wild-Type p53. Proc. Natl. Acad. Sci. USA 1990, 87, 6166–6170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, S.J.; Markowitz, S.; Fearon, E.R.; Willson, J.K.; Vogelstein, B. Suppression of Human Colorectal Carcinoma Cell Growth by Wild-Type p53. Science 1990, 249, 912–915. [Google Scholar] [CrossRef]
- Donehower, L.A.; Harvey, M.; Slagle, B.L.; McArthur, M.J.; Montgomery, C.A.; Butel, J.S.; Bradley, A. Mice Deficient for p53 Are Developmentally Normal but Susceptible to Spontaneous Tumours. Nature 1992, 356, 215–221. [Google Scholar] [CrossRef]
- Nigro, J.M.; Baker, S.J.; Preisinger, A.C.; Jessup, J.M.; Hosteller, R.; Cleary, K.; Signer, S.H.; Davidson, N.; Baylin, S.; Devilee, P.; et al. Mutations in the p53 Gene Occur in Diverse Human Tumour Types. Nature 1989, 342, 705–708. [Google Scholar] [CrossRef] [Green Version]
- Baker, S.J.; Fearon, E.R.; Nigro, J.M.; Hamilton, S.R.; Preisinger, A.C.; Jessup, J.M.; vanTuinen, P.; Ledbetter, D.H.; Barker, D.F.; Nakamura, Y.; et al. Chromosome 17 Deletions and p53 Gene Mutations in Colorectal Carcinomas. Science 1989, 244, 217–221. [Google Scholar] [CrossRef]
- Roy, B.; Beamon, J.; Balint, E.; Reisman, D. Transactivation of the Human p53 Tumor Suppressor Gene by C-Myc/Max Contributes to Elevated Mutant p53 Expression in Some Tumors. Mol. Cell Biol. 1994, 14, 7805–7815. [Google Scholar] [CrossRef] [Green Version]
- Raman, V.; Martensen, S.A.; Reisman, D.; Evron, E.; Odenwald, W.F.; Jaffee, E.; Marks, J.; Sukumar, S. Compromised HOXA5 Function Can Limit p53 Expression in Human Breast Tumours. Nature 2000, 405, 974–978. [Google Scholar] [CrossRef]
- Gabay, M.; Li, Y.; Felsher, D.W. MYC Activation Is a Hallmark of Cancer Initiation and Maintenance. Cold Spring Harb. Perspect. Med. 2014, 4, a014241. [Google Scholar] [CrossRef] [Green Version]
- Ordonez-Moran, P.; Dafflon, C.; Imajo, M.; Nishida, E.; Huelsken, J. HOXA5 Counteracts Stem Cell Traits by Inhibiting Wnt Signaling in Colorectal Cancer. Cancer Cell 2015, 28, 815–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teo, W.W.; Merino, V.F.; Cho, S.; Korangath, P.; Liang, X.; Wu, R.C.; Neumann, N.M.; Ewald, A.J.; Sukumar, S. HOXA5 Determines Cell Fate Transition and Impedes Tumor Initiation and Progression in Breast Cancer through Regulation of E-Cadherin and CD24. Oncogene 2016, 35, 5539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, J.R.F.; Bateman, A.C.; Hanson, H.; An, Q.; Evans, G.; Rahman, N.; Jones, J.L.; Eccles, D.M. A Novel HER2-Positive Breast Cancer Phenotype Arising from Germline TP53 Mutations. J. Med. Genet. 2010, 47, 771–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eastham, J.A.; Stapleton, A.M.; Gousse, A.E.; Timme, T.L.; Yang, G.; Slawin, K.M.; Wheeler, T.M.; Scardino, P.T.; Thompson, T.C. Association of p53 Mutations with Metastatic Prostate Cancer. Clin. Cancer Res. 1995, 1, 1111–1118. [Google Scholar] [PubMed]
- Grignon, D.J.; Sarkar, F.H.; Forman, J.D.; Caplan, R.; Pajak, T.F.; Lawton, C.A.; Hammond, E.H.; Pilepich, M.V.; Mesic, J.; Fu, K.K.; et al. p53 Status and Prognosis of Locally Advanced Prostatic Adenocarcinoma: A Study Based on RTOG 8610. JNCI J. Natl. Cancer Inst. 1997, 89, 158–165. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Zhang, C.; Feng, Z. Tumor Suppressor p53 and Its Gain-of-Function Mutants in Cancer. Acta Biochim. Biophys. Sin. 2014, 46, 170–179. [Google Scholar] [CrossRef] [Green Version]
- Baliou, E.; Nonni, A.; Keramopoulos, D.; Ragos, V.; Tsiambas, E.; Patsouris, E.; Pavlakis, K. Deregulation of p53-MDM2 Auto-Regulatory Pathway in Breast Carcinoma. J. BUON 2016, 21, 1099–1103. [Google Scholar]
- Oren, M.; Rotter, V. Mutant p53 Gain-of-Function in Cancer. Cold Spring Harb. Perspect. Biol. 2010, 2, a001107. [Google Scholar] [CrossRef]
- Mello, S.S.; Attardi, L.D. Not All p53 Gain-of-Function Mutants Are Created Equal. Cell Death Differ. 2013, 20, 855–857. [Google Scholar] [CrossRef] [Green Version]
- Gaiddon, C.; Lokshin, M.; Ahn, J.; Zhang, T.; Prives, C. A Subset of Tumor-Derived Mutant Forms of p53 down-Regulate P63 and P73 through a Direct Interaction with the p53 Core Domain. Mol. Cell Biol. 2001, 21, 1874–1887. [Google Scholar] [CrossRef]
- Dötsch, V.; Bernassola, F.; Coutandin, D.; Candi, E.; Melino, G. P63 and P73, the Ancestors of p53. Cold Spring Harb. Perspect. Biol. 2010, 2, a004887. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; McDonnell, T.J.; Montes de Oca Luna, R.; Kapoor, M.; Mims, B.; El-Naggar, A.K.; Lozano, G. Solo, MHigh Metastatic Potential in Mice Inheriting a Targeted p53 Missense Mutation. Proc. Natl. Acad. Sci. USA 2000, 97, 4174–4179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olive, K.P.; Tuveson, D.A.; Ruhe, Z.C.; Yin, B.; Willis, N.A.; Bronson, R.T.; Crowley, D.; Jacks, T. Mutant p53 Gain of Function in Two Mouse Models of Li-Fraumeni Syndrome. Cell 2004, 119, 847–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, G.A.; Iwakuma, T.; Suh, Y.A.; Liu, G.; Rao, V.A.; Parant, J.M.; Valentin-Vega, Y.A.; Terzian, T.; Caldwell, L.C.; Strong, L.C.; et al. Gain of Function of a p53 Hot Spot Mutation in a Mouse Model of Li-Fraumeni Syndrome. Cell 2004, 119, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Solomon, H.; Dinowitz, N.; Pateras, I.S.; Cooks, T.; Shetzer, Y.; Molchadsky, A.; Charni, M.; Rabani, S.; Koifman, G.; Tarcic, O.; et al. Mutant p53 Gain of Function Underlies High Expression Levels of Colorectal Cancer Stem Cells Markers. Oncogene 2018, 37, 1669–1684. [Google Scholar] [CrossRef] [Green Version]
