
Metabolic Pathways, Enzymes, and Metabolites: Opportunities in Cancer Therapy

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
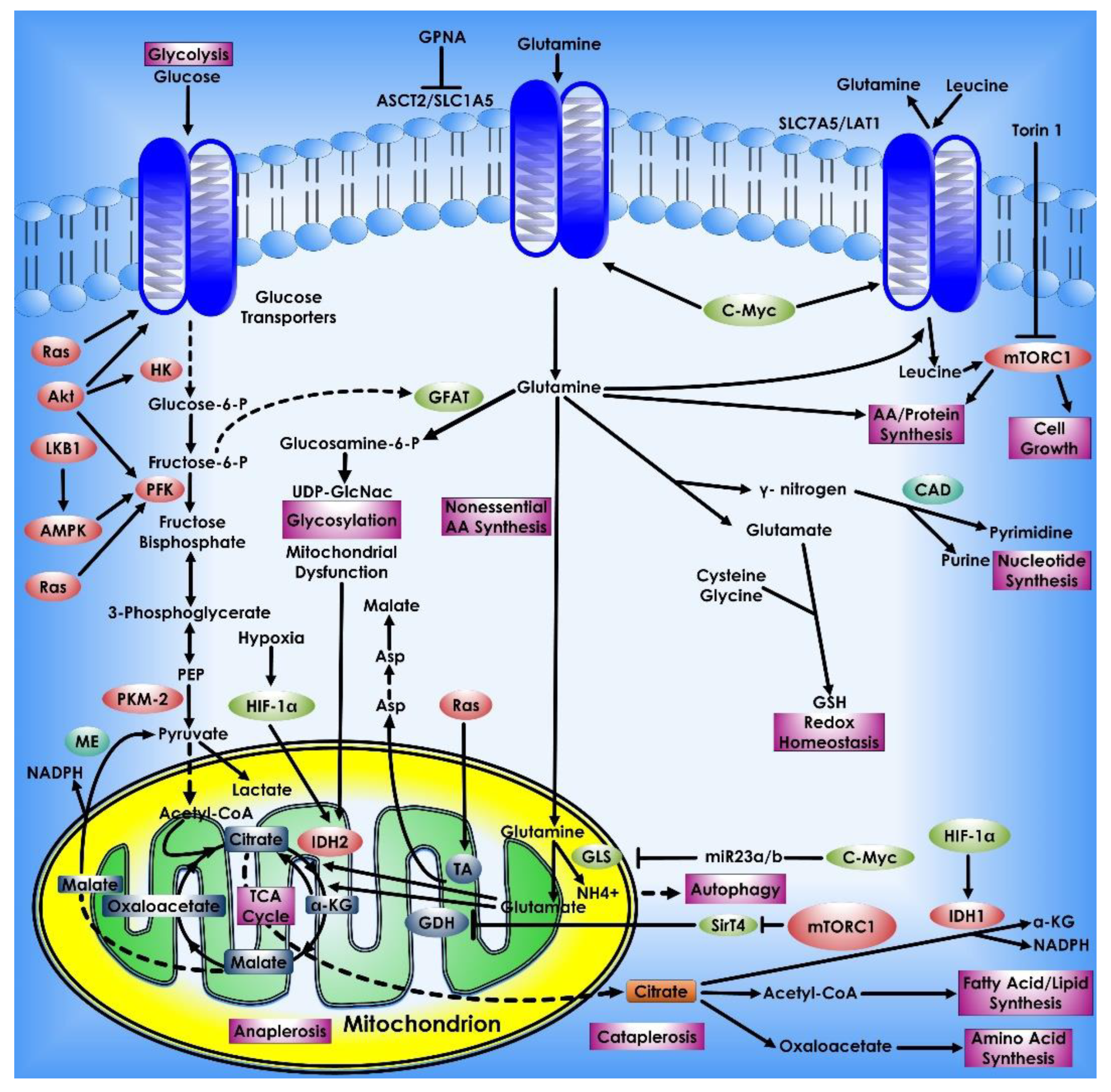
2. Glucose Metabolism in Cancer Cells
3. Role of Oncogenes in Cancer Metabolism
4. Role of Tumour Suppressor Genes and miRNAs in Cancer Metabolism
5. c-Myc and Cancer Metabolism
6. Fatty Acid Metabolism in Cancer Cells
7. Enzyme of the Branched Chain Amino Acid Metabolism in Cancer Cells
8. Glutamine Metabolism in Cancer Cells
9. Targeting Cancer Metabolism for Therapeutic Purposes
10. Conclusions and Challenges
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhang, W.; Zhang, S.-L.; Hu, X.; Tam, K.Y. Targeting Tumor Metabolism for Cancer Treatment: Is Pyruvate Dehydrogenase Kinases (PDKs) a Viable Anticancer Target? Int. J. Biol. Sci. 2015, 11, 1390–1400. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O. The Metabolism of Carcinoma Cells1. J. Cancer Res. 1925, 9, 148–163. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chae, Y.C.; Kim, J.H. Cancer Stem Cell Metabolism: Target for Cancer Therapy. BMB Rep. 2018, 51, 319–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Outschoorn, U.E.; Peiris-Pagés, M.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer Metabolism: A Therapeutic Perspective. Nat. Rev. Clin. Oncol. 2017, 14, 11–31. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, J.; Lin, C.; Liu, W.; Huo, Y.; Yang, M.; Jiang, S.-H.; Sun, Y.; Hua, R. Endoplasmic Reticulum Stress-Dependent Expression of ERO1L Promotes Aerobic Glycolysis in Pancreatic Cancer. Theranostics 2020, 10, 8400–8414. [Google Scholar] [CrossRef] [PubMed]
- Domiński, A.; Krawczyk, M.; Konieczny, T.; Kasprów, M.; Foryś, A.; Pastuch-Gawołek, G.; Kurcok, P. Biodegradable PH-Responsive Micelles Loaded with 8-Hydroxyquinoline Glycoconjugates for Warburg Effect Based Tumor Targeting. Eur. J. Pharm. Biopharm. 2020, 154, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Boroughs, L.K.; DeBerardinis, R.J. Metabolic Pathways Promoting Cancer Cell Survival and Growth. Nat. Cell Biol. 2015, 17, 351–359. [Google Scholar] [CrossRef] [Green Version]
- Hurley, H.J.; Dewald, H.; Rothkopf, Z.S.; Singh, S.; Jenkins, F.; Deb, P.; De, S.; Barnes, B.J.; Fitzgerald-Bocarsly, P. Frontline Science: AMPK Regulates Metabolic Reprogramming Necessary for Interferon Production in Human Plasmacytoid Dendritic Cells. J. Leukoc. Biol. 2021, 109, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Guerra, L.; Bonetti, L.; Brenner, D. Metabolic Modulation of Immunity: A New Concept in Cancer Immunotherapy. Cell Rep. 2020, 32, 107848. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, A.; Deshpande, K.; Arfuso, F.; Newsholme, P.; Dharmarajan, A. Cancer Stem Cell Metabolism: A Potential Target for Cancer Therapy. Mol. Cancer 2016, 15, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villalba, M.; Rathore, M.G.; Lopez-Royuela, N.; Krzywinska, E.; Garaude, J.; Allende-Vega, N. From Tumor Cell Metabolism to Tumor Immune Escape. Int. J. Biochem. Cell Biol. 2013, 45, 106–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; DeBerardinis, R.J. Mechanisms and Implications of Metabolic Heterogeneity in Cancer. Cell Metab. 2019, 30, 434–446. [Google Scholar] [CrossRef] [PubMed]
- Brahimi-Horn, M.C.; Chiche, J.; Pouysségur, J. Hypoxia Signalling Controls Metabolic Demand. Curr. Opin. Cell Biol. 2007, 19, 223–229. [Google Scholar] [CrossRef]
- Casazza, A.; Di Conza, G.; Wenes, M.; Finisguerra, V.; Deschoemaeker, S.; Mazzone, M. Tumor Stroma: A Complexity Dictated by the Hypoxic Tumor Microenvironment. Oncogene 2014, 33, 1743–1754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pouysségur, J.; Mechta-Grigoriou, F. Redox Regulation of the Hypoxia-Inducible Factor. Biol. Chem. 2006, 387, 1337–1346. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Pavlova, N.N.; Thompson, C.B. Cancer Cell Metabolism: The Essential Role of the Nonessential Amino Acid, Glutamine. EMBO J. 2017, 36, 1302–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patra, K.C.; Hay, N. The Pentose Phosphate Pathway and Cancer. Trends Biochem. Sci. 2014, 39, 347–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeon, S.-M.; Chandel, N.S.; Hay, N. AMPK Regulates NADPH Homeostasis to Promote Tumour Cell Survival during Energy Stress. Nature 2012, 485, 661–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alfarouk, K.O.; Ahmed, S.B.M.; Elliott, R.L.; Benoit, A.; Alqahtani, S.S.; Ibrahim, M.E.; Bashir, A.H.H.; Alhoufie, S.T.S.; Elhassan, G.O.; Wales, C.C.; et al. The Pentose Phosphate Pathway Dynamics in Cancer and Its Dependency on Intracellular PH. Metabolites 2020, 10, 285. [Google Scholar] [CrossRef] [PubMed]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of Cancer Cell Metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iurlaro, R.; León-Annicchiarico, C.L.; Muñoz-Pinedo, C. Regulation of Cancer Metabolism by Oncogenes and Tumor Suppressors. Methods Enzymol. 2014, 542, 59–80. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, A.; Malvi, P.; Wajapeyee, N. Oncogene-Directed Alterations in Cancer Cell Metabolism. Trends Cancer 2016, 2, 365–377. [Google Scholar] [CrossRef] [Green Version]
- Kerr, E.M.; Martins, C.P. Metabolic Rewiring in Mutant Kras Lung Cancer. FEBS J. 2018, 285, 28–41. [Google Scholar] [CrossRef] [Green Version]
- Pusch, O.; Soucek, T.; Hengstschläger-Ottnad, E.; Bernaschek, G.; Hengstschläger, M. Cellular Targets for Activation by C-Myc Include the DNA Metabolism Enzyme Thymidine Kinase. DNA Cell Biol. 1997, 16, 737–747. [Google Scholar] [CrossRef]
- Li, F.; Wang, Y.; Zeller, K.I.; Potter, J.J.; Wonsey, D.R.; O’Donnell, K.A.; Kim, J.-W.; Yustein, J.T.; Lee, L.A.; Dang, C.V. Myc Stimulates Nuclearly Encoded Mitochondrial Genes and Mitochondrial Biogenesis. Mol. Cell Biol. 2005, 25, 6225–6234. [Google Scholar] [CrossRef] [Green Version]
- Mannava, S.; Grachtchouk, V.; Wheeler, L.J.; Im, M.; Zhuang, D.; Slavina, E.G.; Mathews, C.K.; Shewach, D.S.; Nikiforov, M.A. Direct Role of Nucleotide Metabolism in C-MYC-Dependent Proliferation of Melanoma Cells. Cell Cycle 2008, 7, 2392–2400. [Google Scholar] [CrossRef]
