Gut Microbiota and Tumor Immune Escape: A New Perspective for Improving Tumor Immunotherapy

 , ,
, , {kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Gut Microbiota Influences Tumor Immune Escape through Tumor Microenvironment
2.1. Gut Microbiota, Th17 Cells, and IL-17 Families
2.2. Gut Microbiota and TAMs
2.3. Gut Microbiota and Other Immune Cells in the TME
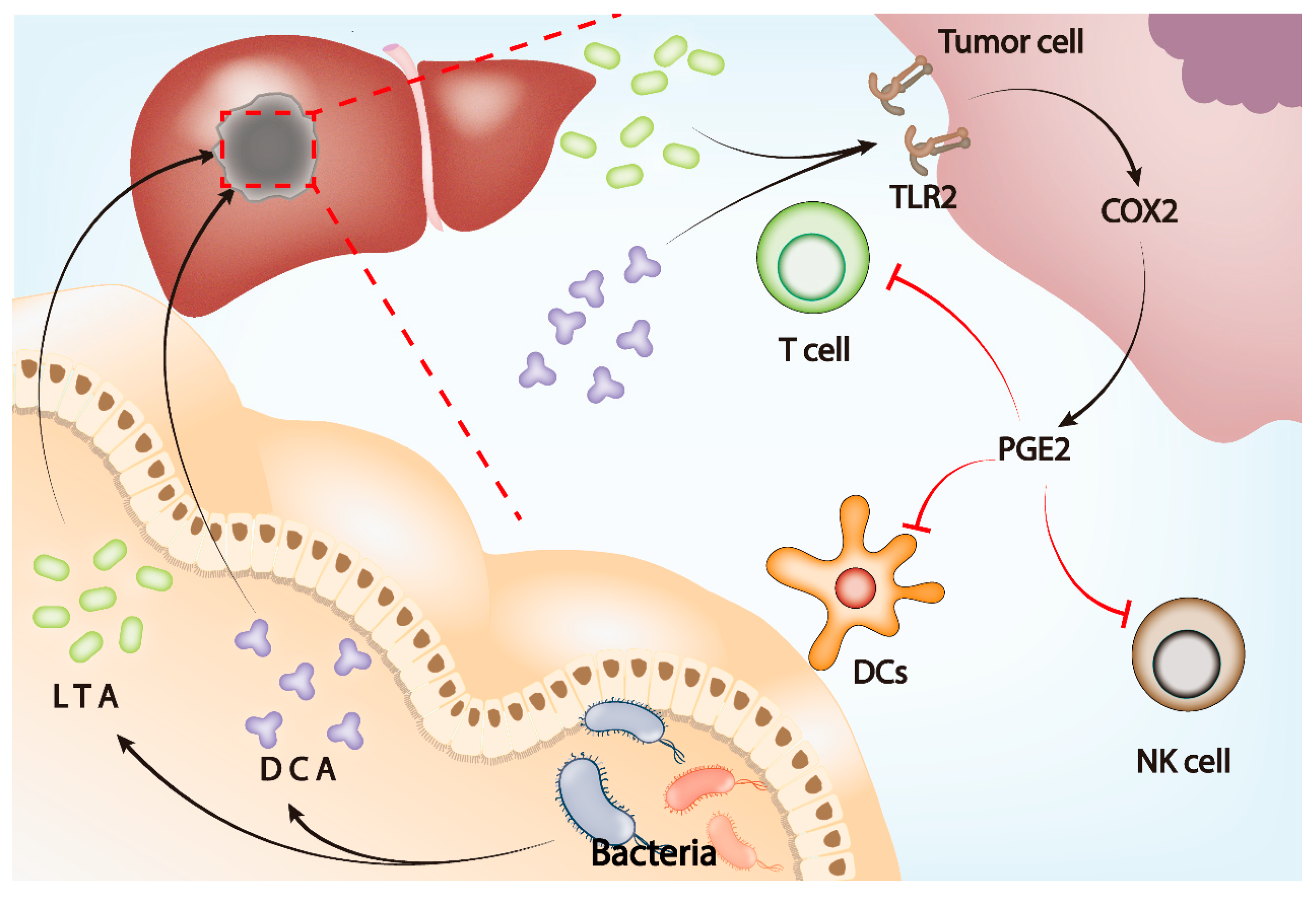
2.4. Gut Microbiota and PGE2
3. Gut Microbiota and Tumor Immunotherapy
3.1. Gut Microbiota Affects the Efficacy of Immune Checkpoint Inhibitors
3.2. Gut Microbiota and Antibiotic Use in ICI Therapy
3.3. Gut Microbiota Affects the Efficacy of CAR T-Cell Therapy
4. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Clemente, J.C.; Ursell, L.K.; Parfrey, L.W.; Knight, R. The impact of the gut microbiota on human health: An integrative view. Cell 2012, 148, 1258–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, J.; Lee, S.M.; Shen, Y.; Khosravi, A.; Mazmanian, S.K. Host-bacterial symbiosis in health and disease. Adv. Immunol. 2010, 107, 243–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen, P.; Molinero-Perez, A.; O’Riordan, K.J.; McCafferty, C.P.; O’Halloran, K.D.; Cryan, J.F. Microbiota and sleep: Awakening the gut feeling. Trends Mol. Med. 2021, 27, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Du, J.; Wu, X.; Abdelrehem, A.; Ren, Y.; Liu, C.; Zhou, X.; Wang, S. Crosstalk between autophagy and microbiota in cancer progression. Mol. Cancer 2021, 20, 163. [Google Scholar] [CrossRef]
- Cullin, N.; Azevedo Antunes, C.; Straussman, R.; Stein-Thoeringer, C.K.; Elinav, E. Microbiome and cancer. Cancer Cell 2021, 39, 1317–1341. [Google Scholar] [CrossRef]
- Schwabe, R.F.; Jobin, C. The microbiome and cancer. Nat. Rev. Cancer 2013, 13, 800–812. [Google Scholar] [CrossRef] [Green Version]
- Agirman, G.; Yu, K.B.; Hsiao, E.Y. Signaling inflammation across the gut-brain axis. Science 2021, 374, 1087–1092. [Google Scholar] [CrossRef]
- Villa, C.R.; Ward, W.E.; Comelli, E.M. Gut microbiota-bone axis. Crit. Rev. Food Sci. Nutr. 2017, 57, 1664–1672. [Google Scholar] [CrossRef]
- Beyaz Coşkun, A.; Sağdiçoğlu Celep, A.G. Therapeutic modulation methods of gut microbiota and gut-liver axis. Crit. Rev. Food Sci. Nutr. 2022, 62, 6505–6515. [Google Scholar] [CrossRef]
- Yang, J.; Wei, H.; Zhou, Y.; Szeto, C.H.; Li, C.; Lin, Y.; Coker, O.O.; Lau, H.C.H.; Chan, A.W.H.; Sung, J.J.Y.; et al. High-Fat Diet Promotes Colorectal Tumorigenesis Through Modulating Gut Microbiota and Metabolites. Gastroenterology 2022, 162, 135–149.e132. [Google Scholar] [CrossRef]
- Ohtani, N.; Hara, E. Gut-liver axis-mediated mechanism of liver cancer: A special focus on the role of gut microbiota. Cancer Sci. 2021, 112, 4433–4443. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xia, Y.; Sun, J. Breast and gut microbiome in health and cancer. Genes Dis. 2021, 8, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Wu, J.; Jin, D.; Wang, B.; Cao, H. Fecal microbiota transplantation in cancer management: Current status and perspectives. Int. J. Cancer 2019, 145, 2021–2031. [Google Scholar] [CrossRef] [Green Version]
- Eslami, M.; Yousefi, B.; Kokhaei, P.; Hemati, M.; Nejad, Z.R.; Arabkari, V.; Namdar, A. Importance of probiotics in the prevention and treatment of colorectal cancer. J. Cell. Physiol. 2019, 234, 17127–17143. [Google Scholar] [CrossRef]
- Zhang, X.; Coker, O.O.; Chu, E.S.; Fu, K.; Lau, H.C.H.; Wang, Y.X.; Chan, A.W.H.; Wei, H.; Yang, X.; Sung, J.J.Y.; et al. Dietary cholesterol drives fatty liver-associated liver cancer by modulating gut microbiota and metabolites. Gut 2021, 70, 761–774. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.L.; Mao, Y.Q.; Zhang, Z.Y.; Li, Z.M.; Kong, C.Y.; Chen, H.L.; Cai, P.R.; Han, B.; Ye, T.; Wang, L.S. Pectin supplement significantly enhanced the anti-PD-1 efficacy in tumor-bearing mice humanized with gut microbiota from patients with colorectal cancer. Theranostics 2021, 11, 4155–4170. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [Green Version]
- Jhunjhunwala, S.; Hammer, C.; Delamarre, L. Antigen presentation in cancer: Insights into tumour immunogenicity and immune evasion. Nat. Rev. Cancer 2021, 21, 298–312. [Google Scholar] [CrossRef]
- Zhang, J.; Huang, D.; Saw, P.E.; Song, E. Turning cold tumors hot: From molecular mechanisms to clinical applications. Trends Immunol. 2022, 43, 523–545. [Google Scholar] [CrossRef] [PubMed]
- Kadosh, E.; Snir-Alkalay, I.; Venkatachalam, A.; May, S.; Lasry, A.; Elyada, E.; Zinger, A.; Shaham, M.; Vaalani, G.; Mernberger, M.; et al. The gut microbiome switches mutant p53 from tumour-suppressive to oncogenic. Nature 2020, 586, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Matson, V.; Chervin, C.S.; Gajewski, T.F. Cancer and the Microbiome-Influence of the Commensal Microbiota on Cancer, Immune Responses, and Immunotherapy. Gastroenterology 2021, 160, 600–613. [Google Scholar] [CrossRef] [PubMed]
- Houot, R.; Schultz, L.M.; Marabelle, A.; Kohrt, H. T-cell-based Immunotherapy: Adoptive Cell Transfer and Checkpoint Inhibition. Cancer Immunol. Res. 2015, 3, 1115–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Salhy, M.; Hatlebakk, J.G.; Gilja, O.H.; Bråthen Kristoffersen, A.; Hausken, T. Efficacy of faecal microbiota transplantation for patients with irritable bowel syndrome in a randomised, double-blind, placebo-controlled study. Gut 2020, 69, 859–867. [Google Scholar] [CrossRef] [Green Version]
- Arias-Borrego, A.; Selma-Royo, M.; Collado, M.C.; Abril, N.; García-Barrera, T. Impact of “chemical cocktails” exposure in shaping mice gut microbiota and the role of selenium supplementation combining metallomics, metabolomics, and metataxonomics. J. Hazard. Mater. 2022, 438, 129444. [Google Scholar] [CrossRef] [PubMed]
- Sanders, M.E.; Merenstein, D.J.; Reid, G.; Gibson, G.R.; Rastall, R.A. Probiotics and prebiotics in intestinal health and disease: From biology to the clinic. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Hinshaw, D.C.; Shevde, L.A. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef] [Green Version]
