
K-RAS Associated Gene-Mutation-Based Algorithm for Prediction of Treatment Response of Patients with Subtypes of Breast Cancer and Especially Triple-Negative Cancer

 ,
,  and
and
Abstract
:Simple Summary
Abstract
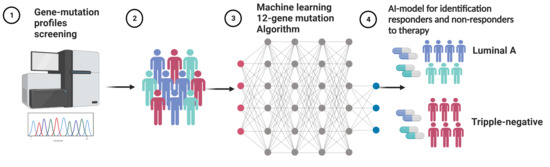
1. Introduction
2. Materials and Methods
2.1. Breast Cancer TCGA Cohort
2.2. Breast Cancer MSK Cohort
2.3. Machine Learning Algorithms
2.4. Statistical Analysis
3. Results
3.1. Development of the 12-Gene Algorithm for Stratification of Responder and Non-Responder Patients to Predict Treatment Response
3.2. Assessment of the 12-Gene Algorithm for Prediction of Progression-Free Survival after First-Line Therapy in the TCGA Cohort
3.3. The 12-Gene Algorithm as a Predictive Biomarker for Treatment Response in the TCGA Cohort
3.4. Validation of the 7-Gene Algorithm in the MSK Cohort
3.5. Assessment of the 12-Gene Algorithm as a Predictive Biomarker for PFS in the MSK Cohort
3.6. Validation of the 12-Gene Algorithm as an Independent PFS Predictor in the MSK Cohort
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lim, E.; Metzger-Filho, O.; Winer, E.P. The natural history of hormone receptor-positive breast cancer. Oncology 2012, 26, 688–694+696. [Google Scholar]
- Sørlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; Van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [Green Version]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; Van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [Green Version]
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-Negative Breast Cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef] [Green Version]
- Solzak, J.P.; Atale, R.V.; Hancock, B.A.; Sinn, A.L.; Pollok, K.E.; Jones, D.R.; Radovich, M. Dual PI3K and Wnt pathway inhibition is a synergistic combination against triple negative breast cancer. NPJ Breast Cancer 2017, 3, 17. [Google Scholar] [CrossRef] [Green Version]
- Hudis, C.A.; Gianni, L. Triple-Negative Breast Cancer: An Unmet Medical Need. Oncologist 2011, 16, 1–11. [Google Scholar] [CrossRef] [Green Version]
- NICE. Early and Locally Advanced Breast Cancer: Diagnosis and Management. NICE Guideline [NG101]. Available online: https://www.nice.org.uk/guidance/ng101 (accessed on 1 January 2022).
- Beck, A.H.; Sangoi, A.R.; Leung, S.; Marinelli, R.J.; Nielsen, T.O.; Van de Vijver, M.J.; West, R.B.; Van de Rijn, M.; Koller, D. Systematic Analysis of Breast Cancer Morphology Uncovers Stromal Features Associated with Survival. Sci. Transl. Med. 2011, 3, 108ra113. [Google Scholar] [CrossRef] [Green Version]
- Desmedt, C.; Haibe-Kains, B.; Wirapati, P.; Buyse, M.; Larsimont, D.; Bontempi, G.; Delorenzi, M.; Piccart, M.; Sotiriou, C. Biological Processes Associated with Breast Cancer Clinical Outcome Depend on the Molecular Subtypes. Clin. Cancer Res. 2008, 14, 5158–5165. [Google Scholar] [CrossRef] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Razavi, P.; Chang, M.T.; Xu, G.; Bandlamudi, C.; Ross, D.S.; Vasan, N.; Cai, Y.; Bielski, C.M.; Donoghue, M.T.A.; Jonsson, P.; et al. The Genomic Landscape of Endocrine-Resistant Advanced Breast Cancers. Cancer Cell 2018, 34, 427–438.e426. [Google Scholar] [CrossRef] [Green Version]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A Comprehensive Survey of Ras Mutations in Cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.-K.; Suh, Y.; Yoo, K.-C.; Cui, Y.-H.; Kim, H.; Kim, M.-J.; Gyu Kim, I.; Lee, S.-J. Activation of KRAS promotes the mesenchymal features of basal-type breast cancer. Exp. Mol. Med. 2015, 47, e137. [Google Scholar] [CrossRef] [Green Version]