- Roman-Rosales, A.A.; Garcia-Villa, E.; Herrera, L.A.; Gariglio, P.; Diaz-Chavez, J. Mutant p53 Gain of Function Induces HER2 Over-Expression in Cancer Cells. BMC Cancer 2018, 18, 709. [Google Scholar] [CrossRef] [Green Version]
- Schwartzenberg-Bar-Yoseph, F.; Armoni, M.; Karnieli, E. The Tumor Suppressor p53 Down-Regulates Glucose Transporters GLUT1and GLUT4 Gene Expression. Cancer Res 2004, 64, 2627–2633. [Google Scholar] [CrossRef] [Green Version]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.C.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. TIGAR, a p53-Inducible Regulator of Glycolysis and Apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.-J.; Chao, C.-H.; Xia, W.; Yang, J.-Y.; Xiong, Y.; Li, C.-W.; Yu, W.-H.; Rehman, S.K.; Hsu, J.L.; Lee, H.-H.; et al. p53 Regulates Epithelial–Mesenchymal Transition and Stem Cell Properties through Modulating MiRNAs. Nat. Cell Biol. 2011, 13, 317–323. [Google Scholar] [CrossRef] [Green Version]
- Ren, D.; Wang, M.; Guo, W.; Zhao, X.; Tu, X.; Huang, S.; Zou, X.; Peng, X. Wild-Type p53 Suppresses the Epithelial-Mesenchymal Transition and Stemness in PC-3 Prostate Cancer Cells by Modulating MiR-145. Int. J. Oncol. 2013, 42, 1473–1481. [Google Scholar] [CrossRef]
- Brighenti, E.; Calabrese, C.; Liguori, G.; Giannone, F.A.; Trerè, D.; Montanaro, L.; Derenzini, M. Interleukin 6 Downregulates p53 Expression and Activity by Stimulating Ribosome Biogenesis: A New Pathway Connecting Inflammation to Cancer. Oncogene 2014, 33, 4396–4406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastor, D.M.; Irby, R.B.; Poritz, L.S. Tumor Necrosis Factor α Induces p53 Up-Regulated Modulator of Apoptosis Expression in Colorectal Cancer Cell Lines. Dis. Colon Rectum 2010, 53, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-ΚB Signaling in Inflammation. Signal Transduct. Target Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.; Burdelya, L.G.; Swiatkowski, S.M.; Boiko, A.D.; Howe, P.H.; Stark, G.R.; Gudkov, A. V Secreted Transforming Growth Factor Beta2 Activates NF-KappaB, Blocks Apoptosis, and Is Essential for the Survival of Some Tumor Cells. Proc. Natl. Acad. Sci. USA 2004, 101, 7112–7117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirch, H.C.; Flaswinkel, S.; Rumpf, H.; Brockmann, D.; Esche, H. Expression of Human p53 Requires Synergistic Activation of Transcription from the p53 Promoter by AP-1, NF-KappaB and Myc/Max. Oncogene 1999, 18, 2728–2738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujiwara, T.; Grimm, E.A.; Mukhopadhyay, T.; Cai, D.W.; Owen-Schaub, L.B.; Roth, J.A. A Retroviral Wild-Type p53 Expression Vector Penetrates Human Lung Cancer Spheroids and Inhibits Growth by Inducing Apoptosis. Cancer Res. 1993, 53, 4129–4133. [Google Scholar]
- Shaw, P.; Bovey, R.; Tardy, S.; Sahli, R.; Sordat, B.; Costa, J. Induction of Apoptosis by Wild-Type p53 in a Human Colon Tumor-Derived Cell Line. Proc. Natl. Acad. Sci. USA 1992, 89, 4495–4499. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, T.; Cai, D.W.; Georges, R.N.; Mukhopadhyay, T.; Grimm, E.A.; Roth, J.A. Therapeutic Effect of a Retroviral Wild-Type p53 Expression Vector in an Orthotopic Lung Cancer Model. JNCI J. Natl. Cancer Inst. 1994, 86, 1458–1462. [Google Scholar] [CrossRef]
- Roth, J.A.; Nguyen, D.; Lawrence, D.D.; Kemp, B.L.; Carrasco, C.H.; Ferson, D.Z.; Hong, W.K.; Komaki, R.; Lee, J.J.; Nesbitt, J.C.; et al. Retrovirus–Mediated Wild–Type p53 Gene Transfer to Tumors of Patients with Lung Cancer. Nat. Med. 1996, 2, 985–991. [Google Scholar] [CrossRef]
- Milone, M.C.; O’Doherty, U. Clinical Use of Lentiviral Vectors. Leukemia 2018, 32, 1529–1541. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.W.; Fang, X.; Mazur, W.; French, B.A.; Georges, R.N.; Roth, J.A. High-Efficiency Gene Transfer and High-Level Expression of Wild-Type p53 in Human Lung Cancer Cells Mediated by Recombinant Adenovirus. Cancer Gene Ther. 1994, 1, 5–13. [Google Scholar] [PubMed]
- Spitz, F.R.; Nguyen, D.; Skibber, J.M.; Meyn, R.E.; Cristiano, R.J.; Roth, J.A. Adenoviral-Mediated Wild-Type p53 Gene Expression Sensitizes Colorectal Cancer Cells to Ionizing Radiation. Clin. Cancer Res. 1996, 2, 1665–1671. [Google Scholar] [PubMed]
- Nielsen, L.L.; Dell, J.; Maxwell, E.; Armstrong, L.; Maneval, D.; Catino, J.J. Efficacy of p53 Adenovirus-Mediated Gene Therapy against Human Breast Cancer Xenografts. Cancer Gene Ther. 1997, 4, 129–138. [Google Scholar] [PubMed]
- Swisher, S.G.; Roth, J.A.; Nemunaitis, J.; Lawrence, D.D.; Kemp, B.L.; Carrasco, C.H.; Connors, D.G.; El-Naggar, A.K.; Fossella, F.; Glisson, B.S.; et al. Adenovirus-Mediated p53 Gene Transfer in Advanced Non-Small-Cell Lung Cancer. JNCI J. Natl. Cancer Inst. 1999, 91, 763–771. [Google Scholar] [CrossRef] [Green Version]
