



Ewing Sarcoma Meets Epigenetics, Immunology and Nanomedicine: Moving Forward into Novel Therapeutic Strategies

 , , , ,
, , , ,  , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Epigenetic and Immunotherapy-Based Treatments in EWS: Moving Forward in Targeted Therapies
2.1. Epigenetic Therapy
2.1.1. DNA Methylation
2.1.2. Nucleosome Remodeling
2.1.3. Histone Modifications and Modifiers
Histone Writers: Polycomb Group and G9a Methyltransferase
Histone Erasers: Deacetylases and Demethylases
Histone Readers: Bromodomains
2.2. Immunotherapy
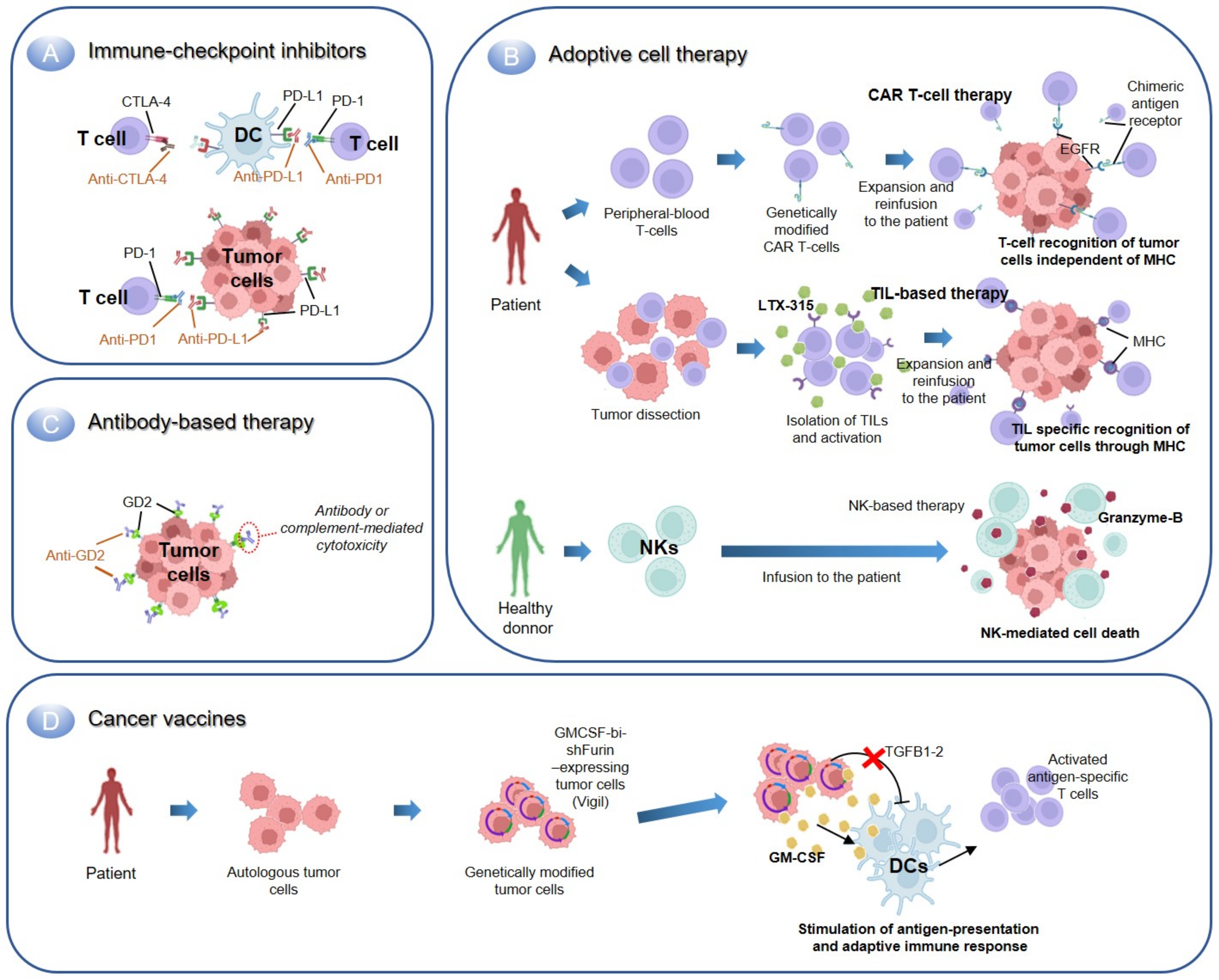
2.2.1. Immune Checkpoint Inhibitors
2.2.2. Adoptive Cell Therapy
Transfer of T-Cells
Transfer of Natural Killer Cells
2.2.3. Antibody-Based Immunotherapy
2.2.4. Cancer Vaccines
3. Nanotherapy: A Refined Target-Specific Drug Delivery System
3.1. Ewing Sarcoma Nano-Systems
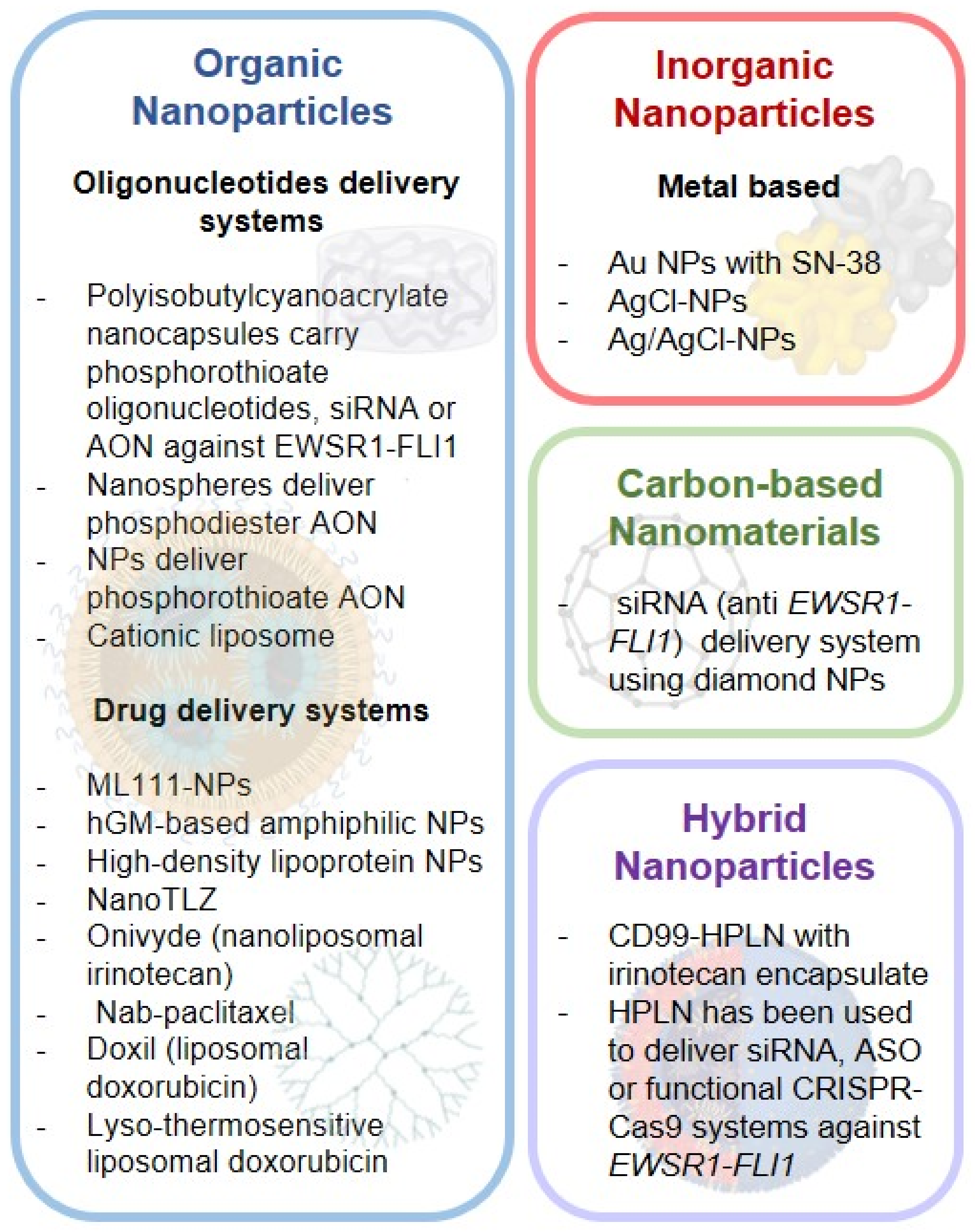
3.1.1. Organic NPS
Oligonucleotide Delivery Systems

{kind=link}
{kind=link}
| Molecular Mechanism | Interventions | Clinical Trial Identifier | Patients | Phase | Status |
|---|---|---|---|---|---|
| Oncogene driver inhibition | Biological: pbi-shRNA™ EWS/FLI1 Type 1 LPX | NCT02736565 | EWS | I | Active, not recruiting |
| DNA damage by topoisomerase inhibition | Onivyde + Talazoparib (Arm A) or Temozolomide (Arm B) | NCT04901702 | Recurrent Solid Tumors: EWS; Hepatoblastoma; Neuroblastoma; Osteosarcoma; Rhabdomyosarcoma; Wilms Tumor. Refractory Solid Tumors: EWS; Hepatoblastoma; Malignant Germ Cell Tumor; Malignant Solid Neoplasm; Neuroblastoma; Osteosarcoma; Peripheral Primitive Neuroectodermal Tumor; Rhabdoid Tumor; Rhabdomyosarcoma | I/II | Active, not recruiting |
| MM-398 (Irinotecan Sucrosofate Liposome) + cyclophosphamide | NCT02013336 | Recurrent or Refractory Solid Tumors: EWS; Rhabdomyosarcoma; Neuroblastoma; Osteosarcoma | I | Recruiting | |
| Depolymerization of microtubules (paclitaxel) | Nab-paclitaxel | NCT03275818 | Desmoplastic Small Round Cell, Adult; Desmoplastic Small Round Cell, childhood; EWS; Desmoid | II | Completed |
| Nab-paclitaxel | NCT01962103 | Neuroblastoma; Rhabdomyosarcoma; EWS; Epitheliod Sarcoma, Soft Tissue Sarcoma, Spindle Cell Melanoma; Melanoma; Osteosarcoma; Histiocytoma; Fibrosarcoma; Dermatofibrosarcoma | I/II | Completed [152] | |
| Nab-paclitaxel + Gemcitabine | NCT03507491 | Cancer | I | Recruiting | |
| Nab-Paclitaxel + Gemcitabine | NCT02945800 | Osteosarcoma; EWS; Rhabdomyosarcoma; Soft Tissue Sarcoma | II | Recruiting | |
| DNA damage by intercalation, disruption of topoisomerase-II and generation of free radicals (doxorubicin) | Disulfiram + Copper Gluconate and Liposomal Doxorubicin | NCT05210374 | Relapsed Sarcomas (including EWS) | I | Not yet recruiting |
| Liposomal Doxorubicin + MR-HIFU Hyperthermia | NCT02557854 | EWS; Rhabdomyosarcoma; Wilms Tumor; Neuroblastoma; Hepatoblastoma; Germ Cell Tumor | I | Withdrawn | |
| Temsirolimus + liposomal doxorubicin | NCT00949325 | Sarcoma (including EWS) | I/II | Completed [153] | |
| Lyso-thermosensitive liposomal doxorubicin (LTLD) + MR-HIFU Hyperthermia | NCT04791228 | EWS; Malignant Epithelial Neoplasm; Rhabdomyosarcoma; Wilms Tumor; Hepatic Tumor; Germ Cell Tumor | II | Not yet recruiting | |
| Lyso-thermosensitive liposomal doxorubicin + Magnetic resonance high intensity focused ultrasound | NCT02536183 | Rhabdomyosarcoma; EWS; Osteosarcoma; Neuroblastoma; Wilms Tumor; Hepatic Tumor; Germ Cell Tumors | I | Recruiting |
Drug Delivery Systems
3.1.2. Inorganic NPS
3.1.3. Carbon-Based Nanomaterials
3.1.4. Hybrid NPS
4. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- De Alava, E.L.S.L.; Stamenkovic, I. Ewing sarcoma. In WHO Classification of Tumours of Soft Tissue and Bone, 5th ed.; IARC Press: Lyon, France, 2020. [Google Scholar]
- Mora, J.; Castaneda, A.; Perez-Jaume, S.; Lopez-Pousa, A.; Maradiegue, E.; Valverde, C.; Martin-Broto, J.; Garcia Del Muro, X.; Cruz, O.; Cruz, J.; et al. GEIS-21: A multicentric phase II study of intensive chemotherapy including gemcitabine and docetaxel for the treatment of Ewing sarcoma of children and adults: A report from the Spanish sarcoma group (GEIS). Br. J. Cancer 2017, 117, 767–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delattre, O.; Zucman, J.; Plougastel, B.; Desmaze, C.; Melot, T.; Peter, M.; Kovar, H.; Joubert, I.; de Jong, P.; Rouleau, G.; et al. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature 1992, 359, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Mitelman, F.; Johansson, B.; Mertens, F. The impact of translocations and gene fusions on cancer causation. Nat. Rev. Cancer 2007, 7, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Grunewald, T.G.P.; Cidre-Aranaz, F.; Surdez, D.; Tomazou, E.M.; de Alava, E.; Kovar, H.; Sorensen, P.H.; Delattre, O.; Dirksen, U. Ewing sarcoma. Nat. Rev. Dis. Primers 2018, 4, 5. [Google Scholar] [CrossRef] [PubMed]
- Nacev, B.A.; Jones, K.B.; Intlekofer, A.M.; Yu, J.S.E.; Allis, C.D.; Tap, W.D.; Ladanyi, M.; Nielsen, T.O. The epigenomics of sarcoma. Nat. Rev. Cancer 2020, 20, 608–623. [Google Scholar] [CrossRef]
- Riggi, N.; Knoechel, B.; Gillespie, S.M.; Rheinbay, E.; Boulay, G.; Suva, M.L.; Rossetti, N.E.; Boonseng, W.E.; Oksuz, O.; Cook, E.B.; et al. EWS-FLI1 Utilizes Divergent Chromatin Remodeling Mechanisms to Directly Activate or Repress Enhancer Elements in Ewing Sarcoma. Cancer Cell 2014, 26, 668–681. [Google Scholar] [CrossRef] [Green Version]
- Tomazou, E.M.; Sheffield, N.C.; Schmidl, C.; Schuster, M.; Schonegger, A.; Datlinger, P.; Kubicek, S.; Bock, C.; Kovar, H. Epigenome mapping reveals distinct modes of gene regulation and widespread enhancer reprogramming by the oncogenic fusion protein EWS-FLI1. Cell Rep. 2015, 10, 1082–1095. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Wallach, M.; Krishna, A.; Kurmasheva, R.; Sridhar, S. Recent Developments in Nanomedicine for Pediatric Cancer. J. Clin. Med. 2021, 10, 1437. [Google Scholar] [CrossRef]
- Crompton, B.D.; Stewart, C.; Taylor-Weiner, A.; Alexe, G.; Kurek, K.C.; Calicchio, M.L.; Kiezun, A.; Carter, S.L.; Shukla, S.A.; Mehta, S.S.; et al. The genomic landscape of pediatric Ewing sarcoma. Cancer Discov. 2014, 4, 1326–1341. [Google Scholar] [CrossRef] [Green Version]
- Riggi, N.; Suva, M.L.; Stamenkovic, I. Ewing’s Sarcoma. N. Engl. J. Med. 2021, 384, 154–164. [Google Scholar] [CrossRef]
- Chen, T.; Dent, S.Y. Chromatin modifiers and remodellers: Regulators of cellular differentiation. Nat. Rev. Genet. 2014, 15, 93–106. [Google Scholar] [CrossRef] [Green Version]
- Suva, M.L.; Riggi, N.; Bernstein, B.E. Epigenetic reprogramming in cancer. Science 2013, 339, 1567–1570. [Google Scholar] [CrossRef] [Green Version]
- Perry, J.A.; Seong, B.K.A.; Stegmaier, K. Biology and Therapy of Dominant Fusion Oncoproteins Involving Transcription Factor and Chromatin Regulators in Sarcomas. Annu. Rev. Cancer Biol. 2019, 3, 299–321. [Google Scholar] [CrossRef]
- Dawson, M.A. The cancer epigenome: Concepts, challenges, and therapeutic opportunities. Science 2017, 355, 1147–1152. [Google Scholar] [CrossRef]
- Chen, Z.; Zhang, Y. Role of Mammalian DNA Methyltransferases in Development. Annu. Rev. Biochem. 2020, 89, 135–158. [Google Scholar] [CrossRef]
- Sheffield, N.C.; Pierron, G.; Klughammer, J.; Datlinger, P.; Schonegger, A.; Schuster, M.; Hadler, J.; Surdez, D.; Guillemot, D.; Lapouble, E.; et al. DNA methylation heterogeneity defines a disease spectrum in Ewing sarcoma. Nat. Med. 2017, 23, 386–395. [Google Scholar] [CrossRef] [Green Version]