- Liu, W.; Le, A.; Hancock, C.; Lane, A.N.; Dang, C.V.; Fan, T.W.-M.; Phang, J.M. Reprogramming of Proline and Glutamine Metabolism Contributes to the Proliferative and Metabolic Responses Regulated by Oncogenic Transcription Factor C-MYC. Proc. Natl. Acad. Sci. USA 2012, 109, 8983–8988. [Google Scholar] [CrossRef] [Green Version]
- Edmunds, L.R.; Sharma, L.; Kang, A.; Lu, J.; Vockley, J.; Basu, S.; Uppala, R.; Goetzman, E.S.; Beck, M.E.; Scott, D.; et al. C-Myc Programs Fatty Acid Metabolism and Dictates Acetyl-CoA Abundance and Fate. J. Biol. Chem. 2014, 289, 25382–25392. [Google Scholar] [CrossRef] [Green Version]
- Wellen, K.E.; Lu, C.; Mancuso, A.; Lemons, J.M.S.; Ryczko, M.; Dennis, J.W.; Rabinowitz, J.D.; Coller, H.A.; Thompson, C.B. The Hexosamine Biosynthetic Pathway Couples Growth Factor-Induced Glutamine Uptake to Glucose Metabolism. Genes Dev. 2010, 24, 2784–2799. [Google Scholar] [CrossRef]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic Glycolysis: Meeting the Metabolic Requirements of Cell Proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeiffer, T.; Schuster, S.; Bonhoeffer, S. Cooperation and Competition in the Evolution of ATP-Producing Pathways. Science 2001, 292, 504–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroemer, G.; Pouyssegur, J. Tumor Cell Metabolism: Cancer’s Achilles’ Heel. Cancer Cell 2008, 13, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.V.; Herst, P.M.; Tan, A.S. Metabolic Flexibility and Cell Hierarchy in Metastatic Cancer. Mitochondrion 2010, 10, 584–588. [Google Scholar] [CrossRef]
- Fantin, V.R.; St-Pierre, J.; Leder, P. Attenuation of LDH-A Expression Uncovers a Link between Glycolysis, Mitochondrial Physiology, and Tumor Maintenance. Cancer Cell 2006, 9, 425–434. [Google Scholar] [CrossRef] [Green Version]
- Gatenby, R.A.; Gillies, R.J. A Microenvironmental Model of Carcinogenesis. Nat. Rev. Cancer 2008, 8, 56–61. [Google Scholar] [CrossRef]
- Vaupel, P. Metabolic Microenvironment of Tumor Cells: A Key Factor in Malignant Progression. Exp. Oncol. 2010, 32, 125–127. [Google Scholar]
- Chen, J.L.-Y.; Lucas, J.E.; Schroeder, T.; Mori, S.; Wu, J.; Nevins, J.; Dewhirst, M.; West, M.; Chi, J.-T. The Genomic Analysis of Lactic Acidosis and Acidosis Response in Human Cancers. PLoS Genet. 2008, 4, e1000293. [Google Scholar] [CrossRef] [Green Version]
- Denko, N.C. Hypoxia, HIF1 and Glucose Metabolism in the Solid Tumour. Nat. Rev. Cancer 2008, 8, 705–713. [Google Scholar] [CrossRef]
- Andreucci, E.; Peppicelli, S.; Ruzzolini, J.; Bianchini, F.; Biagioni, A.; Papucci, L.; Magnelli, L.; Mazzanti, B.; Stecca, B.; Calorini, L. The Acidic Tumor Microenvironment Drives a Stem-like Phenotype in Melanoma Cells. J. Mol. Med. 2020, 98, 1431–1446. [Google Scholar] [CrossRef]
- Nogueira, V.; Park, Y.; Chen, C.-C.; Xu, P.-Z.; Chen, M.-L.; Tonic, I.; Unterman, T.; Hay, N. Akt Determines Replicative Senescence and Oxidative or Oncogenic Premature Senescence and Sensitizes Cells to Oxidative Apoptosis. Cancer Cell 2008, 14, 458–470. [Google Scholar] [CrossRef] [PubMed]
- Chandra, D.; Singh, K.K. Genetic Insights into OXPHOS Defect and Its Role in Cancer. Biochim. Biophys. Acta 2011, 1807, 620–625. [Google Scholar] [CrossRef] [Green Version]
- Owens, K.M.; Kulawiec, M.; Desouki, M.M.; Vanniarajan, A.; Singh, K.K. Impaired OXPHOS Complex III in Breast Cancer. PLoS ONE 2011, 6, e23846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno-Sánchez, R.; Rodríguez-Enríquez, S.; Marín-Hernández, A.; Saavedra, E. Energy Metabolism in Tumor Cells. FEBS J. 2007, 274, 1393–1418. [Google Scholar] [CrossRef] [PubMed]
- López-Ríos, F.; Sánchez-Aragó, M.; García-García, E.; Ortega, A.D.; Berrendero, J.R.; Pozo-Rodríguez, F.; López-Encuentra, A.; Ballestín, C.; Cuezva, J.M. Loss of the Mitochondrial Bioenergetic Capacity Underlies the Glucose Avidity of Carcinomas. Cancer Res. 2007, 67, 9013–9017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anastasiou, D.; Poulogiannis, G.; Asara, J.M.; Boxer, M.B.; Jiang, J.; Shen, M.; Bellinger, G.; Sasaki, A.T.; Locasale, J.W.; Auld, D.S.; et al. Inhibition of Pyruvate Kinase M2 by Reactive Oxygen Species Contributes to Cellular Antioxidant Responses. Science 2011, 334, 1278–1283. [Google Scholar] [CrossRef] [Green Version]
- Hamanaka, R.B.; Chandel, N.S. Cell Biology. Warburg Effect and Redox Balance. Science 2011, 334, 1219–1220. [Google Scholar] [CrossRef]
- Patra, K.C.; Hay, N. Hexokinase 2 as Oncotarget. Oncotarget 2013, 4, 1862–1863. [Google Scholar] [CrossRef]
- Shinohara, Y.; Yamamoto, K.; Kogure, K.; Ichihara, J.; Terada, H. Steady State Transcript Levels of the Type II Hexokinase and Type 1 Glucose Transporter in Human Tumor Cell Lines. Cancer Lett. 1994, 82, 27–32. [Google Scholar] [CrossRef]
- Mathupala, S.P.; Rempel, A.; Pedersen, P.L. Glucose Catabolism in Cancer Cells: Identification and Characterization of a Marked Activation Response of the Type II Hexokinase Gene to Hypoxic Conditions. J. Biol. Chem. 2001, 276, 43407–43412. [Google Scholar] [CrossRef] [Green Version]
- Israelsen, W.J.; Vander Heiden, M.G. Pyruvate Kinase: Function, Regulation and Role in Cancer. Semin. Cell Dev. Biol. 2015, 43, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Valvona, C.J.; Fillmore, H.L.; Nunn, P.B.; Pilkington, G.J. The Regulation and Function of Lactate Dehydrogenase A: Therapeutic Potential in Brain Tumor. Brain Pathol. 2015, 26, 3–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doherty, J.R.; Cleveland, J.L. Targeting Lactate Metabolism for Cancer Therapeutics. J. Clin. Investig. 2013, 123, 3685–3692. [Google Scholar] [CrossRef] [PubMed]
- Feron, O. Pyruvate into Lactate and Back: From the Warburg Effect to Symbiotic Energy Fuel Exchange in Cancer Cells. Radiother. Oncol. 2009, 92, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Hirschhaeuser, F.; Sattler, U.G.A.; Mueller-Klieser, W. Lactate: A Metabolic Key Player in Cancer. Cancer Res. 2011, 71, 6921–6925. [Google Scholar] [CrossRef] [Green Version]