- Clayman, G.L.; el-Naggar, A.K.; Lippman, S.M.; Henderson, Y.C.; Frederick, M.; Merritt, J.A.; Zumstein, L.A.; Timmons, T.M.; Liu, T.J.; Ginsberg, L.; et al. Adenovirus-Mediated p53 Gene Transfer in Patients with Advanced Recurrent Head and Neck Squamous Cell Carcinoma. J. Clin. Oncol. 1998, 16, 2221–2232. [Google Scholar] [CrossRef]
- Peng, Z.; Han, D.; Zhang, S.; Pan, J.; Tang, P.; Xiao, S.; Chen, C.; Huang, Z.; Zhang, W.; Zhang, X.; et al. Clinical Evaluation of Safety and Efficacy of Intratumoral Administration of a Recombinant Adenoviral-p53 Anticancer Agent (Genkaxin). Mol. Ther. 2003, 7, S422–S423. [Google Scholar] [CrossRef]
- Swisher, S.G.; Roth, J.A.; Komaki, R.; Gu, J.; Lee, J.J.; Hicks, M.; Ro, J.Y.; Hong, W.K.; Merritt, J.A.; Ahrar, K.; et al. Induction of p53-Regulated Genes and Tumor Regression in Lung Cancer Patients after Intratumoral Delivery of Adenoviral p53 (INGN 201) and Radiation Therapy. Clin. Cancer Res. 2003, 9, 93–101. [Google Scholar]
- Zhang, W.-W.; Li, L.; Li, D.; Liu, J.; Li, X.; Li, W.; Xu, X.; Zhang, M.J.; Chandler, L.A.; Lin, H.; et al. The First Approved Gene Therapy Product for Cancer Ad-p53 (Gendicine): 12 Years in the Clinic. Hum. Gene Ther. 2018, 29, 160–179. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.-X.; Zhang, S.; He, X.-H.; Liu, S.-Y.; Ma, C.; Zou, X.-P. Clinical Utility of Recombinant Adenoviral Human p53 Gene Therapy: Current Perspectives. Onco Targets Ther. 2014, 7, 1901–1909. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Suh, Y.-A.; Fuller, M.Y.; Jackson, J.G.; Xiong, S.; Terzian, T.; Quintás-Cardama, A.; Bankson, J.A.; El-Naggar, A.K.; Lozano, G. Restoring Expression of Wild-Type p53 Suppresses Tumor Growth but Does Not Cause Tumor Regression in Mice with a p53 Missense Mutation. J. Clin. Investig. 2011, 121, 893–904. [Google Scholar] [CrossRef]
- Monti, P.; Campomenosi, P.; Ciribilli, Y.; Iannone, R.; Inga, A.; Abbondandolo, A.; Resnick, M.A.; Fronza, G. Tumour p53 Mutations Exhibit Promoter Selective Dominance over Wild Type p53. Oncogene 2002, 21, 1641–1648. [Google Scholar] [CrossRef] [Green Version]
- Clayman, G.L.; El-Naggar, A.K.; Roth, J.A.; Zhang, W.-W.; Goepfert, H.; Taylor, D.L.; Liu, T.-J. In Vivo Molecular Therapy with p53 Adenovirus for Microscopic Residual Head and Neck Squamous Carcinoma. Cancer Res. 1995, 55, 1–6. [Google Scholar] [PubMed]
- Zhang, W.-W.; Alemany, R.; Wang, J.; Koch, P.E.; Ordonez, N.G.; Roth, J.A. Safety Evaluation of Ad5CMY-p53 In Vitro and In Vivo. Hum. Gene Ther. 1995, 6, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Schirmbeck, R.; Reimann, J.; Kochanek, S.; Kreppel, F. The Immunogenicity of Adenovirus Vectors Limits the Multispecificity of CD8 T-Cell Responses to Vector-Encoded Transgenic Antigens. Mol. Ther. 2008, 16, 1609–1616. [Google Scholar] [CrossRef] [PubMed]
- Coughlan, L. Factors Which Contribute to the Immunogenicity of Non-Replicating Adenoviral Vectored Vaccines. Front. Immunol. 2020, 11, 909. [Google Scholar] [CrossRef] [PubMed]
- Tesniere, A.; Schlemmer, F.; Boige, V.; Kepp, O.; Martins, I.; Ghiringhelli, F.; Aymeric, L.; Michaud, M.; Apetoh, L.; Barault, L.; et al. Immunogenic Death of Colon Cancer Cells Treated with Oxaliplatin. Oncogene 2010, 29, 482–491. [Google Scholar] [CrossRef] [Green Version]
- Casares, N.; Pequignot, M.O.; Tesniere, A.; Ghiringhelli, F.; Roux, S.; Chaput, N.; Schmitt, E.; Hamai, A.; Hervas-Stubbs, S.; Obeid, M.; et al. Caspase-Dependent Immunogenicity of Doxorubicin-Induced Tumor Cell Death. J. Exp. Med. 2005, 202, 1691–1701. [Google Scholar] [CrossRef]
- Sobol, R.E.; Menander, K.B.; Chada, S.; Wiederhold, D.; Sellman, B.; Talbott, M.; Nemunaitis, J.J. Analysis of Adenoviral p53 Gene Therapy Clinical Trials in Recurrent Head and Neck Squamous Cell Carcinoma. Front. Oncol. 2021, 11, 1223. [Google Scholar] [CrossRef]
- Chada, S.; Wiederhold, D.; Menander, K.B.; Sellman, B.; Talbott, M.; Nemunaitis, J.J.; Ahn, H.M.; Jung, B.K.; Yun, C.O.; Sobol, R.E. Tumor Suppressor Immune Gene Therapy to Reverse Immunotherapy Resistance. Cancer Gene Ther. 2022, 29, 825–834. [Google Scholar] [CrossRef]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In Vivo Activation of the p53 Pathway by Small-Molecule Antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [Green Version]
- Vu, B.; Wovkulich, P.; Pizzolato, G.; Lovey, A.; Ding, Q.; Jiang, N.; Liu, J.-J.; Zhao, C.; Glenn, K.; Wen, Y.; et al. Discovery of RG7112: A Small-Molecule MDM2 Inhibitor in Clinical Development. ACS Med. Chem. Lett. 2013, 4, 466–469. [Google Scholar] [CrossRef] [Green Version]