- Park, H.R.; Jung, W.W.; Kim, H.S.; Park, Y.K. Microarray-based DNA methylation study of Ewing’s sarcoma of the bone. Oncol. Lett. 2014, 8, 1613–1617. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.; Liu, X.; Zeng, Y.; Liu, J.; Wu, F. DNA methyltransferase inhibitors combination therapy for the treatment of solid tumor: Mechanism and clinical application. Clin. Epigenetics 2021, 13, 166. [Google Scholar] [CrossRef]
- Cristalli, C.; Manara, M.C.; Valente, S.; Pellegrini, E.; Bavelloni, A.; De Feo, A.; Blalock, W.; Di Bello, E.; Pineyro, D.; Merkel, A.; et al. Novel Targeting of DNA Methyltransferase Activity Inhibits Ewing Sarcoma Cell Proliferation and Enhances Tumor Cell Sensitivity to DNA Damaging Drugs by Activating the DNA Damage Response. Front. Endocrinol. 2022, 13, 876602. [Google Scholar] [CrossRef]
- Sankar, S.; Bell, R.; Stephens, B.; Zhuo, R.; Sharma, S.; Bearss, D.J.; Lessnick, S.L. Mechanism and relevance of EWS/FLI-mediated transcriptional repression in Ewing sarcoma. Oncogene 2013, 32, 5089–5100. [Google Scholar] [CrossRef]
- Sankar, S.; Theisen, E.R.; Bearss, J.; Mulvihill, T.; Hoffman, L.M.; Sorna, V.; Beckerle, M.C.; Sharma, S.; Lessnick, S.L. Reversible LSD1 inhibition interferes with global EWS/ETS transcriptional activity and impedes Ewing sarcoma tumor growth. Clin. Cancer Res. 2014, 20, 4584–4597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pishas, K.I.; Drenberg, C.D.; Taslim, C.; Theisen, E.R.; Johnson, K.M.; Saund, R.S.; Pop, I.L.; Crompton, B.D.; Lawlor, E.R.; Tirode, F.; et al. Therapeutic Targeting of KDM1A/LSD1 in Ewing Sarcoma with SP-2509 Engages the Endoplasmic Reticulum Stress Response. Mol. Cancer 2018, 17, 1902–1916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theisen, E.R.; Selich-Anderson, J.; Miller, K.R.; Tanner, J.M.; Taslim, C.; Pishas, K.I.; Sharma, S.; Lessnick, S.L. Chromatin profiling reveals relocalization of lysine-specific demethylase 1 by an oncogenic fusion protein. Epigenetics 2021, 16, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Kurmasheva, R.T.; Erickson, S.W.; Han, R.; Teicher, B.A.; Smith, M.A.; Roth, M.; Gorlick, R.; Houghton, P.J. In vivo evaluation of the lysine-specific demethylase (KDM1A/LSD1) inhibitor SP-2577 (Seclidemstat) against pediatric sarcoma preclinical models: A report from the Pediatric Preclinical Testing Consortium (PPTC). Pediatr. Blood Cancer 2021, 68, e29304. [Google Scholar] [CrossRef] [PubMed]
- Grohar, P.J.; Griffin, L.B.; Yeung, C.; Chen, Q.R.; Pommier, Y.; Khanna, C.; Khan, J.; Helman, L.J. Ecteinascidin 743 interferes with the activity of EWS-FLI1 in Ewing sarcoma cells. Neoplasia 2011, 13, 145–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boulay, G.; Sandoval, G.J.; Riggi, N.; Iyer, S.; Buisson, R.; Naigles, B.; Awad, M.E.; Rengarajan, S.; Volorio, A.; McBride, M.J.; et al. Cancer-Specific Retargeting of BAF Complexes by a Prion-like Domain. Cell 2017, 171, 163–178.e19. [Google Scholar] [CrossRef] [Green Version]
- Harlow, M.L.; Chasse, M.H.; Boguslawski, E.A.; Sorensen, K.M.; Gedminas, J.M.; Kitchen-Goosen, S.M.; Rothbart, S.B.; Taslim, C.; Lessnick, S.L.; Peck, A.S.; et al. Trabectedin Inhibits EWS-FLI1 and Evicts SWI/SNF from Chromatin in a Schedule-dependent Manner. Clin. Cancer Res. 2019, 25, 3417–3429. [Google Scholar] [CrossRef] [Green Version]
- Baruchel, S.; Pappo, A.; Krailo, M.; Baker, K.S.; Wu, B.; Villaluna, D.; Lee-Scott, M.; Adamson, P.C.; Blaney, S.M. A phase 2 trial of trabectedin in children with recurrent rhabdomyosarcoma, Ewing sarcoma and non-rhabdomyosarcoma soft tissue sarcomas: A report from the Children’s Oncology Group. Eur. J. Cancer 2012, 48, 579–585. [Google Scholar] [CrossRef]
- Lau, L.; Supko, J.G.; Blaney, S.; Hershon, L.; Seibel, N.; Krailo, M.; Qu, W.; Malkin, D.; Jimeno, J.; Bernstein, M.; et al. A phase I and pharmacokinetic study of ecteinascidin-743 (Yondelis) in children with refractory solid tumors. A Children’s Oncology Group study. Clin. Cancer Res. 2005, 11, 672–677. [Google Scholar] [CrossRef]
- Grohar, P.J.; Segars, L.E.; Yeung, C.; Pommier, Y.; D’Incalci, M.; Mendoza, A.; Helman, L.J. Dual targeting of EWS-FLI1 activity and the associated DNA damage response with trabectedin and SN38 synergistically inhibits Ewing sarcoma cell growth. Clin. Cancer Res. 2014, 20, 1190–1203. [Google Scholar] [CrossRef]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef] [Green Version]
- Chan, H.L.; Beckedorff, F.; Zhang, Y.; Garcia-Huidobro, J.; Jiang, H.; Colaprico, A.; Bilbao, D.; Figueroa, M.E.; LaCava, J.; Shiekhattar, R.; et al. Polycomb complexes associate with enhancers and promote oncogenic transcriptional programs in cancer through multiple mechanisms. Nat. Commun. 2018, 9, 3377. [Google Scholar] [CrossRef] [Green Version]
- Rai, K.; Akdemir, K.C.; Kwong, L.N.; Fiziev, P.; Wu, C.J.; Keung, E.Z.; Sharma, S.; Samant, N.S.; Williams, M.; Axelrad, J.B.; et al. Dual Roles of RNF2 in Melanoma Progression. Cancer Discov. 2015, 5, 1314–1327. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Molina, S.; Figuerola-Bou, E.; Blanco, E.; Sanchez-Jimenez, M.; Taboas, P.; Gomez, S.; Ballare, C.; Garcia-Dominguez, D.J.; Prada, E.; Hontecillas-Prieto, L.; et al. RING1B recruits EWSR1-FLI1 and cooperates in the remodeling of chromatin necessary for Ewing sarcoma tumorigenesis. Sci. Adv. 2020, 6, eaba3058. [Google Scholar] [CrossRef]
- Wang, S.; Hwang, E.E.; Guha, R.; O’Neill, A.F.; Melong, N.; Veinotte, C.J.; Conway Saur, A.; Wuerthele, K.; Shen, M.; McKnight, C.; et al. High-throughput Chemical Screening Identifies Focal Adhesion Kinase and Aurora Kinase B Inhibition as a Synergistic Treatment Combination in Ewing Sarcoma. Clin. Cancer Res. 2019, 25, 4552–4566. [Google Scholar] [CrossRef] [Green Version]
- Richter, G.H.; Plehm, S.; Fasan, A.; Rossler, S.; Unland, R.; Bennani-Baiti, I.M.; Hotfilder, M.; Lowel, D.; von Luettichau, I.; Mossbrugger, I.; et al. EZH2 is a mediator of EWS/FLI1 driven tumor growth and metastasis blocking endothelial and neuro-ectodermal differentiation. Proc. Natl. Acad. Sci. USA 2009, 106, 5324–5329. [Google Scholar] [CrossRef] [Green Version]
- Riggi, N.; Suva, M.L.; Suva, D.; Cironi, L.; Provero, P.; Tercier, S.; Joseph, J.M.; Stehle, J.C.; Baumer, K.; Kindler, V.; et al. EWS-FLI-1 expression triggers a Ewing’s sarcoma initiation program in primary human mesenchymal stem cells. Cancer Res. 2008, 68, 2176–2185. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, M.; Yamazaki, Y.; Kanno, Y.; Igarashi, K.; Aisaki, K.; Kanno, J.; Nakamura, T. Ewing’s sarcoma precursors are highly enriched in embryonic osteochondrogenic progenitors. J. Clin. Investig. 2014, 124, 3061–3074. [Google Scholar] [CrossRef] [Green Version]
- Kurmasheva, R.T.; Sammons, M.; Favours, E.; Wu, J.; Kurmashev, D.; Cosmopoulos, K.; Keilhack, H.; Klaus, C.R.; Houghton, P.J.; Smith, M.A. Initial testing (stage 1) of tazemetostat (EPZ-6438), a novel EZH2 inhibitor, by the Pediatric Preclinical Testing Program. Pediatr. Blood Cancer 2017, 64, e26218. [Google Scholar] [CrossRef] [Green Version]
- Kailayangiri, S.; Altvater, B.; Lesch, S.; Balbach, S.; Gottlich, C.; Kuhnemundt, J.; Mikesch, J.H.; Schelhaas, S.; Jamitzky, S.; Meltzer, J.; et al. EZH2 Inhibition in Ewing Sarcoma Upregulates GD2 Expression for Targeting with Gene-Modified T Cells. Mol. Ther. 2019, 27, 933–946. [Google Scholar] [CrossRef]
