
An Update on the Metabolic Landscape of Oncogenic Viruses


Abstract
:Simple Summary
Abstract
1. Introduction
2. Reprogramming of Central Carbon Metabolism in Cancer Cells
2.1. Glucose Uptake
2.2. Glycolysis
2.3. Pentose Phosphate Pathway (PPP)
2.4. Tricarboxylic Acid Cycle (TCA)
3. Reprogramming of Fatty Acid Metabolism
4. Reprogramming of Amino Acid Metabolism
5. Role of Oncogenes, Tumor Suppressors, and Cell Signaling in Regulating Cancer Cell Metabolism
5.1. PI3K/Akt/mTOR Pathway
5.2. Hypoxia-Inducible Factor (HIF1α)
5.3. cMyc
6. Metabolic Reprogramming in Oncoviruses
6.1. Epstein–Barr Virus (EBV)
6.2. Human Papilloma Virus (HPV)
6.3. Human T Cell Leukemia Virus 1 (HTLV-1)
6.4. Hepatitis B Virus (HBV)
6.5. Hepatitis C Virus (HCV)
6.6. Kaposi Sarcoma-Associated Herpesvirus (KSHV)
6.7. Merkel Cell Polyomavirus (MCV)
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warburg, O. About the formation of cancer cells. Strahlentherapie 1956, 34, 3–13; discussion 41–57. [Google Scholar] [PubMed]
- Warburg, O. Origin of cancer cells. Oncologia 1956, 9, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, R.G.; Thompson, C.B. Tumor suppressors and cell metabolism: A recipe for cancer growth. Genes Dev. 2009, 23, 537–548. [Google Scholar] [CrossRef] [Green Version]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef] [Green Version]
- Hsu, P.P.; Sabatini, D.M. Cancer cell metabolism: Warburg and beyond. Cell 2008, 134, 703–707. [Google Scholar] [CrossRef] [Green Version]
- Swierczynski, J.; Hebanowska, A.; Sledzinski, T. Role of abnormal lipid metabolism in development, progression, diagnosis and therapy of pancreatic cancer. World J. Gastroenterol. 2014, 20, 2279–2303. [Google Scholar] [CrossRef]
- Hassanein, M.; Hoeksema, M.D.; Shiota, M.; Qian, J.; Harris, B.K.; Chen, H.; Clark, J.E.; Alborn, W.E.; Eisenberg, R.; Massion, P.P. Slc1a5 mediates glutamine transport required for lung cancer cell growth and survival. Clin. Cancer Res. 2013, 19, 560–570. [Google Scholar] [CrossRef] [Green Version]
- Mates, J.M.; Segura, J.A.; Martin-Rufian, M.; Campos-Sandoval, J.A.; Alonso, F.J.; Marquez, J. Glutaminase isoenzymes as key regulators in metabolic and oxidative stress against cancer. Curr. Mol. Med. 2013, 13, 514–534. [Google Scholar] [CrossRef]
- Taheri, F.; Goudarzi, H.; Faghihloo, E. Aneuploidy and oncoviruses. Rev. Med. Virol. 2019, 29, e2076. [Google Scholar] [CrossRef] [PubMed]
- White, M.K.; Pagano, J.S.; Khalili, K. Viruses and human cancers: A long road of discovery of molecular paradigms. Clin. Microbiol. Rev. 2014, 27, 463–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiller, J.T.; Lowy, D.R. An introduction to virus infections and human cancer. Recent Results Cancer Res. 2021, 217, 1–11. [Google Scholar] [PubMed]
- Racker, E. Bioenergetics and the problem of tumor growth. Am. Sci. 1972, 60, 56–63. [Google Scholar]
- Vander Heiden, M.G.; Lunt, S.Y.; Dayton, T.L.; Fiske, B.P.; Israelsen, W.J.; Mattaini, K.R.; Vokes, N.I.; Stephanopoulos, G.; Cantley, L.C.; Metallo, C.M.; et al. Metabolic pathway alterations that support cell proliferation. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 325–334. [Google Scholar] [CrossRef] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Weinhouse, S. On respiratory impairment in cancer cells. Science 1956, 124, 267–269. [Google Scholar] [CrossRef]
- Zu, X.L.; Guppy, M. Cancer metabolism: Facts, fantasy, and fiction. Biochem. Biophys. Res. Commun. 2004, 313, 459–465. [Google Scholar] [CrossRef]
- Moreno-Sanchez, R.; Rodriguez-Enriquez, S.; Marin-Hernandez, A.; Saavedra, E. Energy metabolism in tumor cells. FEBS J. 2007, 274, 1393–1418. [Google Scholar] [CrossRef]
- Fantin, V.R.; St-Pierre, J.; Leder, P. Attenuation of ldh-a expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell 2006, 9, 425–434. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.; Kamphorst, J.J.; Mathew, R.; Chung, M.K.; White, E.; Shlomi, T.; Rabinowitz, J.D. Glutamine-driven oxidative phosphorylation is a major atp source in transformed mammalian cells in both normoxia and hypoxia. Mol. Syst. Biol. 2013, 9, 712. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Pouyssegur, J. Tumor cell metabolism: Cancer’s achilles’ heel. Cancer Cell 2008, 13, 472–482. [Google Scholar] [CrossRef]
- Halama, A.; Suhre, K. Advancing cancer treatment by targeting glutamine metabolism-a roadmap. Cancers 2022, 14, 553. [Google Scholar] [CrossRef] [PubMed]
- Adekola, K.; Rosen, S.T.; Shanmugam, M. Glucose transporters in cancer metabolism. Curr. Opin. Oncol. 2012, 24, 650–654. [Google Scholar] [CrossRef]
- Rodriguez-Enriquez, S.; Marin-Hernandez, A.; Gallardo-Perez, J.C.; Moreno-Sanchez, R. Kinetics of transport and phosphorylation of glucose in cancer cells. J. Cell Physiol. 2009, 221, 552–559. [Google Scholar] [CrossRef]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osthus, R.C.; Shim, H.; Kim, S.; Li, Q.; Reddy, R.; Mukherjee, M.; Xu, Y.; Wonsey, D.; Lee, L.A.; Dang, C.V. Deregulation of glucose transporter 1 and glycolytic gene expression by c-myc. J. Biol. Chem. 2000, 275, 21797–21800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boroughs, L.K.; DeBerardinis, R.J. Metabolic pathways promoting cancer cell survival and growth. Nat. Cell Biol. 2015, 17, 351–359. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Tang, H.; Liu, X.; Liu, P.; Yang, L.; Xie, X.; Ye, F.; Song, C.; Xie, X.; Wei, W. Mir-22 as a prognostic factor targets glucose transporter protein type 1 in breast cancer. Cancer Lett. 2015, 356, 410–417. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, T.; Kusuyama, J.; Bandow, K.; Matsuguchi, T. Glut1 expression is increased by p53 reduction to switch metabolism to glycolysis during osteoblast differentiation. Biochem. J. 2020, 477, 1795–1811. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Wen, J.; Tian, T.; Lu, Z.; Wang, Y.; Wang, Z.; Wang, X.; Yang, Y. Glut-1 overexpression as an unfavorable prognostic biomarker in patients with colorectal cancer. Oncotarget 2017, 8, 11788–11796. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Yongzhi, H.; Chen, S.; Luo, X.; Lin, Y.; Zhou, Y.; Jin, H.; Hou, B.; Deng, Y.; Tu, L.; et al. The prognostic value of glut1 in cancers: A systematic review and meta-analysis. Oncotarget 2017, 8, 43356–43367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Lu, P.; Zhou, S.; Zhang, L.; Zhao, J.H.; Tang, J.H. Predictive value of glucose transporter-1 and glucose transporter-3 for survival of cancer patients: A meta-analysis. Oncotarget 2017, 8, 13206–13213. [Google Scholar] [CrossRef] [Green Version]
- Chan, D.A.; Sutphin, P.D.; Nguyen, P.; Turcotte, S.; Lai, E.W.; Banh, A.; Reynolds, G.E.; Chi, J.T.; Wu, J.; Solow-Cordero, D.E.; et al. Targeting glut1 and the warburg effect in renal cell carcinoma by chemical synthetic lethality. Sci. Transl. Med. 2011, 3, 94ra70. [Google Scholar] [CrossRef] [Green Version]
- Kraus, D.; Reckenbeil, J.; Veit, N.; Kuerpig, S.; Meisenheimer, M.; Beier, I.; Stark, H.; Winter, J.; Probstmeier, R. Targeting glucose transport and the nad pathway in tumor cells with stf-31: A re-evaluation. Cell. Oncol. 2018, 41, 485–494. [Google Scholar] [CrossRef]
- Kawauchi, K.; Araki, K.; Tobiume, K.; Tanaka, N. P53 regulates glucose metabolism through an ikk-nf-kappab pathway and inhibits cell transformation. Nat. Cell Biol. 2008, 10, 611–618. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Lin, X.; Cai, M.; Zheng, X.; Lian, L.; Fan, D.; Wu, X.; Lan, P.; Wang, J. Overexpression of hexokinase 1 as a poor prognosticator in human colorectal cancer. Tumour. Biol. 2016, 37, 3887–3895. [Google Scholar] [CrossRef]
- Mathupala, S.P.; Rempel, A.; Pedersen, P.L. Glucose catabolism in cancer cells: Identification and characterization of a marked activation response of the type ii hexokinase gene to hypoxic conditions. J. Biol. Chem. 2001, 276, 43407–43412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Xiong, H.; Wu, F.; Zhang, Y.; Wang, J.; Zhao, L.; Guo, X.; Chang, L.J.; Zhang, Y.; You, M.J.; et al. Hexokinase 2-mediated warburg effect is required for pten- and p53-deficiency-driven prostate cancer growth. Cell Rep. 2014, 8, 1461–1474. [Google Scholar] [CrossRef] [Green Version]
- Peschiaroli, A.; Giacobbe, A.; Formosa, A.; Markert, E.K.; Bongiorno-Borbone, L.; Levine, A.J.; Candi, E.; D’Alessandro, A.; Zolla, L.; Finazzi Agro, A.; et al. Mir-143 regulates hexokinase 2 expression in cancer cells. Oncogene 2013, 32, 797–802. [Google Scholar] [CrossRef]
- Lauterwasser, J.; Fimm-Todt, F.; Oelgeklaus, A.; Schreiner, A.; Funk, K.; Falquez-Medina, H.; Klesse, R.; Jahreis, G.; Zerbes, R.M.; O’Neill, K.; et al. Hexokinases inhibit death receptor-dependent apoptosis on the mitochondria. Proc. Natl. Acad. Sci. USA 2021, 118, e2021175118. [Google Scholar] [CrossRef] [PubMed]
- De Jesus, A.; Keyhani-Nejad, F.; Pusec, C.M.; Goodman, L.; Geier, J.A.; Stoolman, J.S.; Stanczyk, P.J.; Nguyen, T.; Xu, K.; Suresh, K.V.; et al. Hexokinase 1 cellular localization regulates the metabolic fate of glucose. Mol. Cell 2022, 82, 1261–1277.e1269. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, H. Reprogramming of glucose, fatty acid and amino acid metabolism for cancer progression. Cell. Mol. Life Sci. 2016, 73, 377–392. [Google Scholar] [CrossRef] [PubMed]
- Al Hasawi, N.; Alkandari, M.F.; Luqmani, Y.A. Phosphofructokinase: A mediator of glycolytic flux in cancer progression. Crit. Rev. Oncol. Hematol. 2014, 92, 312–321. [Google Scholar] [CrossRef]
- Fu, Q.; Yu, Z. Phosphoglycerate kinase 1 (pgk1) in cancer: A promising target for diagnosis and therapy. Life Sci. 2020, 256, 117863. [Google Scholar] [CrossRef]
- Chang, Y.C.; Chiou, J.; Yang, Y.F.; Su, C.Y.; Lin, Y.F.; Yang, C.N.; Lu, P.J.; Huang, M.S.; Yang, C.J.; Hsiao, M. Therapeutic targeting of aldolase a interactions inhibits lung cancer metastasis and prolongs survival. Cancer Res. 2019, 79, 4754–4766. [Google Scholar] [CrossRef] [Green Version]