- Hegde, P.S.; Chen, D.S. Top 10 Challenges in Cancer Immunotherapy. Immunity 2020, 52, 17–35. [Google Scholar] [CrossRef]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Wang, L.; Jiang, Y.; Zhang, Y.; Wang, Y.; Huang, S.; Wang, Z.; Tian, B.; Yang, Y.; Jiang, W.; Pang, D. Association analysis of IL-17A and IL-17F polymorphisms in Chinese Han women with breast cancer. PLoS ONE 2012, 7, e34400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grote, V.A.; Kaaks, R.; Nieters, A.; Tjønneland, A.; Halkjær, J.; Overvad, K.; Skjelbo Nielsen, M.R.; Boutron-Ruault, M.C.; Clavel-Chapelon, F.; Racine, A.; et al. Inflammation marker and risk of pancreatic cancer: A nested case-control study within the EPIC cohort. Br. J. Cancer 2012, 106, 1866–1874. [Google Scholar] [CrossRef] [PubMed]
- Kipanyula, M.J.; Seke Etet, P.F.; Vecchio, L.; Farahna, M.; Nukenine, E.N.; Nwabo Kamdje, A.H. Signaling pathways bridging microbial-triggered inflammation and cancer. Cell Signal 2013, 25, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Ohkusa, T.; Yoshida, T.; Sato, N.; Watanabe, S.; Tajiri, H.; Okayasu, I. Commensal bacteria can enter colonic epithelial cells and induce proinflammatory cytokine secretion: A possible pathogenic mechanism of ulcerative colitis. J. Med. Microbiol 2009, 58, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.; Xie, G.; Jia, W. Bile acid-microbiota crosstalk in gastrointestinal inflammation and carcinogenesis. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 111–128. [Google Scholar] [CrossRef] [Green Version]
- Man, S.M. Inflammasomes in the gastrointestinal tract: Infection, cancer and gut microbiota homeostasis. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 721–737. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Karki, R.; Kanneganti, T.D. Diverging inflammasome signals in tumorigenesis and potential targeting. Nat. Rev. Cancer 2019, 19, 197–214. [Google Scholar] [CrossRef]
- Stappenbeck, T.S.; Hooper, L.V.; Gordon, J.I. Developmental regulation of intestinal angiogenesis by indigenous microbes via Paneth cells. Proc. Natl. Acad. Sci. USA 2002, 99, 15451–15455. [Google Scholar] [CrossRef] [Green Version]
- Reinhardt, C.; Bergentall, M.; Greiner, T.U.; Schaffner, F.; Ostergren-Lundén, G.; Petersen, L.C.; Ruf, W.; Bäckhed, F. Tissue factor and PAR1 promote microbiota-induced intestinal vascular remodelling. Nature 2012, 483, 627–631. [Google Scholar] [CrossRef]
- Eisenreich, A.; Zakrzewicz, A.; Huber, K.; Thierbach, H.; Pepke, W.; Goldin-Lang, P.; Schultheiss, H.P.; Pries, A.; Rauch, U. Regulation of pro-angiogenic tissue factor expression in hypoxia-induced human lung cancer cells. Oncol. Rep. 2013, 30, 462–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van den Berg, Y.W.; van den Hengel, L.G.; Myers, H.R.; Ayachi, O.; Jordanova, E.; Ruf, W.; Spek, C.A.; Reitsma, P.H.; Bogdanov, V.Y.; Versteeg, H.H. Alternatively spliced tissue factor induces angiogenesis through integrin ligation. Proc. Natl. Acad. Sci. USA 2009, 106, 19497–19502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schirbel, A.; Kessler, S.; Rieder, F.; West, G.; Rebert, N.; Asosingh, K.; McDonald, C.; Fiocchi, C. Pro-angiogenic activity of TLRs and NLRs: A novel link between gut microbiota and intestinal angiogenesis. Gastroenterology 2013, 144, 613–623.e619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prorok-Hamon, M.; Friswell, M.K.; Alswied, A.; Roberts, C.L.; Song, F.; Flanagan, P.K.; Knight, P.; Codling, C.; Marchesi, J.R.; Winstanley, C.; et al. Colonic mucosa-associated diffusely adherent afaC+ Escherichia coli expressing lpfA and pks are increased in inflammatory bowel disease and colon cancer. Gut 2014, 63, 761–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cane, G.; Ginouvès, A.; Marchetti, S.; Buscà, R.; Pouysségur, J.; Berra, E.; Hofman, P.; Vouret-Craviari, V. HIF-1alpha mediates the induction of IL-8 and VEGF expression on infection with Afa/Dr diffusely adhering E. coli and promotes EMT-like behaviour. Cell. Microbiol. 2010, 12, 640–653. [Google Scholar] [CrossRef]
- Waldner, M.J.; Wirtz, S.; Jefremow, A.; Warntjen, M.; Neufert, C.; Atreya, R.; Becker, C.; Weigmann, B.; Vieth, M.; Rose-John, S.; et al. VEGF receptor signaling links inflammation and tumorigenesis in colitis-associated cancer. J. Exp. Med. 2010, 207, 2855–2868. [Google Scholar] [CrossRef]
- Lin, S.-C.; Lo, Y.-C.; Wu, H. Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature 2010, 465, 885–890. [Google Scholar] [CrossRef] [Green Version]
- Valkov, E.; Stamp, A.; Dimaio, F.; Baker, D.; Verstak, B.; Roversi, P.; Kellie, S.; Sweet, M.J.; Mansell, A.; Gay, N.J.; et al. Crystal structure of Toll-like receptor adaptor MAL/TIRAP reveals the molecular basis for signal transduction and disease protection. Proc. Natl. Acad. Sci. USA 2011, 108, 14879–14884. [Google Scholar] [CrossRef] [Green Version]
- Del Prete, A.; Allavena, P.; Santoro, G.; Fumarulo, R.; Corsi, M.M.; Mantovani, A. Molecular pathways in cancer-related inflammation. Biochem. Med. 2011, 21, 264–275. [Google Scholar] [CrossRef]
- Ozdemir, H.; Ciftçi, E.; Karbuz, A.; Oktay, G.; Aysev, D.; Yavuz, G.; Ince, E. Successful treatment of multidrug-resistant Escherichia coli bacteremia with tigecycline in an acute myeloid leukemia child. Turk. J. Pediatr. 2012, 54, 59–60. [Google Scholar]
- Kurucu, N.; Kul, S.; Tosun, I.; Erduran, E.; Köksal, I. Fungemia and renal fungus ball formation with Candida norvegensis in a child with acute lymphoblastic leukemia. Turk. J. Pediatr. 2011, 53, 448–451. [Google Scholar] [PubMed]
- De Carvalho Parahym, A.M.R.; da Silva, C.M.; Leão, M.P.C.; Macario, M.C.; Filho, G.A.d.T.M.H.; de Oliveira, N.T.; Neves, R.P. Invasive infection in an acute myeloblastic leukemia patient due to triazole-resistant Candida tropicalis. Diagn. Microbiol. Infect. Dis. 2011, 71, 291–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gogia, A.; Bakhshi, S. Plasmablastic lymphoma of oral cavity in a HIV-negative child. Pediatr. Blood Cancer 2010, 55, 390–391. [Google Scholar] [CrossRef] [PubMed]
- Marteau, P.; Chaput, U. Bacteria as trigger for chronic gastrointestinal disorders. Dig. Dis. 2011, 29, 166–171. [Google Scholar] [CrossRef]
- Khan, M.A.; Jakate, S.; Komanduri, S. Rare AIDS-associated plasmablastic lymphoma as the initial presentation of AIDS. Clin. Adv. Hematol. Oncol. 2010, 8, 55–57. [Google Scholar]