- Hay, N. Reprogramming Glucose Metabolism in Cancer: Can It Be Exploited for Cancer Therapy? Nat. Rev. Cancer 2016, 16, 635–649. [Google Scholar] [CrossRef] [Green Version]
- Gao, P.; Sun, L.; He, X.; Cao, Y.; Zhang, H. MicroRNAs and the Warburg Effect: New Players in an Old Arena. Curr. Gene Ther. 2012, 12, 285–291. [Google Scholar] [CrossRef]
- Mikawa, T.; LLeonart, M.E.; Takaori-Kondo, A.; Inagaki, N.; Yokode, M.; Kondoh, H. Dysregulated Glycolysis as an Oncogenic Event. Cell Mol. Life Sci. 2015, 72, 1881–1892. [Google Scholar] [CrossRef] [PubMed]
- Kress, S.; Stein, A.; Maurer, P.; Weber, B.; Reichert, J.; Buchmann, A.; Huppert, P.; Schwarz, M. Expression of Hypoxia-Inducible Genes in Tumor Cells. J. Cancer Res. Clin. Oncol. 1998, 124, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Li, C.; Zhang, H. Hypoxia and Cancer Cell Metabolism. Acta Biochim. Biophys. Sin. 2014, 46, 214–219. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Gong, H.-J.; Xiao, H. Somatic TP53 mutations and comparison of different TP53 functional domains in human cancers: Data analysis from the IARC TP53 database and the National Cancer Institute GDC data portal. Med. Data Min. 2021, 4, 3. [Google Scholar]
- Malhotra, L.; Singh, A.; Kaur, P.; Ethayathulla, A.S. Comprehensive Omics Studies of P53 Mutants in Human Cancer. Brief. Funct. Genom. 2022, elac015. [Google Scholar] [CrossRef] [PubMed]
- Zwaans, B.M.M.; Lombard, D.B. Interplay between Sirtuins, MYC and Hypoxia-Inducible Factor in Cancer-Associated Metabolic Reprogramming. Dis. Model. Mech. 2014, 7, 1023–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallows, W.C.; Yu, W.; Denu, J.M. Regulation of Glycolytic Enzyme Phosphoglycerate Mutase-1 by Sirt1 Protein-Mediated Deacetylation. J. Biol. Chem. 2012, 287, 3850–3858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsusaka, T.; Guo, T.; Yagura, T.; Inoue, T.; Yokode, M.; Inagaki, N.; Kondoh, H. Deacetylation of Phosphoglycerate Mutase in Its Distinct Central Region by SIRT2 Down-Regulates Its Enzymatic Activity. Genes Cells 2014, 19, 766–777. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Li, F.; Lv, L.; Li, T.; Zhou, X.; Deng, C.-X.; Guan, K.-L.; Lei, Q.-Y.; Xiong, Y. Oxidative Stress Activates SIRT2 to Deacetylate and Stimulate Phosphoglycerate Mutase. Cancer Res. 2014, 74, 3630–3642. [Google Scholar] [CrossRef] [Green Version]
- Houtkooper, R.H.; Pirinen, E.; Auwerx, J. Sirtuins as Regulators of Metabolism and Healthspan. Nat. Rev. Mol. Cell Biol. 2012, 13, 225–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; He, X.; Cao, Y.; Gao, P.; Zhang, H. MicroRNAs and Energy Metabolism in Cancer Cells. In MicroRNAs: Key Regulators of Oncogenesis; Babashah, S., Ed.; Springer International Publishing: Cham, Switzerland, 2014; pp. 83–95. ISBN 978-3-319-03725-7. [Google Scholar]
- Hung, C.-L.; Wang, L.-Y.; Yu, Y.-L.; Chen, H.-W.; Srivastava, S.; Petrovics, G.; Kung, H.-J. A Long Noncoding RNA Connects C-Myc to Tumor Metabolism. Proc. Natl. Acad. Sci. USA 2014, 111, 18697–18702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, L.; Thomson, J.M.; Hemann, M.T.; Hernando-Monge, E.; Mu, D.; Goodson, S.; Powers, S.; Cordon-Cardo, C.; Lowe, S.W.; Hannon, G.J.; et al. A MicroRNA Polycistron as a Potential Human Oncogene. Nature 2005, 435, 828–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diosdado, B.; van de Wiel, M.A.; Terhaar Sive Droste, J.S.; Mongera, S.; Postma, C.; Meijerink, W.J.H.J.; Carvalho, B.; Meijer, G.A. MiR-17-92 Cluster Is Associated with 13q Gain and c-Myc Expression during Colorectal Adenoma to Adenocarcinoma Progression. Br. J. Cancer 2009, 101, 707–714. [Google Scholar] [CrossRef]
- Mu, P.; Han, Y.-C.; Betel, D.; Yao, E.; Squatrito, M.; Ogrodowski, P.; de Stanchina, E.; D’Andrea, A.; Sander, C.; Ventura, A. Genetic Dissection of the MiR-17~92 Cluster of MicroRNAs in Myc-Induced B-Cell Lymphomas. Genes Dev. 2009, 23, 2806–2811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garofalo, M.; Di Leva, G.; Romano, G.; Nuovo, G.; Suh, S.-S.; Ngankeu, A.; Taccioli, C.; Pichiorri, F.; Alder, H.; Secchiero, P.; et al. MiR-221&222 Regulate TRAIL Resistance and Enhance Tumorigenicity through PTEN and TIMP3 Downregulation. Cancer Cell 2009, 16, 498–509. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Chan, C.S.; Wu, R.; Zhang, C.; Sun, Y.; Song, J.S.; Tang, L.H.; Levine, A.J.; Feng, Z. Negative Regulation of Tumor Suppressor P53 by MicroRNA MiR-504. Mol. Cell 2010, 38, 689–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, M.T.N.; Teh, C.; Shyh-Chang, N.; Xie, H.; Zhou, B.; Korzh, V.; Lodish, H.F.; Lim, B. MicroRNA-125b Is a Novel Negative Regulator of P53. Genes Dev. 2009, 23, 862–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, B.C.; Shim, H.; Li, Q.; Wu, C.S.; Lee, L.A.; Maity, A.; Dang, C.V. Identification of Putative C-Myc-Responsive Genes: Characterization of Rcl, a Novel Growth-Related Gene. Mol. Cell Biol. 1997, 17, 4967–4978. [Google Scholar] [CrossRef] [Green Version]
- Ramanathan, A.; Wang, C.; Schreiber, S.L. Perturbational Profiling of a Cell-Line Model of Tumorigenesis by Using Metabolic Measurements. Proc. Natl. Acad. Sci. USA 2005, 102, 5992–5997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, C.V.; Le, A.; Gao, P. MYC-Induced Cancer Cell Energy Metabolism and Therapeutic Opportunities. Clin. Cancer Res. 2009, 15, 6479–6483. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Gao, P.; Liu, Y.-C.; Semenza, G.L.; Dang, C.V. Hypoxia-Inducible Factor 1 and Dysregulated c-Myc Cooperatively Induce Vascular Endothelial Growth Factor and Metabolic Switches Hexokinase 2 and Pyruvate Dehydrogenase Kinase 1. Mol. Cell Biol. 2007, 27, 7381–7393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osthus, R.C.; Shim, H.; Kim, S.; Li, Q.; Reddy, R.; Mukherjee, M.; Xu, Y.; Wonsey, D.; Lee, L.A.; Dang, C.V. Deregulation of Glucose Transporter 1 and Glycolytic Gene Expression by C-Myc. J. Biol. Chem. 2000, 275, 21797–21800. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-Mediated Expression of Pyruvate Dehydrogenase Kinase: A Metabolic Switch Required for Cellular Adaptation to Hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, A.; Miller, D.M. Structural Analysis of Alpha-Enolase. Mapping the Functional Domains Involved in down-Regulation of the c-Myc Protooncogene. J. Biol. Chem. 2000, 275, 5958–5965. [Google Scholar] [CrossRef] [Green Version]