- Fruman, D.A.; Rommel, C. PI3K and cancer: Lessons, challenges and opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefort, S.; Tan, S.; Balani, S.; Rafn, B.; Pellacani, D.; Hirst, M.; Sorensen, P.H.; Eaves, C.J. Initiation of human mammary cell tumorigenesis by mutant KRAS requires YAP inactivation. Oncogene 2020, 39, 1957–1968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the undruggable RAS: Mission Possible? Nat. Rev. Drug Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGlynn, L.M.; Kirkegaard, T.; Edwards, J.; Tovey, S.; Cameron, D.; Twelves, C.; Bartlett, J.M.S.; Cooke, T.G. Ras/Raf-1/MAPK Pathway Mediates Response to Tamoxifen but not Chemotherapy in Breast Cancer Patients. Clin. Cancer Res. 2009, 15, 1487–1495. [Google Scholar] [CrossRef] [Green Version]
- Rani, A.; Stebbing, J.; Giamas, G.; Murphy, J. Endocrine Resistance in Hormone Receptor Positive Breast Cancer–From Mechanism to Therapy. Front. Endocrinol. 2019, 10, 245. [Google Scholar] [CrossRef] [Green Version]
- Mendes-Pereira, A.M.; Sims, D.; Dexter, T.; Fenwick, K.; Assiotis, I.; Kozarewa, I.; Mitsopoulos, C.; Hakas, J.; Zvelebil, M.; Lord, C.J.; et al. Genome-wide functional screen identifies a compendium of genes affecting sensitivity to tamoxifen. Proc. Natl. Acad. Sci. USA 2012, 109, 2730–2735. [Google Scholar] [CrossRef] [Green Version]
- Ciriello, G.; Gatza Michael, L.; Beck Andrew, H.; Wilkerson Matthew, D.; Rhie Suhn, K.; Pastore, A.; Zhang, H.; McLellan, M.; Yau, C.; Kandoth, C.; et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell 2015, 163, 506–519. [Google Scholar] [CrossRef] [Green Version]
- Augello, M.A.; Hickey, T.E.; Knudsen, K.E. FOXA1: Master of steroid receptor function in cancer. EMBO J. 2011, 30, 3885–3894. [Google Scholar] [CrossRef] [Green Version]
- Djuric, U.; Zadeh, G.; Aldape, K.; Diamandis, P. Precision histology: How deep learning is poised to revitalize histomorphology for personalized cancer care. NPJ Precis. Oncol. 2017, 1, 22. [Google Scholar] [CrossRef] [PubMed]
- Johnson, H.; Guo, J.; Zhang, X.; Zhang, H.; Simoulis, A.; Wu, A.H.B.; Xia, T.; Li, F.; Tan, W.; Johnson, A.; et al. Development and validation of a 25-Gene Panel urine test for prostate cancer diagnosis and potential treatment follow-up. BMC Med. 2020, 18, 376. [Google Scholar] [CrossRef] [PubMed]
- Sammut, S.-J.; Crispin-Ortuzar, M.; Chin, S.-F.; Provenzano, E.; Bardwell, H.A.; Ma, W.; Cope, W.; Dariush, A.; Dawson, S.-J.; Abraham, J.E.; et al. Multi-omic machine learning predictor of breast cancer therapy response. Nature 2022, 601, 623–629. [Google Scholar] [CrossRef]
- Ehteshami Bejnordi, B.; Veta, M.; Van Diest, P.J.; Van Ginneken, B.; Karssemeijer, N.; Litjens, G.; Van der Laak, J.A.W.M.; CAMELYON16 Consortium. Diagnostic Assessment of Deep Learning Algorithms for Detection of Lymph Node Metastases in Women with Breast Cancer. JAMA 2017, 318, 2199–2210. [Google Scholar] [CrossRef] [Green Version]