- McAllister, F.; Bailey, J.M.; Alsina, J.; Nirschl, C.J.; Sharma, R.; Fan, H.; Rattigan, Y.; Roeser, J.C.; Lankapalli, R.H.; Zhang, H.; et al. Oncogenic Kras activates a hematopoietic-to-epithelial IL-17 signaling axis in preinvasive pancreatic neoplasia. Cancer Cell 2014, 25, 621–637. [Google Scholar] [CrossRef] [Green Version]
- Tartour, E.; Fossiez, F.; Joyeux, I.; Galinha, A.; Gey, A.; Claret, E.; Sastre-Garau, X.; Couturier, J.; Mosseri, V.; Vives, V.; et al. Interleukin 17, a T-cell-derived cytokine, promotes tumorigenicity of human cervical tumors in nude mice. Cancer Res. 1999, 59, 3698–3704. [Google Scholar]
- Gaffen, S.L.; Jain, R.; Garg, A.V.; Cua, D.J. The IL-23-IL-17 immune axis: From mechanisms to therapeutic testing. Nat. Rev. Immunol. 2014, 14, 585–600. [Google Scholar] [CrossRef] [Green Version]
- Tosolini, M.; Kirilovsky, A.; Mlecnik, B.; Fredriksen, T.; Mauger, S.; Bindea, G.; Berger, A.; Bruneval, P.; Fridman, W.-H.; Pagès, F.; et al. Clinical impact of different classes of infiltrating T cytotoxic and helper cells (Th1, th2, treg, th17) in patients with colorectal cancer. Cancer Res. 2011, 71, 1263–1271. [Google Scholar] [CrossRef] [Green Version]
- Cui, G.; Yuan, A.; Goll, R.; Florholmen, J. IL-17A in the tumor microenvironment of the human colorectal adenoma-carcinoma sequence. Scand. J. Gastroenterol. 2012, 47, 1304–1312. [Google Scholar] [CrossRef]
- Hurtado, C.G.; Wan, F.; Housseau, F.; Sears, C.L. Roles for Interleukin 17 and Adaptive Immunity in Pathogenesis of Colorectal Cancer. Gastroenterology 2018, 155, 1706–1715. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.I.; Atarashi, K.; Manel, N.; Brodie, E.L.; Shima, T.; Karaoz, U.; Wei, D.; Goldfarb, K.C.; Santee, C.A.; Lynch, S.V.; et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 2009, 139, 485–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaboriau-Routhiau, V.; Rakotobe, S.; Lécuyer, E.; Mulder, I.; Lan, A.; Bridonneau, C.; Rochet, V.; Pisi, A.; De Paepe, M.; Brandi, G.; et al. The key role of segmented filamentous bacteria in the coordinated maturation of gut helper T cell responses. Immunity 2009, 31, 677–689. [Google Scholar] [CrossRef] [PubMed]
- Hang, S.; Paik, D.; Yao, L.; Kim, E.; Trinath, J.; Lu, J.; Ha, S.; Nelson, B.N.; Kelly, S.P.; Wu, L.; et al. Bile acid metabolites control T(H)17 and T(reg) cell differentiation. Nature 2019, 576, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Jiang, X.; Xiao, Y.; Zhou, T.; Guo, Y.; Wang, R.; Zhao, Z.; Xiao, H.; Hou, C.; Ma, L.; et al. IL-17A signaling in colonic epithelial cells inhibits pro-inflammatory cytokine production by enhancing the activity of ERK and PI3K. PLoS ONE 2014, 9, e89714. [Google Scholar] [CrossRef] [Green Version]
- Chung, L.; Thiele Orberg, E.; Geis, A.L.; Chan, J.L.; Fu, K.; DeStefano Shields, C.E.; Dejea, C.M.; Fathi, P.; Chen, J.; Finard, B.B.; et al. Bacteroides fragilis Toxin Coordinates a Pro-carcinogenic Inflammatory Cascade via Targeting of Colonic Epithelial Cells. Cell Host Microbe 2018, 23, 203–214.e205. [Google Scholar] [CrossRef] [Green Version]
- Rutkowski, M.R.; Stephen, T.L.; Svoronos, N.; Allegrezza, M.J.; Tesone, A.J.; Perales-Puchalt, A.; Brencicova, E.; Escovar-Fadul, X.; Nguyen, J.M.; Cadungog, M.G.; et al. Microbially driven TLR5-dependent signaling governs distal malignant progression through tumor-promoting inflammation. Cancer Cell 2015, 27, 27–40. [Google Scholar] [CrossRef] [Green Version]
- Grivennikov, S.I.; Wang, K.; Mucida, D.; Stewart, C.A.; Schnabl, B.; Jauch, D.; Taniguchi, K.; Yu, G.-Y.; Osterreicher, C.H.; Hung, K.E.; et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature 2012, 491, 254–258. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Rhee, K.-J.; Albesiano, E.; Rabizadeh, S.; Wu, X.; Yen, H.-R.; Huso, D.L.; Brancati, F.L.; Wick, E.; McAllister, F.; et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat. Med. 2009, 15, 1016–1022. [Google Scholar] [CrossRef]
- Yang, Z.; Zhang, B.; Li, D.; Lv, M.; Huang, C.; Shen, G.X.; Huang, B. Mast cells mobilize myeloid-derived suppressor cells and Treg cells in tumor microenvironment via IL-17 pathway in murine hepatocarcinoma model. PLoS ONE 2010, 5, e8922. [Google Scholar] [CrossRef]
- De Aquino, S.G.; Abdollahi-Roodsaz, S.; Koenders, M.I.; van de Loo, F.A.J.; Pruijn, G.J.M.; Marijnissen, R.J.; Walgreen, B.; Helsen, M.M.; van den Bersselaar, L.A.; de Molon, R.S.; et al. Periodontal pathogens directly promote autoimmune experimental arthritis by inducing a TLR2- and IL-1-driven Th17 response. J. Immunol. 2014, 192, 4103–4111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calcinotto, A.; Brevi, A.; Chesi, M.; Ferrarese, R.; Garcia Perez, L.; Grioni, M.; Kumar, S.; Garbitt, V.M.; Sharik, M.E.; Henderson, K.J.; et al. Microbiota-driven interleukin-17-producing cells and eosinophils synergize to accelerate multiple myeloma progression. Nat. Commun. 2018, 9, 4832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hideshima, T.; Mitsiades, C.; Tonon, G.; Richardson, P.G.; Anderson, K.C. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat. Rev. Cancer 2007, 7, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yi, T.; Kortylewski, M.; Pardoll, D.M.; Zeng, D.; Yu, H. IL-17 can promote tumor growth through an IL-6-Stat3 signaling pathway. J. Exp. Med. 2009, 206, 1457–1464. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Ma, Y.; Wang, X.; Jiang, J.; Zhang, C.; Wang, X.; Jiang, Y.; Huang, H.; Hong, L. IL-17A Increases Multiple Myeloma Cell Viability by Positively Regulating Syk Expression. Transl. Oncol. 2019, 12, 1086–1091. [Google Scholar] [CrossRef]
- Ramirez-Carrozzi, V.; Sambandam, A.; Luis, E.; Lin, Z.; Jeet, S.; Lesch, J.; Hackney, J.; Kim, J.; Zhou, M.; Lai, J.; et al. IL-17C regulates the innate immune function of epithelial cells in an autocrine manner. Nat. Immunol. 2011, 12, 1159–1166. [Google Scholar] [CrossRef]
- Song, X.; Gao, H.; Lin, Y.; Yao, Y.; Zhu, S.; Wang, J.; Liu, Y.; Yao, X.; Meng, G.; Shen, N.; et al. Alterations in the microbiota drive interleukin-17C production from intestinal epithelial cells to promote tumorigenesis. Immunity 2014, 40, 140–152. [Google Scholar] [CrossRef] [Green Version]