- Sedoris, K.C.; Thomas, S.D.; Miller, D.M. Hypoxia Induces Differential Translation of Enolase/MBP-1. BMC Cancer 2010, 10, 157. [Google Scholar] [CrossRef] [PubMed]
- Valera, A.; Pujol, A.; Gregori, X.; Riu, E.; Visa, J.; Bosch, F. Evidence from Transgenic Mice That Myc Regulates Hepatic Glycolysis. FASEB J. 1995, 9, 1067–1078. [Google Scholar] [CrossRef] [PubMed]
- Menssen, A.; Hermeking, H. Characterization of the C-MYC-Regulated Transcriptome by SAGE: Identification and Analysis of c-MYC Target Genes. Proc. Natl. Acad. Sci. USA 2002, 99, 6274–6279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coller, H.A.; Grandori, C.; Tamayo, P.; Colbert, T.; Lander, E.S.; Eisenman, R.N.; Golub, T.R. Expression Analysis with Oligonucleotide Microarrays Reveals That MYC Regulates Genes Involved in Growth, Cell Cycle, Signaling, and Adhesion. Proc. Natl. Acad. Sci. USA 2000, 97, 3260–3265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Q.M.; Malek, R.L.; Kim, S.; Chiao, C.; He, M.; Ruffy, M.; Sanka, K.; Lee, N.H.; Dang, C.V.; Liu, E.T. Identification of C-Myc Responsive Genes Using Rat CDNA Microarray. Cancer Res. 2000, 60, 5922–5928. [Google Scholar] [PubMed]
- Fernandez, P.C.; Frank, S.R.; Wang, L.; Schroeder, M.; Liu, S.; Greene, J.; Cocito, A.; Amati, B. Genomic Targets of the Human C-Myc Protein. Genes Dev. 2003, 17, 1115–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrish, F.; Noonan, J.; Perez-Olsen, C.; Gafken, P.R.; Fitzgibbon, M.; Kelleher, J.; VanGilst, M.; Hockenbery, D. Myc-Dependent Mitochondrial Generation of Acetyl-CoA Contributes to Fatty Acid Biosynthesis and Histone Acetylation during Cell Cycle Entry. J. Biol. Chem. 2010, 285, 36267–36274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrish, F.; Hockenbery, D. Myc’s Mastery of Mitochondrial Mischief. Cell Cycle 2003, 2, 11–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Donnell, K.A.; Wentzel, E.A.; Zeller, K.I.; Dang, C.V.; Mendell, J.T. C-Myc-Regulated MicroRNAs Modulate E2F1 Expression. Nature 2005, 435, 839–843. [Google Scholar] [CrossRef]
- Yuneva, M.; Zamboni, N.; Oefner, P.; Sachidanandam, R.; Lazebnik, Y. Deficiency in Glutamine but Not Glucose Induces MYC-Dependent Apoptosis in Human Cells. J. Cell Biol. 2007, 178, 93–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swierczynski, J.; Hebanowska, A.; Sledzinski, T. Role of Abnormal Lipid Metabolism in Development, Progression, Diagnosis and Therapy of Pancreatic Cancer. World J. Gastroenterol. 2014, 20, 2279–2303. [Google Scholar] [CrossRef] [PubMed]
- Catalina-Rodriguez, O.; Kolukula, V.K.; Tomita, Y.; Preet, A.; Palmieri, F.; Wellstein, A.; Byers, S.; Giaccia, A.J.; Glasgow, E.; Albanese, C.; et al. The Mitochondrial Citrate Transporter, CIC, Is Essential for Mitochondrial Homeostasis. Oncotarget 2012, 3, 1220–1235. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Bollu, L.R.; Tozzi, F.; Ye, X.; Bhattacharya, R.; Gao, G.; Dupre, E.; Xia, L.; Lu, J.; Fan, F.; et al. ATP Citrate Lyase Mediates Resistance of Colorectal Cancer Cells to SN38. Mol. Cancer Ther. 2013, 12, 2782–2791. [Google Scholar] [CrossRef] [Green Version]
- Szutowicz, A.; Kwiatkowski, J.; Angielski, S. Lipogenetic and Glycolytic Enzyme Activities in Carcinoma and Nonmalignant Diseases of the Human Breast. Br. J. Cancer 1979, 39, 681–687. [Google Scholar] [CrossRef] [PubMed]
- Beckner, M.E.; Fellows-Mayle, W.; Zhang, Z.; Agostino, N.R.; Kant, J.A.; Day, B.W.; Pollack, I.F. Identification of ATP Citrate Lyase as a Positive Regulator of Glycolytic Function in Glioblastomas. Int. J. Cancer 2010, 126, 2282–2295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Wang, Y.; Shen, L.; Pang, Y.; Qiao, Z.; Liu, P. Prognostic and Therapeutic Implications of Increased ATP Citrate Lyase Expression in Human Epithelial Ovarian Cancer. Oncol. Rep. 2012, 27, 1156–1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, R.; Tao, R.; Gao, X.; Li, T.; Zhou, X.; Guan, K.-L.; Xiong, Y.; Lei, Q.-Y. Acetylation Stabilizes ATP-Citrate Lyase to Promote Lipid Biosynthesis and Tumor Growth. Mol. Cell 2013, 51, 506–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.-H.; Jang, H.; Lee, S.-M.; Lee, J.-E.; Choi, J.; Kim, T.W.; Cho, E.-J.; Youn, H.-D. ATP-Citrate Lyase Regulates Cellular Senescence via an AMPK- and P53-Dependent Pathway. FEBS J. 2015, 282, 361–371. [Google Scholar] [CrossRef]
- Bourbeau, M.P.; Bartberger, M.D. Recent Advances in the Development of Acetyl-CoA Carboxylase (ACC) Inhibitors for the Treatment of Metabolic Disease. J. Med. Chem. 2015, 58, 525–536. [Google Scholar] [CrossRef] [PubMed]
- Laurent, G.; German, N.J.; Saha, A.K.; de Boer, V.C.J.; Davies, M.; Koves, T.R.; Dephoure, N.; Fischer, F.; Boanca, G.; Vaitheesvaran, B.; et al. SIRT4 Coordinates the Balance between Lipid Synthesis and Catabolism by Repressing Malonyl CoA Decarboxylase. Mol. Cell 2013, 50, 686–698. [Google Scholar] [CrossRef] [Green Version]
- Menendez, J.A.; Lupu, R. Fatty Acid Synthase and the Lipogenic Phenotype in Cancer Pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef] [PubMed]
- Hopperton, K.E.; Duncan, R.E.; Bazinet, R.P.; Archer, M.C. Fatty Acid Synthase Plays a Role in Cancer Metabolism beyond Providing Fatty Acids for Phospholipid Synthesis or Sustaining Elevations in Glycolytic Activity. Exp. Cell Res. 2014, 320, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Feng, X.; Ren, C.; Jiang, X.; Liu, W.; Huang, W.; Liu, Z.; Li, Z.; Zeng, L.; Wang, L.; et al. Over-Expression of BCAT1, a c-Myc Target Gene, Induces Cell Proliferation, Migration and Invasion in Nasopharyngeal Carcinoma. Mol. Cancer 2013, 12, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.-H.; Hu, W.-J.; Chen, B.-C.; Grahn, T.-H.-M.; Zhao, Y.-R.; Bao, H.-L.; Zhu, Y.-F.; Zhang, Q.-Y. BCAT1, a Key Prognostic Predictor of Hepatocellular Carcinoma, Promotes Cell Proliferation and Induces Chemoresistance to Cisplatin. Liver Int. 2016, 36, 1836–1847. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Liu, Q.; Jia, Y.; Tu, K.; Yao, Y.; Liu, Q.; Guo, C. BCAT1 Promotes Tumor Cell Migration and Invasion in Hepatocellular Carcinoma. Oncol. Lett. 2016, 12, 2648–2656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Z.; Achreja, A.; Meurs, N.; Animasahun, O.; Owen, S.; Mittal, A.; Parikh, P.; Lo, T.-W.; Franco-Barraza, J.; Shi, J.; et al. Tumour-Reprogrammed Stromal BCAT1 Fuels Branched-Chain Ketoacid Dependency in Stromal-Rich PDAC Tumours. Nat. Metab. 2020, 2, 775–792. [Google Scholar] [CrossRef]