- Ji, S.; Zhang, B.; Liu, J.; Qin, Y.; Liang, C.; Shi, S.; Jin, K.; Liang, D.; Xu, W.; Xu, H.; et al. Aldoa functions as an oncogene in the highly metastatic pancreatic cancer. Cancer Lett. 2016, 374, 127–135. [Google Scholar] [CrossRef]
- Du, S.; Guan, Z.; Hao, L.; Song, Y.; Wang, L.; Gong, L.; Liu, L.; Qi, X.; Hou, Z.; Shao, S. Fructose-bisphosphate aldolase a is a potential metastasis-associated marker of lung squamous cell carcinoma and promotes lung cell tumorigenesis and migration. PLoS ONE 2014, 9, e85804. [Google Scholar] [CrossRef] [Green Version]
- Kuang, Q.; Liang, Y.; Zhuo, Y.; Cai, Z.; Jiang, F.; Xie, J.; Zheng, Y.; Zhong, W. The aldoa metabolism pathway as a potential target for regulation of prostate cancer proliferation. Oncol. Targets Ther. 2021, 14, 3353–3366. [Google Scholar] [CrossRef]
- Gao, Q.; Zhu, H.; Dong, L.; Shi, W.; Chen, R.; Song, Z.; Huang, C.; Li, J.; Dong, X.; Zhou, Y.; et al. Integrated proteogenomic characterization of hbv-related hepatocellular carcinoma. Cell 2019, 179, 1240. [Google Scholar] [CrossRef]
- Niu, Y.; Lin, Z.; Wan, A.; Sun, L.; Yan, S.; Liang, H.; Zhan, S.; Chen, D.; Bu, X.; Liu, P.; et al. Loss-of-function genetic screening identifies aldolase a as an essential driver for liver cancer cell growth under hypoxia. Hepatology 2021, 74, 1461–1479. [Google Scholar] [CrossRef] [PubMed]
- Mazurek, S.; Boschek, C.B.; Hugo, F.; Eigenbrodt, E. Pyruvate kinase type m2 and its role in tumor growth and spreading. Semin. Cancer Biol. 2005, 15, 300–308. [Google Scholar] [CrossRef] [PubMed]
- Christofk, H.R.; Vander Heiden, M.G.; Harris, M.H.; Ramanathan, A.; Gerszten, R.E.; Wei, R.; Fleming, M.D.; Schreiber, S.L.; Cantley, L.C. The m2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008, 452, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. Hif-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, S.; Archer, S.L.; Allalunis-Turner, J.; Haromy, A.; Beaulieu, C.; Thompson, R.; Lee, C.T.; Lopaschuk, G.D.; Puttagunta, L.; Bonnet, S.; et al. A mitochondria-k+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 2007, 11, 37–51. [Google Scholar] [CrossRef] [Green Version]
- Yao, F.; Zhao, T.; Zhong, C.; Zhu, J.; Zhao, H. Ldha is necessary for the tumorigenicity of esophageal squamous cell carcinoma. Tumour. Biol. 2013, 34, 25–31. [Google Scholar] [CrossRef]
- Le, A.; Cooper, C.R.; Gouw, A.M.; Dinavahi, R.; Maitra, A.; Deck, L.M.; Royer, R.E.; Vander Jagt, D.L.; Semenza, G.L.; Dang, C.V. Inhibition of lactate dehydrogenase a induces oxidative stress and inhibits tumor progression. Proc. Natl. Acad. Sci. USA 2010, 107, 2037–2042. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Yang, Z.; Chen, Z.; Chen, R.; Zhao, D.; Zhou, Y.; Qiao, L. Effects of the suppression of lactate dehydrogenase a on the growth and invasion of human gastric cancer cells. Oncol. Rep. 2015, 33, 157–162. [Google Scholar] [CrossRef] [Green Version]
- San-Millan, I.; Brooks, G.A. Reexamining cancer metabolism: Lactate production for carcinogenesis could be the purpose and explanation of the warburg effect. Carcinogenesis 2017, 38, 119–133. [Google Scholar] [CrossRef] [Green Version]
- Vegran, F.; Boidot, R.; Michiels, C.; Sonveaux, P.; Feron, O. Lactate influx through the endothelial cell monocarboxylate transporter mct1 supports an nf-kappab/il-8 pathway that drives tumor angiogenesis. Cancer Res. 2011, 71, 2550–2560. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.Y.; Collins, C.C.; Gout, P.W.; Wang, Y. Cancer-generated lactic acid: A regulatory, immunosuppressive metabolite? J. Pathol. 2013, 230, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Goetze, K.; Walenta, S.; Ksiazkiewicz, M.; Kunz-Schughart, L.A.; Mueller-Klieser, W. Lactate enhances motility of tumor cells and inhibits monocyte migration and cytokine release. Int. J. Oncol. 2011, 39, 453–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory effect of tumor cell-derived lactic acid on human t cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, P.; Du, W.; Wu, M. Regulation of the pentose phosphate pathway in cancer. Protein. Cell 2014, 5, 592–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nogueira, V.; Hay, N. Molecular pathways: Reactive oxygen species homeostasis in cancer cells and implications for cancer therapy. Clin. Cancer Res. 2013, 19, 4309–4314. [Google Scholar] [CrossRef] [Green Version]
- Lu, S.C. Glutathione synthesis. Biochim. Biophys. Acta 2013, 1830, 3143–3153. [Google Scholar] [CrossRef] [Green Version]
- Kowalik, M.A.; Columbano, A.; Perra, A. Emerging role of the pentose phosphate pathway in hepatocellular carcinoma. Front. Oncol. 2017, 7, 87. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Marcos, P.J.; Nobrega-Pereira, S. Nadph: New oxygen for the ros theory of aging. Oncotarget 2016, 7, 50814–50815. [Google Scholar] [CrossRef]
- Chen, M.; Shen, M.; Li, Y.; Liu, C.; Zhou, K.; Hu, W.; Xu, B.; Xia, Y.; Tang, W. Gc-ms-based metabolomic analysis of human papillary thyroid carcinoma tissue. Int. J. Mol. Med. 2015, 36, 1607–1614. [Google Scholar] [CrossRef] [Green Version]
- Langbein, S.; Frederiks, W.M.; zur Hausen, A.; Popa, J.; Lehmann, J.; Weiss, C.; Alken, P.; Coy, J.F. Metastasis is promoted by a bioenergetic switch: New targets for progressive renal cell cancer. Int. J. Cancer 2008, 122, 2422–2428. [Google Scholar] [CrossRef]
- Zampella, E.J.; Bradley, E.L., Jr.; Pretlow, T.G., 2nd. Glucose-6-phosphate dehydrogenase: A possible clinical indicator for prostatic carcinoma. Cancer 1982, 49, 384–387. [Google Scholar] [CrossRef]
- Kuo, W.; Lin, J.; Tang, T.K. Human glucose-6-phosphate dehydrogenase (g6pd) gene transforms nih 3t3 cells and induces tumors in nude mice. Int. J. Cancer 2000, 85, 857–864. [Google Scholar] [CrossRef]
- Patra, K.C.; Hay, N. The pentose phosphate pathway and cancer. Trends Biochem. Sci. 2014, 39, 347–354. [Google Scholar] [CrossRef] [Green Version]
- Stanton, R.C.; Seifter, J.L.; Boxer, D.C.; Zimmerman, E.; Cantley, L.C. Rapid release of bound glucose-6-phosphate dehydrogenase by growth factors. Correlation with increased enzymatic activity. J. Biol. Chem. 1991, 266, 12442–12448. [Google Scholar] [CrossRef]
- Tian, W.N.; Pignatare, J.N.; Stanton, R.C. Signal transduction proteins that associate with the platelet-derived growth factor (pdgf) receptor mediate the pdgf-induced release of glucose-6-phosphate dehydrogenase from permeabilized cells. J. Biol. Chem. 1994, 269, 14798–14805. [Google Scholar] [CrossRef]
- Pan, S.; World, C.J.; Kovacs, C.J.; Berk, B.C. Glucose 6-phosphate dehydrogenase is regulated through c-src-mediated tyrosine phosphorylation in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 895–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Zhang, X.; Li, Y.; Shao, Y.; Xiao, J.; Zhu, G.; Li, F. Pak4 regulates g6pd activity by p53 degradation involving colon cancer cell growth. Cell Death Dis. 2017, 8, e2820. [Google Scholar] [CrossRef] [Green Version]
- Jiang, P.; Du, W.; Wang, X.; Mancuso, A.; Gao, X.; Wu, M.; Yang, X. P53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat. Cell Biol. 2011, 13, 310–316. [Google Scholar] [CrossRef] [Green Version]
- Hong, X.; Song, R.; Song, H.; Zheng, T.; Wang, J.; Liang, Y.; Qi, S.; Lu, Z.; Song, X.; Jiang, H.; et al. Pten antagonises tcl1/hnrnpk-mediated g6pd pre-mrna splicing which contributes to hepatocarcinogenesis. Gut 2014, 63, 1635–1647. [Google Scholar] [CrossRef]
- Ge, T.; Yang, J.; Zhou, S.; Wang, Y.; Li, Y.; Tong, X. The role of the pentose phosphate pathway in diabetes and cancer. Front. Endocrinol. 2020, 11, 365. [Google Scholar] [CrossRef]
- Stincone, A.; Prigione, A.; Cramer, T.; Wamelink, M.M.; Campbell, K.; Cheung, E.; Olin-Sandoval, V.; Gruning, N.M.; Kruger, A.; Tauqeer Alam, M.; et al. The return of metabolism: Biochemistry and physiology of the pentose phosphate pathway. Biol. Rev. Camb. Philos. Soc. 2015, 90, 927–963. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Zhou, Y. Crucial role of the pentose phosphate pathway in malignant tumors. Oncol. Lett. 2019, 17, 4213–4221. [Google Scholar] [CrossRef] [Green Version]
- Basta, P.V.; Bensen, J.T.; Tse, C.K.; Perou, C.M.; Sullivan, P.F.; Olshan, A.F. Genetic variation in transaldolase 1 and risk of squamous cell carcinoma of the head and neck. Cancer Detect Prev. 2008, 32, 200–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Guo, K.; Gao, D.; Kang, X.; Jiang, K.; Li, Y.; Sun, L.; Zhang, S.; Sun, C.; Liu, X.; et al. Identification of transaldolase as a novel serum biomarker for hepatocellular carcinoma metastasis using xenografted mouse model and clinic samples. Cancer Lett. 2011, 313, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Langbein, S.; Zerilli, M.; Zur Hausen, A.; Staiger, W.; Rensch-Boschert, K.; Lukan, N.; Popa, J.; Ternullo, M.P.; Steidler, A.; Weiss, C.; et al. Expression of transketolase tktl1 predicts colon and urothelial cancer patient survival: Warburg effect reinterpreted. Br. J. Cancer 2006, 94, 578–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owen, O.E.; Kalhan, S.C.; Hanson, R.W. The key role of anaplerosis and cataplerosis for citric acid cycle function. J. Biol. Chem. 2002, 277, 30409–30412. [Google Scholar] [CrossRef] [Green Version]
- Catalina-Rodriguez, O.; Kolukula, V.K.; Tomita, Y.; Preet, A.; Palmieri, F.; Wellstein, A.; Byers, S.; Giaccia, A.J.; Glasgow, E.; Albanese, C.; et al. The mitochondrial citrate transporter, cic, is essential for mitochondrial homeostasis. Oncotarget 2012, 3, 1220–1235. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Bollu, L.R.; Tozzi, F.; Ye, X.; Bhattacharya, R.; Gao, G.; Dupre, E.; Xia, L.; Lu, J.; Fan, F.; et al. Atp citrate lyase mediates resistance of colorectal cancer cells to sn38. Mol. Cancer Ther. 2013, 12, 2782–2791. [Google Scholar] [CrossRef] [Green Version]
- Beckner, M.E.; Fellows-Mayle, W.; Zhang, Z.; Agostino, N.R.; Kant, J.A.; Day, B.W.; Pollack, I.F. Identification of atp citrate lyase as a positive regulator of glycolytic function in glioblastomas. Int. J. Cancer 2010, 126, 2282–2295. [Google Scholar] [CrossRef] [Green Version]