- Johnson, H.; El-Schich, Z.; Ali, A.; Zhang, X.; Simoulis, A.; Wingren, A.G.; Persson, J.L. Gene-Mutation-Based Algorithm for Prediction of Treatment Response in Colorectal Cancer Patients. Cancers 2022, 14, 2045. [Google Scholar] [CrossRef]
- Guo, J.; Johnson, H.; Zhang, X.; Feng, X.; Zhang, H.; Simoulis, A.; Wu, A.H.; Xia, T.; Li, F.; Tan, W.; et al. A 23-Gene Classifier urine test for prostate cancer prognosis. Clin. Transl. Med. 2021, 11, e340. [Google Scholar] [CrossRef] [PubMed]
- Koboldt, D.C.; Fulton, R.S.; McLellan, M.D.; Schmidt, H.; Kalicki-Veizer, J.; McMichael, J.F.; Fulton, L.L.; Dooling, D.J.; Ding, L.; Mardis, E.R.; et al. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef] [Green Version]
- Stephens, P.J.; Tarpey, P.S.; Davies, H.; Van Loo, P.; Greenman, C.; Wedge, D.C.; Nik-Zainal, S.; Martin, S.; Varela, I.; Bignell, G.R.; et al. The landscape of cancer genes and mutational processes in breast cancer. Nature 2012, 486, 400–404. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TCGA Cohort | MSK Cohort | |
|---|---|---|
| No of patients | 399 | 807 |
| Median age at diagnosis (Q1, Q3) | 59 (49, 68) | 54 (46, 65) |
| Cancers stage at diagnosis (%) | ||
| Stage I | 50 (13%) | 342 (42%) |
| Stage II | 240 (60%) | 232 (29%) |
| Stage III | 98 (25%) | 99 (12%) |
| Stage IV | 5 (1%) | 134 (17%) |
| Unknown | 6 (2%) | 0 |
| Tumor laterality (%) | ||
| Left side | 213 (53%) | 420 (52%) |
| Right side | 186 (47%) | 387 (48%) |
| Triple-negative cancer (%) | 42 (11%) | 75 (9%) |
| Luminal A cancer (%) | 155 (39%) | 501 (62%) |
| Luminal B cancer (%) | 14 (4%) | 6 (0.7%) |
| HER2+ cancer (%) | 14 (4%) | 15 (1.9%) |
| Cancer type unknown | 174 (44%) | 210 (26%) |
| Overall survival (%) | ||
| Living | 377 (94%) | 713 (88%) |
| Diseased | 22 (6%) | 94 (12%) |
| Progression/recurrence after treatment (%) | ||
| Progressed/recurrent | 46 (12%) | 314 (39%) |
| Non-progressed/non-recurrent | 353 (88%) | 493 (61%) |
| Sensitivity (95% CI) | Specificity (95% CI) | PPV (95% CI) | NPV (95% CI) | |
|---|---|---|---|---|
| TCGA Cohort (n = 399) | ||||
| 12-Gene Algorithm | 72% (59–85%) | 97% (95–98%) | 73% (60–86%) | 97% (95–98%) |
| Cancer stage | 11% (1.9–20%) | 99% (99–100%) | 71% (38–105%) | 89% (86–92%) |
| Combination | 78% (66–90%) | 96% (94–98%) | 73% (61–86%) | 97% (95–99%) |
| Triple-negative breast cancer in the TCGA Cohort (n = 42) | ||||
| 12-Gene Algorithm | 71% (38–105%) | 97% (92–103%) | 83% (54–113%) | 94% (87–102%) |
| Cancer stage | 0% (0–0%) | 100% (100–100%) | 0% (0–0%) | 83% (72–95%) |
| Combination | 91% (82–101%) | 100% (100–100%) | 70% (42–98%) | 100% (100–100%) |
| Luminal A breast cancer in the TCGA Cohort (n = 155) | ||||
| 12-Gene Algorithm | 85% (65–104%) | 96% (93–100%) | 69% (46–91%) | 99% (97–101%) |
| Cancer stage | 0% (0–0%) | 100% (100–100%) | 0% (0–0%) | 91% (87–96%) |
| Combination | 85% (65–104%) | 97% (94–100%) | 73% (51–96%) | 99% (97–101%) |
| Univariate | Multivariate | |||
|---|---|---|---|---|
| HR (95% CI) | p Value | HR (95% CI) | p Value | |
| TCGA Cohort (n = 399) | ||||
| 12-Gene Algorithm | 21.6 (11.3–41.5) | <0.0001 | 19.7 (10.2–38.1) | <0.0001 |
| Cancer stage | 2.8 (1.6–5.1) | <0.001 | 1.9 (1.1–3.5) | 0.031 |
| Triple-negative breast cancer in the TCGA Cohort (n = 42) | ||||
| 12-Gene Algorithm | 19.3 (3.7–101.3) | 0.000 | 22.3 (4.0–125.7) | 0.000 |