- Ding, K.; Lu, Y.; Nikolovska-Coleska, Z.; Qiu, S.; Ding, Y.; Gao, W.; Stuckey, J.; Krajewski, K.; Roller, P.P.; Tomita, Y.; et al. Structure-Based Design of Potent Non-Peptide MDM2 Inhibitors. J. Am. Chem. Soc. 2005, 127, 10130–10131. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Sun, W.; Zhao, Y.; McEachern, D.; Meaux, I.; Barrière, C.; Stuckey, J.A.; Meagher, J.L.; Bai, L.; Liu, L.; et al. SAR405838: An Optimized Inhibitor of MDM2–p53 Interaction That Induces Complete and Durable Tumor Regression. Cancer Res. 2014, 74, 5855–5865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Liu, L.; Sun, W.; Lu, J.; McEachern, D.; Li, X.; Yu, S.; Bernard, D.; Ochsenbein, P.; Ferey, V.; et al. Diastereomeric Spirooxindoles as Highly Potent and Efficacious MDM2 Inhibitors. J. Am. Chem. Soc. 2013, 135, 7223–7234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, D.; Li, Z.; Rew, Y.; Gribble, M.; Bartberger, M.D.; Beck, H.P.; Canon, J.; Chen, A.; Chen, X.; Chow, D.; et al. Discovery of AMG 232, a Potent, Selective, and Orally Bioavailable MDM2–p53 Inhibitor in Clinical Development. J. Med. Chem. 2014, 57, 1454–1472. [Google Scholar] [CrossRef] [PubMed]
- Ray-Coquard, I.; Blay, J.-Y.; Italiano, A.; Le Cesne, A.; Penel, N.; Zhi, J.; Heil, F.; Rueger, R.; Graves, B.; Ding, M.; et al. Effect of the MDM2 Antagonist RG7112 on the p53 Pathway in Patients with MDM2-Amplified, Well-Differentiated or Dedifferentiated Liposarcoma: An Exploratory Proof-of-Mechanism Study. Lancet Oncol. 2012, 13, 1133–1140. [Google Scholar] [CrossRef]
- Mahfoudhi, E.; Lordier, L.; Marty, C.; Pan, J.; Roy, A.; Roy, L.; Rameau, P.; Abbes, S.; Debili, N.; Raslova, H.; et al. p53 Activation Inhibits All Types of Hematopoietic Progenitors and All Stages of Megakaryopoiesis. Oncotarget 2016, 7, 31980–31992. [Google Scholar] [CrossRef]
- Haronikova, L.; Bonczek, O.; Zatloukalova, P.; Kokas-Zavadil, F.; Kucerikova, M.; Coates, P.J.; Fahraeus, R.; Vojtesek, B. Resistance Mechanisms to Inhibitors of p53-MDM2 Interactions in Cancer Therapy: Can We Overcome Them? Cell Mol. Biol. Lett. 2021, 26, 53. [Google Scholar] [CrossRef] [PubMed]
- Wagner, A.J.; Banerji, U.; Mahipal, A.; Somaiah, N.; Hirsch, H.; Fancourt, C.; Johnson-Levonas, A.O.; Lam, R.; Meister, A.K.; Russo, G.; et al. Phase I Trial of the Human Double Minute 2 Inhibitor MK-8242 in Patients with Advanced Solid Tumors. J. Clin. Oncol. 2017, 35, 1304–1311. [Google Scholar] [CrossRef]
- Efeyan, A.; Ortega-Molina, A.; Velasco-Miguel, S.; Herranz, D.; Vassilev, L.T.; Serrano, M. Induction of p53-Dependent Senescence by the MDM2 Antagonist Nutlin-3a in Mouse Cells of Fibroblast Origin. Cancer Res. 2007, 67, 7350–7357. [Google Scholar] [CrossRef] [Green Version]
- Coppé, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borthakur, G.; Duvvuri, S.; Ruvolo, V.; Tripathi, D.N.; Piya, S.; Burks, J.; Jacamo, R.; Kojima, K.; Ruvolo, P.; Fueyo-Margareto, J.; et al. MDM2 Inhibitor, Nutlin 3a, Induces p53 Dependent Autophagy in Acute Leukemia by AMP Kinase Activation. PLoS ONE 2015, 10, e0139254. [Google Scholar] [CrossRef] [PubMed]
- Pechackova, S.; Burdova, K.; Benada, J.; Kleiblova, P.; Jenikova, G.; Macurek, L. Inhibition of WIP1 Phosphatase Sensitizes Breast Cancer Cells to Genotoxic Stress and to MDM2 Antagonist Nutlin-3. Oncotarget 2016, 7, 14458. [Google Scholar] [CrossRef] [Green Version]
- Puszynski, K.; Gandolfi, A.; d’Onofrio, A. The Pharmacodynamics of the p53-Mdm2 Targeting Drug Nutlin: The Role of Gene-Switching Noise. PLoS Comput. Biol. 2014, 10, e1003991. [Google Scholar] [CrossRef] [Green Version]
- Garcia, D.; Warr, M.R.; Martins, C.P.; Brown Swigart, L.; Passegué, E.; Evan, G.I. Validation of MdmX as a Therapeutic Target for Reactivating p53 in Tumors. Genes Dev. 2011, 25, 1746–1757. [Google Scholar] [CrossRef] [Green Version]
- Yu, D.-H.; Xu, Z.-Y.; Mo, S.; Yuan, L.; Cheng, X.-D.; Qin, J.-J. Targeting MDMX for Cancer Therapy: Rationale, Strategies, and Challenges. Front. Oncol. 2020, 10, 1389. [Google Scholar] [CrossRef]
- Reed, D.; Shen, Y.; Shelat, A.A.; Arnold, L.A.; Ferreira, A.M.; Zhu, F.; Mills, N.; Smithson, D.C.; Regni, C.A.; Bashford, D.; et al. Identification and Characterization of the First Small Molecule Inhibitor of MDMX. J. Biol. Chem. 2010, 285, 10786–10796. [Google Scholar] [CrossRef] [Green Version]
- Karan, G.; Wang, H.; Chakrabarti, A.; Karan, S.; Liu, Z.; Xia, Z.; Gundluru, M.; Moreton, S.; Saunthararajah, Y.; Jackson, M.W.; et al. Identification of a Small Molecule That Overcomes HdmX-Mediated Suppression of p53. Mol. Cancer Ther. 2016, 15, 574–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dewaele, M.; Tabaglio, T.; Willekens, K.; Bezzi, M.; Teo, S.X.; Low, D.H.P.; Koh, C.M.; Rambow, F.; Fiers, M.; Rogiers, A.; et al. Antisense Oligonucleotide-Mediated MDM4 Exon 6 Skipping Impairs Tumor Growth. J. Clin. Investig. 2016, 126, 68–84. [Google Scholar] [CrossRef]
- Bykov, V.J.N.; Issaeva, N.; Shilov, A.; Hultcrantz, M.; Pugacheva, E.; Chumakov, P.; Bergman, J.; Wiman, K.G.; Selivanova, G. Restoration of the Tumor Suppressor Function to Mutant p53 by a Low-Molecular-Weight Compound. Nat. Med. 2002, 8, 282–288. [Google Scholar] [CrossRef]
- Lambert, J.M.R.; Gorzov, P.; Veprintsev, D.B.; Söderqvist, M.; Segerbäck, D.; Bergman, J.; Fersht, A.R.; Hainaut, P.; Wiman, K.G.; Bykov, V.J.N. PRIMA-1 Reactivates Mutant p53 by Covalent Binding to the Core Domain. Cancer Cell 2009, 15, 376–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Degtjarik, O.; Golovenko, D.; Diskin-Posner, Y.; Abrahmsén, L.; Rozenberg, H.; Shakked, Z. Structural Basis of Reactivation of Oncogenic p53 Mutants by a Small Molecule: Methylene Quinuclidinone (MQ). Nat. Commun. 2021, 12, 7057. [Google Scholar] [CrossRef] [PubMed]