- Szutowicz, A.; Kwiatkowski, J.; Angielski, S. Lipogenetic and glycolytic enzyme activities in carcinoma and nonmalignant diseases of the human breast. Br. J. Cancer 1979, 39, 681–687. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Y.; Shen, L.; Pang, Y.; Qiao, Z.; Liu, P. Prognostic and therapeutic implications of increased atp citrate lyase expression in human epithelial ovarian cancer. Oncol. Rep. 2012, 27, 1156–1162. [Google Scholar] [CrossRef] [PubMed]
- Bourbeau, M.P.; Bartberger, M.D. Recent advances in the development of acetyl-coa carboxylase (acc) inhibitors for the treatment of metabolic disease. J. Med. Chem. 2015, 58, 525–536. [Google Scholar] [CrossRef] [PubMed]
- Carling, D.; Zammit, V.A.; Hardie, D.G. A common bicyclic protein kinase cascade inactivates the regulatory enzymes of fatty acid and cholesterol biosynthesis. FEBS Lett. 1987, 223, 217–222. [Google Scholar] [CrossRef] [Green Version]
- Menendez, J.A.; Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.A.; Han, W.F.; Morin, P.J.; Chrest, F.J.; Pizer, E.S. Activation of fatty acid synthesis during neoplastic transformation: Role of mitogen-activated protein kinase and phosphatidylinositol 3-kinase. Exp. Cell Res. 2002, 279, 80–90. [Google Scholar] [CrossRef]
- Jin, L.; Alesi, G.N.; Kang, S. Glutaminolysis as a target for cancer therapy. Oncogene 2016, 35, 3619–3625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by idh1 mediates lipogenesis under hypoxia. Nature 2011, 481, 380–384. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Zhou, L.; Shi, Q.; Zhao, Y.; Lin, H.; Zhang, M.; Zhao, S.; Yang, Y.; Ling, Z.Q.; Guan, K.L.; et al. Sirt3-dependent got2 acetylation status affects the malate-aspartate nadh shuttle activity and pancreatic tumor growth. EMBO J. 2015, 34, 1110–1125. [Google Scholar] [CrossRef] [Green Version]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Pavlova, N.N.; Thompson, C.B. Cancer cell metabolism: The essential role of the nonessential amino acid, glutamine. EMBO J. 2017, 36, 1302–1315. [Google Scholar] [CrossRef] [Green Version]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Tchernyshyov, I.; Chang, T.C.; Lee, Y.S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. C-myc suppression of mir-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef] [Green Version]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, A.; Lane, A.N.; Hamaker, M.; Bose, S.; Gouw, A.; Barbi, J.; Tsukamoto, T.; Rojas, C.J.; Slusher, B.S.; Zhang, H.; et al. Glucose-independent glutamine metabolism via tca cycling for proliferation and survival in b cells. Cell Metab. 2012, 15, 110–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicklin, P.; Bergman, P.; Zhang, B.; Triantafellow, E.; Wang, H.; Nyfeler, B.; Yang, H.; Hild, M.; Kung, C.; Wilson, C.; et al. Bidirectional transport of amino acids regulates mtor and autophagy. Cell 2009, 136, 521–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.H.; Qiu, Y.; Stamatatos, O.; Janowitz, T.; Lukey, M.J. Enhancing the efficacy of glutamine metabolism inhibitors in cancer therapy. Trends Cancer 2021, 7, 790–804. [Google Scholar] [CrossRef]
- Qu, X.; Sun, J.; Zhang, Y.; Li, J.; Hu, J.; Li, K.; Gao, L.; Shen, L. C-myc-driven glycolysis via txnip suppression is dependent on glutaminase-mondoa axis in prostate cancer. Biochem. Biophys. Res. Commun. 2018, 504, 415–421. [Google Scholar] [CrossRef]
- Lampa, M.; Arlt, H.; He, T.; Ospina, B.; Reeves, J.; Zhang, B.; Murtie, J.; Deng, G.; Barberis, C.; Hoffmann, D.; et al. Glutaminase is essential for the growth of triple-negative breast cancer cells with a deregulated glutamine metabolism pathway and its suppression synergizes with mtor inhibition. PLoS ONE 2017, 12, e0185092. [Google Scholar] [CrossRef] [Green Version]
- Mates, J.M.; Campos-Sandoval, J.A.; Santos-Jimenez, J.L.; Marquez, J. Dysregulation of glutaminase and glutamine synthetase in cancer. Cancer Lett. 2019, 467, 29–39. [Google Scholar] [CrossRef]
- Tannir, N.M.; Agarwal, N.; Porta, C.; Lawrence, N.J.; Motzer, R.; McGregor, B.; Lee, R.J.; Jain, R.K.; Davis, N.; Appleman, L.J.; et al. Efficacy and safety of telaglenastat plus cabozantinib vs placebo plus cabozantinib in patients with advanced renal cell carcinoma: The cantata randomized clinical trial. JAMA Oncol. 2022, 8, 1411–1418. [Google Scholar] [CrossRef]
- Amelio, I.; Cutruzzola, F.; Antonov, A.; Agostini, M.; Melino, G. Serine and glycine metabolism in cancer. Trends Biochem. Sci. 2014, 39, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Maddocks, O.D.; Labuschagne, C.F.; Adams, P.D.; Vousden, K.H. Serine metabolism supports the methionine cycle and DNA/rna methylation through de novo atp synthesis in cancer cells. Mol. Cell 2016, 61, 210–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahu, N.; Dela Cruz, D.; Gao, M.; Sandoval, W.; Haverty, P.M.; Liu, J.; Stephan, J.P.; Haley, B.; Classon, M.; Hatzivassiliou, G.; et al. Proline starvation induces unresolved er stress and hinders mtorc1-dependent tumorigenesis. Cell Metab. 2016, 24, 753–761. [Google Scholar] [CrossRef] [Green Version]
- Tang, L.; Zeng, J.; Geng, P.; Fang, C.; Wang, Y.; Sun, M.; Wang, C.; Wang, J.; Yin, P.; Hu, C.; et al. Global metabolic profiling identifies a pivotal role of proline and hydroxyproline metabolism in supporting hypoxic response in hepatocellular carcinoma. Clin. Cancer Res. 2018, 24, 474–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Togashi, Y.; Arao, T.; Kato, H.; Matsumoto, K.; Terashima, M.; Hayashi, H.; de Velasco, M.A.; Fujita, Y.; Kimura, H.; Yasuda, T.; et al. Frequent amplification of oraov1 gene in esophageal squamous cell cancer promotes an aggressive phenotype via proline metabolism and ros production. Oncotarget 2014, 5, 2962–2973. [Google Scholar] [CrossRef] [Green Version]
- Elia, I.; Broekaert, D.; Christen, S.; Boon, R.; Radaelli, E.; Orth, M.F.; Verfaillie, C.; Grunewald, T.G.P.; Fendt, S.M. Proline metabolism supports metastasis formation and could be inhibited to selectively target metastasizing cancer cells. Nat. Commun. 2017, 8, 15267. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Hancock, C.N.; Fischer, J.W.; Harman, M.; Phang, J.M. Proline biosynthesis augments tumor cell growth and aerobic glycolysis: Involvement of pyridine nucleotides. Sci. Rep. 2015, 5, 17206. [Google Scholar] [CrossRef] [Green Version]
- Pegg, A.E.; Casero, R.A., Jr. Current status of the polyamine research field. Methods Mol. Biol. 2011, 720, 3–35. [Google Scholar]
- Terui, Y.; Yoshida, T.; Sakamoto, A.; Saito, D.; Oshima, T.; Kawazoe, M.; Yokoyama, S.; Igarashi, K.; Kashiwagi, K. Polyamines protect nucleic acids against depurination. Int. J. Biochem. Cell Biol. 2018, 99, 147–153. [Google Scholar] [CrossRef]
- Kurata, H.T.; Akrouh, A.; Li, J.B.; Marton, L.J.; Nichols, C.G. Scanning the topography of polyamine blocker binding in an inwardly rectifying potassium channel. J. Biol. Chem. 2013, 288, 6591–6601. [Google Scholar] [CrossRef] [Green Version]
- Rao, J.N.; Rathor, N.; Zhuang, R.; Zou, T.; Liu, L.; Xiao, L.; Turner, D.J.; Wang, J.Y. Polyamines regulate intestinal epithelial restitution through trpc1-mediated Ca2+ signaling by differentially modulating stim1 and stim2. Am. J. Physiol. Cell Physiol. 2012, 303, C308–C317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray-Stewart, T.R.; Woster, P.M.; Casero, R.A., Jr. Targeting polyamine metabolism for cancer therapy and prevention. Biochem. J. 2016, 473, 2937–2953. [Google Scholar] [CrossRef] [PubMed]
- Iurlaro, R.; Leon-Annicchiarico, C.L.; Munoz-Pinedo, C. Regulation of cancer metabolism by oncogenes and tumor suppressors. Methods Enzymol. 2014, 542, 59–80. [Google Scholar] [PubMed]
- Nagarajan, A.; Malvi, P.; Wajapeyee, N. Oncogene-directed alterations in cancer cell metabolism. Trends Cancer 2016, 2, 365–377. [Google Scholar] [CrossRef] [Green Version]
- Shaw, R.J.; Cantley, L.C. Ras, pi(3)k and mtor signalling controls tumour cell growth. Nature 2006, 441, 424–430. [Google Scholar] [CrossRef]
- Barata, J.T.; Silva, A.; Brandao, J.G.; Nadler, L.M.; Cardoso, A.A.; Boussiotis, V.A. Activation of pi3k is indispensable for interleukin 7-mediated viability, proliferation, glucose use, and growth of t cell acute lymphoblastic leukemia cells. J. Exp. Med. 2004, 200, 659–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edinger, A.L.; Thompson, C.B. Akt maintains cell size and survival by increasing mtor-dependent nutrient uptake. Mol. Biol. Cell 2002, 13, 2276–2288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roos, S.; Jansson, N.; Palmberg, I.; Saljo, K.; Powell, T.L.; Jansson, T. Mammalian target of rapamycin in the human placenta regulates leucine transport and is down-regulated in restricted fetal growth. J. Physiol. 2007, 582, 449–459. [Google Scholar] [CrossRef]
- Wieman, H.L.; Wofford, J.A.; Rathmell, J.C. Cytokine stimulation promotes glucose uptake via phosphatidylinositol-3 kinase/akt regulation of glut1 activity and trafficking. Mol. Biol. Cell 2007, 18, 1437–1446. [Google Scholar] [CrossRef] [Green Version]
- Xu, R.H.; Pelicano, H.; Zhang, H.; Giles, F.J.; Keating, M.J.; Huang, P. Synergistic effect of targeting mtor by rapamycin and depleting atp by inhibition of glycolysis in lymphoma and leukemia cells. Leukemia 2005, 19, 2153–2158. [Google Scholar] [CrossRef]
- Elstrom, R.L.; Bauer, D.E.; Buzzai, M.; Karnauskas, R.; Harris, M.H.; Plas, D.R.; Zhuang, H.; Cinalli, R.M.; Alavi, A.; Rudin, C.M.; et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004, 64, 3892–3899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plas, D.R.; Talapatra, S.; Edinger, A.L.; Rathmell, J.C.; Thompson, C.B. Akt and bcl-xl promote growth factor-independent survival through distinct effects on mitochondrial physiology. J. Biol. Chem. 2001, 276, 12041–12048. [Google Scholar] [CrossRef] [PubMed]