- Elorza, A.; Soro-Arnáiz, I.; Meléndez-Rodríguez, F.; Rodríguez-Vaello, V.; Marsboom, G.; de Cárcer, G.; Acosta-Iborra, B.; Albacete-Albacete, L.; Ordóñez, A.; Serrano-Oviedo, L.; et al. HIF2α Acts as an MTORC1 Activator through the Amino Acid Carrier SLC7A5. Mol. Cell 2012, 48, 681–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Chen, Y.; Shi, X.; Zhou, M.; Bao, L.; Hatanpaa, K.J.; Patel, T.; DeBerardinis, R.J.; Wang, Y.; Luo, W. Regulation of Branched-Chain Amino Acid Metabolism by Hypoxia-Inducible Factor in Glioblastoma. Cell Mol. Life Sci. 2021, 78, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Hattori, A.; Tsunoda, M.; Konuma, T.; Kobayashi, M.; Nagy, T.; Glushka, J.; Tayyari, F.; McSkimming, D.; Kannan, N.; Tojo, A.; et al. Cancer Progression by Reprogrammed BCAA Metabolism in Myeloid Leukaemia. Nature 2017, 545, 500–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dey, P.; Baddour, J.; Muller, F.; Wu, C.C.; Wang, H.; Liao, W.-T.; Lan, Z.; Chen, A.; Gutschner, T.; Kang, Y.; et al. Genomic Deletion of Malic Enzyme 2 Confers Collateral Lethality in Pancreatic Cancer. Nature 2017, 542, 119–123. [Google Scholar] [CrossRef] [Green Version]
- Li, J.-T.; Yin, M.; Wang, D.; Wang, J.; Lei, M.-Z.; Zhang, Y.; Liu, Y.; Zhang, L.; Zou, S.-W.; Hu, L.-P.; et al. BCAT2-Mediated BCAA Catabolism Is Critical for Development of Pancreatic Ductal Adenocarcinoma. Nat. Cell Biol. 2020, 22, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Hatazawa, Y.; Tadaishi, M.; Nagaike, Y.; Morita, A.; Ogawa, Y.; Ezaki, O.; Takai-Igarashi, T.; Kitaura, Y.; Shimomura, Y.; Kamei, Y.; et al. PGC-1α-Mediated Branched-Chain Amino Acid Metabolism in the Skeletal Muscle. PLoS ONE 2014, 9, e91006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, N.; Yoshikawa, N.; Ito, N.; Maruyama, T.; Suzuki, Y.; Takeda, S.; Nakae, J.; Tagata, Y.; Nishitani, S.; Takehana, K.; et al. Crosstalk between Glucocorticoid Receptor and Nutritional Sensor MTOR in Skeletal Muscle. Cell Metab. 2011, 13, 170–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimomura, Y.; Murakami, T.; Nakai, N.; Nagasaki, M.; Harris, R.A. Exercise Promotes BCAA Catabolism: Effects of BCAA Supplementation on Skeletal Muscle during Exercise. J. Nutr. 2004, 134, 1583S–1587S. [Google Scholar] [CrossRef] [Green Version]
- Terakura, D.; Shimizu, M.; Iwasa, J.; Baba, A.; Kochi, T.; Ohno, T.; Kubota, M.; Shirakami, Y.; Shiraki, M.; Takai, K.; et al. Preventive Effects of Branched-Chain Amino Acid Supplementation on the Spontaneous Development of Hepatic Preneoplastic Lesions in C57BL/KsJ-Db/Db Obese Mice. Carcinogenesis 2012, 33, 2499–2506. [Google Scholar] [CrossRef] [Green Version]
- Arakawa, M.; Masaki, T.; Nishimura, J.; Seike, M.; Yoshimatsu, H. The Effects of Branched-Chain Amino Acid Granules on the Accumulation of Tissue Triglycerides and Uncoupling Proteins in Diet-Induced Obese Mice. Endocr. J. 2011, 58, 161–170. [Google Scholar] [CrossRef] [Green Version]
- Zou, H.; Liao, M.; Xu, W.; Yao, R.; Liao, W. Data Mining of the Expression and Regulatory Role of BCAT1 in Hepatocellular Carcinoma. Oncol. Lett. 2019, 18, 5879–5888. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, J.; Ren, S.; Sun, D.; Huang, H.-Y.; Wang, H.; Jin, Y.; Li, F.; Zheng, C.; Yang, L.; et al. Branched-Chain Amino Acid Metabolic Reprogramming Orchestrates Drug Resistance to EGFR Tyrosine Kinase Inhibitors. Cell Rep. 2019, 28, 512–525.e6. [Google Scholar] [CrossRef]
- Gu, Z.; Liu, Y.; Cai, F.; Patrick, M.; Zmajkovic, J.; Cao, H.; Zhang, Y.; Tasdogan, A.; Chen, M.; Qi, L.; et al. Loss of EZH2 Reprograms BCAA Metabolism to Drive Leukemic Transformation. Cancer Discov. 2019, 9, 1228–1247. [Google Scholar] [CrossRef]
- McBrayer, S.K.; Mayers, J.R.; DiNatale, G.J.; Shi, D.D.; Khanal, J.; Chakraborty, A.A.; Sarosiek, K.A.; Briggs, K.J.; Robbins, A.K.; Sewastianik, T.; et al. Transaminase Inhibition by 2-Hydroxyglutarate Impairs Glutamate Biosynthesis and Redox Homeostasis in Glioma. Cell 2018, 175, 101–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phang, J.M.; Liu, W.; Hancock, C.; Christian, K.J. The Proline Regulatory Axis and Cancer. Front. Oncol. 2012, 2, 60. [Google Scholar] [CrossRef]
- Scott, L.; Lamb, J.; Smith, S.; Wheatley, D.N. Single Amino Acid (Arginine) Deprivation: Rapid and Selective Death of Cultured Transformed and Malignant Cells. Br. J. Cancer 2000, 83, 800–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feun, L.G.; Kuo, M.T.; Savaraj, N. Arginine Deprivation in Cancer Therapy. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Erez, A.; DeBerardinis, R.J. Metabolic Dysregulation in Monogenic Disorders and Cancer—Finding Method in Madness. Nat. Rev. Cancer 2015, 15, 440–448. [Google Scholar] [CrossRef]
- Pearce, E.L.; Poffenberger, M.C.; Chang, C.-H.; Jones, R.G. Fueling Immunity: Insights into Metabolism and Lymphocyte Function. Science 2013, 342, 1242454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, K.; Suda, T. Metabolic Requirements for the Maintenance of Self-Renewing Stem Cells. Nat. Rev. Mol. Cell Biol. 2014, 15, 243–256. [Google Scholar] [CrossRef] [Green Version]
- Vander Heiden, M.G. Targeting Cancer Metabolism: A Therapeutic Window Opens. Nat. Rev. Drug Discov. 2011, 10, 671–684. [Google Scholar] [CrossRef] [Green Version]