- McGeachy, M.J.; Cua, D.J.; Gaffen, S.L. The IL-17 Family of Cytokines in Health and Disease. Immunity 2019, 50, 892–906. [Google Scholar] [CrossRef]
- Brevi, A.; Cogrossi, L.L.; Grazia, G.; Masciovecchio, D.; Impellizzieri, D.; Lacanfora, L.; Grioni, M.; Bellone, M. Much More Than IL-17A: Cytokines of the IL-17 Family Between Microbiota and Cancer. Front. Immunol. 2020, 11, 565470. [Google Scholar] [CrossRef]
- Miossec, P.; Kolls, J.K. Targeting IL-17 and TH17 cells in chronic inflammation. Nat. Rev. Drug Discov. 2012, 11, 763–776. [Google Scholar] [CrossRef]
- Chanmee, T.; Ontong, P.; Konno, K.; Itano, N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers 2014, 6, 1670–1690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, F.; Qin, X.; Wang, B.; Li, Q.; Hu, J.; Cheng, X.; Guo, D.; Cheng, F.; Fang, C.; Tan, Y.; et al. ALKBH5 Facilitates Hypoxia-Induced Paraspeckle Assembly and IL8 Secretion to Generate an Immunosuppressive Tumor Microenvironment. Cancer Res. 2021, 81, 5876–5888. [Google Scholar] [CrossRef] [PubMed]
- Wenes, M.; Shang, M.; Di Matteo, M.; Goveia, J.; Martín-Pérez, R.; Serneels, J.; Prenen, H.; Ghesquière, B.; Carmeliet, P.; Mazzone, M. Macrophage Metabolism Controls Tumor Blood Vessel Morphogenesis and Metastasis. Cell Metab. 2016, 24, 701–715. [Google Scholar] [CrossRef] [PubMed]
- Ahirwar, D.K.; Charan, M.; Mishra, S.; Verma, A.K.; Shilo, K.; Ramaswamy, B.; Ganju, R.K. Slit2 Inhibits Breast Cancer Metastasis by Activating M1-Like Phagocytic and Antifibrotic Macrophages. Cancer Res. 2021, 81, 5255–5267. [Google Scholar] [CrossRef]
- Espindola, M.S.; Habiel, D.M.; Narayanan, R.; Jones, I.; Coelho, A.L.; Murray, L.A.; Jiang, D.; Noble, P.W.; Hogaboam, C.M. Targeting of TAM Receptors Ameliorates Fibrotic Mechanisms in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2018, 197, 1443–1456. [Google Scholar] [CrossRef]
- Torres, S.; Bartolomé, R.A.; Mendes, M.; Barderas, R.; Fernandez-Aceñero, M.J.; Peláez-García, A.; Peña, C.; Lopez-Lucendo, M.; Villar-Vázquez, R.; de Herreros, A.G.; et al. Proteome profiling of cancer-associated fibroblasts identifies novel proinflammatory signatures and prognostic markers for colorectal cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 6006–6019. [Google Scholar] [CrossRef] [Green Version]
- Ruffell, B.; Coussens, L.M. Macrophages and therapeutic resistance in cancer. Cancer Cell 2015, 27, 462–472. [Google Scholar] [CrossRef] [Green Version]
- Italiani, P.; Boraschi, D. From Monocytes to M1/M2 Macrophages: Phenotypical vs. Functional Differentiation. Front. Immunol. 2014, 5, 514. [Google Scholar] [CrossRef] [Green Version]
- Bruns, H.; Büttner, M.; Fabri, M.; Mougiakakos, D.; Bittenbring, J.T.; Hoffmann, M.H.; Beier, F.; Pasemann, S.; Jitschin, R.; Hofmann, A.D.; et al. Vitamin D-dependent induction of cathelicidin in human macrophages results in cytotoxicity against high-grade B cell lymphoma. Sci. Transl. Med. 2015, 7, 282ra247. [Google Scholar] [CrossRef]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Yu, Y.; Wang, X.; Zhang, T. Tumor-Associated Macrophages in Tumor Immunity. Front. Immunol. 2020, 11, 583084. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Helming, L.; Gordon, S. Alternative activation of macrophages: An immunologic functional perspective. Annu. Rev. Immunol. 2009, 27, 451–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Dyken, S.J.; Locksley, R.M. Interleukin-4- and interleukin-13-mediated alternatively activated macrophages: Roles in homeostasis and disease. Annu. Rev. Immunol. 2013, 31, 317–343. [Google Scholar] [CrossRef] [PubMed]
- Redente, E.F.; Dwyer-Nield, L.D.; Merrick, D.T.; Raina, K.; Agarwal, R.; Pao, W.; Rice, P.L.; Shroyer, K.R.; Malkinson, A.M. Tumor progression stage and anatomical site regulate tumor-associated macrophage and bone marrow-derived monocyte polarization. Am. J. Pathol. 2010, 176, 2972–2985. [Google Scholar] [CrossRef]
- Kuang, D.M.; Zhao, Q.; Peng, C.; Xu, J.; Zhang, J.P.; Wu, C.; Zheng, L. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J. Exp. Med. 2009, 206, 1327–1337. [Google Scholar] [CrossRef] [Green Version]
- Curiel, T.J.; Coukos, G.; Zou, L.; Alvarez, X.; Cheng, P.; Mottram, P.; Evdemon-Hogan, M.; Conejo-Garcia, J.R.; Zhang, L.; Burow, M.; et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 2004, 10, 942–949. [Google Scholar] [CrossRef]
- Torroella-Kouri, M.; Silvera, R.; Rodriguez, D.; Caso, R.; Shatry, A.; Opiela, S.; Ilkovitch, D.; Schwendener, R.A.; Iragavarapu-Charyulu, V.; Cardentey, Y.; et al. Identification of a subpopulation of macrophages in mammary tumor-bearing mice that are neither M1 nor M2 and are less differentiated. Cancer Res. 2009, 69, 4800–4809. [Google Scholar] [CrossRef] [Green Version]
- Sami, E.; Paul, B.T.; Koziol, J.A.; ElShamy, W.M. The Immunosuppressive Microenvironment in BRCA1-IRIS-Overexpressing TNBC Tumors Is Induced by Bidirectional Interaction with Tumor-Associated Macrophages. Cancer Res. 2020, 80, 1102–1117. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Zhang, X.; Zhu, L.; Yang, X.; He, F.; Wang, T.; Bao, T.; Lu, H.; Wang, H.; Yang, S. Inulin alleviates inflammation of alcoholic liver disease via SCFAs-inducing suppression of M1 and facilitation of M2 macrophages in mice. Int. Immunopharmacol. 2020, 78, 106062. [Google Scholar] [CrossRef]
- Wang, L.; Gong, Z.; Zhang, X.; Zhu, F.; Liu, Y.; Jin, C.; Du, X.; Xu, C.; Chen, Y.; Cai, W.; et al. Gut microbial bile acid metabolite skews macrophage polarization and contributes to high-fat diet-induced colonic inflammation. Gut Microbes 2020, 12, 1819155. [Google Scholar] [CrossRef]
- Long, J.; Liu, X.K.; Kang, Z.P.; Wang, M.X.; Zhao, H.M.; Huang, J.Q.; Xiao, Q.P.; Liu, D.Y.; Zhong, Y.B. Ginsenoside Rg1 ameliorated experimental colitis by regulating the balance of M1/M2 macrophage polarization and the homeostasis of intestinal flora. Eur. J. Pharmacol. 2022, 917, 174742. [Google Scholar] [CrossRef] [PubMed]