- Garcia-Dominguez, D.J.; Hajji, N.; Lopez-Alemany, R.; Sanchez-Molina, S.; Figuerola-Bou, E.; Moron Civanto, F.J.; Rello-Varona, S.; Andres-Leon, E.; Benito, A.; Keun, H.C.; et al. Selective histone methyltransferase G9a inhibition reduces metastatic development of Ewing sarcoma through the epigenetic regulation of NEU1. Oncogene 2022, 41, 2638–2650. [Google Scholar] [CrossRef] [PubMed]
- Sakimura, R.; Tanaka, K.; Nakatani, F.; Matsunobu, T.; Li, X.; Hanada, M.; Okada, T.; Nakamura, T.; Matsumoto, Y.; Iwamoto, Y. Antitumor effects of histone deacetylase inhibitor on Ewing’s family tumors. Int. J. Cancer 2005, 116, 784–792. [Google Scholar] [CrossRef] [PubMed]
- Jaboin, J.; Wild, J.; Hamidi, H.; Khanna, C.; Kim, C.J.; Robey, R.; Bates, S.E.; Thiele, C.J. MS-27-275, an inhibitor of histone deacetylase, has marked in vitro and in vivo antitumor activity against pediatric solid tumors. Cancer Res. 2002, 62, 6108–6115. [Google Scholar]
- Sonnemann, J.; Dreyer, L.; Hartwig, M.; Palani, C.D.; Hongle, T.T.; Klier, U.; Broker, B.; Volker, U.; Beck, J.F. Histone deacetylase inhibitors induce cell death and enhance the apoptosis-inducing activity of TRAIL in Ewing’s sarcoma cells. J. Cancer Res. Clin. Oncol. 2007, 133, 847–858. [Google Scholar] [CrossRef] [PubMed]
- Keshelava, N.; Houghton, P.J.; Morton, C.L.; Lock, R.B.; Carol, H.; Keir, S.T.; Maris, J.M.; Reynolds, C.P.; Gorlick, R.; Kolb, E.A.; et al. Initial testing (stage 1) of vorinostat (SAHA) by the pediatric preclinical testing program. Pediatr. Blood Cancer 2009, 53, 505–508. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Baltezor, M.; Rajewski, L.; Crow, J.; Samuel, G.; Staggs, V.S.; Chastain, K.M.; Toretsky, J.A.; Weir, S.J.; Godwin, A.K. Targeted inhibition of histone deacetylase leads to suppression of Ewing sarcoma tumor growth through an unappreciated EWS-FLI1/HDAC3/HSP90 signaling axis. J. Mol. Med. 2019, 97, 957–972. [Google Scholar] [CrossRef]
- Garcia-Dominguez, D.J.; Hajji, N.; Sanchez-Molina, S.; Figuerola-Bou, E.; de Pablos, R.M.; Espinosa-Oliva, A.M.; Andres-Leon, E.; Terron-Camero, L.C.; Flores-Campos, R.; Pascual-Pasto, G.; et al. Selective inhibition of HDAC6 regulates expression of the oncogenic driver EWSR1-FLI1 through the EWSR1 promoter in Ewing sarcoma. Oncogene 2021, 40, 5843–5853. [Google Scholar] [CrossRef]
- Schmidt, O.; Nehls, N.; Prexler, C.; von Heyking, K.; Groll, T.; Pardon, K.; Garcia, H.D.; Hensel, T.; Gurgen, D.; Henssen, A.G.; et al. Class I histone deacetylases (HDAC) critically contribute to Ewing sarcoma pathogenesis. J. Exp. Clin. Cancer Res. 2021, 40, 322. [Google Scholar] [CrossRef]
- Garcia-Dominguez, D.J.; Hontecillas-Prieto, L.; Rodriguez-Nunez, P.; Pascual-Pasto, G.; Vila-Ubach, M.; Garcia-Mejias, R.; Robles, M.J.; Tirado, O.M.; Mora, J.; Carcaboso, A.M.; et al. The combination of epigenetic drugs SAHA and HCI-2509 synergistically inhibits EWS-FLI1 and tumor growth in Ewing sarcoma. Oncotarget 2018, 9, 31397–31410. [Google Scholar] [CrossRef] [Green Version]
- Welch, D.; Kahen, E.; Fridley, B.; Brohl, A.S.; Cubitt, C.L.; Reed, D.R. Small molecule inhibition of lysine-specific demethylase 1 (LSD1) and histone deacetylase (HDAC) alone and in combination in Ewing sarcoma cell lines. PLoS ONE 2019, 14, e0222228. [Google Scholar] [CrossRef]
- Pedot, G.; Marques, J.G.; Ambuhl, P.P.; Wachtel, M.; Kasper, S.; Ngo, Q.A.; Niggli, F.K.; Schafer, B.W. Inhibition of HDACs reduces Ewing sarcoma tumor growth through EWS-FLI1 protein destabilization. Neoplasia 2022, 27, 100784. [Google Scholar] [CrossRef]
- Parrish, J.K.; Sechler, M.; Winn, R.A.; Jedlicka, P. The histone demethylase KDM3A is a microRNA-22-regulated tumor promoter in Ewing Sarcoma. Oncogene 2015, 34, 257–262. [Google Scholar] [CrossRef] [Green Version]
- Sechler, M.; Parrish, J.K.; Birks, D.K.; Jedlicka, P. The histone demethylase KDM3A, and its downstream target MCAM, promote Ewing Sarcoma cell migration and metastasis. Oncogene 2017, 36, 4150–4160. [Google Scholar] [CrossRef] [Green Version]
- Parrish, J.K.; McCann, T.S.; Sechler, M.; Sobral, L.M.; Ren, W.; Jones, K.L.; Tan, A.C.; Jedlicka, P. The Jumonji-domain histone demethylase inhibitor JIB-04 deregulates oncogenic programs and increases DNA damage in Ewing Sarcoma, resulting in impaired cell proliferation and survival, and reduced tumor growth. Oncotarget 2018, 9, 33110–33123. [Google Scholar] [CrossRef] [Green Version]
- Heisey, D.A.R.; Jacob, S.; Lochmann, T.L.; Kurupi, R.; Ghotra, M.S.; Calbert, M.L.; Shende, M.; Maves, Y.K.; Koblinski, J.E.; Dozmorov, M.G.; et al. Pharmaceutical Interference of the EWS-FLI1-driven Transcriptome By Cotargeting H3K27ac and RNA Polymerase Activity in Ewing Sarcoma. Mol. Cancer 2021, 20, 1868–1879. [Google Scholar] [CrossRef]
- Alqahtani, A.; Choucair, K.; Ashraf, M.; Hammouda, D.M.; Alloghbi, A.; Khan, T.; Senzer, N.; Nemunaitis, J. Bromodomain and extra-terminal motif inhibitors: A review of preclinical and clinical advances in cancer therapy. Future Sci. OA 2019, 5, FSO372. [Google Scholar] [CrossRef] [Green Version]
- Shorstova, T.; Foulkes, W.D.; Witcher, M. Achieving clinical success with BET inhibitors as anti-cancer agents. Br. J. Cancer 2021, 124, 1478–1490. [Google Scholar] [CrossRef]
- Bid, H.K.; Phelps, D.A.; Xaio, L.; Guttridge, D.C.; Lin, J.; London, C.; Baker, L.H.; Mo, X.; Houghton, P.J. The Bromodomain BET Inhibitor JQ1 Suppresses Tumor Angiogenesis in Models of Childhood Sarcoma. Mol. Cancer 2016, 15, 1018–1028. [Google Scholar] [CrossRef] [Green Version]
- Gollavilli, P.N.; Pawar, A.; Wilder-Romans, K.; Natesan, R.; Engelke, C.G.; Dommeti, V.L.; Krishnamurthy, P.M.; Nallasivam, A.; Apel, I.J.; Xu, T.; et al. EWS/ETS-Driven Ewing Sarcoma Requires BET Bromodomain Proteins. Cancer Res. 2018, 78, 4760–4773. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Huang, D.; Saw, P.E.; Song, E. Turning cold tumors hot: From molecular mechanisms to clinical applications. Trends. Immunol. 2022, 43, 523–545. [Google Scholar] [CrossRef]
- Altvater, B.; Kailayangiri, S.; Theimann, N.; Ahlmann, M.; Farwick, N.; Chen, C.; Pscherer, S.; Neumann, I.; Mrachatz, G.; Hansmeier, A.; et al. Common Ewing sarcoma-associated antigens fail to induce natural T cell responses in both patients and healthy individuals. Cancer Immunol. Immunother. CII 2014, 63, 1047–1060. [Google Scholar] [CrossRef] [PubMed]
- Berghuis, D.; de Hooge, A.S.; Santos, S.J.; Horst, D.; Wiertz, E.J.; van Eggermond, M.C.; van den Elsen, P.J.; Taminiau, A.H.; Ottaviano, L.; Schaefer, K.L.; et al. Reduced human leukocyte antigen expression in advanced-stage Ewing sarcoma: Implications for immune recognition. J. Pathol. 2009, 218, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Spurny, C.; Kailayangiri, S.; Altvater, B.; Jamitzky, S.; Hartmann, W.; Wardelmann, E.; Ranft, A.; Dirksen, U.; Amler, S.; Hardes, J.; et al. T cell infiltration into Ewing sarcomas is associated with local expression of immune-inhibitory HLA-G. Oncotarget 2018, 9, 6536–6549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, A.H.; Highfill, S.L.; Cui, Y.; Smith, J.P.; Walker, A.J.; Ramakrishna, S.; El-Etriby, R.; Galli, S.; Tsokos, M.G.; Orentas, R.J.; et al. Reduction of MDSCs with All-trans Retinoic Acid Improves CAR Therapy Efficacy for Sarcomas. Cancer Immunol. Res. 2016, 4, 869–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zheng, J. Functions of Immune Checkpoint Molecules Beyond Immune Evasion. Adv. Exp. Med. Biol. 2020, 1248, 201–226. [Google Scholar] [CrossRef]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef]