- Amelio, I.; Cutruzzolá, F.; Antonov, A.; Agostini, M.; Melino, G. Serine and Glycine Metabolism in Cancer. Trends Biochem. Sci. 2014, 39, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Phang, J.M.; Liu, W.; Hancock, C.N.; Fischer, J.W. Proline Metabolism and Cancer: Emerging Links to Glutamine and Collagen. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 71–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, A.; Cooper, C.R.; Gouw, A.M.; Dinavahi, R.; Maitra, A.; Deck, L.M.; Royer, R.E.; Vander Jagt, D.L.; Semenza, G.L.; Dang, C.V. Inhibition of Lactate Dehydrogenase A Induces Oxidative Stress and Inhibits Tumor Progression. Proc. Natl. Acad. Sci. USA 2010, 107, 2037–2042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, H.; Hanai, J.-I.; Ren, J.-G.; Kats, L.; Burgess, K.; Bhargava, P.; Signoretti, S.; Billiard, J.; Duffy, K.J.; Grant, A.; et al. Targeting Lactate Dehydrogenase—A Inhibits Tumorigenesis and Tumor Progression in Mouse Models of Lung Cancer and Impacts Tumor-Initiating Cells. Cell Metab. 2014, 19, 795–809. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-H.; Israelsen, W.J.; Lee, D.; Yu, V.W.C.; Jeanson, N.T.; Clish, C.B.; Cantley, L.C.; Vander Heiden, M.G.; Scadden, D.T. Cell-State-Specific Metabolic Dependency in Hematopoiesis and Leukemogenesis. Cell 2014, 158, 1309–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, R.; Smith, J.; Rocher-Ros, V.; Nadkarni, S.; Montero-Melendez, T.; D’Acquisto, F.; Bland, E.J.; Bombardieri, M.; Pitzalis, C.; Perretti, M.; et al. Lactate Regulates Metabolic and Pro-Inflammatory Circuits in Control of T Cell Migration and Effector Functions. PLoS Biol. 2015, 13, e1002202. [Google Scholar] [CrossRef] [PubMed]
- Colegio, O.R.; Chu, N.-Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional Polarization of Tumour-Associated Macrophages by Tumour-Derived Lactic Acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef] [Green Version]
- Possemato, R.; Marks, K.M.; Shaul, Y.D.; Pacold, M.E.; Kim, D.; Birsoy, K.; Sethumadhavan, S.; Woo, H.-K.; Jang, H.G.; Jha, A.K.; et al. Functional Genomics Reveal That the Serine Synthesis Pathway Is Essential in Breast Cancer. Nature 2011, 476, 346–350. [Google Scholar] [CrossRef] [Green Version]
- Maddocks, O.D.K.; Berkers, C.R.; Mason, S.M.; Zheng, L.; Blyth, K.; Gottlieb, E.; Vousden, K.H. Serine Starvation Induces Stress and P53-Dependent Metabolic Remodelling in Cancer Cells. Nature 2013, 493, 542–546. [Google Scholar] [CrossRef]
- Lewis, C.A.; Parker, S.J.; Fiske, B.P.; McCloskey, D.; Gui, D.Y.; Green, C.R.; Vokes, N.I.; Feist, A.M.; Vander Heiden, M.G.; Metallo, C.M. Tracing Compartmentalized NADPH Metabolism in the Cytosol and Mitochondria of Mammalian Cells. Mol. Cell 2014, 55, 253–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labuschagne, C.F.; van den Broek, N.J.F.; Mackay, G.M.; Vousden, K.H.; Maddocks, O.D.K. Serine, but Not Glycine, Supports One-Carbon Metabolism and Proliferation of Cancer Cells. Cell Rep. 2014, 7, 1248–1258. [Google Scholar] [CrossRef] [Green Version]
- Das, K.; Roychoudhury, A. Reactive Oxygen Species (ROS) and Response of Antioxidants as ROS-Scavengers during Environmental Stress in Plants. Front. Environ. Sci. 2014, 2, 53. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Fiske, B.P.; Birsoy, K.; Freinkman, E.; Kami, K.; Possemato, R.L.; Chudnovsky, Y.; Pacold, M.E.; Chen, W.W.; Cantor, J.R.; et al. SHMT2 Drives Glioma Cell Survival in Ischaemia but Imposes a Dependence on Glycine Clearance. Nature 2015, 520, 363–367. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, R.; Jain, M.; Madhusudhan, N.; Sheppard, N.G.; Strittmatter, L.; Kampf, C.; Huang, J.; Asplund, A.; Mootha, V.K. Metabolic Enzyme Expression Highlights a Key Role for MTHFD2 and the Mitochondrial Folate Pathway in Cancer. Nat. Commun. 2014, 5, 3128. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, S.E.; Chandel, N.S. Targeting Mitochondria Metabolism for Cancer Therapy. Nat. Chem. Biol. 2015, 11, 9–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, J.M.M.; Donnelly, L.A.; Emslie-Smith, A.M.; Alessi, D.R.; Morris, A.D. Metformin and Reduced Risk of Cancer in Diabetic Patients. BMJ 2005, 330, 1304–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Memmott, R.M.; Mercado, J.R.; Maier, C.R.; Kawabata, S.; Fox, S.D.; Dennis, P.A. Metformin Prevents Tobacco Carcinogen-Induced Lung Tumorigenesis. Cancer Prev. Res. 2010, 3, 1066–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomimoto, A.; Endo, H.; Sugiyama, M.; Fujisawa, T.; Hosono, K.; Takahashi, H.; Nakajima, N.; Nagashima, Y.; Wada, K.; Nakagama, H.; et al. Metformin Suppresses Intestinal Polyp Growth in ApcMin/+ Mice. Cancer Sci. 2008, 99, 2136–2141. [Google Scholar] [CrossRef]
- Bridges, H.R.; Jones, A.J.Y.; Pollak, M.N.; Hirst, J. Effects of Metformin and Other Biguanides on Oxidative Phosphorylation in Mitochondria. Biochem. J. 2014, 462, 475–487. [Google Scholar] [CrossRef] [Green Version]
- El-Mir, M.Y.; Nogueira, V.; Fontaine, E.; Avéret, N.; Rigoulet, M.; Leverve, X. Dimethylbiguanide Inhibits Cell Respiration via an Indirect Effect Targeted on the Respiratory Chain Complex I. J. Biol. Chem. 2000, 275, 223–228. [Google Scholar] [CrossRef] [Green Version]
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence That Metformin Exerts Its Anti-Diabetic Effects through Inhibition of Complex 1 of the Mitochondrial Respiratory Chain. Biochem. J. 2000, 348 Pt 3, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin Inhibits Mitochondrial Complex I of Cancer Cells to Reduce Tumorigenesis. eLife 2014, 3, e02242. [Google Scholar] [CrossRef]
- Griss, T.; Vincent, E.E.; Egnatchik, R.; Chen, J.; Ma, E.H.; Faubert, B.; Viollet, B.; DeBerardinis, R.J.; Jones, R.G. Metformin Antagonizes Cancer Cell Proliferation by Suppressing Mitochondrial-Dependent Biosynthesis. PLoS Biol. 2015, 13, e1002309. [Google Scholar] [CrossRef] [Green Version]
- Emami Riedmaier, A.; Fisel, P.; Nies, A.T.; Schaeffeler, E.; Schwab, M. Metformin and Cancer: From the Old Medicine Cabinet to Pharmacological Pitfalls and Prospects. Trends Pharmacol. Sci. 2013, 34, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Pollak, M. Overcoming Drug Development Bottlenecks with Repurposing: Repurposing Biguanides to Target Energy Metabolism for Cancer Treatment. Nat. Med. 2014, 20, 591–593. [Google Scholar] [CrossRef]