- Cluzeau, T.; Sebert, M.; Rahmé, R.; Cuzzubbo, S.; Lehmann-Che, J.; Madelaine, I.; Peterlin, P.; Bève, B.; Attalah, H.; Chermat, F.; et al. Eprenetapopt Plus Azacitidine in TP53-Mutated Myelodysplastic Syndromes and Acute Myeloid Leukemia: A Phase II Study by the Groupe Francophone des Myélodysplasies (GFM). J. Clin. Oncol. 2021, 39, 1575–1583. [Google Scholar] [CrossRef] [PubMed]
- Aprea Therapeutics. Aprea Therapeutics Announces Results of Primary Endpoint from Phase 3 Trial of Eprenetapopt in TP53 Mutant Myelodysplastic Syndromes (MDS); Aprea Therapeutics: Stockholm, Sweden, 2020. [Google Scholar]
- Boeckler, F.M.; Joerger, A.C.; Jaggi, G.; Rutherford, T.J.; Veprintsev, D.B.; Fersht, A.R. Targeted Rescue of a Destabilized Mutant of p53 by an in Silico Screened Drug. Proc. Natl. Acad. Sci. USA 2008, 105, 10360–10365. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Wilcken, R.; Joerger, A.C.; Chuckowree, I.S.; Amin, J.; Spencer, J.; Fersht, A.R. Small Molecule Induced Reactivation of Mutant p53 in Cancer Cells. Nucleic Acids Res. 2013, 41, 6034–6044. [Google Scholar] [CrossRef]
- Hiraki, M.; Hwang, S.-Y.; Cao, S.; Ramadhar, T.R.; Byun, S.; Yoon, K.W.; Lee, J.H.; Chu, K.; Gurkar, A.U.; Kolev, V.; et al. Small-Molecule Reactivation of Mutant p53 to Wild-Type-like p53 through the p53-Hsp40 Regulatory Axis. Chem. Biol. 2015, 22, 1206–1216. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, D.; Liao, W.; Zeng, S.X.; Lu, H. Reviving the Guardian of the Genome: Small Molecule Activators of p53. Pharmacol. Ther. 2017, 178, 92–108. [Google Scholar] [CrossRef]
- Schulz-Heddergott, R.; Stark, N.; Edmunds, S.J.; Li, J.; Conradi, L.-C.; Bohnenberger, H.; Ceteci, F.; Greten, F.R.; Dobbelstein, M.; Moll, U.M. Therapeutic Ablation of Gain-of-Function Mutant p53 in Colorectal Cancer Inhibits Stat3-Mediated Tumor Growth and Invasion. Cancer Cell 2018, 34, 298–314. [Google Scholar] [CrossRef] [Green Version]
- Bossi, G.; Lapi, E.; Strano, S.; Rinaldo, C.; Blandino, G.; Sacchi, A. Mutant p53 Gain of Function: Reduction of Tumor Malignancy of Human Cancer Cell Lines through Abrogation of Mutant p53 Expression. Oncogene 2006, 25, 304–309. [Google Scholar] [CrossRef] [Green Version]
- Yan, W.; Liu, G.; Scoumanne, A.; Chen, X. Suppression of Inhibitor of Differentiation 2, a Target of Mutant p53, Is Required for Gain-of-Function Mutations. Cancer Res. 2008, 68, 6789–6796. [Google Scholar] [CrossRef] [Green Version]
- Alexandrova, E.M.; Yallowitz, A.R.; Li, D.; Xu, S.; Schulz, R.; Proia, D.A.; Lozano, G.; Dobbelstein, M.; Moll, U.M. Improving Survival by Exploiting Tumour Dependence on Stabilized Mutant p53 for Treatment. Nature 2015, 523, 352–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Marchenko, N.D.; Moll, U.M. SAHA Shows Preferential Cytotoxicity in Mutant p53 Cancer Cells by Destabilizing Mutant p53 through Inhibition of the HDAC6-Hsp90 Chaperone Axis. Cell Death Differ. 2011, 18, 1904–1913. [Google Scholar] [CrossRef] [PubMed]
- Pillai, R.N.; Fennell, D.A.; Kovcin, V.; Ciuleanu, T.-E.; Ramlau, R.; Kowalski, D.; Schenker, M.; Yalcin, I.; Teofilovici, F.; Vukovic, V.M.; et al. Randomized Phase III Study of Ganetespib, a Heat Shock Protein 90 Inhibitor, with Docetaxel Versus Docetaxel in Advanced Non–Small-Cell Lung Cancer (GALAXY-2). J. Clin. Oncol. 2019, 38, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Blumenschein, G.R.; Kies, M.S.; Papadimitrakopoulou, V.A.; Lu, C.; Kumar, A.J.; Ricker, J.L.; Chiao, J.H.; Chen, C.; Frankel, S.R. Phase II Trial of the Histone Deacetylase Inhibitor Vorinostat (ZolinzaTM, Suberoylanilide Hydroxamic Acid, SAHA) in Patients with Recurrent and/or Metastatic Head and Neck Cancer. Invest. New Drugs 2008, 26, 81–87. [Google Scholar] [CrossRef]
- Martinez, L.A.; Naguibneva, I.; Lehrmann, H.; Vervisch, A.; Tchénio, T.; Lozano, G.; Harel-Bellan, A. Synthetic Small Inhibiting RNAs: Efficient Tools to Inactivate Oncogenic Mutations and Restore p53 Pathways. Proc. Natl. Acad. Sci. USA 2002, 99, 14849–14854. [Google Scholar] [CrossRef] [Green Version]
- Ubby, I.; Krueger, C.; Rosato, R.; Qian, W.; Chang, J.; Sabapathy, K. Cancer Therapeutic Targeting Using Mutant–p53-Specific SiRNAs. Oncogene 2019, 38, 3415–3427. [Google Scholar] [CrossRef] [Green Version]
- Debbas, M.; White, E. Wild-Type p53 Mediates Apoptosis by E1A, Which Is Inhibited by E1B. Genes Dev. 1993, 7, 546–554. [Google Scholar] [CrossRef] [Green Version]
- Bischoff, J.R.; Kirn, D.H.; Williams, A.; Heise, C.; Horn, S.; Muna, M.; Ng, L.; Nye, J.A.; Sampson-Johannes, A.; Fattaey, A.; et al. An Adenovirus Mutant That Replicates Selectively in p53-Deficient Human Tumor Cells. Science 1996, 274, 373–376. [Google Scholar] [CrossRef]
- Heise, C.; Sampson-Johannes, A.; Williams, A.; Mccormick, F.; Von Hoff, D.D.; Kirn, D.H. ONYX-015, an E1B Gene-Attenuated Adenovirus, Causes Tumor-Specific Cytolysis and Antitumoral Efficacy That Can Be Augmented by Standard Chemotherapeutic Agents. Nat. Med. 1997, 3, 639–645. [Google Scholar] [CrossRef]