- Weber, J.S.; D’Angelo, S.P.; Minor, D.; Hodi, F.S.; Gutzmer, R.; Neyns, B.; Hoeller, C.; Khushalani, N.I.; Miller, W.H., Jr.; Lao, C.D.; et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): A randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015, 16, 375–384. [Google Scholar] [CrossRef]
- Merchant, M.S.; Wright, M.; Baird, K.; Wexler, L.H.; Rodriguez-Galindo, C.; Bernstein, D.; Delbrook, C.; Lodish, M.; Bishop, R.; Wolchok, J.D.; et al. Phase I Clinical Trial of Ipilimumab in Pediatric Patients with Advanced Solid Tumors. Clin. Cancer Res. 2016, 22, 1364–1370. [Google Scholar] [CrossRef] [Green Version]
- Tawbi, H.A.; Burgess, M.; Bolejack, V.; Van Tine, B.A.; Schuetze, S.M.; Hu, J.; D’Angelo, S.; Attia, S.; Riedel, R.F.; Priebat, D.A.; et al. Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): A multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 1493–1501. [Google Scholar] [CrossRef]
- D’Angelo, S.P.; Mahoney, M.R.; Van Tine, B.A.; Atkins, J.; Milhem, M.M.; Jahagirdar, B.N.; Antonescu, C.R.; Horvath, E.; Tap, W.D.; Schwartz, G.K.; et al. Nivolumab with or without ipilimumab treatment for metastatic sarcoma (Alliance A091401): Two open-label, non-comparative, randomised, phase 2 trials. Lancet Oncol. 2018, 19, 416–426. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [Green Version]
- McGranahan, N.; Furness, A.J.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef] [Green Version]
- Machado, I.; Lopez-Guerrero, J.A.; Scotlandi, K.; Picci, P.; Llombart-Bosch, A. Immunohistochemical analysis and prognostic significance of PD-L1, PD-1, and CD8+ tumor-infiltrating lymphocytes in Ewing’s sarcoma family of tumors (ESFT). Virchows Arch. 2018, 472, 815–824. [Google Scholar] [CrossRef]
- Kim, C.; Kim, E.K.; Jung, H.; Chon, H.J.; Han, J.W.; Shin, K.H.; Hu, H.; Kim, K.S.; Choi, Y.D.; Kim, S.; et al. Prognostic implications of PD-L1 expression in patients with soft tissue sarcoma. BMC Cancer 2016, 16, 434. [Google Scholar] [CrossRef] [Green Version]
- Lapeyre-Prost, A.; Terme, M.; Pernot, S.; Pointet, A.L.; Voron, T.; Tartour, E.; Taieb, J. Immunomodulatory Activity of VEGF in Cancer. Int. Rev. Cell Mol. Biol. 2017, 330, 295–342. [Google Scholar] [CrossRef]
- Wilky, B.A.; Trucco, M.M.; Subhawong, T.K.; Florou, V.; Park, W.; Kwon, D.; Wieder, E.D.; Kolonias, D.; Rosenberg, A.E.; Kerr, D.A.; et al. Axitinib plus pembrolizumab in patients with advanced sarcomas including alveolar soft-part sarcoma: A single-centre, single-arm, phase 2 trial. Lancet Oncol. 2019, 20, 837–848. [Google Scholar] [CrossRef]
- Chua-Alcala, V.S.; Kim, K.; Assudani, N.; Al-Shihabi, A.; Quon, D.; Wong, S.; Chawla, S.P.; Gordon, E.M. Initial results of a phase III investigation of safety/efficacy of nivolumab and ABI-009 (nab-sirolimus) in advanced undifferentiated pleomorphic sarcoma (UPS), liposarcoma (LPS), chondrosarcoma (CS), osteosarcoma (OS), and Ewing sarcoma. J. Clin. Oncol. 2019, 37, 21. [Google Scholar] [CrossRef]
- Charych, D.H.; Hoch, U.; Langowski, J.L.; Lee, S.R.; Addepalli, M.K.; Kirk, P.B.; Sheng, D.; Liu, X.; Sims, P.W.; VanderVeen, L.A.; et al. NKTR-214, an Engineered Cytokine with Biased IL2 Receptor Binding, Increased Tumor Exposure, and Marked Efficacy in Mouse Tumor Models. Clin. Cancer Res. 2016, 22, 680–690. [Google Scholar] [CrossRef] [Green Version]
- Sharma, M.; Khong, H.; Fa’ak, F.; Bentebibel, S.E.; Janssen, L.M.E.; Chesson, B.C.; Creasy, C.A.; Forget, M.A.; Kahn, L.M.S.; Pazdrak, B.; et al. Bempegaldesleukin selectively depletes intratumoral Tregs and potentiates T cell-mediated cancer therapy. Nat. Commun. 2020, 11, 661. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Zhang, Q.; Chen, W.; Shan, B.; Ding, Y.; Zhang, G.; Cao, N.; Liu, L.; Zhang, Y. B7-H3 is overexpressed in patients suffering osteosarcoma and associated with tumor aggressiveness and metastasis. PLoS ONE 2013, 8, e70689. [Google Scholar] [CrossRef] [PubMed]
- Paijens, S.T.; Vledder, A.; de Bruyn, M.; Nijman, H.W. Tumor-infiltrating lymphocytes in the immunotherapy era. Cell. Mol. Immunol. 2021, 18, 842–859. [Google Scholar] [CrossRef] [PubMed]
- Yabe, H.; Tsukahara, T.; Kawaguchi, S.; Wada, T.; Torigoe, T.; Sato, N.; Terai, C.; Aoki, M.; Hirose, S.; Morioka, H.; et al. Prognostic significance of HLA class I expression in Ewing’s sarcoma family of tumors. J. Surg. Oncol. 2011, 103, 380–385. [Google Scholar] [CrossRef] [PubMed]
- Mullinax, J.E.; Hall, M.; Beatty, M.; Weber, A.M.; Sannasardo, Z.; Svrdlin, T.; Hensel, J.; Bui, M.; Richards, A.; Gonzalez, R.J.; et al. Expanded Tumor-infiltrating Lymphocytes From Soft Tissue Sarcoma Have Tumor-specific Function. J. Immunother. 2021, 44, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.; Monberg, T.; Albieri, B.; Sundvold, V.; Rekdal, O.; Junker, N.; Svane, I.M. LTX-315 and adoptive cell therapy using tumor-infiltrating lymphocytes in patients with metastatic soft tissue sarcoma. J. Clin. Oncol. 2022, 40, 11567. [Google Scholar] [CrossRef]
- Von Heyking, K.; Calzada-Wack, J.; Gollner, S.; Neff, F.; Schmidt, O.; Hensel, T.; Schirmer, D.; Fasan, A.; Esposito, I.; Muller-Tidow, C.; et al. The endochondral bone protein CHM1 sustains an undifferentiated, invasive phenotype, promoting lung metastasis in Ewing sarcoma. Mol. Oncol. 2017, 11, 1288–1301. [Google Scholar] [CrossRef] [Green Version]
- Thiel, U.; Schober, S.J.; Einspieler, I.; Kirschner, A.; Thiede, M.; Schirmer, D.; Gall, K.; Blaeschke, F.; Schmidt, O.; Jabar, S.; et al. Ewing sarcoma partial regression without GvHD by chondromodulin-I/HLA-A*02:01-specific allorestricted T cell receptor transgenic T cells. Oncoimmunology 2017, 6, e1312239. [Google Scholar] [CrossRef]
- Thiel, U.; Wawer, A.; von Luettichau, I.; Bender, H.U.; Blaeschke, F.; Grunewald, T.G.; Steinborn, M.; Roper, B.; Bonig, H.; Klingebiel, T.; et al. Bone marrow involvement identifies a subgroup of advanced Ewing sarcoma patients with fatal outcome irrespective of therapy in contrast to curable patients with multiple bone metastases but unaffected marrow. Oncotarget 2016, 7, 70959–70968. [Google Scholar] [CrossRef] [Green Version]
- D’Angelo, S.P.; Melchiori, L.; Merchant, M.S.; Bernstein, D.; Glod, J.; Kaplan, R.; Grupp, S.; Tap, W.D.; Chagin, K.; Binder, G.K.; et al. Antitumor Activity Associated with Prolonged Persistence of Adoptively Transferred NY-ESO-1 (c259)T Cells in Synovial Sarcoma. Cancer Discov. 2018, 8, 944–957. [Google Scholar] [CrossRef] [Green Version]
- Robbins, P.F.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; Dudley, M.E.; Wunderlich, J.R.; Nahvi, A.V.; Helman, L.J.; Mackall, C.L.; et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J. Clin. Oncol. 2011, 29, 917–924. [Google Scholar] [CrossRef] [Green Version]
- Feins, S.; Kong, W.; Williams, E.F.; Milone, M.C.; Fraietta, J.A. An introduction to chimeric antigen receptor (CAR) T-cell immunotherapy for human cancer. Am. J. Hematol. 2019, 94, S3–S9. [Google Scholar] [CrossRef]
- Brentjens, R.J.; Davila, M.L.; Riviere, I.; Park, J.; Wang, X.; Cowell, L.G.; Bartido, S.; Stefanski, J.; Taylor, C.; Olszewska, M.; et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci. Transl. Med. 2013, 5, 177ra138. [Google Scholar] [CrossRef] [Green Version]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [Green Version]
- Brown, C.E.; Mackall, C.L. CAR T cell therapy: Inroads to response and resistance. Nat. Rev. Immunol. 2019, 19, 73–74. [Google Scholar] [CrossRef]