- Kleer, C.G.; Bloushtain-Qimron, N.; Chen, Y.H.; Carrasco, D.; Hu, M.; Yao, J.; Kraeft, S.K.; Collins, L.C.; Sabel, M.S.; Argani, P.; et al. Epithelial and stromal cathepsin K and CXCL14 expression in breast tumor progression. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 5357–5367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novinec, M.; Lenarčič, B. Cathepsin K: A unique collagenolytic cysteine peptidase. Biol. Chem. 2013, 394, 1163–1179. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Ma, Y.; Zhang, Y.; Pan, Z.; Lu, Y.; Liu, P.; Lu, B. Identification of Cathepsin K in the Peritoneal Metastasis of Ovarian Carcinoma Using In-silico, Gene Expression Analysis. J. Cancer 2016, 7, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Zhou, R.; Wang, H.; Li, W.; Pan, M.; Yao, X.; Zhan, W.; Yang, S.; Xu, L.; Ding, Y.; et al. Gut microbiota-stimulated cathepsin K secretion mediates TLR4-dependent M2 macrophage polarization and promotes tumor metastasis in colorectal cancer. Cell Death Differ. 2019, 26, 2447–2463. [Google Scholar] [CrossRef]
- Chai, N.; Xiong, Y.; Zhang, Y.; Cheng, Y.; Shi, W.; Yao, Y.; Sui, H.; Zhu, H. YYFZBJS inhibits colorectal tumorigenesis by remodeling gut microbiota and influence on M2 macrophage polarization in vivo and in vitro. Am. J. Cancer Res. 2021, 11, 5338–5357. [Google Scholar] [CrossRef]
- Zmora, N.; Suez, J.; Elinav, E. You are what you eat: Diet, health and the gut microbiota. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 35–56. [Google Scholar] [CrossRef] [Green Version]
- O’Keefe, S.J.; Li, J.V.; Lahti, L.; Ou, J.; Carbonero, F.; Mohammed, K.; Posma, J.M.; Kinross, J.; Wahl, E.; Ruder, E.; et al. Fat, fibre and cancer risk in African Americans and rural Africans. Nat. Commun. 2015, 6, 6342. [Google Scholar] [CrossRef] [Green Version]
- Flint, H.J.; Duncan, S.H.; Scott, K.P.; Louis, P. Links between diet, gut microbiota composition and gut metabolism. Proc. Nutr. Soc. 2015, 74, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Guo, Z.; Song, X.; Liu, L.; Dong, W.; Wang, S.; Xu, M.; Yang, C.; Wang, B.; Cao, H. High-fat diet-induced dysbiosis mediates MCP-1/CCR2 axis-dependent M2 macrophage polarization and promotes intestinal adenoma-adenocarcinoma sequence. J. Cell. Mol. Med. 2020, 24, 2648–2662. [Google Scholar] [CrossRef] [Green Version]
- Hezaveh, K.; Shinde, R.S.; Klötgen, A.; Halaby, M.J.; Lamorte, S.; Ciudad, M.T.; Quevedo, R.; Neufeld, L.; Liu, Z.Q.; Jin, R.; et al. Tryptophan-derived microbial metabolites activate the aryl hydrocarbon receptor in tumor-associated macrophages to suppress anti-tumor immunity. Immunity 2022, 55, 324–340.e328. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Zitvogel, L.; Palucka, A.K. Neutralizing tumor-promoting chronic inflammation: A magic bullet? Science 2013, 339, 286–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchem, J.B.; Brennan, D.J.; Knolhoff, B.L.; Belt, B.A.; Zhu, Y.; Sanford, D.E.; Belaygorod, L.; Carpenter, D.; Collins, L.; Piwnica-Worms, D.; et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013, 73, 1128–1141. [Google Scholar] [CrossRef] [PubMed]
- DeNardo, D.G.; Brennan, D.J.; Rexhepaj, E.; Ruffell, B.; Shiao, S.L.; Madden, S.F.; Gallagher, W.M.; Wadhwani, N.; Keil, S.D.; Junaid, S.A.; et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011, 1, 54–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, S.; Niu, M.; O’Mary, H.; Cui, Z. Targeting of tumor-associated macrophages made possible by PEG-sheddable, mannose-modified nanoparticles. Mol. Pharm. 2013, 10, 3525–3530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagai, T.; Tanaka, M.; Tsuneyoshi, Y.; Xu, B.; Michie, S.A.; Hasui, K.; Hirano, H.; Arita, K.; Matsuyama, T. Targeting tumor-associated macrophages in an experimental glioma model with a recombinant immunotoxin to folate receptor beta. Cancer Immunol. Immunother. CII 2009, 58, 1577–1586. [Google Scholar] [CrossRef]
- Lin, Y.; Wei, C.; Liu, Y.; Qiu, Y.; Liu, C.; Guo, F. Selective ablation of tumor-associated macrophages suppresses metastasis and angiogenesis. Cancer Sci. 2013, 104, 1217–1225. [Google Scholar] [CrossRef]
- Cieslewicz, M.; Tang, J.; Yu, J.L.; Cao, H.; Zavaljevski, M.; Motoyama, K.; Lieber, A.; Raines, E.W.; Pun, S.H. Targeted delivery of proapoptotic peptides to tumor-associated macrophages improves survival. Proc. Natl. Acad. Sci. USA 2013, 110, 15919–15924. [Google Scholar] [CrossRef] [Green Version]
- Fan, L.; Xu, C.; Ge, Q.; Lin, Y.; Wong, C.C.; Qi, Y.; Ye, B.; Lian, Q.; Zhuo, W.; Si, J.; et al. A. Muciniphila Suppresses Colorectal Tumorigenesis by Inducing TLR2/NLRP3-Mediated M1-Like TAMs. Cancer Immunol. Res. 2021, 9, 1111–1124. [Google Scholar] [CrossRef]
- Yang, Y.; Weng, W.; Peng, J.; Hong, L.; Yang, L.; Toiyama, Y.; Gao, R.; Liu, M.; Yin, M.; Pan, C.; et al. Fusobacterium nucleatum Increases Proliferation of Colorectal Cancer Cells and Tumor Development in Mice by Activating Toll-Like Receptor 4 Signaling to Nuclear Factor-κB, and Up-regulating Expression of MicroRNA-21. Gastroenterology 2017, 152, 851–866.e824. [Google Scholar] [CrossRef] [Green Version]
- Hong, J.; Guo, F.; Lu, S.Y.; Shen, C.; Ma, D.; Zhang, X.; Xie, Y.; Yan, T.; Yu, T.; Sun, T.; et al. F. nucleatum targets lncRNA ENO1-IT1 to promote glycolysis and oncogenesis in colorectal cancer. Gut 2021, 70, 2123–2137. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Fan, L.; Lin, Y.; Shen, W.; Qi, Y.; Zhang, Y.; Chen, Z.; Wang, L.; Long, Y.; Hou, T.; et al. Fusobacterium nucleatum promotes colorectal cancer metastasis through miR-1322/CCL20 axis and M2 polarization. Gut Microbes 2021, 13, 1980347. [Google Scholar] [CrossRef]
- Mima, K.; Sukawa, Y.; Nishihara, R.; Qian, Z.R.; Yamauchi, M.; Inamura, K.; Kim, S.A.; Masuda, A.; Nowak, J.A.; Nosho, K.; et al. Fusobacterium nucleatum and T Cells in Colorectal Carcinoma. JAMA Oncol. 2015, 1, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Nosho, K.; Sukawa, Y.; Adachi, Y.; Ito, M.; Mitsuhashi, K.; Kurihara, H.; Kanno, S.; Yamamoto, I.; Ishigami, K.; Igarashi, H.; et al. Association of Fusobacterium nucleatum with immunity and molecular alterations in colorectal cancer. World J. Gastroenterol. 2016, 22, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Borowsky, J.; Haruki, K.; Lau, M.C.; Dias Costa, A.; Väyrynen, J.P.; Ugai, T.; Arima, K.; da Silva, A.; Felt, K.D.; Zhao, M.; et al. Association of Fusobacterium nucleatum with Specific T-cell Subsets in the Colorectal Carcinoma Microenvironment. Clin. Cancer Res. 2021, 27, 2816–2826. [Google Scholar] [CrossRef] [PubMed]