- Rathmell, J.C.; Fox, C.J.; Plas, D.R.; Hammerman, P.S.; Cinalli, R.M.; Thompson, C.B. Akt-directed glucose metabolism can prevent bax conformation change and promote growth factor-independent survival. Mol. Cell Biol. 2003, 23, 7315–7328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, D.E.; Hatzivassiliou, G.; Zhao, F.; Andreadis, C.; Thompson, C.B. Atp citrate lyase is an important component of cell growth and transformation. Oncogene 2005, 24, 6314–6322. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.; Wang, J.; Lu, X.; Thewke, D.P.; Mason, R.J. Kgf induces lipogenic genes through a pi3k and jnk/srebp-1 pathway in h292 cells. J. Lipid. Res. 2005, 46, 2624–2635. [Google Scholar] [CrossRef] [Green Version]
- Gingras, A.C.; Raught, B.; Sonenberg, N. Regulation of translation initiation by frap/mtor. Genes Dev. 2001, 15, 807–826. [Google Scholar] [CrossRef] [Green Version]
- Duvel, K.; Yecies, J.L.; Menon, S.; Raman, P.; Lipovsky, A.I.; Souza, A.L.; Triantafellow, E.; Ma, Q.; Gorski, R.; Cleaver, S.; et al. Activation of a metabolic gene regulatory network downstream of mtor complex 1. Mol. Cell 2010, 39, 171–183. [Google Scholar] [CrossRef] [Green Version]
- Peterson, T.R.; Sengupta, S.S.; Harris, T.E.; Carmack, A.E.; Kang, S.A.; Balderas, E.; Guertin, D.A.; Madden, K.L.; Carpenter, A.E.; Finck, B.N.; et al. Mtor complex 1 regulates lipin 1 localization to control the srebp pathway. Cell 2011, 146, 408–420. [Google Scholar] [CrossRef] [Green Version]
- Ben-Sahra, I.; Howell, J.J.; Asara, J.M.; Manning, B.D. Stimulation of de novo pyrimidine synthesis by growth signaling through mtor and s6k1. Science 2013, 339, 1323–1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, M.S.; Salmena, L.; Pandolfi, P.P. The functions and regulation of the pten tumour suppressor. Nat. Rev. Mol. Cell Biol. 2012, 13, 283–296. [Google Scholar] [CrossRef]
- Garcia-Cao, I.; Song, M.S.; Hobbs, R.M.; Laurent, G.; Giorgi, C.; de Boer, V.C.; Anastasiou, D.; Ito, K.; Sasaki, A.T.; Rameh, L.; et al. Systemic elevation of pten induces a tumor-suppressive metabolic state. Cell 2012, 149, 49–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bieging, K.T.; Mello, S.S.; Attardi, L.D. Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer 2014, 14, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Berkers, C.R.; Maddocks, O.D.; Cheung, E.C.; Mor, I.; Vousden, K.H. Metabolic regulation by p53 family members. Cell Metab. 2013, 18, 617–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vousden, K.H.; Ryan, K.M. P53 and metabolism. Nat. Rev. Cancer 2009, 9, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Gatenby, R.A.; Smallbone, K.; Maini, P.K.; Rose, F.; Averill, J.; Nagle, R.B.; Worrall, L.; Gillies, R.J. Cellular adaptations to hypoxia and acidosis during somatic evolution of breast cancer. Br. J. Cancer 2007, 97, 646–653. [Google Scholar] [CrossRef] [Green Version]
- Keith, B.; Johnson, R.S.; Simon, M.C. Hif1alpha and hif2alpha: Sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer 2011, 12, 9–22. [Google Scholar] [CrossRef] [Green Version]
- O’Rourke, J.F.; Pugh, C.W.; Bartlett, S.M.; Ratcliffe, P.J. Identification of hypoxically inducible mrnas in hela cells using differential-display pcr. Role of hypoxia-inducible factor-1. Eur. J. Biochem. 1996, 241, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L.; Roth, P.H.; Fang, H.M.; Wang, G.L. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J. Biol. Chem. 1994, 269, 23757–23763. [Google Scholar] [CrossRef]
- Schodel, J.; Oikonomopoulos, S.; Ragoussis, J.; Pugh, C.W.; Ratcliffe, P.J.; Mole, D.R. High-resolution genome-wide mapping of hif-binding sites by chip-seq. Blood 2011, 117, e207–e217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wise, D.R.; Ward, P.S.; Shay, J.E.; Cross, J.R.; Gruber, J.J.; Sachdeva, U.M.; Platt, J.M.; DeMatteo, R.G.; Simon, M.C.; Thompson, C.B. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of alpha-ketoglutarate to citrate to support cell growth and viability. Proc. Natl. Acad. Sci. USA 2011, 108, 19611–19616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wierstra, I.; Alves, J. The c-myc promoter: Still mystery and challenge. Adv. Cancer Res. 2008, 99, 113–333. [Google Scholar] [PubMed]
- Kim, J.W.; Zeller, K.I.; Wang, Y.; Jegga, A.G.; Aronow, B.J.; O’Donnell, K.A.; Dang, C.V. Evaluation of myc e-box phylogenetic footprints in glycolytic genes by chromatin immunoprecipitation assays. Mol. Cell Biol. 2004, 24, 5923–5936. [Google Scholar] [CrossRef] [PubMed]
- David, C.J.; Chen, M.; Assanah, M.; Canoll, P.; Manley, J.L. Hnrnp proteins controlled by c-myc deregulate pyruvate kinase mrna splicing in cancer. Nature 2010, 463, 364–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuneva, M.; Zamboni, N.; Oefner, P.; Sachidanandam, R.; Lazebnik, Y. Deficiency in glutamine but not glucose induces myc-dependent apoptosis in human cells. J. Cell Biol. 2007, 178, 93–105. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Dillon, C.P.; Shi, L.Z.; Milasta, S.; Carter, R.; Finkelstein, D.; McCormick, L.L.; Fitzgerald, P.; Chi, H.; Munger, J.; et al. The transcription factor myc controls metabolic reprogramming upon t lymphocyte activation. Immunity 2011, 35, 871–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Q.M.; Malek, R.L.; Kim, S.; Chiao, C.; He, M.; Ruffy, M.; Sanka, K.; Lee, N.H.; Dang, C.V.; Liu, E.T. Identification of c-myc responsive genes using rat cdna microarray. Cancer Res. 2000, 60, 5922–5928. [Google Scholar] [PubMed]
- Nikiforov, M.A.; Chandriani, S.; O’Connell, B.; Petrenko, O.; Kotenko, I.; Beavis, A.; Sedivy, J.M.; Cole, M.D. A functional screen for myc-responsive genes reveals serine hydroxymethyltransferase, a major source of the one-carbon unit for cell metabolism. Mol. Cell Biol. 2002, 22, 5793–5800. [Google Scholar] [CrossRef] [Green Version]
- O’Connell, B.C.; Cheung, A.F.; Simkevich, C.P.; Tam, W.; Ren, X.; Mateyak, M.K.; Sedivy, J.M. A large scale genetic analysis of c-myc-regulated gene expression patterns. J. Biol. Chem. 2003, 278, 12563–12573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Song, L.; Wan, Q.; Wu, G.; Li, X.; Wang, Y.; Wang, J.; Liu, Z.; Zhong, X.; He, X.; et al. Cmyc-mediated activation of serine biosynthesis pathway is critical for cancer progression under nutrient deprivation conditions. Cell Res. 2015, 25, 429–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labuschagne, C.F.; van den Broek, N.J.; Mackay, G.M.; Vousden, K.H.; Maddocks, O.D. Serine, but not glycine, supports one-carbon metabolism and proliferation of cancer cells. Cell Rep. 2014, 7, 1248–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Kenyon, W.J.; Li, Q.; Mullberg, J.; Hutt-Fletcher, L.M. Epstein-barr virus uses different complexes of glycoproteins gh and gl to infect b lymphocytes and epithelial cells. J. Virol. 1998, 72, 5552–5558. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Xie, L.; Shi, F.; Tang, M.; Li, Y.; Hu, J.; Zhao, L.; Zhao, L.; Yu, X.; Luo, X.; et al. Targeting the signaling in epstein-barr virus-associated diseases: Mechanism, regulation, and clinical study. Signal Transduct. Target Ther. 2021, 6, 15. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y. Ebv based cancer prevention and therapy in nasopharyngeal carcinoma. NPJ Precis. Oncol. 2017, 1, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murata, T.; Tsurumi, T. Switching of ebv cycles between latent and lytic states. Rev. Med. Virol. 2014, 24, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Germini, D.; Sall, F.B.; Shmakova, A.; Wiels, J.; Dokudovskaya, S.; Drouet, E.; Vassetzky, Y. Oncogenic properties of the ebv zebra protein. Cancers 2020, 12, 1479. [Google Scholar] [CrossRef] [PubMed]
- Young, L.S.; Yap, L.F.; Murray, P.G. Epstein-barr virus: More than 50 years old and still providing surprises. Nat. Rev. Cancer 2016, 16, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Munz, C. Latency and lytic replication in epstein-barr virus-associated oncogenesis. Nat. Rev. Microbiol. 2019, 17, 691–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McFadden, K.; Hafez, A.Y.; Kishton, R.; Messinger, J.E.; Nikitin, P.A.; Rathmell, J.C.; Luftig, M.A. Metabolic stress is a barrier to epstein-barr virus-mediated b-cell immortalization. Proc. Natl. Acad. Sci. USA 2016, 113, E782–E790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Jia, L.; Liu, T.; Yip, Y.L.; Tang, W.C.; Lin, W.; Deng, W.; Lo, K.W.; You, C.; Lung, M.L.; et al. Mtorc2-mediated pdhe1alpha nuclear translocation links ebv-lmp1 reprogrammed glucose metabolism to cancer metastasis in nasopharyngeal carcinoma. Oncogene 2019, 38, 4669–4684. [Google Scholar] [CrossRef] [Green Version]
- Lo, A.K.; Dawson, C.W.; Young, L.S.; Ko, C.W.; Hau, P.M.; Lo, K.W. Activation of the fgfr1 signalling pathway by the epstein-barr virus-encoded lmp1 promotes aerobic glycolysis and transformation of human nasopharyngeal epithelial cells. J. Pathol. 2015, 237, 238–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, L.; Hu, Z.Y.; Dong, X.; Tan, Z.; Li, W.; Tang, M.; Chen, L.; Yang, L.; Tao, Y.; Jiang, Y.; et al. Targeting epstein-barr virus oncoprotein lmp1-mediated glycolysis sensitizes nasopharyngeal carcinoma to radiation therapy. Oncogene 2014, 33, 4568–4578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Yan, B.; Lai, W.; Shi, Y.; Xiao, D.; Jia, J.; Liu, S.; Li, H.; Lu, J.; Li, Z.; et al. Repression of hox genes by lmp1 in nasopharyngeal carcinoma and modulation of glycolytic pathway genes by hoxc8. Oncogene 2015, 34, 6079–6091. [Google Scholar] [CrossRef] [PubMed]
- Bonglack, E.N.; Messinger, J.E.; Cable, J.M.; Ch’ng, J.; Parnell, K.M.; Reinoso-Vizcaino, N.M.; Barry, A.P.; Russell, V.S.; Dave, S.S.; Christofk, H.R.; et al. Monocarboxylate transporter antagonism reveals metabolic vulnerabilities of viral-driven lymphomas. Proc. Natl. Acad. Sci. USA 2021, 118, e2022495118. [Google Scholar] [CrossRef]