- Shah, N.N.; Fry, T.J. Mechanisms of resistance to CAR T cell therapy. Nat. Rev. Clin. Oncol. 2019, 16, 372–385. [Google Scholar] [CrossRef]
- Dobrenkov, K.; Ostrovnaya, I.; Gu, J.; Cheung, I.Y.; Cheung, N.K. Oncotargets GD2 and GD3 are highly expressed in sarcomas of children, adolescents, and young adults. Pediatr. Blood Cancer 2016, 63, 1780–1785. [Google Scholar] [CrossRef] [Green Version]
- Kailayangiri, S.; Altvater, B.; Meltzer, J.; Pscherer, S.; Luecke, A.; Dierkes, C.; Titze, U.; Leuchte, K.; Landmeier, S.; Hotfilder, M.; et al. The ganglioside antigen G(D2) is surface-expressed in Ewing sarcoma and allows for MHC-independent immune targeting. Br. J. Cancer 2012, 106, 1123–1133. [Google Scholar] [CrossRef]
- Yu, A.L.; Gilman, A.L.; Ozkaynak, M.F.; London, W.B.; Kreissman, S.G.; Chen, H.X.; Smith, M.; Anderson, B.; Villablanca, J.G.; Matthay, K.K.; et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N. Engl. J. Med. 2010, 363, 1324–1334. [Google Scholar] [CrossRef] [Green Version]
- Majzner, R.G.; Theruvath, J.L.; Nellan, A.; Heitzeneder, S.; Cui, Y.; Mount, C.W.; Rietberg, S.P.; Linde, M.H.; Xu, P.; Rota, C.; et al. CAR T Cells Targeting B7-H3, a Pan-Cancer Antigen, Demonstrate Potent Preclinical Activity Against Pediatric Solid Tumors and Brain Tumors. Clin. Cancer Res. 2019, 25, 2560–2574. [Google Scholar] [CrossRef]
- Kersting, N.; Kunzler Souza, B.; Araujo Vieira, I.; Pereira Dos Santos, R.; Brufatto Olguins, D.; Jose Gregianin, L.; Tesainer Brunetto, A.; Lunardi Brunetto, A.; Roesler, R.; Brunetto de Farias, C.; et al. Epidermal Growth Factor Receptor Regulation of Ewing Sarcoma Cell Function. Oncology 2018, 94, 383–393. [Google Scholar] [CrossRef]
- Albert, C.M.; Pinto, N.R.; Taylor, M.; Wilson, A.; Rawlings-Rhea, S.; Mgebroff, S.; Brown, C.; Lindgren, C.; Huang, W.; Seidel, K.; et al. STRIvE-01: Phase I study of EGFR806 CAR T-cell immunotherapy for recurrent/refractory solid tumors in children and young adults. J. Clin. Oncol. 2022, 40, 2541. [Google Scholar] [CrossRef]
- Vivier, E.; Ugolini, S.; Blaise, D.; Chabannon, C.; Brossay, L. Targeting natural killer cells and natural killer T cells in cancer. Nat. Reviews. Immunol. 2012, 12, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Cho, D.; Shook, D.R.; Shimasaki, N.; Chang, Y.H.; Fujisaki, H.; Campana, D. Cytotoxicity of activated natural killer cells against pediatric solid tumors. Clin. Cancer Res. 2010, 16, 3901–3909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhoeven, D.H.; de Hooge, A.S.; Mooiman, E.C.; Santos, S.J.; ten Dam, M.M.; Gelderblom, H.; Melief, C.J.; Hogendoorn, P.C.; Egeler, R.M.; van Tol, M.J.; et al. NK cells recognize and lyse Ewing sarcoma cells through NKG2D and DNAM-1 receptor dependent pathways. Mol. Immunol. 2008, 45, 3917–3925. [Google Scholar] [CrossRef] [PubMed]
- Kailayangiri, S.; Altvater, B.; Spurny, C.; Jamitzky, S.; Schelhaas, S.; Jacobs, A.H.; Wiek, C.; Roellecke, K.; Hanenberg, H.; Hartmann, W.; et al. Targeting Ewing sarcoma with activated and GD2-specific chimeric antigen receptor-engineered human NK cells induces upregulation of immune-inhibitory HLA-G. Oncoimmunology 2017, 6, e1250050. [Google Scholar] [CrossRef] [Green Version]
- Thakar, M.S.; Browning, M.; Hari, P.; Charlson, J.A.; Margolis, D.A.; Logan, B.; Schloemer, N.; Kelly, M.E.; Newman, A.; Johnson, B.; et al. Phase II trial using haploidentical hematopoietic cell transplantation (HCT) followed by donor natural killer (NK) cell infusion and sirolimus maintenance for patients with high-risk solid tumors. J. Clin. Oncol. 2020, 38, e23551. [Google Scholar] [CrossRef]
- Denman, C.J.; Senyukov, V.V.; Somanchi, S.S.; Phatarpekar, P.V.; Kopp, L.M.; Johnson, J.L.; Singh, H.; Hurton, L.; Maiti, S.N.; Huls, M.H.; et al. Membrane-bound IL-21 promotes sustained ex vivo proliferation of human natural killer cells. PLoS ONE 2012, 7, e30264. [Google Scholar] [CrossRef]
- Nayyar, G.; Chu, Y.; Cairo, M.S. Overcoming Resistance to Natural Killer Cell Based Immunotherapies for Solid Tumors. Front. Oncol. 2019, 9, 51. [Google Scholar] [CrossRef] [Green Version]
- Early Breast Cancer Trialists’ Collaborative Group. Trastuzumab for early-stage, HER2-positive breast cancer: A meta-analysis of 13 864 women in seven randomised trials. Lancet Oncol. 2021, 22, 1139–1150. [Google Scholar] [CrossRef]
- Weiner, G.J. Monoclonal antibody mechanisms of action in cancer. Immunol. Res. 2007, 39, 271–278. [Google Scholar] [CrossRef]
- O’Neill, A.; Shah, N.; Zitomersky, N.; Ladanyi, M.; Shukla, N.; Uren, A.; Loeb, D.; Toretsky, J. Insulin-like growth factor 1 receptor as a therapeutic target in ewing sarcoma: Lack of consistent upregulation or recurrent mutation and a review of the clinical trial literature. Sarcoma 2013, 2013, 450478. [Google Scholar] [CrossRef]
- Toretsky, J.A.; Kalebic, T.; Blakesley, V.; LeRoith, D.; Helman, L.J. The insulin-like growth factor-I receptor is required for EWS/FLI-1 transformation of fibroblasts. J. Biol. Chem. 1997, 272, 30822–30827. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, G.; Mallinger, R.; Hofbauer, S.; Havel, M. The monoclonal HBA-71 antibody modulates proliferation of thymocytes and Ewing’s sarcoma cells by interfering with the action of insulin-like growth factor I. Thymus 1991, 18, 33–41. [Google Scholar]
- Scotlandi, K.; Benini, S.; Nanni, P.; Lollini, P.L.; Nicoletti, G.; Landuzzi, L.; Serra, M.; Manara, M.C.; Picci, P.; Baldini, N. Blockage of insulin-like growth factor-I receptor inhibits the growth of Ewing’s sarcoma in athymic mice. Cancer Res. 1998, 58, 4127–4131. [Google Scholar]
- Juergens, H.; Daw, N.C.; Geoerger, B.; Ferrari, S.; Villarroel, M.; Aerts, I.; Whelan, J.; Dirksen, U.; Hixon, M.L.; Yin, D.; et al. Preliminary efficacy of the anti-insulin-like growth factor type 1 receptor antibody figitumumab in patients with refractory Ewing sarcoma. J. Clin. Oncol. 2011, 29, 4534–4540. [Google Scholar] [CrossRef] [Green Version]
- Malempati, S.; Weigel, B.; Ingle, A.M.; Ahern, C.H.; Carroll, J.M.; Roberts, C.T.; Reid, J.M.; Schmechel, S.; Voss, S.D.; Cho, S.Y.; et al. Phase I/II trial and pharmacokinetic study of cixutumumab in pediatric patients with refractory solid tumors and Ewing sarcoma: A report from the Children’s Oncology Group. J. Clin. Oncol. 2012, 30, 256–262. [Google Scholar] [CrossRef]
- Pappo, A.S.; Patel, S.R.; Crowley, J.; Reinke, D.K.; Kuenkele, K.P.; Chawla, S.P.; Toner, G.C.; Maki, R.G.; Meyers, P.A.; Chugh, R.; et al. R1507, a monoclonal antibody to the insulin-like growth factor 1 receptor, in patients with recurrent or refractory Ewing sarcoma family of tumors: Results of a phase II Sarcoma Alliance for Research through Collaboration study. J. Clin. Oncol. 2011, 29, 4541–4547. [Google Scholar] [CrossRef]
- Zollner, S.K.; Amatruda, J.F.; Bauer, S.; Collaud, S.; de Alava, E.; DuBois, S.G.; Hardes, J.; Hartmann, W.; Kovar, H.; Metzler, M.; et al. Ewing Sarcoma-Diagnosis, Treatment, Clinical Challenges and Future Perspectives. J. Clin. Med. 2021, 10, 1685. [Google Scholar] [CrossRef]
- Shulman, D.S.; Merriam, P.; Choy, E.; Guenther, L.M.; Cavanaugh, K.; Kao, P.-C.; Posner, A.; Fairchild, G.; Barker, E.; Stegmaier, K.; et al. Phase 2 trial of palbociclib and ganitumab in patients with relapsed Ewing sarcoma. J. Clin. Oncol. 2022, 40, e23507. [Google Scholar] [CrossRef]
- Casey, D.L.; Lin, T.Y.; Cheung, N.V. Exploiting Signaling Pathways and Immune Targets Beyond the Standard of Care for Ewing Sarcoma. Front. Oncol. 2019, 9, 537. [Google Scholar] [CrossRef] [PubMed]