- Goodrum, F.D.; Ornelles, D.A. p53 Status Does Not Determine Outcome of E1B 55-Kilodalton Mutant Adenovirus Lytic Infection. J. Virol. 1998, 72, 9479–9490. [Google Scholar] [CrossRef] [Green Version]
- Rothmann, T.; Hengstermann, A.; Whitaker, N.J.; Scheffner, M.; zur Hausen, H. Replication of ONYX-015, a Potential Anticancer Adenovirus, Is Independent of p53 Status in Tumor Cells. J. Virol. 1998, 72, 9470–9478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogulski, K.R.; Freytag, S.O.; Zhang, K.; Gilbert, J.D.; Paielli, D.L.; Kim, J.H.; Heise, C.C.; Kirn, D.H. In Vivo Antitumor Activity of ONYX-015 Is Influenced by p53 Status and Is Augmented by Radiotherapy. Cancer Res. 2000, 60, 1193–1196. [Google Scholar] [PubMed]
- Nemunaitis, J.; Khuri, F.; Ganly, I.; Arseneau, J.; Posner, M.; Vokes, E.; Kuhn, J.; McCarty, T.; Landers, S.; Blackburn, A.; et al. Phase II Trial of Intratumoral Administration of ONYX-015, a Replication-Selective Adenovirus, in Patients with Refractory Head and Neck Cancer. J. Clin. Oncol. 2001, 19, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Kirn, D. Clinical Research Results with Dl1520 (Onyx-015), a Replication-Selective Adenovirus for the Treatment of Cancer: What Have We Learned? Gene Ther. 2001, 8, 89–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reid, T.; Galanis, E.; Abbruzzese, J.; Sze, D.; Wein, L.M.; Andrews, J.; Randlev, B.; Heise, C.; Uprichard, M.; Hatfield, M.; et al. Hepatic Arterial Infusion of a Replication-Selective Oncolytic Adenovirus (Dl1520). Cancer Res. 2002, 62, 6070–6079. [Google Scholar] [PubMed]
- Khuri, F.R.; Nemunaitis, J.; Ganly, I.; Arseneau, J.; Tannock, I.F.; Romel, L.; Gore, M.; Ironside, J.; MacDougall, R.H.; Heise, C.; et al. A Controlled Trial of Intratumoral ONYX-015, a Selectively-Replicating Adenovirus, in Combination with Cisplatin and 5-Fluorouracil in Patients with Recurrent Head and Neck Cancer. Nat. Med. 2000, 6, 879–885. [Google Scholar] [CrossRef]
- Garber, K. China Approves World’s First Oncolytic Virus Therapy for Cancer Treatment. JNCI J. Natl. Cancer Inst. 2006, 98, 298–300. [Google Scholar] [CrossRef] [Green Version]
- Weissmueller, S.; Manchado, E.; Saborowski, M.; Morris, J.P., 4th; Wagenblast, E.; Davis, C.A.; Moon, S.-H.; Pfister, N.T.; Tschaharganeh, D.F.; Kitzing, T.; et al. Mutant p53 Drives Pancreatic Cancer Metastasis through Cell-Autonomous PDGF Receptor β Signaling. Cell 2014, 157, 382–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Agostino, S.; Strano, S.; Emiliozzi, V.; Zerbini, V.; Mottolese, M.; Sacchi, A.; Blandino, G.; Piaggio, G. Gain of Function of Mutant p53: The Mutant p53/NF-Y Protein Complex Reveals an Aberrant Transcriptional Mechanism of Cell Cycle Regulation. Cancer Cell 2006, 10, 191–202. [Google Scholar] [CrossRef] [Green Version]
- Welti, J.; Sharp, A.; Brooks, N.; Yuan, W.; McNair, C.; Chand, S.N.; Pal, A.; Figueiredo, I.; Riisnaes, R.; Gurel, B.; et al. Targeting the P300/CBP Axis in Lethal Prostate Cancer. Cancer Discov. 2021, 11, 1118–1137. [Google Scholar] [CrossRef]
- Lasko, L.M.; Jakob, C.G.; Edalji, R.P.; Qiu, W.; Montgomery, D.; Digiammarino, E.L.; Hansen, T.M.; Risi, R.M.; Frey, R.; Manaves, V.; et al. Discovery of a Selective Catalytic P300/CBP Inhibitor That Targets Lineage-Specific Tumours. Nature 2017, 550, 128–132. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, R.; Li, Z.; Mei, L.; Wan, S.; Ding, H.; Chen, Z.; Xing, J.; Feng, H.; Han, J.; et al. Discovery of Highly Potent, Selective, and Orally Efficacious P300/CBP Histone Acetyltransferases Inhibitors. J. Med. Chem. 2020, 63, 1337–1360. [Google Scholar] [CrossRef] [PubMed]
- Capaci, V.; Mantovani, F.; Del Sal, G. Amplifying Tumor–Stroma Communication: An Emerging Oncogenic Function of Mutant p53. Front. Oncol. 2021, 10, 2869. [Google Scholar] [CrossRef]
- Dong, Z.-Y.; Zhong, W.-Z.; Zhang, X.-C.; Su, J.; Xie, Z.; Liu, S.-Y.; Tu, H.-Y.; Chen, H.-J.; Sun, Y.-L.; Zhou, Q.; et al. Potential Predictive Value of TP53 and KRAS Mutation Status for Response to PD-1 Blockade Immunotherapy in Lung Adenocarcinoma. Clin. Cancer Res. 2017, 23, 3012–3024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, H.; Liu, S.-Y.; Zhou, J.-Y.; Xu, J.-T.; Zhang, H.-K.; Yan, H.-H.; Huan, J.-J.; Dai, P.-P.; Xu, C.-R.; Su, J.; et al. Specific TP53 Subtype as Biomarker for Immune Checkpoint Inhibitors in Lung Adenocarcinoma. eBioMedicine 2020, 60, 102990. [Google Scholar] [CrossRef] [PubMed]
- Asgari, A.; Lesyk, G.; Poitras, E.; Govindasamy, N.; Terry, K.; To, R.; Back, V.; Rudzinski, J.K.; Lewis, J.D.; Jurasz, P. Platelets Stimulate Programmed Death-Ligand 1 Expression by Cancer Cells: Inhibition by Anti-Platelet Drugs. J. Thromb. Haemost. 2021, 19, 2862–2872. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, D.W.; Raturi, A.; Bhandari, P.; Sosnowski, D.; Grin, L.; Wee, P.; Vega, H.; Gyoba, J.; Hejazi, M.; Ablack, J.; et al. Selective Ablation of Solid Tumors Using a p53-Targeted FAST-LNP Gene Therapy. Cancer Res. 2020, 80, 4069. [Google Scholar] [CrossRef]
- Urist, M.; Tanaka, T.; Poyurovsky, M.V.; Prives, C. P73 Induction after DNA Damage Is Regulated by Checkpoint Kinases Chk1 and Chk2. Genes Dev. 2004, 18, 3041–3054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottifredi, V.; Karni-Schmidt, O.; Shieh, S.S.; Prives, C. p53 Down-Regulates CHK1 through P21 and the Retinoblastoma Protein. Mol. Cell Biol. 2001, 21, 1066–1076. [Google Scholar] [CrossRef] [Green Version]