- Chen, Q.; Feng, J.; Wu, J.; Yu, Z.; Zhang, W.; Chen, Y.; Yao, P.; Zhang, H. Hkdc1 c-terminal based peptides inhibit extranodal natural killer/t-cell lymphoma by modulation of mitochondrial function and ebv suppression. Leukemia 2020, 34, 2736–2748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Jia, L.; Tsang, C.M.; Tsao, S.W. Ebv infection and glucose metabolism in nasopharyngeal carcinoma. Adv. Exp. Med. Biol. 2017, 1018, 75–90. [Google Scholar] [PubMed]
- Cai, L.M.; Lyu, X.M.; Luo, W.R.; Cui, X.F.; Ye, Y.F.; Yuan, C.C.; Peng, Q.X.; Wu, D.H.; Liu, T.F.; Wang, E.; et al. Ebv-mir-bart7-3p promotes the emt and metastasis of nasopharyngeal carcinoma cells by suppressing the tumor suppressor pten. Oncogene 2015, 34, 2156–2166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyu, X.; Wang, J.; Guo, X.; Wu, G.; Jiao, Y.; Faleti, O.D.; Liu, P.; Liu, T.; Long, Y.; Chong, T.; et al. Ebv-mir-bart1-5p activates ampk/mtor/hif1 pathway via a pten independent manner to promote glycolysis and angiogenesis in nasopharyngeal carcinoma. PLoS Pathog. 2018, 14, e1007484. [Google Scholar] [CrossRef] [Green Version]
- Krishna, G.; Soman Pillai, V.; Valiya Veettil, M. Upregulation of gls1 isoforms kga and gac facilitates mitochondrial metabolism and cell proliferation in epstein-barr virus infected cells. Viruses 2020, 12, 811. [Google Scholar] [CrossRef]
- Wang, L.W.; Shen, H.; Nobre, L.; Ersing, I.; Paulo, J.A.; Trudeau, S.; Wang, Z.; Smith, N.A.; Ma, Y.; Reinstadler, B.; et al. Epstein-barr-virus-induced one-carbon metabolism drives b cell transformation. Cell Metab. 2019, 30, 539–555.e511. [Google Scholar] [CrossRef] [Green Version]
- Guo, R.; Liang, J.H.; Zhang, Y.; Lutchenkov, M.; Li, Z.; Wang, Y.; Trujillo-Alonso, V.; Puri, R.; Giulino-Roth, L.; Gewurz, B.E. Methionine metabolism controls the b cell ebv epigenome and viral latency. Cell Metab. 2022, 34, 1280–1297.e1289. [Google Scholar] [CrossRef]
- Adamson, A.L.; Le, B.T.; Siedenburg, B.D. Inhibition of mtorc1 inhibits lytic replication of epstein-barr virus in a cell-type specific manner. Virol. J. 2014, 11, 110. [Google Scholar] [CrossRef] [PubMed]
- Hafez, A.Y.; Messinger, J.E.; McFadden, K.; Fenyofalvi, G.; Shepard, C.N.; Lenzi, G.M.; Kim, B.; Luftig, M.A. Limited nucleotide pools restrict epstein-barr virus-mediated b-cell immortalization. Oncogenesis 2017, 6, e349. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.H.; Wang, C.; Yiu, S.P.T.; Zhao, B.; Guo, R.; Gewurz, B.E. Epstein-barr virus induced cytidine metabolism roles in transformed b-cell growth and survival. mBio 2021, 12, e0153021. [Google Scholar] [CrossRef]
- Young, L.S.; Rickinson, A.B. Epstein-barr virus: 40 years on. Nat. Rev. Cancer 2004, 4, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Darekar, S.; Georgiou, K.; Yurchenko, M.; Yenamandra, S.P.; Chachami, G.; Simos, G.; Klein, G.; Kashuba, E. Epstein-barr virus immortalization of human b-cells leads to stabilization of hypoxia-induced factor 1 alpha, congruent with the warburg effect. PLoS ONE 2012, 7, e42072. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, K.; Das, P.; Chattopadhyay, N.R.; Mal, S.; Choudhuri, T. The interplay between epstein-bar virus (ebv) with the p53 and its homologs during ebv associated malignancies. Heliyon 2019, 5, e02624. [Google Scholar] [CrossRef]
- Li, Y.; Webster-Cyriaque, J.; Tomlinson, C.C.; Yohe, M.; Kenney, S. Fatty acid synthase expression is induced by the epstein-barr virus immediate-early protein brlf1 and is required for lytic viral gene expression. J. Virol. 2004, 78, 4197–4206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.W.; Wang, Z.; Ersing, I.; Nobre, L.; Guo, R.; Jiang, S.; Trudeau, S.; Zhao, B.; Weekes, M.P.; Gewurz, B.E. Epstein-barr virus subverts mevalonate and fatty acid pathways to promote infected b-cell proliferation and survival. PLoS Pathog. 2019, 15, e1008030. [Google Scholar] [CrossRef] [Green Version]
- Doorbar, J.; Quint, W.; Banks, L.; Bravo, I.G.; Stoler, M.; Broker, T.R.; Stanley, M.A. The biology and life-cycle of human papillomaviruses. Vaccine 2012, 30 (Suppl. 5), F55–F70. [Google Scholar] [CrossRef]
- Crosbie, E.J.; Einstein, M.H.; Franceschi, S.; Kitchener, H.C. Human papillomavirus and cervical cancer. Lancet 2013, 382, 889–899. [Google Scholar] [CrossRef]
- Satterwhite, C.L.; Torrone, E.; Meites, E.; Dunne, E.F.; Mahajan, R.; Ocfemia, M.C.; Su, J.; Xu, F.; Weinstock, H. Sexually transmitted infections among us women and men: Prevalence and incidence estimates, 2008. Sex Transm. Dis. 2013, 40, 187–193. [Google Scholar] [CrossRef] [PubMed]
- de Martel, C.; Plummer, M.; Vignat, J.; Franceschi, S. Worldwide burden of cancer attributable to hpv by site, country and hpv type. Int. J. Cancer 2017, 141, 664–670. [Google Scholar] [CrossRef] [PubMed]
- Mazurek, S.; Zwerschke, W.; Jansen-Durr, P.; Eigenbrodt, E. Effects of the human papilloma virus hpv-16 e7 oncoprotein on glycolysis and glutaminolysis: Role of pyruvate kinase type m2 and the glycolytic-enzyme complex. Biochem. J. 2001, 356, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Crook, T.; Morgenstern, J.P.; Crawford, L.; Banks, L. Continued expression of hpv-16 e7 protein is required for maintenance of the transformed phenotype of cells co-transformed by hpv-16 plus ej-ras. EMBO J. 1989, 8, 513–519. [Google Scholar] [CrossRef]
- Anastasiou, D.; Poulogiannis, G.; Asara, J.M.; Boxer, M.B.; Jiang, J.K.; Shen, M.; Bellinger, G.; Sasaki, A.T.; Locasale, J.W.; Auld, D.S.; et al. Inhibition of pyruvate kinase m2 by reactive oxygen species contributes to cellular antioxidant responses. Science 2011, 334, 1278–1283. [Google Scholar] [CrossRef] [Green Version]
- Darnell, G.A.; Schroder, W.A.; Antalis, T.M.; Lambley, E.; Major, L.; Gardner, J.; Birrell, G.; Cid-Arregui, A.; Suhrbier, A. Human papillomavirus e7 requires the protease calpain to degrade the retinoblastoma protein. J. Biol. Chem. 2007, 282, 37492–37500. [Google Scholar] [CrossRef] [Green Version]
- Spangle, J.M.; Munger, K. The human papillomavirus type 16 e6 oncoprotein activates mtorc1 signaling and increases protein synthesis. J. Virol. 2010, 84, 9398–9407. [Google Scholar] [CrossRef] [Green Version]
- Pim, D.; Massimi, P.; Dilworth, S.M.; Banks, L. Activation of the protein kinase b pathway by the hpv-16 e7 oncoprotein occurs through a mechanism involving interaction with pp2a. Oncogene 2005, 24, 7830–7838. [Google Scholar] [CrossRef] [Green Version]
- Fischer, M.; Uxa, S.; Stanko, C.; Magin, T.M.; Engeland, K. Human papilloma virus e7 oncoprotein abrogates the p53-p21-dream pathway. Sci. Rep. 2017, 7, 2603. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Wu, J.; Ling, M.T.; Zhao, L.; Zhao, K.N. The role of the pi3k/akt/mtor signalling pathway in human cancers induced by infection with human papillomaviruses. Mol. Cancer 2015, 14, 87. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Meng, X.; Ma, J.; Zheng, Y.; Wang, Q.; Wang, Y.; Shang, H. Human papillomavirus 16 e6 contributes hif-1alpha induced warburg effect by attenuating the vhl-hif-1alpha interaction. Int. J. Mol. Sci. 2014, 15, 7974–7986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veldman, T.; Liu, X.; Yuan, H.; Schlegel, R. Human papillomavirus e6 and myc proteins associate in vivo and bind to and cooperatively activate the telomerase reverse transcriptase promoter. Proc. Natl. Acad. Sci. USA 2003, 100, 8211–8216. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V.; O’Donnell, K.A.; Zeller, K.I.; Nguyen, T.; Osthus, R.C.; Li, F. The c-myc target gene network. Semin. Cancer Biol. 2006, 16, 253–264. [Google Scholar] [CrossRef]
- Cruz-Gregorio, A.; Aranda-Rivera, A.K.; Aparicio-Trejo, O.E.; Coronado-Martinez, I.; Pedraza-Chaverri, J.; Lizano, M. E6 oncoproteins from high-risk human papillomavirus induce mitochondrial metabolism in a head and neck squamous cell carcinoma model. Biomolecules 2019, 9, 351. [Google Scholar] [CrossRef] [Green Version]
- Crusius, K.; Auvinen, E.; Alonso, A. Enhancement of egf- and pma-mediated map kinase activation in cells expressing the human papillomavirus type 16 e5 protein. Oncogene 1997, 15, 1437–1444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romanczuk, H.; Howley, P.M. Disruption of either the e1 or the e2 regulatory gene of human papillomavirus type 16 increases viral immortalization capacity. Proc. Natl. Acad. Sci. USA 1992, 89, 3159–3163. [Google Scholar] [CrossRef] [Green Version]
- Lai, D.; Tan, C.L.; Gunaratne, J.; Quek, L.S.; Nei, W.; Thierry, F.; Bellanger, S. Localization of hpv-18 e2 at mitochondrial membranes induces ros release and modulates host cell metabolism. PLoS ONE 2013, 8, e75625. [Google Scholar] [CrossRef] [Green Version]
- Pappa, K.I.; Daskalakis, G.; Anagnou, N.P. Metabolic rewiring is associated with hpv-specific profiles in cervical cancer cell lines. Sci. Rep. 2021, 11, 17718. [Google Scholar] [CrossRef]
- Prusinkiewicz, M.A.; Gameiro, S.F.; Ghasemi, F.; Dodge, M.J.; Zeng, P.Y.F.; Maekebay, H.; Barrett, J.W.; Nichols, A.C.; Mymryk, J.S. Survival-associated metabolic genes in human papillomavirus-positive head and neck cancers. Cancers 2020, 12, 253. [Google Scholar] [CrossRef] [Green Version]
- Poiesz, B.J.; Ruscetti, F.W.; Gazdar, A.F.; Bunn, P.A.; Minna, J.D.; Gallo, R.C. Detection and isolation of type c retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous t-cell lymphoma. Proc. Natl. Acad. Sci. USA 1980, 77, 7415–7419. [Google Scholar] [CrossRef] [Green Version]
- Ratner, L. Molecular biology of human t cell leukemia virus. Semin. Diagn. Pathol. 2020, 37, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.; Bangham, C.R.M. Htlv-1: Regulating the balance between proviral latency and reactivation. Front. Microbiol. 2018, 9, 449. [Google Scholar] [CrossRef]