- Wagner, L.; Turpin, B.; Nagarajan, R.; Weiss, B.; Cripe, T.; Geller, J. Pilot study of vincristine, oral irinotecan, and temozolomide (VOIT regimen) combined with bevacizumab in pediatric patients with recurrent solid tumors or brain tumors. Pediatr. Blood Cancer 2013, 60, 1447–1451. [Google Scholar] [CrossRef] [PubMed]
- Lowery, C.D.; Blosser, W.; Dowless, M.; Renschler, M.; Perez, L.V.; Stephens, J.; Pytowski, B.; Wasserstrom, H.; Stancato, L.F.; Falcon, B. Anti-VEGFR2 therapy delays growth of preclinical pediatric tumor models and enhances anti-tumor activity of chemotherapy. Oncotarget 2019, 10, 5523–5533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tap, W.D.; Jones, R.L.; Van Tine, B.A.; Chmielowski, B.; Elias, A.D.; Adkins, D.; Agulnik, M.; Cooney, M.M.; Livingston, M.B.; Pennock, G.; et al. Olaratumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: An open-label phase 1b and randomised phase 2 trial. Lancet 2016, 388, 488–497. [Google Scholar] [CrossRef]
- Tap, W.D.; Wagner, A.J.; Schoffski, P.; Martin-Broto, J.; Krarup-Hansen, A.; Ganjoo, K.N.; Yen, C.C.; Abdul Razak, A.R.; Spira, A.; Kawai, A.; et al. Effect of Doxorubicin Plus Olaratumab vs Doxorubicin Plus Placebo on Survival in Patients With Advanced Soft Tissue Sarcomas: The ANNOUNCE Randomized Clinical Trial. JAMA 2020, 323, 1266–1276. [Google Scholar] [CrossRef] [PubMed]
- Attia, S.; Villalobos, V.M.; Hindi, N.; Tine, B.A.V.; Wagner, A.J.; Chmielowski, B.; Levy, D.E.; Ceccarelli, M.; Jones, R.L.; Dickson, M.A. Phase (Ph) 1b/2 evaluation of olaratumab in combination with gemcitabine and docetaxel in advanced soft tissue sarcoma (STS). J. Clin. Oncol. 2021, 39, 11517. [Google Scholar] [CrossRef]
- Schöffski, P.; Bahleda, R.; Wagner, A.J.; Burgess, M.; Junker, N.C.; More, M.; Peterson, P.; Ceccarelli, M.; William, T. Results of an open-label, phase 1a/1b study of olaratumab plus pembrolizumab in patients with unresectable, locally advanced or metastatic soft tissue sarcoma. Ann. Oncol. 2021, 32 (Suppl. 7), S1428–S1457. [Google Scholar] [CrossRef]
- Kramer, K.; Pandit-Taskar, N.; Donzelli, M.; Wolden, S.L.; Zanzonico, P.; Humm, J.; Haque, S.; Souweidane, M.M.; Lewis, J.; Lyashchenko, S.K.; et al. Intraventricular radioimmunotherapy targeting B7H3 for CNS malignancies. J. Clin. Oncol 2019, 37, e13592. [Google Scholar] [CrossRef]
- Jones, R.L.; Chawla, S.P.; Attia, S.; Schoffski, P.; Gelderblom, H.; Chmielowski, B.; Le Cesne, A.; Van Tine, B.A.; Trent, J.C.; Patel, S.; et al. A phase 1 and randomized controlled phase 2 trial of the safety and efficacy of the combination of gemcitabine and docetaxel with ontuxizumab (MORAb-004) in metastatic soft-tissue sarcomas. Cancer 2019, 125, 2445–2454. [Google Scholar] [CrossRef] [Green Version]
- Chau, C.H.; Steeg, P.S.; Figg, W.D. Antibody-drug conjugates for cancer. Lancet 2019, 394, 793–804. [Google Scholar] [CrossRef]
- Pardali, E.; van der Schaft, D.W.; Wiercinska, E.; Gorter, A.; Hogendoorn, P.C.; Griffioen, A.W.; ten Dijke, P. Critical role of endoglin in tumor cell plasticity of Ewing sarcoma and melanoma. Oncogene 2011, 30, 334–345. [Google Scholar] [CrossRef] [Green Version]
- Puerto-Camacho, P.; Amaral, A.T.; Lamhamedi-Cherradi, S.E.; Menegaz, B.A.; Castillo-Ecija, H.; Ordonez, J.L.; Dominguez, S.; Jordan-Perez, C.; Diaz-Martin, J.; Romero-Perez, L.; et al. Preclinical Efficacy of Endoglin-Targeting Antibody-Drug Conjugates for the Treatment of Ewing Sarcoma. Clin. Cancer Res. 2019, 25, 2228–2240. [Google Scholar] [CrossRef]
- Fleuren, E.D.; Hillebrandt-Roeffen, M.H.; Flucke, U.E.; Te Loo, D.M.; Boerman, O.C.; van der Graaf, W.T.; Versleijen-Jonkers, Y.M. The role of AXL and the in vitro activity of the receptor tyrosine kinase inhibitor BGB324 in Ewing sarcoma. Oncotarget 2014, 5, 12753–12768. [Google Scholar] [CrossRef] [Green Version]
- Sgouros, G.; Bodei, L.; McDevitt, M.R.; Nedrow, J.R. Radiopharmaceutical therapy in cancer: Clinical advances and challenges. Nat. Rev. Drug Discov. 2020, 19, 589–608. [Google Scholar] [CrossRef]
- Rouleau, C.; Smale, R.; Fu, Y.S.; Hui, G.; Wang, F.; Hutto, E.; Fogle, R.; Jones, C.M.; Krumbholz, R.; Roth, S.; et al. Endosialin is expressed in high grade and advanced sarcomas: Evidence from clinical specimens and preclinical modeling. Int. J. Oncol. 2011, 39, 73–89. [Google Scholar] [CrossRef]
- Norris, R.E.; Fox, E.; Reid, J.M.; Ralya, A.; Liu, X.W.; Minard, C.; Weigel, B.J. Phase 1 trial of ontuxizumab (MORAb-004) in children with relapsed or refractory solid tumors: A report from the Children’s Oncology Group Phase 1 Pilot Consortium (ADVL1213). Pediatr. Blood Cancer 2018, 65, e26944. [Google Scholar] [CrossRef]
- Cicone, F.; Denoel, T.; Gnesin, S.; Riggi, N.; Irving, M.; Jakka, G.; Schaefer, N.; Viertl, D.; Coukos, G.; Prior, J.O. Preclinical Evaluation and Dosimetry of [(111)In]CHX-DTPA-scFv78-Fc Targeting Endosialin/Tumor Endothelial Marker 1 (TEM1). Mol. Imaging Biol. 2020, 22, 979–991. [Google Scholar] [CrossRef] [Green Version]
- Fedorova, L.; Mudry, P.; Pilatova, K.; Selingerova, I.; Merhautova, J.; Rehak, Z.; Valik, D.; Hlavackova, E.; Cerna, D.; Faberova, L.; et al. Assessment of Immune Response Following Dendritic Cell-Based Immunotherapy in Pediatric Patients With Relapsing Sarcoma. Front. Oncol. 2019, 9, 1169. [Google Scholar] [CrossRef]
- Ghisoli, M.; Barve, M.; Mennel, R.; Lenarsky, C.; Horvath, S.; Wallraven, G.; Pappen, B.O.; Whiting, S.; Rao, D.; Senzer, N.; et al. Three-year Follow up of GMCSF/bi-shRNA(furin) DNA-transfected Autologous Tumor Immunotherapy (Vigil) in Metastatic Advanced Ewing’s Sarcoma. Mol. Ther. 2016, 24, 1478–1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navya, P.N.; Kaphle, A.; Srinivas, S.P.; Bhargava, S.K.; Rotello, V.M.; Daima, H.K. Current trends and challenges in cancer management and therapy using designer nanomaterials. Nano Converg. 2019, 6, 23. [Google Scholar] [CrossRef] [Green Version]
- Tan, K.X.; Barhoum, A.; Pan, S.; Danquah, M.K. Chapter 5—Risks and toxicity of nanoparticles and nanostructured materials. In Emerging Applications of Nanoparticles and Architecture Nanostructures; Barhoum, A., Makhlouf, A.S.H., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 121–139. [Google Scholar] [CrossRef]
- Harish, V.; Tewari, D.; Gaur, M.; Yadav, A.B.; Swaroop, S.; Bechelany, M.; Barhoum, A. Review on Nanoparticles and Nanostructured Materials: Bioimaging, Biosensing, Drug Delivery, Tissue Engineering, Antimicrobial, and Agro-Food Applications. Nanomaterials 2022, 12, 457. [Google Scholar] [CrossRef] [PubMed]
- Jeevanandam, J.; Barhoum, A.; Chan, Y.S.; Dufresne, A.; Danquah, M.K. Review on nanoparticles and nanostructured materials: History, sources, toxicity and regulations. Beilstein J. Nanotechnol. 2018, 9, 1050–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kemp, J.A.; Kwon, Y.J. Cancer nanotechnology: Current status and perspectives. Nano Converg. 2021, 8, 34. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Kantoff, P.W.; Wooster, R.; Farokhzad, O.C. Cancer nanomedicine: Progress, challenges and opportunities. Nat. Rev. Cancer 2017, 17, 20–37. [Google Scholar] [CrossRef]
- Anu Mary Ealia, S.; Saravanakumar, M.P. A review on the classification, characterisation, synthesis of nanoparticles and their application. IOP Conf. Ser. Mater. Sci. Eng. 2017, 263, 032019. [Google Scholar] [CrossRef]
- Lambert, G.; Bertrand, J.R.; Fattal, E.; Subra, F.; Pinto-Alphandary, H.; Malvy, C.; Auclair, C.; Couvreur, P. EWS fli-1 antisense nanocapsules inhibits ewing sarcoma-related tumor in mice. Biochem. Biophys. Res. Commun. 2000, 279, 401–406. [Google Scholar] [CrossRef]