- Sachdeva, M.; Zhu, S.; Wu, F.; Wu, H.; Walia, V.; Kumar, S.; Elble, R.; Watabe, K.; Mo, Y.-Y. p53 Represses C-Myc through Induction of the Tumor Suppressor MiR-145. Proc. Natl. Acad. Sci. USA 2009, 106, 3207–3212. [Google Scholar] [CrossRef] [Green Version]
- Shao, J.; Fujiwara, T.; Kadowaki, Y.; Fukazawa, T.; Waku, T.; Itoshima, T.; Yamatsuji, T.; Nishizaki, M.; Roth, J.A.; Tanaka, N. Overexpression of the Wild-Type p53 Gene Inhibits NF-ΚB Activity and Synergizes with Aspirin to Induce Apoptosis in Human Colon Cancer Cells. Oncogene 2000, 19, 726–736. [Google Scholar] [CrossRef] [PubMed]
- Bos, J.L. Review Ras Oncogenes in Human Cancer: A Review. Cancer Res. 1989, 49, 4682–4689. [Google Scholar] [PubMed]
- Arpaia, E.; Blaser, H.; Quintela-Fandino, M.; Duncan, G.; Leong, H.S.; Ablack, A.; Nambiar, S.C.; Lind, E.F.; Silvester, J.; Fleming, C.K.; et al. The Interaction between Caveolin-1 and Rho-GTPases Promotes Metastasis by Controlling the Expression of Alpha5-Integrin and the Activation of Src, Ras and Erk. Oncogene 2012, 31, 884–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grzes, M.; Oron, M.; Staszczak, Z.; Jaiswar, A.; Nowak-Niezgoda, M.; Walerych, D. A Driver Never Works Alone—Interplay Networks of Mutant p53, MYC, RAS, and Other Universal Oncogenic Drivers in Human Cancer. Cancers 2020, 12, 1532. [Google Scholar] [CrossRef]
- Ho, J.S.L.; Ma, W.; Mao, D.Y.L.; Benchimol, S. p53-Dependent Transcriptional Repression of c-Myc Is Required for G 1 Cell Cycle Arrest. Mol. Cell Biol. 2005, 25, 7423–7431. [Google Scholar] [CrossRef] [Green Version]
- Santoro, A.; Vlachou, T.; Luzi, L.; Melloni, G.; Mazzarella, L.; D’Elia, E.; Aobuli, X.; Pasi, C.E.; Reavie, L.; Bonetti, P.; et al. p53 Loss in Breast Cancer Leads to Myc Activation, Increased Cell Plasticity, and Expression of a Mitotic Signature with Prognostic Value. Cell Rep. 2019, 26, 624–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buganim, Y.; Solomon, H.; Rais, Y.; Kistner, D.; Nachmany, I.; Brait, M.; Madar, S.; Goldstein, I.; Kalo, E.; Adam, N.; et al. p53 Regulates the Ras Circuit to Inhibit the Expression of a Cancer-Related Gene Signature by Various Molecular Pathways. Cancer Res. 2010, 70, 2274–2284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X. Tied up in Loops: Positive and Negative Autoregulation of p53. Cold Spring Harb. Perspect. Biol. 2010, 2, a000984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeimet, A.G.; Marth, C. Why Did p53 Gene Therapy Fail in Ovarian Cancer? Lancet Oncol. 2003, 4, 415–422. [Google Scholar] [CrossRef]
- Bessis, N.; GarciaCozar, F.J.; Boissier, M.-C. Immune Responses to Gene Therapy Vectors: Influence on Vector Function and Effector Mechanisms. Gene Ther. 2004, 11, S10–S17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halbert, C.L.; Rutledge, E.A.; Allen, J.M.; Russell, D.W.; Miller, A.D. Repeat Transduction in the Mouse Lung by Using Adeno-Associated Virus Vectors with Different Serotypes. J. Virol. 2000, 74, 1524–1532. [Google Scholar] [CrossRef]
- Boutin, S.; Monteilhet, V.; Veron, P.; Leborgne, C.; Benveniste, O.; Montus, M.F.; Masurier, C. Prevalence of Serum IgG and Neutralizing Factors against Adeno-Associated Virus (AAV) Types 1, 2, 5, 6, 8, and 9 in the Healthy Population: Implications for Gene Therapy Using AAV Vectors. Hum. Gene Ther. 2010, 21, 704–712. [Google Scholar] [CrossRef] [PubMed]
- Nayak, S.; Herzog, R.W. Progress and Prospects: Immune Responses to Viral Vectors. Gene Ther. 2010, 17, 295–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, V.; Twisk, J.; Kwikkers, K.; Aronica, E.; Brisson, D.; Methot, J.; Petry, H.; Gaudet, D. Immune Responses to Intramuscular Administration of Alipogene Tiparvovec (AAV1-LPL(S447X)) in a Phase II Clinical Trial of Lipoprotein Lipase Deficiency Gene Therapy. Hum. Gene Ther. 2014, 25, 180–188. [Google Scholar] [CrossRef] [Green Version]
- Masat, E.; Pavani, G.; Mingozzi, F. Humoral Immunity to AAV Vectors in Gene Therapy: Challenges and Potential Solutions. Discov. Med. 2013, 15, 379–389. [Google Scholar]
- Mingozzi, F.; Maus, M.V.; Hui, D.J.; Sabatino, D.E.; Murphy, S.L.; Rasko, J.E.J.; Ragni, M.V.; Manno, C.S.; Sommer, J.; Jiang, H.; et al. CD8+ T-Cell Responses to Adeno-Associated Virus Capsid in Humans. Nat. Med. 2007, 13, 419–422. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Kanasty, R.L.; Eltoukhy, A.A.; Vegas, A.J.; Dorkin, J.R.; Anderson, D.G. Non-Viral Vectors for Gene-Based Therapy. Nat. Rev. Genet. 2014, 15, 541–555. [Google Scholar] [CrossRef]
- Akinc, A.; Maier, M.A.; Manoharan, M.; Fitzgerald, K.; Jayaraman, M.; Barros, S.; Ansell, S.; Du, X.; Hope, M.J.; Madden, T.D.; et al. The Onpattro Story and the Clinical Translation of Nanomedicines Containing Nucleic Acid-Based Drugs. Nat. Nanotechnol. 2019, 14, 1084–1087. [Google Scholar] [CrossRef] [PubMed]