- Manel, N.; Kim, F.J.; Kinet, S.; Taylor, N.; Sitbon, M.; Battini, J.L. The ubiquitous glucose transporter glut-1 is a receptor for htlv. Cell 2003, 115, 449–459. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, A.; Mateus, M.; Thinnes, C.C.; McCullagh, J.S.; Schofield, C.J.; Taylor, G.P.; Bangham, C.R.M. Glucose metabolism and oxygen availability govern reactivation of the latent human retrovirus htlv-1. Cell Chem. Biol. 2017, 24, 1377–1387.e1373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuoka, M.; Jeang, K.T. Human t-cell leukemia virus type 1 (htlv-1) and leukemic transformation: Viral infectivity, tax, hbz and therapy. Oncogene 2011, 30, 1379–1389. [Google Scholar] [CrossRef] [Green Version]
- Mahgoub, M.; Yasunaga, J.I.; Iwami, S.; Nakaoka, S.; Koizumi, Y.; Shimura, K.; Matsuoka, M. Sporadic on/off switching of htlv-1 tax expression is crucial to maintain the whole population of virus-induced leukemic cells. Proc. Natl. Acad. Sci. USA 2018, 115, E1269–E1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, W.; Cao, X.; Pan, G.; Niu, Q.; Feng, X.; Xu, M.; Li, M.; Huang, Y.; Yi, Q.; Yan, D. Orp4l is a prerequisite for the induction of t-cell leukemogenesis associated with human t-cell leukemia virus 1. Blood 2022, 139, 1052–1065. [Google Scholar] [CrossRef]
- Ho, Y.K.; Zhi, H.; DeBiaso, D.; Philip, S.; Shih, H.M.; Giam, C.Z. Htlv-1 tax-induced rapid senescence is driven by the transcriptional activity of nf-kappab and depends on chronically activated ikkalpha and p65/rela. J. Virol. 2012, 86, 9474–9483. [Google Scholar] [CrossRef] [Green Version]
- Kawatsuki, A.; Yasunaga, J.I.; Mitobe, Y.; Green, P.L.; Matsuoka, M. Htlv-1 bzip factor protein targets the rb/e2f-1 pathway to promote proliferation and apoptosis of primary CD4+ t cells. Oncogene 2016, 35, 4509–4517. [Google Scholar] [CrossRef] [Green Version]
- Hutchison, T.; Malu, A.; Yapindi, L.; Bergeson, R.; Peck, K.; Romeo, M.; Harrod, C.; Pope, J.; Smitherman, L.; Gwinn, W.; et al. The tp53-induced glycolysis and apoptosis regulator mediates cooperation between htlv-1 p30(ii) and the retroviral oncoproteins tax and hbz and is highly expressed in an in vivo xenograft model of htlv-1-induced lymphoma. Virology 2018, 520, 39–58. [Google Scholar] [CrossRef]
- Ehtiati, S.; Youssefi, M.; Rafatpanah, H.; Mashkani, B.; Khadem-Rezaiyan, M.; Zahedi Avval, F. Glutathione reductase system changes in htlv-1 infected patients. Virusdisease 2022, 33, 32–38. [Google Scholar] [CrossRef]
- Zhao, T.; Wang, Z.; Fang, J.; Cheng, W.; Zhang, Y.; Huang, J.; Xu, L.; Gou, H.; Zeng, L.; Jin, Z.; et al. Htlv-1 activates yap via nf-kappab/p65 to promote oncogenesis. Proc. Natl. Acad. Sci. USA 2022, 119, e2115316119. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology 2012, 142, 1264–1273.e1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsukuda, S.; Watashi, K. Hepatitis b virus biology and life cycle. Antiviral Res. 2020, 182, 104925. [Google Scholar] [CrossRef] [PubMed]
- Karayiannis, P. Hepatitis b virus: Virology, molecular biology, life cycle and intrahepatic spread. Hepatol. Int. 2017, 11, 500–508. [Google Scholar] [CrossRef] [PubMed]
- Levrero, M.; Zucman-Rossi, J. Mechanisms of hbv-induced hepatocellular carcinoma. J. Hepatol. 2016, 64, S84–S101. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, S.; Lau, K.C.; Coffin, C.S.; Patel, T.R. Molecular mechanisms of viral hepatitis induced hepatocellular carcinoma. World J. Gastroenterol. 2020, 26, 5759–5783. [Google Scholar] [CrossRef]
- Xie, Y. Hepatitis b virus-associated hepatocellular carcinoma. Adv. Exp. Med. Biol. 2017, 1018, 11–21. [Google Scholar]
- Arzumanyan, A.; Reis, H.M.; Feitelson, M.A. Pathogenic mechanisms in hbv- and hcv-associated hepatocellular carcinoma. Nat. Rev. Cancer 2013, 13, 123–135. [Google Scholar] [CrossRef]
- Shin, H.J.; Park, Y.H.; Kim, S.U.; Moon, H.B.; Park, D.S.; Han, Y.H.; Lee, C.H.; Lee, D.S.; Song, I.S.; Lee, D.H.; et al. Hepatitis b virus x protein regulates hepatic glucose homeostasis via activation of inducible nitric oxide synthase. J. Biol. Chem. 2011, 286, 29872–29881. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Zhu, W.; Zhang, L.; Lei, H.; Wu, X.; Guo, L.; Chen, X.; Wang, Y.; Tang, H. The metabolic responses to hepatitis b virus infection shed new light on pathogenesis and targets for treatment. Sci. Rep. 2015, 5, 8421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.Y.; Wang, W.H.; Che, L.; Lan, Y.; Zhang, L.Y.; Zhan, D.L.; Huang, Z.Y.; Lin, Z.N.; Lin, Y.C. Bnip3l-dependent mitophagy promotes hbx-induced cancer stemness of hepatocellular carcinoma cells via glycolysis metabolism reprogramming. Cancers 2020, 12, 655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Fang, M.; He, Z.; Cui, D.; Jia, S.; Lin, X.; Xu, X.; Zhou, T.; Liu, W. Hepatitis b virus stimulates g6pd expression through hbx-mediated nrf2 activation. Cell Death Dis. 2015, 6, e1980. [Google Scholar] [CrossRef] [Green Version]
- Teng, C.F.; Hsieh, W.C.; Wu, H.C.; Lin, Y.J.; Tsai, H.W.; Huang, W.; Su, I.J. Hepatitis b virus pre-s2 mutant induces aerobic glycolysis through mammalian target of rapamycin signal cascade. PLoS ONE 2015, 10, e0122373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teng, C.F.; Wu, H.C.; Hsieh, W.C.; Tsai, H.W.; Su, I.J. Activation of atp citrate lyase by mtor signal induces disturbed lipid metabolism in hepatitis b virus pre-s2 mutant tumorigenesis. J. Virol. 2015, 89, 605–614. [Google Scholar] [CrossRef] [Green Version]
- Xie, Q.; Fan, F.; Wei, W.; Liu, Y.; Xu, Z.; Zhai, L.; Qi, Y.; Ye, B.; Zhang, Y.; Basu, S.; et al. Multi-omics analyses reveal metabolic alterations regulated by hepatitis b virus core protein in hepatocellular carcinoma cells. Sci. Rep. 2017, 7, 41089. [Google Scholar] [CrossRef]
- Carroll, P.A.; Diolaiti, D.; McFerrin, L.; Gu, H.; Djukovic, D.; Du, J.; Cheng, P.F.; Anderson, S.; Ulrich, M.; Hurley, J.B.; et al. Deregulated myc requires mondoa/mlx for metabolic reprogramming and tumorigenesis. Cancer Cell 2015, 27, 271–285. [Google Scholar] [CrossRef] [Green Version]
- Moudi, B.; Heidari, Z.; Mahmoudzadeh-Sagheb, H.; Alavian, S.M.; Lankarani, K.B.; Farrokh, P.; Randel Nyengaard, J. Concomitant use of heat-shock protein 70, glutamine synthetase and glypican-3 is useful in diagnosis of hbv-related hepatocellular carcinoma with higher specificity and sensitivity. Eur. J. Histochem. 2018, 62, 2859. [Google Scholar] [CrossRef]
- Kim, S.J.; Khan, M.; Quan, J.; Till, A.; Subramani, S.; Siddiqui, A. Hepatitis b virus disrupts mitochondrial dynamics: Induces fission and mitophagy to attenuate apoptosis. PLoS Pathog. 2013, 9, e1003722. [Google Scholar] [CrossRef] [Green Version]
- Hwang, K.B.; Kyaw, Y.Y.; Kang, H.R.; Seong, M.S.; Cheong, J. Mitochondrial dysfunction stimulates hbv gene expression through lipogenic transcription factor activation. Virus Res. 2020, 277, 197842. [Google Scholar] [CrossRef]
- Hajjou, M.; Norel, R.; Carver, R.; Marion, P.; Cullen, J.; Rogler, L.E.; Rogler, C.E. Cdna microarray analysis of hbv transgenic mouse liver identifies genes in lipid biosynthetic and growth control pathways affected by hbv. J. Med. Virol. 2005, 77, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Yan, S.; He, Y.; Wang, F.; Song, S.; Guo, Y.; Zhou, Q.; Wang, Y.; Lin, Z.; Yang, Y.; et al. Expression of hepatitis b virus proteins in transgenic mice alters lipid metabolism and induces oxidative stress in the liver. J. Hepatol. 2008, 48, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Okamura, H.; Nio, Y.; Akahori, Y.; Kim, S.; Watashi, K.; Wakita, T.; Hijikata, M. Fatty acid biosynthesis is involved in the production of hepatitis b virus particles. Biochem. Biophys. Res. Commun. 2016, 475, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Hyrina, A.; Burdette, D.; Song, Z.; Ramirez, R.; Okesli-Armlovich, A.; Vijayakumar, A.; Bates, J.; Trevaskis, J.L.; Fletcher, S.P.; Lee, W.A.; et al. Targeting lipid biosynthesis pathways for hepatitis b virus cure. PLoS ONE 2022, 17, e0270273. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.D.; Wu, H.; Huang, S.; Zhang, H.L.; Qin, C.J.; Zhao, L.H.; Fu, G.B.; Zhou, X.; Wang, X.M.; Tang, L.; et al. Hbx regulates fatty acid oxidation to promote hepatocellular carcinoma survival during metabolic stress. Oncotarget 2016, 7, 6711–6726. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.L.; Peng, X.E.; Zhu, Y.B.; Yan, X.L.; Chen, W.N.; Lin, X. Hepatitis b virus x protein induces hepatic steatosis by enhancing the expression of liver fatty acid binding protein. J. Virol. 2016, 90, 1729–1740. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Liang, G.; Ou, J.; Goldstein, J.L.; Brown, M.S. Central role for liver x receptor in insulin-mediated activation of srebp-1c transcription and stimulation of fatty acid synthesis in liver. Proc. Natl. Acad. Sci. USA 2004, 101, 11245–11250. [Google Scholar] [CrossRef] [Green Version]
- Seo, J.B.; Moon, H.M.; Kim, W.S.; Lee, Y.S.; Jeong, H.W.; Yoo, E.J.; Ham, J.; Kang, H.; Park, M.G.; Steffensen, K.R.; et al. Activated liver x receptors stimulate adipocyte differentiation through induction of peroxisome proliferator-activated receptor gamma expression. Mol. Cell Biol. 2004, 24, 3430–3444. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.H.; Shin, H.J.; Kim, K.; Choi, H.M.; Rhee, S.H.; Moon, H.B.; Kim, H.H.; Yang, U.S.; Yu, D.Y.; Cheong, J. Hepatitis b virus x protein induces hepatic steatosis via transcriptional activation of srebp1 and ppargamma. Gastroenterology 2007, 132, 1955–1967. [Google Scholar] [CrossRef]
- Kang, S.K.; Chung, T.W.; Lee, J.Y.; Lee, Y.C.; Morton, R.E.; Kim, C.H. The hepatitis b virus x protein inhibits secretion of apolipoprotein b by enhancing the expression of n-acetylglucosaminyltransferase iii. J. Biol. Chem. 2004, 279, 28106–28112. [Google Scholar] [CrossRef] [Green Version]
- Oehler, N.; Volz, T.; Bhadra, O.D.; Kah, J.; Allweiss, L.; Giersch, K.; Bierwolf, J.; Riecken, K.; Pollok, J.M.; Lohse, A.W.; et al. Binding of hepatitis b virus to its cellular receptor alters the expression profile of genes of bile acid metabolism. Hepatology 2014, 60, 1483–1493. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.J.; Zhu, P.; Liang, Y.; Yin, W.G.; Xiao, J.H. Hepatitis b virus induces expression of cholesterol metabolism-related genes via tlr2 in hepg2 cells. World J. Gastroenterol. 2013, 19, 2262–2269. [Google Scholar] [CrossRef] [PubMed]