- Toub, N.; Bertrand, J.R.; Tamaddon, A.; Elhamess, H.; Hillaireau, H.; Maksimenko, A.; Maccario, J.; Malvy, C.; Fattal, E.; Couvreur, P. Efficacy of siRNA nanocapsules targeted against the EWS-Fli1 oncogene in Ewing sarcoma. Pharm. Res. 2006, 23, 892–900. [Google Scholar] [CrossRef]
- Maksimenko, A.; Malvy, C.; Lambert, G.; Bertrand, J.R.; Fattal, E.; Maccario, J.; Couvreur, P. Oligonucleotides targeted against a junction oncogene are made efficient by nanotechnologies. Pharm. Res. 2003, 20, 1565–1567. [Google Scholar] [CrossRef]
- Rao, D.D.; Jay, C.; Wang, Z.; Luo, X.; Kumar, P.; Eysenbach, H.; Ghisoli, M.; Senzer, N.; Nemunaitis, J. Preclinical Justification of pbi-shRNA EWS/FLI1 Lipoplex (LPX) Treatment for Ewing’s Sarcoma. Mol. Ther. 2016, 24, 1412–1422. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Stewart, D.J.; Lee, J.J.; Ji, L.; Ramesh, R.; Jayachandran, G.; Nunez, M.I.; Wistuba, I.I.; Erasmus, J.J.; Hicks, M.E.; et al. Phase I clinical trial of systemically administered TUSC2(FUS1)-nanoparticles mediating functional gene transfer in humans. PLoS ONE 2012, 7, e34833. [Google Scholar] [CrossRef] [Green Version]
- Moreno, L.; Casanova, M.; Chisholm, J.C.; Berlanga, P.; Chastagner, P.B.; Baruchel, S.; Amoroso, L.; Gallego Melcon, S.; Gerber, N.U.; Bisogno, G.; et al. Phase I results of a phase I/II study of weekly nab-paclitaxel in paediatric patients with recurrent/refractory solid tumours: A collaboration with innovative therapies for children with cancer. Eur. J. Cancer 2018, 100, 27–34. [Google Scholar] [CrossRef] [Green Version]
- Thornton, K.A.; Chen, A.R.; Trucco, M.M.; Shah, P.; Wilky, B.A.; Gul, N.; Carrera-Haro, M.A.; Ferreira, M.F.; Shafique, U.; Powell, J.D.; et al. A dose-finding study of temsirolimus and liposomal doxorubicin for patients with recurrent and refractory bone and soft tissue sarcoma. Int. J. Cancer 2013, 133, 997–1005. [Google Scholar] [CrossRef] [Green Version]
- Esfandiari Nazzaro, E.; Sabei, F.Y.; Vogel, W.K.; Nazari, M.; Nicholson, K.S.; Gafken, P.R.; Taratula, O.; Taratula, O.; Davare, M.A.; Leid, M. Discovery and Validation of a Compound to Target Ewing’s Sarcoma. Pharmaceutics 2021, 13, 1553. [Google Scholar] [CrossRef]
- Sabei, F.Y.; Taratula, O.; Albarqi, H.A.; Al-Fatease, A.M.; Moses, A.S.; Demessie, A.A.; Park, Y.; Vogel, W.K.; Esfandiari Nazzaro, E.; Davare, M.A.; et al. A targeted combinatorial therapy for Ewing’s sarcoma. Nanomedicine 2021, 37, 102446. [Google Scholar] [CrossRef]
- Wojcik, B.; Sawosz, E.; Szczepaniak, J.; Strojny, B.; Sosnowska, M.; Daniluk, K.; Zielinska-Gorska, M.; Balaban, J.; Chwalibog, A.; Wierzbicki, M. Effects of Metallic and Carbon-Based Nanomaterials on Human Pancreatic Cancer Cell Lines AsPC-1 and BxPC-3. Int. J. Mol. Sci. 2021, 22, 12100. [Google Scholar] [CrossRef]
- Zaritski, A.; Castillo-Ecija, H.; Kumarasamy, M.; Peled, E.; Sverdlov Arzi, R.; Carcaboso, A.M.; Sosnik, A. Selective Accumulation of Galactomannan Amphiphilic Nanomaterials in Pediatric Solid Tumor Xenografts Correlates with GLUT1 Gene Expression. ACS Appl. Mater. Interfaces 2019, 11, 38483–38496. [Google Scholar] [CrossRef]
- Bell, J.B.; Rink, J.S.; Eckerdt, F.; Clymer, J.; Goldman, S.; Thaxton, C.S.; Platanias, L.C. HDL nanoparticles targeting sonic hedgehog subtype medulloblastoma. Sci. Rep. 2018, 8, 1211. [Google Scholar] [CrossRef] [Green Version]
- Baldwin, P.; Likhotvorik, R.; Baig, N.; Cropper, J.; Carlson, R.; Kurmasheva, R.; Sridhar, S. Nanoformulation of Talazoparib Increases Maximum Tolerated Doses in Combination With Temozolomide for Treatment of Ewing Sarcoma. Front. Oncol. 2019, 9, 1416. [Google Scholar] [CrossRef]
- Choy, E.; Butrynski, J.E.; Harmon, D.C.; Morgan, J.A.; George, S.; Wagner, A.J.; D’Adamo, D.; Cote, G.M.; Flamand, Y.; Benes, C.H.; et al. Phase II study of olaparib in patients with refractory Ewing sarcoma following failure of standard chemotherapy. BMC Cancer 2014, 14, 813. [Google Scholar] [CrossRef] [Green Version]
- Fontaine, S.D.; Ashley, G.W.; Houghton, P.J.; Kurmasheva, R.T.; Diolaiti, M.; Ashworth, A.; Peer, C.J.; Nguyen, R.; Figg, W.D., Sr.; Beckford-Vera, D.R.; et al. A Very Long-Acting PARP Inhibitor Suppresses Cancer Cell Growth in DNA Repair-Deficient Tumor Models. Cancer Res. 2021, 81, 1076–1086. [Google Scholar] [CrossRef]
- Kang, M.H.; Wang, J.; Makena, M.R.; Lee, J.S.; Paz, N.; Hall, C.P.; Song, M.M.; Calderon, R.I.; Cruz, R.E.; Hindle, A.; et al. Activity of MM-398, nanoliposomal irinotecan (nal-IRI), in Ewing’s family tumor xenografts is associated with high exposure of tumor to drug and high SLFN11 expression. Clin. Cancer Res. 2015, 21, 1139–1150. [Google Scholar] [CrossRef] [Green Version]
- Pascual-Pasto, G.; Castillo-Ecija, H.; Unceta, N.; Aschero, R.; Resa-Pares, C.; Gomez-Caballero, A.; Vila-Ubach, M.; Munoz-Aznar, O.; Sunol, M.; Burgueno, V.; et al. SPARC-mediated long-term retention of nab-paclitaxel in pediatric sarcomas. J. Control. Release 2022, 342, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Amoroso, L.; Castel, V.; Bisogno, G.; Casanova, M.; Marquez-Vega, C.; Chisholm, J.C.; Doz, F.; Moreno, L.; Ruggiero, A.; Gerber, N.U.; et al. Phase II results from a phase I/II study to assess the safety and efficacy of weekly nab-paclitaxel in paediatric patients with recurrent or refractory solid tumours: A collaboration with the European Innovative Therapies for Children with Cancer Network. Eur. J. Cancer 2020, 135, 89–97. [Google Scholar] [CrossRef]
- Oesterheld, J.E.; Reed, D.R.; Setty, B.A.; Isakoff, M.S.; Thompson, P.; Yin, H.; Hayashi, M.; Loeb, D.M.; Smith, T.; Makanji, R.; et al. Phase II trial of gemcitabine and nab-paclitaxel in patients with recurrent Ewing sarcoma: A report from the National Pediatric Cancer Foundation. Pediatr. Blood Cancer 2020, 67, e28370. [Google Scholar] [CrossRef] [PubMed]
- Trucco, M.M.; Meyer, C.F.; Thornton, K.A.; Shah, P.; Chen, A.R.; Wilky, B.A.; Carrera-Haro, M.A.; Boyer, L.C.; Ferreira, M.F.; Shafique, U.; et al. A phase II study of temsirolimus and liposomal doxorubicin for patients with recurrent and refractory bone and soft tissue sarcomas. Clin. Sarcoma Res. 2018, 8, 21. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Li, J.; Zhang, X.; Lu, Y.; Wang, J.; Lyu, X.; Chen, Y.; Liu, J.; Cai, H.; Wang, Y.; et al. Gold nano-particles (AuNPs) carrying anti-EBV-miR-BART7-3p inhibit growth of EBV-positive nasopharyngeal carcinoma. Oncotarget 2015, 6, 7838–7850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovacs, D.; Igaz, N.; Marton, A.; Ronavari, A.; Belteky, P.; Bodai, L.; Spengler, G.; Tiszlavicz, L.; Razga, Z.; Hegyi, P.; et al. Core-shell nanoparticles suppress metastasis and modify the tumour-supportive activity of cancer-associated fibroblasts. J. Nanobiotechnol. 2020, 18, 18. [Google Scholar] [CrossRef]
- De Jong, W.H.; Van Der Ven, L.T.; Sleijffers, A.; Park, M.V.; Jansen, E.H.; Van Loveren, H.; Vandebriel, R.J. Systemic and immunotoxicity of silver nanoparticles in an intravenous 28 days repeated dose toxicity study in rats. Biomaterials 2013, 34, 8333–8343. [Google Scholar] [CrossRef] [Green Version]
- Naumann, J.A.; Widen, J.C.; Jonart, L.A.; Ebadi, M.; Tang, J.; Gordon, D.J.; Harki, D.A.; Gordon, P.M. SN-38 Conjugated Gold Nanoparticles Activated by Ewing Sarcoma Specific mRNAs Exhibit In Vitro and In Vivo Efficacy. Bioconjug. Chem. 2018, 29, 1111–1118. [Google Scholar] [CrossRef]