- Birsoy, K.; Possemato, R.; Lorbeer, F.K.; Bayraktar, E.C.; Thiru, P.; Yucel, B.; Wang, T.; Chen, W.W.; Clish, C.B.; Sabatini, D.M. Metabolic Determinants of Cancer Cell Sensitivity to Glucose Limitation and Biguanides. Nature 2014, 508, 108–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shackelford, D.B.; Abt, E.; Gerken, L.; Vasquez, D.S.; Seki, A.; Leblanc, M.; Wei, L.; Fishbein, M.C.; Czernin, J.; Mischel, P.S.; et al. LKB1 Inactivation Dictates Therapeutic Response of Non-Small Cell Lung Cancer to the Metabolism Drug Phenformin. Cancer Cell 2013, 23, 143–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gravel, S.-P.; Hulea, L.; Toban, N.; Birman, E.; Blouin, M.-J.; Zakikhani, M.; Zhao, Y.; Topisirovic, I.; St-Pierre, J.; Pollak, M. Serine Deprivation Enhances Antineoplastic Activity of Biguanides. Cancer Res. 2014, 74, 7521–7533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karsli-Uzunbas, G.; Guo, J.Y.; Price, S.; Teng, X.; Laddha, S.V.; Khor, S.; Kalaany, N.Y.; Jacks, T.; Chan, C.S.; Rabinowitz, J.D.; et al. Autophagy Is Required for Glucose Homeostasis and Lung Tumor Maintenance. Cancer Discov. 2014, 4, 914–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shroff, E.H.; Eberlin, L.S.; Dang, V.M.; Gouw, A.M.; Gabay, M.; Adam, S.J.; Bellovin, D.I.; Tran, P.T.; Philbrick, W.M.; Garcia-Ocana, A.; et al. MYC Oncogene Overexpression Drives Renal Cell Carcinoma in a Mouse Model through Glutamine Metabolism. Proc. Natl. Acad. Sci. USA 2015, 112, 6539–6544. [Google Scholar] [CrossRef] [Green Version]
- Xiang, Y.; Stine, Z.E.; Xia, J.; Lu, Y.; O’Connor, R.S.; Altman, B.J.; Hsieh, A.L.; Gouw, A.M.; Thomas, A.G.; Gao, P.; et al. Targeted Inhibition of Tumor-Specific Glutaminase Diminishes Cell-Autonomous Tumorigenesis. J. Clin. Investig. 2015, 125, 2293–2306. [Google Scholar] [CrossRef] [Green Version]
- Schug, Z.T.; Peck, B.; Jones, D.T.; Zhang, Q.; Grosskurth, S.; Alam, I.S.; Goodwin, L.M.; Smethurst, E.; Mason, S.; Blyth, K.; et al. Acetyl-CoA Synthetase 2 Promotes Acetate Utilization and Maintains Cancer Cell Growth under Metabolic Stress. Cancer Cell 2015, 27, 57–71. [Google Scholar] [CrossRef] [Green Version]
- Comerford, S.A.; Huang, Z.; Du, X.; Wang, Y.; Cai, L.; Witkiewicz, A.K.; Walters, H.; Tantawy, M.N.; Fu, A.; Manning, H.C.; et al. Acetate Dependence of Tumors. Cell 2014, 159, 1591–1602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mashimo, T.; Pichumani, K.; Vemireddy, V.; Hatanpaa, K.J.; Singh, D.K.; Sirasanagandla, S.; Nannepaga, S.; Piccirillo, S.G.; Kovacs, Z.; Foong, C.; et al. Acetate Is a Bioenergetic Substrate for Human Glioblastoma and Brain Metastases. Cell 2014, 159, 1603–1614. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Chen, L.; Tan, X.; Crosby, K.; Guimaraes, A.R.; Upadhyay, R.; Maira, M.; McNamara, K.; Perera, S.A.; Song, Y.; et al. Effective Use of PI3K and MEK Inhibitors to Treat Mutant Kras G12D and PIK3CA H1047R Murine Lung Cancers. Nat. Med. 2008, 14, 1351–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herranz, D.; Ambesi-Impiombato, A.; Sudderth, J.; Sánchez-Martín, M.; Belver, L.; Tosello, V.; Xu, L.; Wendorff, A.A.; Castillo, M.; Haydu, J.E.; et al. Metabolic Reprogramming Induces Resistance to Anti-NOTCH1 Therapies in T Cell Acute Lymphoblastic Leukemia. Nat. Med. 2015, 21, 1182–1189. [Google Scholar] [CrossRef] [PubMed]
- Viale, A.; Pettazzoni, P.; Lyssiotis, C.A.; Ying, H.; Sánchez, N.; Marchesini, M.; Carugo, A.; Green, T.; Seth, S.; Giuliani, V.; et al. Oncogene Ablation-Resistant Pancreatic Cancer Cells Depend on Mitochondrial Function. Nature 2014, 514, 628–632. [Google Scholar] [CrossRef] [Green Version]
- Yuan, P.; Ito, K.; Perez-Lorenzo, R.; Del Guzzo, C.; Lee, J.H.; Shen, C.-H.; Bosenberg, M.W.; McMahon, M.; Cantley, L.C.; Zheng, B. Phenformin Enhances the Therapeutic Benefit of BRAF(V600E) Inhibition in Melanoma. Proc. Natl. Acad. Sci. USA 2013, 110, 18226–18231. [Google Scholar] [CrossRef] [Green Version]
- Roesch, A.; Vultur, A.; Bogeski, I.; Wang, H.; Zimmermann, K.M.; Speicher, D.; Körbel, C.; Laschke, M.W.; Gimotty, P.A.; Philipp, S.E.; et al. Overcoming Intrinsic Multidrug Resistance in Melanoma by Blocking the Mitochondrial Respiratory Chain of Slow-Cycling JARID1B(High) Cells. Cancer Cell 2013, 23, 811–825. [Google Scholar] [CrossRef] [Green Version]
- Janzer, A.; German, N.J.; Gonzalez-Herrera, K.N.; Asara, J.M.; Haigis, M.C.; Struhl, K. Metformin and Phenformin Deplete Tricarboxylic Acid Cycle and Glycolytic Intermediates during Cell Transformation and NTPs in Cancer Stem Cells. Proc. Natl. Acad. Sci. USA 2014, 111, 10574–10579. [Google Scholar] [CrossRef] [Green Version]
- Chandel, N.S.; Tuveson, D.A. The Promise and Perils of Antioxidants for Cancer Patients. N. Engl. J. Med. 2014, 371, 177–178. [Google Scholar] [CrossRef]
- Yun, J.; Mullarky, E.; Lu, C.; Bosch, K.N.; Kavalier, A.; Rivera, K.; Roper, J.; Chio, I.I.C.; Giannopoulou, E.G.; Rago, C.; et al. Vitamin C Selectively Kills KRAS and BRAF Mutant Colorectal Cancer Cells by Targeting GAPDH. Science 2015, 350, 1391–1396. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Espey, M.G.; Sun, A.Y.; Pooput, C.; Kirk, K.L.; Krishna, M.C.; Khosh, D.B.; Drisko, J.; Levine, M. Pharmacologic Doses of Ascorbate Act as a Prooxidant and Decrease Growth of Aggressive Tumor Xenografts in Mice. Proc. Natl. Acad. Sci. USA 2008, 105, 11105–11109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Chapman, J.; Levine, M.; Polireddy, K.; Drisko, J.; Chen, Q. High-Dose Parenteral Ascorbate Enhanced Chemosensitivity of Ovarian Cancer and Reduced Toxicity of Chemotherapy. Sci. Transl. Med. 2014, 6, 222ra18. [Google Scholar] [CrossRef] [PubMed]