- Wan, C.; Allen, T.M.; Cullis, P.R. Lipid Nanoparticle Delivery Systems for SiRNA-Based Therapeutics. Drug Deliv. Transl. Res. 2014, 4, 74–83. [Google Scholar] [CrossRef]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Pérez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 MRNA Covid-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef] [PubMed]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the MRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Dumont, A.; Lohard, S.; Maillet, L.; Juin, P.P.; Barillé-Nion, S. NOXA the BCL-2 Family Member behind the Scenes in Cancer Treatment. J. Cell. Signal. 2020, 1, 127. [Google Scholar]
- Montaño-Samaniego, M.; Bravo-Estupiñan, D.M.; Méndez-Guerrero, O.; Alarcón-Hernández, E.; Ibáñez-Hernández, M. Strategies for Targeting Gene Therapy in Cancer Cells with Tumor-Specific Promoters. Front. Oncol. 2020, 10, 2671. [Google Scholar] [CrossRef]
- Liu, J.; Fu, M.; Wang, M.; Wan, D.; Wei, Y.; Wei, X. Cancer Vaccines as Promising Immuno-Therapeutics: Platforms and Current Progress. J. Hematol. Oncol. 2022, 15, 28. [Google Scholar] [CrossRef]
- Otto, T.; Sicinski, P. Cell Cycle Proteins as Promising Targets in Cancer Therapy. Nat. Rev. Cancer 2017, 17, 93–115. [Google Scholar] [CrossRef] [Green Version]
- Ayoub, N.M. Editorial: Novel Combination Therapies for the Treatment of Solid Cancers. Front. Oncol. 2021, 11, 2377. [Google Scholar] [CrossRef]
- Malone, E.R.; Oliva, M.; Sabatini, P.J.B.; Stockley, T.L.; Siu, L.L. Molecular Profiling for Precision Cancer Therapies. Genome Med. 2020, 12, 8. [Google Scholar] [CrossRef] [Green Version]
- Döhner, H.; Wei, A.H.; Löwenberg, B. Towards Precision Medicine for AML. Nat. Rev. Clin. Oncol. 2021, 18, 577–590. [Google Scholar] [CrossRef]
- Di Nicolantonio, F.; Vitiello, P.P.; Marsoni, S.; Siena, S.; Tabernero, J.; Trusolino, L.; Bernards, R.; Bardelli, A. Precision Oncology in Metastatic Colorectal Cancer—From Biology to Medicine. Nat. Rev. Clin. Oncol. 2021, 18, 506–525. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| MDM2 Inhibitors | |||
|---|---|---|---|
| Compound | Indication | Phase | Clinical Trial Identifier |
| RG7112 | Hematological neoplasms | I | NCT00623870 |
| Acute Myeloid Leukemia | Ib/II | NCT03850535 | |
| RG7388 | Acute Myeloid Leukemia | III | NCT02545283 |
| Solid Tumors | I | NCT00559533 | |
| Multiple Myeloma | I/II | NCT02633059 | |
| Follicular Lymphoma and Large B-Cell Lymphoma | Ib/II | NCT03135262 | |
| Non-Hodgkin’s Lymphoma | I/II | NCT02624986 | |
| Acute Myeloid Leukemia | I | NCT02670044 | |
| Breast Cancer (Stage IV, Estrogen Receptor +) | I/II | NCT03566485 | |
| MK-8242 | Acute Myeloid Leukemia | I | NCT01451437 |
| Solid Tumors | I | NCT01463696 | |
| AMG-232 | Solid Tumors or Multiple Myeloma | I | NCT01723020 |
| Acute Myeloid Leukemia | I | NCT02016729 | |
| Metastatic Melanoma | Ib/IIa | NCT02110355 | |
| Multiple Myeloma | I | NCT03031730 | |
| Soft Tissue Sarcoma | Ib | NCT03217266 | |
| Acute Myeloid Leukemia | Ib | NCT04190550 | |
| MDM2/MDMX Inhibitor | |||
| ALRN-6924 | Hematological neoplasms and Small Tumors | I | NCT03654716 |
| Metastatic Solid Tumors | I | NCT03725436 | |
| Solid Tumors or Lymphoma | I/IIa | NCT02264613 | |
| Mutant p53 Reactivators | |||
| APR-246 | Acute Myeloid Leukemia or Myelodysplastic Syndromes | II | NCT03931291 |
| Myeloid Malignancy | I | NCT04214860 | |
| Solid Tumors | I/II | NCT04383938 | |
| Esophageal Cancer | Ib/II | NCT02999893 | |
| Non-Hodgkin’s Lymphoma, Mantle Cell Lymphoma, Chronic Lymphocytic Leukemia | I/II | NCT04419389 | |
| Myeloid Malignancy | Ib/II | NCT03588078 | |
| Myelodysplastic Syndromes | III | NCT03745716 | |
| High Grade Serous Ovarian Cancer | II | NCT03268382 | |
| Melanoma (BRAF/V600) | Ib/II | NCT03391050 | |
| Myeloid Malignancy | Ib/II | NCT03072043 | |
| High Grade Serous Ovarian Cancer | Ib/II | NCT02098343 | |
| Hematological neoplasms and Prostate Cancer | I | NCT00900614 | |
| Ganetespib | Metastatic Ovarian Cancer (Mutant p53) | I/II | NCT02012192 |
| SAHA | Advanced Cancer (Mutant p53) | I | NCT02042989 |
| Metastatic Melanoma | II | NCT00121225 | |
| Advanced Cancer | I | NCT00324480 | |
| p53 Gene Therapy | |||
| Ad-p53 + Immune Checkpoint Inhibitor | Solid Tumors or Lymphoma | II | NCT03544723 |
| Head and Neck Cancer or Metastatic Solid Tumors | I/II | NCT02842125 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brown, D.W.; Beatty, P.H.; Lewis, J.D. Molecular Targeting of the Most Functionally Complex Gene in Precision Oncology: p53. Cancers 2022, 14, 5176. https://doi.org/10.3390/cancers14215176
Brown DW, Beatty PH, Lewis JD. Molecular Targeting of the Most Functionally Complex Gene in Precision Oncology: p53. Cancers. 2022; 14(21):5176. https://doi.org/10.3390/cancers14215176
Chicago/Turabian StyleBrown, Douglas W., Perrin H. Beatty, and John D. Lewis. 2022. "Molecular Targeting of the Most Functionally Complex Gene in Precision Oncology: p53" Cancers 14, no. 21: 5176. https://doi.org/10.3390/cancers14215176
APA StyleBrown, D. W., Beatty, P. H., & Lewis, J. D. (2022). Molecular Targeting of the Most Functionally Complex Gene in Precision Oncology: p53. Cancers, 14(21), 5176. https://doi.org/10.3390/cancers14215176