- Parhi, L.; Alon-Maimon, T.; Sol, A.; Nejman, D.; Shhadeh, A.; Fainsod-Levi, T.; Yajuk, O.; Isaacson, B.; Abed, J.; Maalouf, N.; et al. Breast cancer colonization by Fusobacterium nucleatum accelerates tumor growth and metastatic progression. Nat. Commun. 2020, 11, 3259. [Google Scholar] [CrossRef] [PubMed]
- Gur, C.; Ibrahim, Y.; Isaacson, B.; Yamin, R.; Abed, J.; Gamliel, M.; Enk, J.; Bar-On, Y.; Stanietsky-Kaynan, N.; Coppenhagen-Glazer, S.; et al. Binding of the Fap2 protein of Fusobacterium nucleatum to human inhibitory receptor TIGIT protects tumors from immune cell attack. Immunity 2015, 42, 344–355. [Google Scholar] [CrossRef] [Green Version]
- Chu, H.; Mazmanian, S.K. Innate immune recognition of the microbiota promotes host-microbial symbiosis. Nat. Immunol. 2013, 14, 668–675. [Google Scholar] [CrossRef] [Green Version]
- Round, J.L.; Lee, S.M.; Li, J.; Tran, G.; Jabri, B.; Chatila, T.A.; Mazmanian, S.K. The Toll-like receptor 2 pathway establishes colonization by a commensal of the human microbiota. Science 2011, 332, 974–977. [Google Scholar] [CrossRef] [Green Version]
- Mazmanian, S.K.; Liu, C.H.; Tzianabos, A.O.; Kasper, D.L. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell 2005, 122, 107–118. [Google Scholar] [CrossRef] [Green Version]
- Jarnicki, A.G.; Lysaght, J.; Todryk, S.; Mills, K.H.G. Suppression of antitumor immunity by IL-10 and TGF-beta-producing T cells infiltrating the growing tumor: Influence of tumor environment on the induction of CD4+ and CD8+ regulatory T cells. J. Immunol. 2006, 177, 896–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wing, K.; Onishi, Y.; Prieto-Martin, P.; Yamaguchi, T.; Miyara, M.; Fehervari, Z.; Nomura, T.; Sakaguchi, S. CTLA-4 control over Foxp3+ regulatory T cell function. Science 2008, 322, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Peuker, K.; Strigli, A.; Tauriello, D.V.F.; Hendricks, A.; von Schönfels, W.; Burmeister, G.; Brosch, M.; Herrmann, A.; Krüger, S.; Nitsche, J.; et al. Microbiota-dependent activation of the myeloid calcineurin-NFAT pathway inhibits B7H3- and B7H4-dependent anti-tumor immunity in colorectal cancer. Immunity 2022, 55, 701–717.e707. [Google Scholar] [CrossRef] [PubMed]
- Uribe-Herranz, M.; Bittinger, K.; Rafail, S.; Guedan, S.; Pierini, S.; Tanes, C.; Ganetsky, A.; Morgan, M.A.; Gill, S.; Tanyi, J.L.; et al. Gut microbiota modulates adoptive cell therapy via CD8α dendritic cells and IL-12. JCI Insight 2018, 3, e94952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uribe-Herranz, M.; Rafail, S.; Beghi, S.; Gil-de-Gómez, L.; Verginadis, I.; Bittinger, K.; Pustylnikov, S.; Pierini, S.; Perales-Linares, R.; Blair, I.A.; et al. Gut microbiota modulate dendritic cell antigen presentation and radiotherapy-induced antitumor immune response. J. Clin. Investig. 2020, 130, 466–479. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Dubois, R.N. Eicosanoids and cancer. Nat. Rev. Cancer 2010, 10, 181–193. [Google Scholar] [CrossRef]
- Mao, Y.; Poschke, I.; Wennerberg, E.; Pico de Coaña, Y.; Egyhazi Brage, S.; Schultz, I.; Hansson, J.; Masucci, G.; Lundqvist, A.; Kiessling, R. Melanoma-educated CD14+ cells acquire a myeloid-derived suppressor cell phenotype through COX-2-dependent mechanisms. Cancer Res. 2013, 73, 3877–3887. [Google Scholar] [CrossRef] [Green Version]
- O’Callaghan, G.; Ryan, A.; Neary, P.; O’Mahony, C.; Shanahan, F.; Houston, A. Targeting the EP1 receptor reduces Fas ligand expression and increases the antitumor immune response in an in vivo model of colon cancer. Int. J. Cancer 2013, 133, 825–834. [Google Scholar] [CrossRef] [Green Version]
- Loo, T.M.; Kamachi, F.; Watanabe, Y.; Yoshimoto, S.; Kanda, H.; Arai, Y.; Nakajima-Takagi, Y.; Iwama, A.; Koga, T.; Sugimoto, Y.; et al. Gut Microbiota Promotes Obesity-Associated Liver Cancer through PGE-Mediated Suppression of Antitumor Immunity. Cancer Discov. 2017, 7, 522–538. [Google Scholar] [CrossRef] [Green Version]
- Mizuno, R.; Kawada, K.; Sakai, Y. Prostaglandin E2/EP Signaling in the Tumor Microenvironment of Colorectal Cancer. Int. J. Mol. Sci. 2019, 20, 6254. [Google Scholar] [CrossRef] [Green Version]
- Fu, A.; Yao, B.; Dong, T.; Chen, Y.; Yao, J.; Liu, Y.; Li, H.; Bai, H.; Liu, X.; Zhang, Y.; et al. Tumor-resident intracellular microbiota promotes metastatic colonization in breast cancer. Cell 2022, 185, 1356–1372.e1326. [Google Scholar] [CrossRef] [PubMed]
- Nejman, D.; Livyatan, I.; Fuks, G.; Gavert, N.; Zwang, Y.; Geller, L.T.; Rotter-Maskowitz, A.; Weiser, R.; Mallel, G.; Gigi, E.; et al. The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science 2020, 368, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Pushalkar, S.; Hundeyin, M.; Daley, D.; Zambirinis, C.P.; Kurz, E.; Mishra, A.; Mohan, N.; Aykut, B.; Usyk, M.; Torres, L.E.; et al. The Pancreatic Cancer Microbiome Promotes Oncogenesis by Induction of Innate and Adaptive Immune Suppression. Cancer Discov. 2018, 8, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Lagoudas, G.K.; Zhao, C.; Bullman, S.; Bhutkar, A.; Hu, B.; Ameh, S.; Sandel, D.; Liang, X.S.; Mazzilli, S.; et al. Commensal Microbiota Promote Lung Cancer Development via γδ T Cells. Cell 2019, 176, 998–1013.e1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Cao, X. Immunosuppressive cells in tumor immune escape and metastasis. J. Mol. Med. 2016, 94, 509–522. [Google Scholar] [CrossRef]
- Minton, K. Immunotherapy: Cytokine boost for CAR T cells. Nat. Rev. Immunol. 2018, 18, 150–151. [Google Scholar] [CrossRef]
- Pulendran, B.; Davis, M.M. The science and medicine of human immunology. Science 2020, 369. [Google Scholar] [CrossRef]
- Demaria, O.; Cornen, S.; Daëron, M.; Morel, Y.; Medzhitov, R.; Vivier, E. Harnessing innate immunity in cancer therapy. Nature 2019, 574, 45–56. [Google Scholar] [CrossRef]
- Gopalakrishnan, V.; Helmink, B.A.; Spencer, C.N.; Reuben, A.; Wargo, J.A. The Influence of the Gut Microbiome on Cancer, Immunity, and Cancer Immunotherapy. Cancer Cell 2018, 33, 570–580. [Google Scholar] [CrossRef] [Green Version]
- Iida, N.; Dzutsev, A.; Stewart, C.A.; Smith, L.; Bouladoux, N.; Weingarten, R.A.; Molina, D.A.; Salcedo, R.; Back, T.; Cramer, S.; et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science 2013, 342, 967–970. [Google Scholar] [CrossRef]