- Tagde, A.; Singh, H.; Kang, M.H.; Reynolds, C.P. The Glutathione Synthesis Inhibitor Buthionine Sulfoximine Synergistically Enhanced Melphalan Activity against Preclinical Models of Multiple Myeloma. Blood Cancer J. 2014, 4, e229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glasauer, A.; Sena, L.A.; Diebold, L.P.; Mazar, A.P.; Chandel, N.S. Targeting SOD1 Reduces Experimental Non–Small-Cell Lung Cancer. J. Clin. Investig. 2014, 124, 117–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dadwal, A.; Baldi, A.; Kumar Narang, R. Nanoparticles as Carriers for Drug Delivery in Cancer. Artif. Cells Nanomed. Biotechnol. 2018, 46, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Palazzolo, S.; Bayda, S.; Hadla, M.; Caligiuri, I.; Corona, G.; Toffoli, G.; Rizzolio, F. The Clinical Translation of Organic Nanomaterials for Cancer Therapy: A Focus on Polymeric Nanoparticles, Micelles, Liposomes and Exosomes. Curr. Med. Chem. 2018, 25, 4224–4268. [Google Scholar] [CrossRef]
- Chen, Y.; Gao, D.-Y.; Huang, L. In Vivo Delivery of MiRNAs for Cancer Therapy: Challenges and Strategies. Adv. Drug Deliv. Rev. 2015, 81, 128–141. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Dirisala, A.; Ge, Z.; Wang, Y.; Yin, W.; Ke, W.; Toh, K.; Xie, J.; Matsumoto, Y.; Anraku, Y.; et al. Therapeutic Vesicular Nanoreactors with Tumor-Specific Activation and Self-Destruction for Synergistic Tumor Ablation. Angew. Chem. 2017, 129, 14213–14218. [Google Scholar] [CrossRef]
- Dirisala, A.; Osada, K.; Chen, Q.; Tockary, T.A.; Machitani, K.; Osawa, S.; Liu, X.; Ishii, T.; Miyata, K.; Oba, M.; et al. Optimized Rod Length of Polyplex Micelles for Maximizing Transfection Efficiency and Their Performance in Systemic Gene Therapy against Stroma-Rich Pancreatic Tumors. Biomaterials 2014, 35, 5359–5368. [Google Scholar] [CrossRef] [Green Version]
- Oba, M.; Vachutinsky, Y.; Miyata, K.; Kano, M.R.; Ikeda, S.; Nishiyama, N.; Itaka, K.; Miyazono, K.; Koyama, H.; Kataoka, K. Antiangiogenic Gene Therapy of Solid Tumor by Systemic Injection of Polyplex Micelles Loading Plasmid DNA Encoding Soluble Flt-1. Mol. Pharm. 2010, 7, 501–509. [Google Scholar] [CrossRef]
- Yu, S.; Chen, Z.; Zeng, X.; Chen, X.; Gu, Z. Advances in Nanomedicine for Cancer Starvation Therapy. Theranostics 2019, 9, 8026–8047. [Google Scholar] [CrossRef]
- Tabish, T.A.; Hamblin, M.R. Mitochondria-Targeted Nanoparticles (MitoNANO): An Emerging Therapeutic Shortcut for Cancer. Biomater. Biosyst. 2021, 3, 100023. [Google Scholar] [CrossRef]
- Chen, P.; Yang, W.; Hong, T.; Miyazaki, T.; Dirisala, A.; Kataoka, K.; Cabral, H. Nanocarriers Escaping from Hyperacidified Endo/Lysosomes in Cancer Cells Allow Tumor-Targeted Intracellular Delivery of Antibodies to Therapeutically Inhibit c-MYC. Biomaterials 2022, 288, 121748. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Target | Effect | Tumour Types Targeted |
|---|---|---|---|
| 2-deoxyglucose | Hexokinase | Inhibits glycolysis | Advanced solid tumors (e.g., lung, breast, prostate, and gastric) |
| Lonidamine | Hexokinase | Inhibits glycolysis | Benign prostatic hyperplasia |
| 3-bromopyruvate | Hexokinase | Inhibits glycolysis | N/A |
| TLN-232 | Pyruvate kinase | Inhibits glycolysis | Metastatic melanoma and renal cell carcinoma |
| Dichloroacetate | PDK1 | Reactivates PDH | Metastatic solid tumors, glioma, and GBM |
| Phenylacetate | Glutamine | Reduces plasma glutamine levels | Brain tumors (e.g., glioma, astrocytoma and medulloblastoma) |
| Asparaginase and Pegasparaginase | Asparagine | Reduces plasmaasparagine levels | ALL, TCL, and BCL |
| Arginine deiminase | Arginine | Reduces plasma asparagine levels | Metastatic melanoma and hepatocellular carcinoma |
| Acetazolamide, Indisulam and other sulfonamides | Carbonic anhydrases | pH regulation | Solid tumors (e.g., pancreatic, lung, melanoma and metastatic breast) |
| Cariporide | NHE1 | pH regulation | N/A |
| SB-204990 | ATP-citrate lyase | Inhibits fatty acid synthesis | N/A |
| Orlistat, GSK837149A, and C75 | FASN | Inhibits fatty acid synthesis | N/A |
| Temsirolimus and Everolimus | mTORC1 | Inhibits mTORC1 | Solid tumors (both metastatic and non-metastatic) |
| Other rapalogues | mTORC1 | Inhibits mTORC1 | Solid tumors (e.g., pancreatic, endometrial and glioblastoma) and lymphoma |
| Torin1 and PP242 | mTORC1 and mTORC2 | Inhibits mTORC1 andmTORC2 | N/A |
| PX-478 | HIF1α | Inhibits HIF signaling | Advanced solid tumors and lymphoma |
| Acriflavine | HIF1α | Inhibits HIF signaling | N/A |
| Tirapazamine and other bioreductivecompounds | Hypoxia | Resensitizes cells to other treatments | Solid tumors (e.g., cervical, SCLC and NSCLC) |
| Bevacizumab and related compounds | Hypoxia, VEGF and VEGFR | Blocks angiogenesis | Solid tumors (e.g., malignant glioma, NSCLC, ovarian, and colorectal) |
| MK-0646, BIIB022, AVE1642, and others | IGF1R | Blocks IGF signaling | Solid tumors (e.g., NSCLC, pancreatic, hepatocellular carcinoma and metastatic breast) |
| BEZ235, XL765, SF1126, and BGT226 | PI3K and mTOR | Inhibits signaling from PI3K andmTORC1 andmTORC2 | Advanced solid tumors (e.g., malignant glioma and NSCLC) |
| GDC-0941 and PX866 | PI3K | Inhibits PI3Ksignaling | Advanced solid tumors (metastatic breast and non-Hodgkin’s lymphoma) |
| Perifosine andGSK690693 | AKT | Inhibits AKT | Solid tumors (e.g., renal cancer and NSCLC) and lymphoma |
| Metformin | AMPK andComplex I(mitochondrial) | Activates AMPK | Solid tumors and lymphoma |
| Antimetabolites (e.g., 5-FU, cytarabine andmethotrexate | Nucleotidebiosyntheticpathway | Inhibits cell proliferation | Many tumor types |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, R.; Mishra, A.; Gautam, P.; Feroz, Z.; Vijayaraghavalu, S.; Likos, E.M.; Shukla, G.C.; Kumar, M. Metabolic Pathways, Enzymes, and Metabolites: Opportunities in Cancer Therapy. Cancers 2022, 14, 5268. https://doi.org/10.3390/cancers14215268
Kumar R, Mishra A, Gautam P, Feroz Z, Vijayaraghavalu S, Likos EM, Shukla GC, Kumar M. Metabolic Pathways, Enzymes, and Metabolites: Opportunities in Cancer Therapy. Cancers. 2022; 14(21):5268. https://doi.org/10.3390/cancers14215268
Chicago/Turabian StyleKumar, Rishabh, Anurag Mishra, Priyanka Gautam, Zainab Feroz, Sivakumar Vijayaraghavalu, Eviania M. Likos, Girish C. Shukla, and Munish Kumar. 2022. "Metabolic Pathways, Enzymes, and Metabolites: Opportunities in Cancer Therapy" Cancers 14, no. 21: 5268. https://doi.org/10.3390/cancers14215268