- Da Silva Ferreira, V.; Eugenio, M.F.C.; Del Nery Dos Santos, E.; de Souza, W.; Sant’Anna, C. Cellular toxicology and mechanism of the response to silver-based nanoparticle exposure in Ewing’s sarcoma cells. Nanotechnology 2021, 32, 115101. [Google Scholar] [CrossRef]
- Alhaddad, A.; Adam, M.P.; Botsoa, J.; Dantelle, G.; Perruchas, S.; Gacoin, T.; Mansuy, C.; Lavielle, S.; Malvy, C.; Treussart, F.; et al. Nanodiamond as a vector for siRNA delivery to Ewing sarcoma cells. Small 2011, 7, 3087–3095. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.; Nagy, J.; Triche, T. Abstract 3705: Targeted anticancer drug delivery to Ewing’s sarcoma using human anti-CD99 targeted hybrid polymerization liposomal nanoparticles. Cancer Res. 2018, 78, 3705. [Google Scholar] [CrossRef]
- Sakpakdeejaroen, I.; Somani, S.; Laskar, P.; Mullin, M.; Dufes, C. Regression of Melanoma Following Intravenous Injection of Plumbagin Entrapped in Transferrin-Conjugated, Lipid-Polymer Hybrid Nanoparticles. Int. J. Nanomed. 2021, 16, 2615–2631. [Google Scholar] [CrossRef]
- Kang, H.; Nagy, J.; Mitra, S.; Triche, T. Abstract 2875: Targeted therapy of Ewing’s sarcoma by human anti CD99 targeted hybrid polymerized liposomal nanoparticles (HPLNs) encapsulating anticancer agents. Cancer Res. 2019, 79, 2875. [Google Scholar] [CrossRef]
- Ginsberg, J.P.; Goodman, P.; Leisenring, W.; Ness, K.K.; Meyers, P.A.; Wolden, S.L.; Smith, S.M.; Stovall, M.; Hammond, S.; Robison, L.L.; et al. Long-term survivors of childhood Ewing sarcoma: Report from the childhood cancer survivor study. J. Natl. Cancer Inst. 2010, 102, 1272–1283. [Google Scholar] [CrossRef] [Green Version]
- Casey, D.L.; Cheung, N.V. Immunotherapy of Pediatric Solid Tumors: Treatments at a Crossroads, with an Emphasis on Antibodies. Cancer Immunol. Res. 2020, 8, 161–166. [Google Scholar] [CrossRef] [Green Version]
- Verduin, M.; Hoeben, A.; De Ruysscher, D.; Vooijs, M. Patient-Derived Cancer Organoids as Predictors of Treatment Response. Front. Oncol. 2021, 11, 641980. [Google Scholar] [CrossRef]


| Molecular Mechanism | Molecular Target | Drug | Clinical Trial Identifier | Patients | Phase | Status/Ref |
|---|---|---|---|---|---|---|
| DNA methylation | IDH | Ivodesinib | NCT04195555 | Advanced Solid Tumors, Lymphoma, or Histiocytic disorders with IDH1 mutations | II | Recruiting |
| Nucleosome remodeling | LSD1/NURD | Seclidemstat + topotecan and cyclophosphamide | NCT03600649 | Ewing Sarcoma (EWS); Myxoid Liposarcoma; Sarcomas with FET-family translocation | I | Recruiting |
| Seclidemstat | NCT05266196 | EWS; Myxoid Liposarcoma; Desmoplastic Small Round Cell Tumor; Extraskeletal Myxoid Chondrosarcoma; Angiomatoid Fibrous Histiocytoma; Clear Cell Sarcoma; Myoepithelial Tumor; Low Grade Fibromyxoid Sarcoma; Sclerosing Epithelioid Fibrosarcoma | I/II | Enrolling | ||
| INCB059872 | NCT03514407 | Refractory or relapsed EWS | Ib | Terminated | ||
| INCB059872 | NCT02712905 | Solid Tumors and Hematologic Malignancy | I/II | Terminated | ||
| SWI/ SNF | Trabectedin + radiation | NCT05131386 | Osteosarcoma; Chondrosarcoma; EWS; Rhabdomyosarcoma; Desmoplastic Small Round Cell Tumor | II | Recruiting | |
| Trabectedin + irinotecan | NCT04067115 | EWS | I | Recruiting | ||
| Lurbinectedin with or without irinotecan | NCT05042934 | Metastatic and recurrent EWS | I/II | Withdrawn | ||
| Lurbinectedin + irinotecan | NCT02611024 | Advanced Solid Tumors; Glioblastoma; Soft Tissue Sarcoma (Excluding GIST) Endometrial Carcinoma; Epithelial Ovarian; Carcinoma; Mesothelioma; Gastroenteropancreatic Neuroendocrine Tumor; SCLC; Gastric Carcinoma; Pancreatic Adenocarcinoma; Colorectal Carcinoma; Neuroendocrine Tumors | I/II | Recruiting | ||
| Histone writer | EZH2 | Tazemetostat | NCT03213665 | Relapsed or refractory: Brain tumors; Solid Tumors; non-Hodgkin Lymphoma; histiocytic disorders with EZH2, SMARCB1, or SMARCA4 gene mutations | II | Active, not recruiting |
| Histone eraser | HDAC | Vorinostat + chemotherapy | NCT04308330 | EWS; Rhabdomyosarcoma; Wilms Tumor; Neuroblastoma; Hepatoblastoma; Germ Cell Tumor | I | Recruiting |
| Histone reader | BET | BMS-986158 and BMS-986378 | NCT03936465 | Pediatric Cancer | I | Recruiting |
| Molecular Target | Molecular Mechanism | Drug | Clinical Trial Identifier | Patients | Phase | Status/Ref |
|---|---|---|---|---|---|---|
| IGF1R | mAb + targeted therapy | Ganitumab + Palbociclib (targets CDK4 and CDK6) | NCT04129151 | EWS; Relapsed EWS | II | Active, not recruiting [120] |
| mAb + chemotherapy | Ganitumab + variouschemotherapy regimens (vincristine, vincristine sulfate, ifosfamide, etoposide, etoposide sulphate, doxorubicin, doxorubicin hydrochloride, cyclophosphamide) | NCT02306161 | Metastatic EWS; Metastatic Bone Malignant neoplasm; Metastatic malignant lung neoplasm; Metastatic and peripheral PNET | III | Active, not recruiting [119] | |
| GD2 | mAb | Hu14.18K322A | NCT02159443 | EWS; Melanoma; Neuroblastoma; Osteosarcoma | I | Completed |
| ADC | 131I-3F8 | NCT00445965 | Brain and CNS tumors; Intraocular melanoma and melanoma; Lung cancer; Metastatic Cancer; Neuroblastoma; Ovarian Cancer; Sarcoma; Small intestine cancer; Retinoblastoma | II | Active not recruiting | |
| AXL | ADC with or without ICI | BA3011 (CAB-AXL-ADC) with or without PD-1 inhibitor | NCT03425279 | Sarcomas and refractory sarcomas; EWS; Non small cell lung cancer; Melanoma; Solid Tumor | I/II | Active, recruiting |
| B7-H3 | ADC | 131I-8H9 | NCT00089245 | Brain and CNS tumors; Sarcoma; Neuroblastoma | I | Active, recruiting [128] |
| Endosialin | mAb + chemotherapy | Ontuxizumab (MORAb-004) + gemcitabine and docetaxel | NCT01574716 | Metastasic soft tissue sarcomas | II | Completed [129] |
| PDGFR | mAb + ICI | Olaratumab (LY3012207) + Pembrolizumab (MK3475) | NCT03126591 | Soft Tissue Sarcoma | I | Active, not recruiting [127] |
| mAb + chemotherapy | Olaratumab (LY3012207) + gemcitabine and docetaxel (ANNOUNCE 2) | NCT02659020 | Soft Tissue Sarcoma | I/II | Completed [126] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez-Molina, S.; Figuerola-Bou, E.; Sánchez-Margalet, V.; de la Cruz-Merino, L.; Mora, J.; de Álava Casado, E.; García-Domínguez, D.J.; Hontecillas-Prieto, L. Ewing Sarcoma Meets Epigenetics, Immunology and Nanomedicine: Moving Forward into Novel Therapeutic Strategies. Cancers 2022, 14, 5473. https://doi.org/10.3390/cancers14215473
Sánchez-Molina S, Figuerola-Bou E, Sánchez-Margalet V, de la Cruz-Merino L, Mora J, de Álava Casado E, García-Domínguez DJ, Hontecillas-Prieto L. Ewing Sarcoma Meets Epigenetics, Immunology and Nanomedicine: Moving Forward into Novel Therapeutic Strategies. Cancers. 2022; 14(21):5473. https://doi.org/10.3390/cancers14215473
Chicago/Turabian StyleSánchez-Molina, Sara, Elisabet Figuerola-Bou, Víctor Sánchez-Margalet, Luis de la Cruz-Merino, Jaume Mora, Enrique de Álava Casado, Daniel José García-Domínguez, and Lourdes Hontecillas-Prieto. 2022. "Ewing Sarcoma Meets Epigenetics, Immunology and Nanomedicine: Moving Forward into Novel Therapeutic Strategies" Cancers 14, no. 21: 5473. https://doi.org/10.3390/cancers14215473
APA StyleSánchez-Molina, S., Figuerola-Bou, E., Sánchez-Margalet, V., de la Cruz-Merino, L., Mora, J., de Álava Casado, E., García-Domínguez, D. J., & Hontecillas-Prieto, L. (2022). Ewing Sarcoma Meets Epigenetics, Immunology and Nanomedicine: Moving Forward into Novel Therapeutic Strategies. Cancers, 14(21), 5473. https://doi.org/10.3390/cancers14215473