- Dubuisson, J. Hepatitis c virus proteins. World J. Gastroenterol. 2007, 13, 2406–2415. [Google Scholar] [CrossRef] [PubMed]
- Negro, F. Abnormalities of lipid metabolism in hepatitis c virus infection. Gut 2010, 59, 1279–1287. [Google Scholar] [CrossRef]
- Sheridan, D.A.; Shawa, I.T.; Thomas, E.L.; Felmlee, D.J.; Bridge, S.H.; Neely, D.; Cobbold, J.F.; Holmes, E.; Bassendine, M.F.; Taylor-Robinson, S.D. Infection with the hepatitis c virus causes viral genotype-specific differences in cholesterol metabolism and hepatic steatosis. Sci. Rep. 2022, 12, 5562. [Google Scholar] [CrossRef]
- Negro, F. Facts and fictions of hcv and comorbidities: Steatosis, diabetes mellitus, and cardiovascular diseases. J. Hepatol. 2014, 61, S69–S78. [Google Scholar] [CrossRef]
- Mannova, P.; Beretta, L. Activation of the n-ras-pi3k-akt-mtor pathway by hepatitis c virus: Control of cell survival and viral replication. J. Virol. 2005, 79, 8742–8749. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, J.K.; Midkiff, B.R.; Israelow, B.; Evans, M.J.; Lanford, R.E.; Walker, C.M.; Lemon, S.M.; McGivern, D.R. Hepatitis c virus indirectly disrupts DNA damage-induced p53 responses by activating protein kinase r. mBio 2017, 8, e00121-17. [Google Scholar] [CrossRef] [Green Version]
- Ramiere, C.; Rodriguez, J.; Enache, L.S.; Lotteau, V.; Andre, P.; Diaz, O. Activity of hexokinase is increased by its interaction with hepatitis c virus protein ns5a. J. Virol. 2014, 88, 3246–3254. [Google Scholar] [CrossRef] [Green Version]
- Ripoli, M.; D’Aprile, A.; Quarato, G.; Sarasin-Filipowicz, M.; Gouttenoire, J.; Scrima, R.; Cela, O.; Boffoli, D.; Heim, M.H.; Moradpour, D.; et al. Hepatitis c virus-linked mitochondrial dysfunction promotes hypoxia-inducible factor 1 alpha-mediated glycolytic adaptation. J. Virol. 2010, 84, 647–660. [Google Scholar] [CrossRef] [Green Version]
- Levy, P.L.; Duponchel, S.; Eischeid, H.; Molle, J.; Michelet, M.; Diserens, G.; Vermathen, M.; Vermathen, P.; Dufour, J.F.; Dienes, H.P.; et al. Hepatitis c virus infection triggers a tumor-like glutamine metabolism. Hepatology 2017, 65, 789–803. [Google Scholar] [CrossRef] [Green Version]
- Diamond, D.L.; Syder, A.J.; Jacobs, J.M.; Sorensen, C.M.; Walters, K.A.; Proll, S.C.; McDermott, J.E.; Gritsenko, M.A.; Zhang, Q.; Zhao, R.; et al. Temporal proteome and lipidome profiles reveal hepatitis c virus-associated reprogramming of hepatocellular metabolism and bioenergetics. PLoS Pathog. 2010, 6, e1000719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrin-Cocon, L.; Kundlacz, C.; Jacquemin, C.; Hanoulle, X.; Aublin-Gex, A.; Figl, M.; Manteca, J.; Andre, P.; Vidalain, P.O.; Lotteau, V.; et al. Domain 2 of hepatitis c virus protein ns5a activates glucokinase and induces lipogenesis in hepatocytes. Int. J. Mol. Sci. 2022, 23, 919. [Google Scholar] [CrossRef]
- Jung, G.S.; Jeon, J.H.; Choi, Y.K.; Jang, S.Y.; Park, S.Y.; Kim, S.W.; Byun, J.K.; Kim, M.K.; Lee, S.; Shin, E.C.; et al. Pyruvate dehydrogenase kinase regulates hepatitis c virus replication. Sci. Rep. 2016, 6, 30846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, T.; Yang, Q.; Tian, F.; Chang, H.; Hu, Z.; Yu, B.; Han, L.; Xing, Y.; Jiu, Y.; He, Y.; et al. Glycometabolism regulates hepatitis c virus release. PLoS Pathog. 2021, 17, e1009746. [Google Scholar] [CrossRef] [PubMed]
- Kasai, D.; Adachi, T.; Deng, L.; Nagano-Fujii, M.; Sada, K.; Ikeda, M.; Kato, N.; Ide, Y.H.; Shoji, I.; Hotta, H. Hcv replication suppresses cellular glucose uptake through down-regulation of cell surface expression of glucose transporters. J. Hepatol. 2009, 50, 883–894. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Liu, Q.; Sun, F.; Qiao, L. Hepatitis c virus nonstructural protein 5a perturbs lipid metabolism by modulating ampk/srebp-1c signaling. Lipids. Health Dis. 2019, 18, 191. [Google Scholar] [CrossRef] [Green Version]
- Shi, Q.; Hoffman, B.; Liu, Q. Pi3k-akt signaling pathway upregulates hepatitis c virus rna translation through the activation of srebps. Virology 2016, 490, 99–108. [Google Scholar] [CrossRef]
- Huang, J.T.; Tseng, C.P.; Liao, M.H.; Lu, S.C.; Yeh, W.Z.; Sakamoto, N.; Chen, C.M.; Cheng, J.C. Hepatitis c virus replication is modulated by the interaction of nonstructural protein ns5b and fatty acid synthase. J. Virol. 2013, 87, 4994–5004. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Hood, B.L.; Chadwick, S.L.; Liu, S.; Watkins, S.C.; Luo, G.; Conrads, T.P.; Wang, T. Fatty acid synthase is up-regulated during hepatitis c virus infection and regulates hepatitis c virus entry and production. Hepatology 2008, 48, 1396–1403. [Google Scholar] [CrossRef] [Green Version]
- Nio, Y.; Hasegawa, H.; Okamura, H.; Miyayama, Y.; Akahori, Y.; Hijikata, M. Liver-specific mono-unsaturated fatty acid synthase-1 inhibitor for anti-hepatitis c treatment. Antiviral Res. 2016, 132, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Sarhan, M.A.; Abdel-Hakeem, M.S.; Mason, A.L.; Tyrrell, D.L.; Houghton, M. Glycogen synthase kinase 3beta inhibitors prevent hepatitis c virus release/assembly through perturbation of lipid metabolism. Sci. Rep. 2017, 7, 2495. [Google Scholar] [CrossRef]
- Zapatero-Belinchon, F.J.; Otjengerdes, R.; Sheldon, J.; Schulte, B.; Carriqui-Madronal, B.; Brogden, G.; Arroyo-Fernandez, L.M.; Vondran, F.W.R.; Maasoumy, B.; von Hahn, T.; et al. Interdependent impact of lipoprotein receptors and lipid-lowering drugs on hcv infectivity. Cells 2021, 10, 1626. [Google Scholar] [CrossRef]
- Gane, E.; Stedman, C.; Dole, K.; Chen, J.; Meyers, C.D.; Wiedmann, B.; Zhang, J.; Raman, P.; Colvin, R.A. A diacylglycerol transferase 1 inhibitor is a potent hepatitis c antiviral in vitro but not in patients in a randomized clinical trial. ACS Infect. Dis. 2017, 3, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Gastaminza, P.; Cheng, G.; Wieland, S.; Zhong, J.; Liao, W.; Chisari, F.V. Cellular determinants of hepatitis c virus assembly, maturation, degradation, and secretion. J. Virol. 2008, 82, 2120–2129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Sun, F.; Owen, D.M.; Li, W.; Chen, Y.; Gale, M., Jr.; Ye, J. Hepatitis c virus production by human hepatocytes dependent on assembly and secretion of very low-density lipoproteins. Proc. Natl. Acad. Sci. USA 2007, 104, 5848–5853. [Google Scholar] [CrossRef] [Green Version]
- Lyn, R.K.; Singaravelu, R.; Kargman, S.; O’Hara, S.; Chan, H.; Oballa, R.; Huang, Z.; Jones, D.M.; Ridsdale, A.; Russell, R.S.; et al. Stearoyl-coa desaturase inhibition blocks formation of hepatitis c virus-induced specialized membranes. Sci. Rep. 2014, 4, 4549. [Google Scholar] [CrossRef] [Green Version]
- Dharancy, S.; Malapel, M.; Perlemuter, G.; Roskams, T.; Cheng, Y.; Dubuquoy, L.; Podevin, P.; Conti, F.; Canva, V.; Philippe, D.; et al. Impaired expression of the peroxisome proliferator-activated receptor alpha during hepatitis c virus infection. Gastroenterology 2005, 128, 334–342. [Google Scholar] [CrossRef] [Green Version]
- Higgs, M.R.; Lerat, H.; Pawlotsky, J.M. Hepatitis c virus-induced activation of beta-catenin promotes c-myc expression and a cascade of pro-carcinogenetic events. Oncogene 2013, 32, 4683–4693. [Google Scholar] [CrossRef] [Green Version]
- Lupberger, J.; Croonenborghs, T.; Roca Suarez, A.A.; Van Renne, N.; Juhling, F.; Oudot, M.A.; Virzi, A.; Bandiera, S.; Jamey, C.; Meszaros, G.; et al. Combined analysis of metabolomes, proteomes, and transcriptomes of hepatitis c virus-infected cells and liver to identify pathways associated with disease development. Gastroenterology 2019, 157, 537–551.e539. [Google Scholar] [CrossRef] [Green Version]
- Uldrick, T.S.; Whitby, D. Update on kshv epidemiology, kaposi sarcoma pathogenesis, and treatment of kaposi sarcoma. Cancer Lett. 2011, 305, 150–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juillard, F.; Tan, M.; Li, S.; Kaye, K.M. Kaposi’s sarcoma herpesvirus genome persistence. Front. Microbiol. 2016, 7, 1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, N.; Shi, F.; Yang, L.; Liao, W.; Cao, Y. Oncogenic viral infection and amino acid metabolism in cancer progression: Molecular insights and clinical implications. Biochim. Biophys. Acta Rev. Cancer 2022, 1877, 188724. [Google Scholar] [CrossRef] [PubMed]
- Friborg, J., Jr.; Kong, W.; Hottiger, M.O.; Nabel, G.J. P53 inhibition by the lana protein of kshv protects against cell death. Nature 1999, 402, 889–894. [Google Scholar] [CrossRef] [PubMed]
- Radkov, S.A.; Kellam, P.; Boshoff, C. The latent nuclear antigen of kaposi sarcoma-associated herpesvirus targets the retinoblastoma-e2f pathway and with the oncogene hras transforms primary rat cells. Nat. Med. 2000, 6, 1121–1127. [Google Scholar] [CrossRef] [PubMed]
- Delgado, T.; Carroll, P.A.; Punjabi, A.S.; Margineantu, D.; Hockenbery, D.M.; Lagunoff, M. Induction of the warburg effect by kaposi’s sarcoma herpesvirus is required for the maintenance of latently infected endothelial cells. Proc. Natl. Acad. Sci. USA 2010, 107, 10696–10701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez, E.L.; Pulliam, T.H.; Dimaio, T.A.; Thalhofer, A.B.; Delgado, T.; Lagunoff, M. Glycolysis, glutaminolysis, and fatty acid synthesis are required for distinct stages of kaposi’s sarcoma-associated herpesvirus lytic replication. J. Virol. 2017, 91, e02237-16. [Google Scholar] [CrossRef] [Green Version]
- Shin, Y.C.; Joo, C.H.; Gack, M.U.; Lee, H.R.; Jung, J.U. Kaposi’s sarcoma-associated herpesvirus viral ifn regulatory factor 3 stabilizes hypoxia-inducible factor-1 alpha to induce vascular endothelial growth factor expression. Cancer Res. 2008, 68, 1751–1759. [Google Scholar] [CrossRef] [Green Version]
- Carroll, P.A.; Kenerson, H.L.; Yeung, R.S.; Lagunoff, M. Latent kaposi’s sarcoma-associated herpesvirus infection of endothelial cells activates hypoxia-induced factors. J. Virol. 2006, 80, 10802–10812. [Google Scholar] [CrossRef] [Green Version]