| Cancer stage | 2.7 (0.52–13.8) | 0.242 | 3.8 (0.62–22.9) | 0.151 |
| Luminal A breast cancer in the TCGA Cohort (n = 155) | ||||
| 12-Gene Algorithm | 47.6 (10.4–217.0) | <0.0001 | 45.4 (9.6–214.5) | <0.0001 |
| Cancer stage | 2.7 (0.89–7.9) | 0.080 | 1.1 (0.36–3.6) | 0.835 |
| Sensitivity (95% CI) | Specificity (95% CI) | PPV (95% CI) | NPV (95% CI) | |
|---|---|---|---|---|
| MSK Cohort (n = 807) | ||||
| 12-Gene Algorithm | 75% (70–79%) | 97% (96–99%) | 95% (92–98%) | 86% (83–89%) |
| Cancer stage | 0% (0–0%) | 100% (100–100%) | 0% (0–0%) | 62% (58–65%) |
| Combination | 75% (70–80%) | 97% (96–99%) | 95% (92–97%) | 86% (83–89%) |
| Triple-negative breast cancer in the MSK Cohort (n = 75) | ||||
| 12-Gene Algorithm | 90% (79–101%) | 91% (83–99%) | 87% (75–99%) | 93% (86–101%) |
| Cancer stage | 0% (0–0%) | 100% (100–100%) | 0% (0–0%) | 61% (50–72%) |
| Combination | 90% (79–101%) | 91% (83–99%) | 87% (75–99%) | 93% (86–101%) |
| Luminal A breast cancer in the MSK Cohort (n = 501) | ||||
| 12-Gene Algorithm | 73% (67–80%) | 99% (97–100%) | 96% (93–99%) | 87% (83–90%) |
| Cancer stage | 0% (0–0%) | 100% (100–100%) | 0% (0–0%) | 65% (61–69%) |
| Combination | 73% (66–80%) | 98% (97–100%) | 96% (93–99%) | 87% (84–91%) |
| Univariate | Multivariate | |||
|---|---|---|---|---|
| HR (95% CI) | p Value | HR (95% CI) | p Value | |
| MSK Cohort (n = 807) | ||||
| 12-Gene Algorithm | 4.4 (3.4–5.7) | <0.0001 | 4.4 (3.3–5.7) | <0.0001 |
| Cancer stage | 1.3 (0.8–1.7) | 0.072 | 1.2 (1.0–1.6) | 0.100 |
| Triple-negative breast cancer in the MSK Cohort (n = 75) | ||||
| 12-Gene Algorithm | 18.6 (4.4–79.2) | <0.0001 | 22.4 (4.9–103.2) | <0.0001 |
| Cancer stage | 0.87 (0.26–2.9) | 0.815 | 2.3 (0.56–10.7) | 0.285 |
| Luminal A breast cancer in the MSK Cohort (n = 501) | ||||
| 12-Gene Algorithm | 3.8 (2.7–5.4) | <0.0001 | 3.7 (2.6–5.4) | <0.0001 |
| Cancer stage | 1.5 (1.0–2.2) | 0.035 | 1.3 (0.94–1.9) | 0.104 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Johnson, H.; Ali, A.; Zhang, X.; Wang, T.; Simoulis, A.; Wingren, A.G.; Persson, J.L. K-RAS Associated Gene-Mutation-Based Algorithm for Prediction of Treatment Response of Patients with Subtypes of Breast Cancer and Especially Triple-Negative Cancer. Cancers 2022, 14, 5322. https://doi.org/10.3390/cancers14215322
Johnson H, Ali A, Zhang X, Wang T, Simoulis A, Wingren AG, Persson JL. K-RAS Associated Gene-Mutation-Based Algorithm for Prediction of Treatment Response of Patients with Subtypes of Breast Cancer and Especially Triple-Negative Cancer. Cancers. 2022; 14(21):5322. https://doi.org/10.3390/cancers14215322
Chicago/Turabian StyleJohnson, Heather, Amjad Ali, Xuhui Zhang, Tianyan Wang, Athanasios Simoulis, Anette Gjörloff Wingren, and Jenny L. Persson. 2022. "K-RAS Associated Gene-Mutation-Based Algorithm for Prediction of Treatment Response of Patients with Subtypes of Breast Cancer and Especially Triple-Negative Cancer" Cancers 14, no. 21: 5322. https://doi.org/10.3390/cancers14215322
APA StyleJohnson, H., Ali, A., Zhang, X., Wang, T., Simoulis, A., Wingren, A. G., & Persson, J. L. (2022). K-RAS Associated Gene-Mutation-Based Algorithm for Prediction of Treatment Response of Patients with Subtypes of Breast Cancer and Especially Triple-Negative Cancer. Cancers, 14(21), 5322. https://doi.org/10.3390/cancers14215322