- Di Modica, M.; Gargari, G.; Regondi, V.; Bonizzi, A.; Arioli, S.; Belmonte, B.; De Cecco, L.; Fasano, E.; Bianchi, F.; Bertolotti, A.; et al. Gut Microbiota Condition the Therapeutic Efficacy of Trastuzumab in HER2-Positive Breast Cancer. Cancer Res. 2021, 81, 2195–2206. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef]
- Carlino, M.S.; Larkin, J.; Long, G.V. Immune checkpoint inhibitors in melanoma. Lancet 2021, 398, 1002–1014. [Google Scholar] [CrossRef]
- Pan, C.; Liu, H.; Robins, E.; Song, W.; Liu, D.; Li, Z.; Zheng, L. Next-generation immuno-oncology agents: Current momentum shifts in cancer immunotherapy. J. Hematol. Oncol. 2020, 13, 29. [Google Scholar] [CrossRef] [Green Version]
- Matson, V.; Fessler, J.; Bao, R.; Chongsuwat, T.; Zha, Y.; Alegre, M.L.; Luke, J.J.; Gajewski, T.F. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 2018, 359, 104–108. [Google Scholar] [CrossRef] [Green Version]
- Davar, D.; Dzutsev, A.K.; McCulloch, J.A.; Rodrigues, R.R.; Chauvin, J.M.; Morrison, R.M.; Deblasio, R.N.; Menna, C.; Ding, Q.; Pagliano, O.; et al. Fecal microbiota transplant overcomes resistance to anti-PD-1 therapy in melanoma patients. Science 2021, 371, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Paulos, C.M.; Wrzesinski, C.; Kaiser, A.; Hinrichs, C.S.; Chieppa, M.; Cassard, L.; Palmer, D.C.; Boni, A.; Muranski, P.; Yu, Z.; et al. Microbial translocation augments the function of adoptively transferred self/tumor-specific CD8+ T cells via TLR4 signaling. J. Clin. Investig. 2007, 117, 2197–2204. [Google Scholar] [CrossRef] [Green Version]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Cho, S.Y.; Yoon, Y.; Park, C.; Sohn, J.; Jeong, J.J.; Jeon, B.N.; Jang, M.; An, C.; Lee, S.; et al. Bifidobacterium bifidum strains synergize with immune checkpoint inhibitors to reduce tumour burden in mice. Nat. Microbiol. 2021, 6, 277–288. [Google Scholar] [CrossRef]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vétizou, M.; Pitt, J.M.; Daillère, R.; Lepage, P.; Waldschmitt, N.; Flament, C.; Rusakiewicz, S.; Routy, B.; Roberti, M.P.; Duong, C.P.; et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 2015, 350, 1079–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clavel, T.; Gomes-Neto, J.C.; Lagkouvardos, I.; Ramer-Tait, A.E. Deciphering interactions between the gut microbiota and the immune system via microbial cultivation and minimal microbiomes. Immunol. Rev. 2017, 279, 8–22. [Google Scholar] [CrossRef]
- Mager, L.F.; Burkhard, R.; Pett, N.; Cooke, N.C.A.; Brown, K.; Ramay, H.; Paik, S.; Stagg, J.; Groves, R.A.; Gallo, M.; et al. Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science 2020, 369, 1481–1489. [Google Scholar] [CrossRef]
- Coutzac, C.; Jouniaux, J.M.; Paci, A.; Schmidt, J.; Mallardo, D.; Seck, A.; Asvatourian, V.; Cassard, L.; Saulnier, P.; Lacroix, L.; et al. Systemic short chain fatty acids limit antitumor effect of CTLA-4 blockade in hosts with cancer. Nat. Commun. 2020, 11, 2168. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Lv, J.; Guo, F.; Li, J.; Jia, Y.; Jiang, D.; Wang, N.; Zhang, C.; Kong, L.; Liu, Y.; et al. Gut Microbiome Influences the Efficacy of PD-1 Antibody Immunotherapy on MSS-Type Colorectal Cancer via Metabolic Pathway. Front. Microbiol. 2020, 11, 814. [Google Scholar] [CrossRef]
- Holland, T.; Fowler, V.G., Jr.; Shelburne, S.A., 3rd. Invasive gram-positive bacterial infection in cancer patients. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2014, 59 (Suppl. 5), S331–S334. [Google Scholar] [CrossRef] [Green Version]
- Bucaneve, G.; Micozzi, A.; Menichetti, F.; Martino, P.; Dionisi, M.S.; Martinelli, G.; Allione, B.; D’Antonio, D.; Buelli, M.; Nosari, A.M.; et al. Levofloxacin to prevent bacterial infection in patients with cancer and neutropenia. N. Engl. J. Med. 2005, 353, 977–987. [Google Scholar] [CrossRef] [Green Version]
- Attiê, R.; Chinen, L.T.; Yoshioka, E.M.; Silva, M.C.; de Lima, V.C. Acute bacterial infection negatively impacts cancer specific survival of colorectal cancer patients. World J. Gastroenterol. 2014, 20, 13930–13935. [Google Scholar] [CrossRef]
- Johanesen, P.A.; Mackin, K.E.; Hutton, M.L.; Awad, M.M.; Larcombe, S.; Amy, J.M.; Lyras, D. Disruption of the Gut Microbiome: Clostridium difficile Infection and the Threat of Antibiotic Resistance. Genes 2015, 6, 1347–1360. [Google Scholar] [CrossRef]
- Leffler, D.A.; Lamont, J.T. Clostridium difficile infection. N. Engl. J. Med. 2015, 372, 1539–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lastours, V.D.; Fantin, B. Resistance to fluoroquinolones in 2013: What are the consequences in internal medicine? Rev. Med. Interne 2014, 35, 601–608. [Google Scholar] [PubMed]
- Galloway-Peña, J.; Brumlow, C.; Shelburne, S. Impact of the Microbiota on Bacterial Infections during Cancer Treatment. Trends Microbiol. 2017, 25, 992–1004. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.B.; Zhou, Y.L.; Fang, J.Y. Gut Microbiota in Cancer Immune Response and Immunotherapy. Trends Cancer 2021, 7, 647–660. [Google Scholar] [CrossRef] [PubMed]
- Lurienne, L.; Cervesi, J.; Duhalde, L.; de Gunzburg, J.; Andremont, A.; Zalcman, G.; Buffet, R.; Bandinelli, P.A. NSCLC Immunotherapy Efficacy and Antibiotic Use: A Systematic Review and Meta-Analysis. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2020, 15, 1147–1159. [Google Scholar] [CrossRef]
- Pinato, D.J.; Howlett, S.; Ottaviani, D.; Urus, H.; Patel, A.; Mineo, T.; Brock, C.; Power, D.; Hatcher, O.; Falconer, A.; et al. Association of Prior Antibiotic Treatment With Survival and Response to Immune Checkpoint Inhibitor Therapy in Patients With Cancer. JAMA Oncol. 2019, 5, 1774–1778. [Google Scholar] [CrossRef]
- Lehrnbecher, T.; Fisher, B.T.; Phillips, B.; Alexander, S.; Ammann, R.A.; Beauchemin, M.; Carlesse, F.; Castagnola, E.; Davis, B.L.; Dupuis, L.L.; et al. Guideline for Antibacterial Prophylaxis Administration in Pediatric Cancer and Hematopoietic Stem Cell Transplantation. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2020, 71, 226–236. [Google Scholar] [CrossRef] [Green Version]