- Cai, Q.; Murakami, M.; Si, H.; Robertson, E.S. A potential alpha-helix motif in the amino terminus of lana encoded by kaposi’s sarcoma-associated herpesvirus is critical for nuclear accumulation of hif-1alpha in normoxia. J. Virol. 2007, 81, 10413–10423. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.K.; Lang, F.; Pei, Y.; Jha, H.C.; Robertson, E.S. Metabolic reprogramming of kaposi’s sarcoma associated herpes virus infected b-cells in hypoxia. PLoS Pathog. 2018, 14, e1007062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatt, A.P.; Damania, B. Aktivation of pi3k/akt/mtor signaling pathway by kshv. Front. Immunol. 2012, 3, 401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatt, A.P.; Jacobs, S.R.; Freemerman, A.J.; Makowski, L.; Rathmell, J.C.; Dittmer, D.P.; Damania, B. Dysregulation of fatty acid synthesis and glycolysis in non-hodgkin lymphoma. Proc. Natl. Acad. Sci. USA 2012, 109, 11818–11823. [Google Scholar] [CrossRef] [PubMed]
- Gjyshi, O.; Bottero, V.; Veettil, M.V.; Dutta, S.; Singh, V.V.; Chikoti, L.; Chandran, B. Kaposi’s sarcoma-associated herpesvirus induces nrf2 during de novo infection of endothelial cells to create a microenvironment conducive to infection. PLoS Pathog. 2014, 10, e1004460. [Google Scholar] [CrossRef] [PubMed]
- Delgado, T.; Sanchez, E.L.; Camarda, R.; Lagunoff, M. Global metabolic profiling of infection by an oncogenic virus: Kshv induces and requires lipogenesis for survival of latent infection. PLoS Pathog. 2012, 8, e1002866. [Google Scholar] [CrossRef] [Green Version]
- Angius, F.; Uda, S.; Piras, E.; Spolitu, S.; Ingianni, A.; Batetta, B.; Pompei, R. Neutral lipid alterations in human herpesvirus 8-infected huvec cells and their possible involvement in neo-angiogenesis. BMC Microbiol. 2015, 15, 74. [Google Scholar] [CrossRef] [Green Version]
- Tso, F.Y.; Kossenkov, A.V.; Lidenge, S.J.; Ngalamika, O.; Ngowi, J.R.; Mwaiselage, J.; Wickramasinghe, J.; Kwon, E.H.; West, J.T.; Lieberman, P.M.; et al. Rna-seq of kaposi’s sarcoma reveals alterations in glucose and lipid metabolism. PLoS Pathog. 2018, 14, e1006844. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, E.L.; Carroll, P.A.; Thalhofer, A.B.; Lagunoff, M. Latent kshv infected endothelial cells are glutamine addicted and require glutaminolysis for survival. PLoS Pathog. 2015, 11, e1005052. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Li, T.; Ramos da Silva, S.; Lee, J.J.; Lu, C.; Eoh, H.; Jung, J.U.; Gao, S.J. A critical role of glutamine and asparagine gamma-nitrogen in nucleotide biosynthesis in cancer cells hijacked by an oncogenic virus. mBio 2017, 8, e01179-17. [Google Scholar] [CrossRef] [Green Version]
- Choi, U.Y.; Lee, J.J.; Park, A.; Zhu, W.; Lee, H.R.; Choi, Y.J.; Yoo, J.S.; Yu, C.; Feng, P.; Gao, S.J.; et al. Oncogenic human herpesvirus hijacks proline metabolism for tumorigenesis. Proc. Natl. Acad. Sci. USA 2020, 117, 8083–8093. [Google Scholar] [CrossRef]
- Fiches, G.N.; Wu, Z.; Zhou, D.; Biswas, A.; Li, T.W.; Kong, W.; Jean, M.; Santoso, N.G.; Zhu, J. Polyamine biosynthesis and eif5a hypusination are modulated by the DNA tumor virus kshv and promote kshv viral infection. PLoS Pathog. 2022, 18, e1010503. [Google Scholar] [CrossRef] [PubMed]
- Choi, U.Y.; Lee, J.J.; Park, A.; Jung, K.L.; Lee, S.A.; Choi, Y.J.; Lee, H.R.; Lai, C.J.; Eoh, H.; Jung, J.U. Herpesvirus-induced spermidine synthesis and eif5a hypusination for viral episomal maintenance. Cell Rep. 2022, 40, 111234. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Shuda, M.; Chang, Y.; Moore, P.S. Clonal integration of a polyomavirus in human merkel cell carcinoma. Science 2008, 319, 1096–1100. [Google Scholar] [CrossRef] [PubMed]
- DeCaprio, J.A. Molecular pathogenesis of merkel cell carcinoma. Annu. Rev. Pathol. 2021, 16, 69–91. [Google Scholar] [CrossRef]
- Pastrana, D.V.; Tolstov, Y.L.; Becker, J.C.; Moore, P.S.; Chang, Y.; Buck, C.B. Quantitation of human seroresponsiveness to merkel cell polyomavirus. PLoS Pathog. 2009, 5, e1000578. [Google Scholar] [CrossRef] [Green Version]
- Paulson, K.G.; Lewis, C.W.; Redman, M.W.; Simonson, W.T.; Lisberg, A.; Ritter, D.; Morishima, C.; Hutchinson, K.; Mudgistratova, L.; Blom, A.; et al. Viral oncoprotein antibodies as a marker for recurrence of merkel cell carcinoma: A prospective validation study. Cancer 2017, 123, 1464–1474. [Google Scholar] [CrossRef] [Green Version]
- Houben, R.; Shuda, M.; Weinkam, R.; Schrama, D.; Feng, H.; Chang, Y.; Moore, P.S.; Becker, J.C. Merkel cell polyomavirus-infected merkel cell carcinoma cells require expression of viral t antigens. J. Virol. 2010, 84, 7064–7072. [Google Scholar] [CrossRef] [Green Version]
- Kwun, H.J.; Shuda, M.; Feng, H.; Camacho, C.J.; Moore, P.S.; Chang, Y. Merkel cell polyomavirus small t antigen controls viral replication and oncoprotein expression by targeting the cellular ubiquitin ligase scffbw7. Cell Host Microbe 2013, 14, 125–135. [Google Scholar] [CrossRef] [Green Version]
- Borchert, S.; Czech-Sioli, M.; Neumann, F.; Schmidt, C.; Wimmer, P.; Dobner, T.; Grundhoff, A.; Fischer, N. High-affinity rb binding, p53 inhibition, subcellular localization, and transformation by wild-type or tumor-derived shortened merkel cell polyomavirus large t antigens. J. Virol. 2014, 88, 3144–3160. [Google Scholar] [CrossRef] [Green Version]
- Berrios, C.; Padi, M.; Keibler, M.A.; Park, D.E.; Molla, V.; Cheng, J.; Lee, S.M.; Stephanopoulos, G.; Quackenbush, J.; DeCaprio, J.A. Merkel cell polyomavirus small t antigen promotes pro-glycolytic metabolic perturbations required for transformation. PLoS Pathog. 2016, 12, e1006020. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Ning, S.; Ghandi, M.; Kryukov, G.V.; Gopal, S.; Deik, A.; Souza, A.; Pierce, K.; Keskula, P.; Hernandez, D.; et al. The landscape of cancer cell line metabolism. Nat. Med. 2019, 25, 850–860. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.H.; Yang, Y.; Chen, C.H.; Hsiao, C.J.; Li, T.N.; Liao, K.J.; Watashi, K.; Chen, B.S.; Wang, L.H. Aerobic glycolysis supports hepatitis b virus protein synthesis through interaction between viral surface antigen and pyruvate kinase isoform m2. PLoS Pathog. 2021, 17, e1008866. [Google Scholar] [CrossRef] [PubMed]
- Su, A.I.; Pezacki, J.P.; Wodicka, L.; Brideau, A.D.; Supekova, L.; Thimme, R.; Wieland, S.; Bukh, J.; Purcell, R.H.; Schultz, P.G.; et al. Genomic analysis of the host response to hepatitis c virus infection. Proc. Natl. Acad. Sci. USA 2002, 99, 15669–15674. [Google Scholar] [CrossRef] [PubMed]
- Ramtohul, Y.K.; Black, C.; Chan, C.C.; Crane, S.; Guay, J.; Guiral, S.; Huang, Z.; Oballa, R.; Xu, L.J.; Zhang, L.; et al. Sar and optimization of thiazole analogs as potent stearoyl-coa desaturase inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 1593–1597. [Google Scholar] [CrossRef] [PubMed]

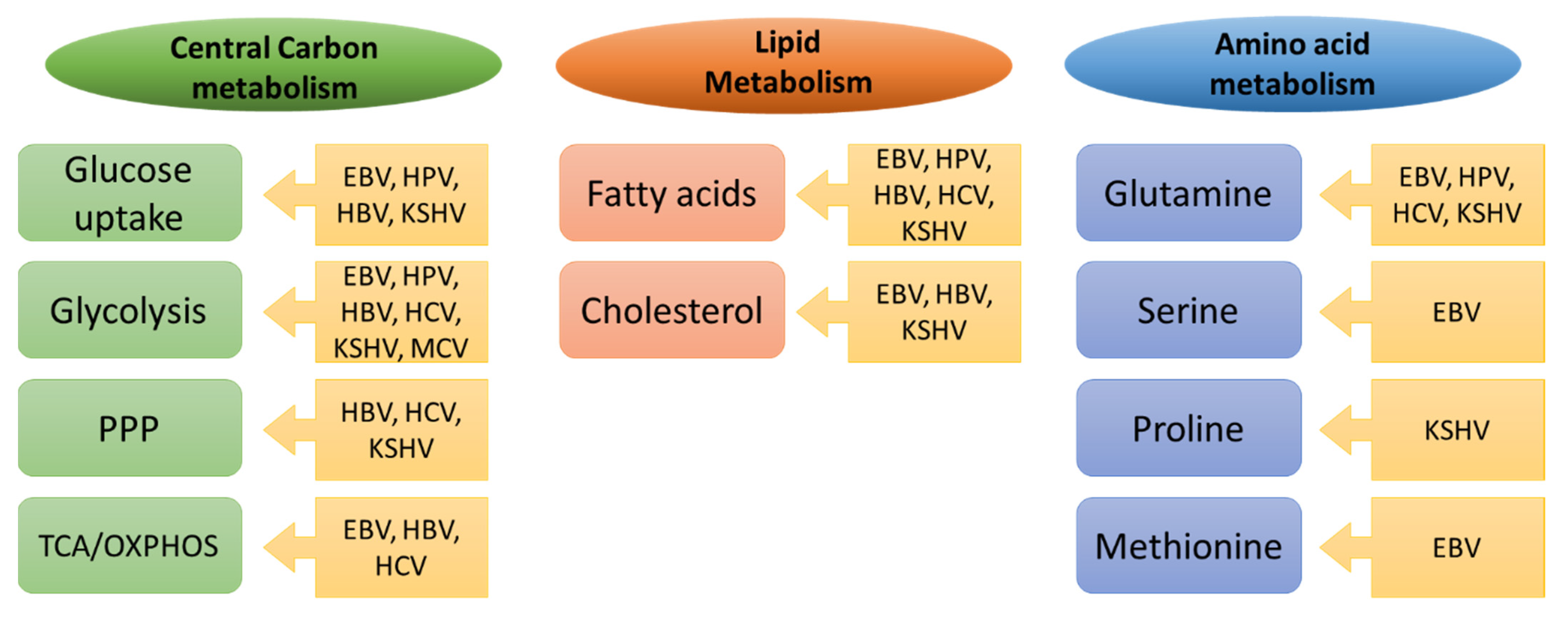
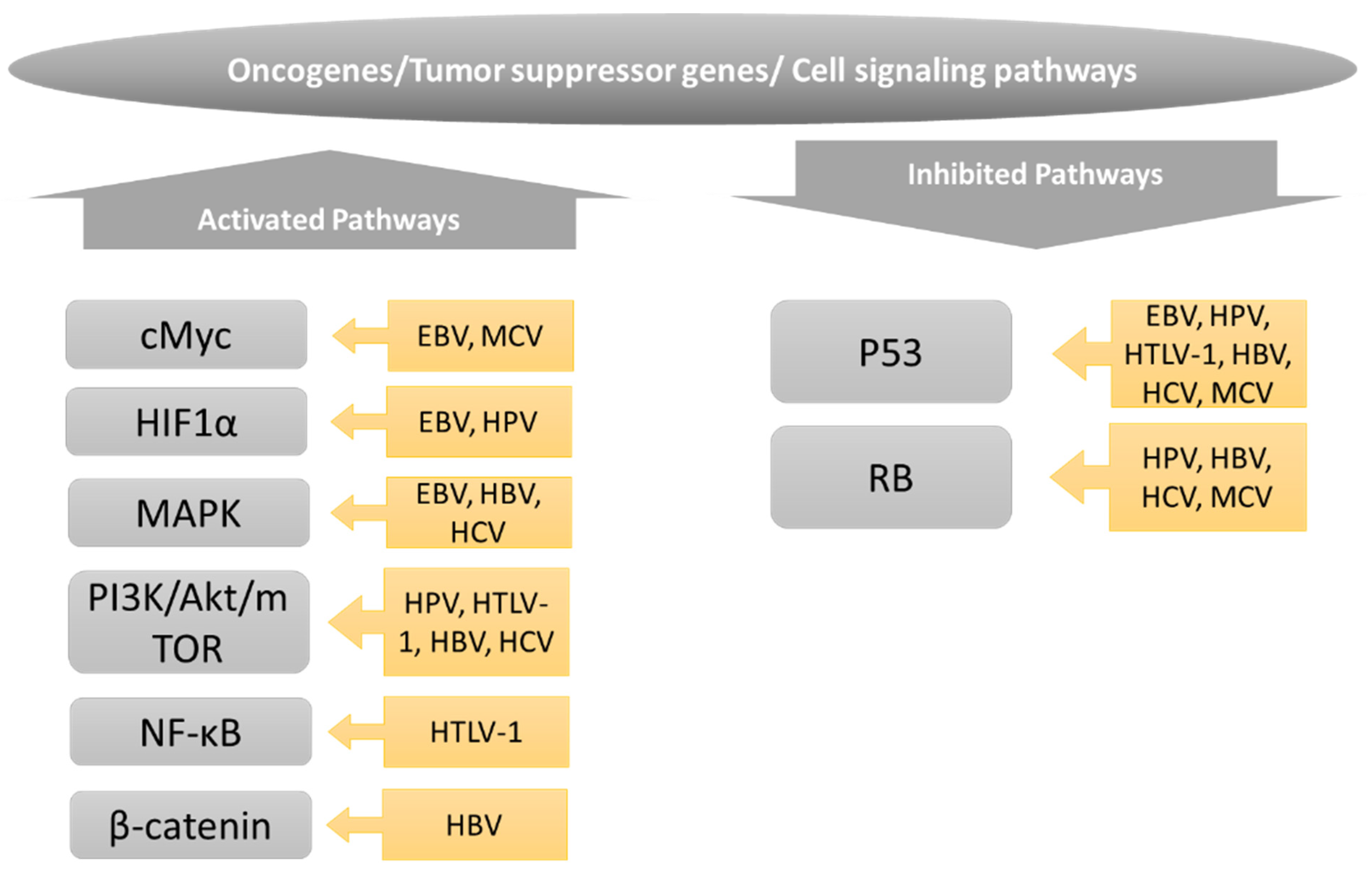

{kind=link}
{kind=link}
{kind=link}
| Metabolic Drug | Target Enzyme | Virus | Model/Cell System | Outcome | Reference |
|---|---|---|---|---|---|
| STF-31 | GLUT1 | EBV | kills NPC cells in vitro | [175] | |
| 2-deoxy-D-Glucose | Hexokinase, PI3K/Akt | HTLV-1 | PBMCs | suppresses viral transcription | [214] |
| HBV | Huh7 | suppresses viral protein production (HBV delivered with adenovirus) | [312] | ||
| oxamate | LDHA | KSHV | TIME | decreases virion production | [287] |
| dichloroacetate | PDK | HCV | Huh7.5 | decreases viral replication | [264] |
| rapamycine | mTOR | EBV | cell type dependent | induces of lytic cycle | [181] |
| CB-839/BPTES | Glutaminase | HCV | Huh7.5 | inhibits replication and infection establishment | [261] |
| KSHV | TIME | decreases virion production | [287] | ||
| difluoromethylornithine | ornithine decarboxylase 1 | KSHV | TIME/BCBL-1 | decreases virion production | [302] |
| N1-guanyl-1,7-diaminoheptane (GC7) | deoxyhypusine synthase | KSHV | TIME/BCBL-1 | decreases virion production | [302] |
| cerulenin | FASN | HCV | Huh7 | decreases viral replication | [313] |
| C75 | FASN | HCV | Huh7 | decreases virion production | [270] |
| GSK1995010 | FASN | HBV | HepG2.2.15.7 | decreases viral replication | [243] |
| 4-(1-(5-(2-cyclopropyl-4-methyl-1H-imidazol-5-yl)- 2,4-dimethylbenzoyl)-3-fluoroazetidin-3- yl)benzonitrile | FASN | HBV | HepG2NTCP | inhibits HBs secretion | [244] |
| LCQ908/pradigastat | diacylglycerol O acyltransferase 1 | HCV | clinical phase trial/Huh7.5 | inhibits replication in vitro but not in vivo | [274] |
| BMS-200150/BMS-20101038 | microsomal transfer protein | HCV | Huh7.5 | decreases virion release | [275,276] |
| 3J [314] | Stearoyl-CoA desaturase | HCV | Huh7.5 | decreases viral replication and assembly | [277] |
| 25-hydroxycholesterol | SREBP | HCV | Huh7 | decreases viral replication | [313] |
| 5-tetradecyloxy-2-furoic acid (TOFA) | acetyl-coA carboxylase | KSHV | TIME | decreases virion production | [287] |
| CP640186 | acetyl-coA carboxylase | HBV | HepG2.2.15.7 | decreases viral replication | [243] |
| 2-(1-((R)-2-(((1s,4S)-4-hydroxycyclohexyl)oxy)-2- (2-methoxyphenyl)ethyl)-5-methyl-6-(oxazol-2-yl)- 2,4-dioxo-1,4-dihydrothieno[2,3-d]pyrimidin-3(2H)- yl)-2-methylpropanoic acid | acetyl-coA carboxylase | HBV | HepG2NTCP | inhibits HBs secretion | [244] |
| (R)-4-((diethylamino)methyl)-N-(2- methoxyphenethyl)-N-(pyrrolidin-3-yl)benzamide | subtilisin kexin isozyme-1/site-1 protease | HBV | HepG2NTCP | inhibits HBs secretion | [244] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaballah, A.; Bartosch, B. An Update on the Metabolic Landscape of Oncogenic Viruses. Cancers 2022, 14, 5742. https://doi.org/10.3390/cancers14235742
Gaballah A, Bartosch B. An Update on the Metabolic Landscape of Oncogenic Viruses. Cancers. 2022; 14(23):5742. https://doi.org/10.3390/cancers14235742
Chicago/Turabian StyleGaballah, Ahmed, and Birke Bartosch. 2022. "An Update on the Metabolic Landscape of Oncogenic Viruses" Cancers 14, no. 23: 5742. https://doi.org/10.3390/cancers14235742