- Goldman, J.D.; Gallaher, A.; Jain, R.; Stednick, Z.; Menon, M.; Boeckh, M.J.; Pottinger, P.S.; Schwartz, S.M.; Casper, C. Infusion-Compatible Antibiotic Formulations for Rapid Administration to Improve Outcomes in Cancer Outpatients With Severe Sepsis and Septic Shock: The Sepsis STAT Pack. J. Natl. Compr. Cancer Netw. JNCCN 2017, 15, 457–464. [Google Scholar] [CrossRef]
- Baruch, E.N.; Youngster, I.; Ben-Betzalel, G.; Ortenberg, R.; Lahat, A.; Katz, L.; Adler, K.; Dick-Necula, D.; Raskin, S.; Bloch, N.; et al. Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science 2021, 371, 602–609. [Google Scholar] [CrossRef]
- Spencer, C.N.; McQuade, J.L.; Gopalakrishnan, V.; McCulloch, J.A.; Vetizou, M.; Cogdill, A.P.; Khan, M.A.W.; Zhang, X.; White, M.G.; Peterson, C.B.; et al. Dietary fiber and probiotics influence the gut microbiome and melanoma immunotherapy response. Science 2021, 374, 1632–1640. [Google Scholar] [CrossRef]
- de Gunzburg, J.; Ghozlane, A.; Ducher, A.; Le Chatelier, E.; Duval, X.; Ruppé, E.; Armand-Lefevre, L.; Sablier-Gallis, F.; Burdet, C.; Alavoine, L.; et al. Protection of the Human Gut Microbiome From Antibiotics. J. Infect. Dis. 2018, 217, 628–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khoder, M.; Tsapis, N.; Domergue-Dupont, V.; Gueutin, C.; Fattal, E. Removal of residual colonic ciprofloxacin in the rat by activated charcoal entrapped within zinc-pectinate beads. Eur. J. Pharm. Sci. Off. J. Eur. Fed. Pharm. Sci. 2010, 41, 281–288. [Google Scholar] [CrossRef]
- de Gunzburg, J.; Ducher, A.; Modess, C.; Wegner, D.; Oswald, S.; Dressman, J.; Augustin, V.; Feger, C.; Andremont, A.; Weitschies, W.; et al. Targeted adsorption of molecules in the colon with the novel adsorbent-based medicinal product, DAV132: A proof of concept study in healthy subjects. J. Clin. Pharmacol. 2015, 55, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Léonard, F.; Andremont, A.; Leclerq, B.; Labia, R.; Tancrède, C. Use of beta-lactamase-producing anaerobes to prevent ceftriaxone from degrading intestinal resistance to colonization. J. Infect. Dis. 1989, 160, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Stiefel, U.; Pultz, N.J.; Harmoinen, J.; Koski, P.; Lindevall, K.; Helfand, M.S.; Donskey, C.J. Oral administration of beta-lactamase preserves colonization resistance of piperacillin-treated mice. J. Infect. Dis. 2003, 188, 1605–1609. [Google Scholar] [CrossRef] [Green Version]
- Stiefel, U.; Nerandzic, M.M.; Koski, P.; Donskey, C.J. Orally administered beta-lactamase enzymes represent a novel strategy to prevent colonization by Clostridium difficile. J. Antimicrob. Chemother. 2008, 62, 1105–1108. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Restifo, N.P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015, 348, 62–68. [Google Scholar] [CrossRef] [Green Version]
- Verdegaal, E.M. Adoptive cell therapy: A highly successful individualized therapy for melanoma with great potential for other malignancies. Curr. Opin. Immunol. 2016, 39, 90–95. [Google Scholar] [CrossRef]
- Luu, M.; Weigand, K.; Wedi, F.; Breidenbend, C.; Leister, H.; Pautz, S.; Adhikary, T.; Visekruna, A. Regulation of the effector function of CD8(+) T cells by gut microbiota-derived metabolite butyrate. Sci. Rep. 2018, 8, 14430. [Google Scholar] [CrossRef] [Green Version]
- Cremonesi, E.; Governa, V.; Garzon, J.F.G.; Mele, V.; Amicarella, F.; Muraro, M.G.; Trella, E.; Galati-Fournier, V.; Oertli, D.; Däster, S.R.; et al. Gut microbiota modulate T cell trafficking into human colorectal cancer. Gut 2018, 67, 1984–1994. [Google Scholar] [CrossRef]
- Snyder, A.; Pamer, E.; Wolchok, J. IMMUNOTHERAPY. Could microbial therapy boost cancer immunotherapy? Science 2015, 350, 1031–1032. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Yin, Q.; Chen, L.; Davis, M.M. Bifidobacterium can mitigate intestinal immunopathology in the context of CTLA-4 blockade. Proc. Natl. Acad. Sci. USA 2018, 115, 157–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinay, D.S.; Ryan, E.P.; Pawelec, G.; Talib, W.H.; Stagg, J.; Elkord, E.; Lichtor, T.; Decker, W.K.; Whelan, R.L.; Kumara, H.; et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin. Cancer Biol. 2015, 35, S185–S198. [Google Scholar] [CrossRef]
- Han, B.; Sivaramakrishnan, P.; Lin, C.J.; Neve, I.A.A.; He, J.; Tay, L.W.R.; Sowa, J.N.; Sizovs, A.; Du, G.; Wang, J.; et al. Microbial Genetic Composition Tunes Host Longevity. Cell 2017, 169, 1249–1262.e1213. [Google Scholar] [CrossRef] [Green Version]
- Giménez-Bastida, J.A.; Ávila-Gálvez, M.; Espín, J.C.; González-Sarrías, A. The gut microbiota metabolite urolithin A, but not other relevant urolithins, induces p53-dependent cellular senescence in human colon cancer cells. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2020, 139, 111260. [Google Scholar] [CrossRef] [PubMed]
- Sugimura, N.; Li, Q.; Chu, E.S.H.; Lau, H.C.H.; Fong, W.; Liu, W.; Liang, C.; Nakatsu, G.; Su, A.C.Y.; Coker, O.O.; et al. Lactobacillus gallinarum modulates the gut microbiota and produces anti-cancer metabolites to protect against colorectal tumourigenesis. Gut 2021, 71, 2011–2021. [Google Scholar] [CrossRef]
- Muir, R.M.; Ibáñez, A.M.; Uratsu, S.L.; Ingham, E.S.; Leslie, C.A.; McGranahan, G.H.; Batra, N.; Goyal, S.; Joseph, J.; Jemmis, E.D.; et al. Mechanism of gallic acid biosynthesis in bacteria (Escherichia coli) and walnut (Juglans regia). Plant Mol. Biol. 2011, 75, 555–565. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, Y.; Huang, J.; Li, Q.; Xia, W.; Zhang, C.; Liu, Z.; Xiao, J.; Yi, Z.; Deng, H.; Xiao, Z.; et al. Gut Microbiota and Tumor Immune Escape: A New Perspective for Improving Tumor Immunotherapy. Cancers 2022, 14, 5317. https://doi.org/10.3390/cancers14215317
He Y, Huang J, Li Q, Xia W, Zhang C, Liu Z, Xiao J, Yi Z, Deng H, Xiao Z, et al. Gut Microbiota and Tumor Immune Escape: A New Perspective for Improving Tumor Immunotherapy. Cancers. 2022; 14(21):5317. https://doi.org/10.3390/cancers14215317
Chicago/Turabian StyleHe, Yunbo, Jinliang Huang, Qiaorong Li, Weiping Xia, Chunyu Zhang, Zhi Liu, Jiatong Xiao, Zhenglin Yi, Hao Deng, Zicheng Xiao, and et al. 2022. "Gut Microbiota and Tumor Immune Escape: A New Perspective for Improving Tumor Immunotherapy" Cancers 14, no. 21: 5317. https://doi.org/10.3390/cancers14215317