
Regulation of RNA Polymerase I Stability and Function
Abstract
:Simple Summary
Abstract
1. Introduction: RNA Polymerase I Is an Essential Enzyme for Ribosome Biogenesis
1.1. Ribosome Biogenesis
1.2. RNA Polymerases Have Diverged RNA Synthetic Targets
1.3. RNA Polymerase I Transcribes the Essential 5.8S, 18S, and 28S Ribosomal RNAs
1.4. Pol I Transcription and Early Steps of Ribosome Biogenesis Are Compartmentalized in the Nucleolus
2. Pol I Transcription Cycle
2.1. Structural Analyses of the Pol I Enzyme in Saccharomyces cerevisiae
2.2. Structural Analyses of the Human Pol I Enzyme
2.3. Key Steps in the Pol I Transcription Cycle
3. Regulation of Pol I Transcription Activity
3.1. Metabolic and Environmental Conditions Regulate Pol I Transcription
3.2. Post-Translational Modification of Transcription Factors
3.3. Post-Translational Modification of Transcription Factors
4. Transcriptional Errors Evoke Cellular Stress Responses
4.1. Pol I Transcription Stress Response
4.2. Pol II and Pol III Transcription Stress Activate Enzyme Destruction
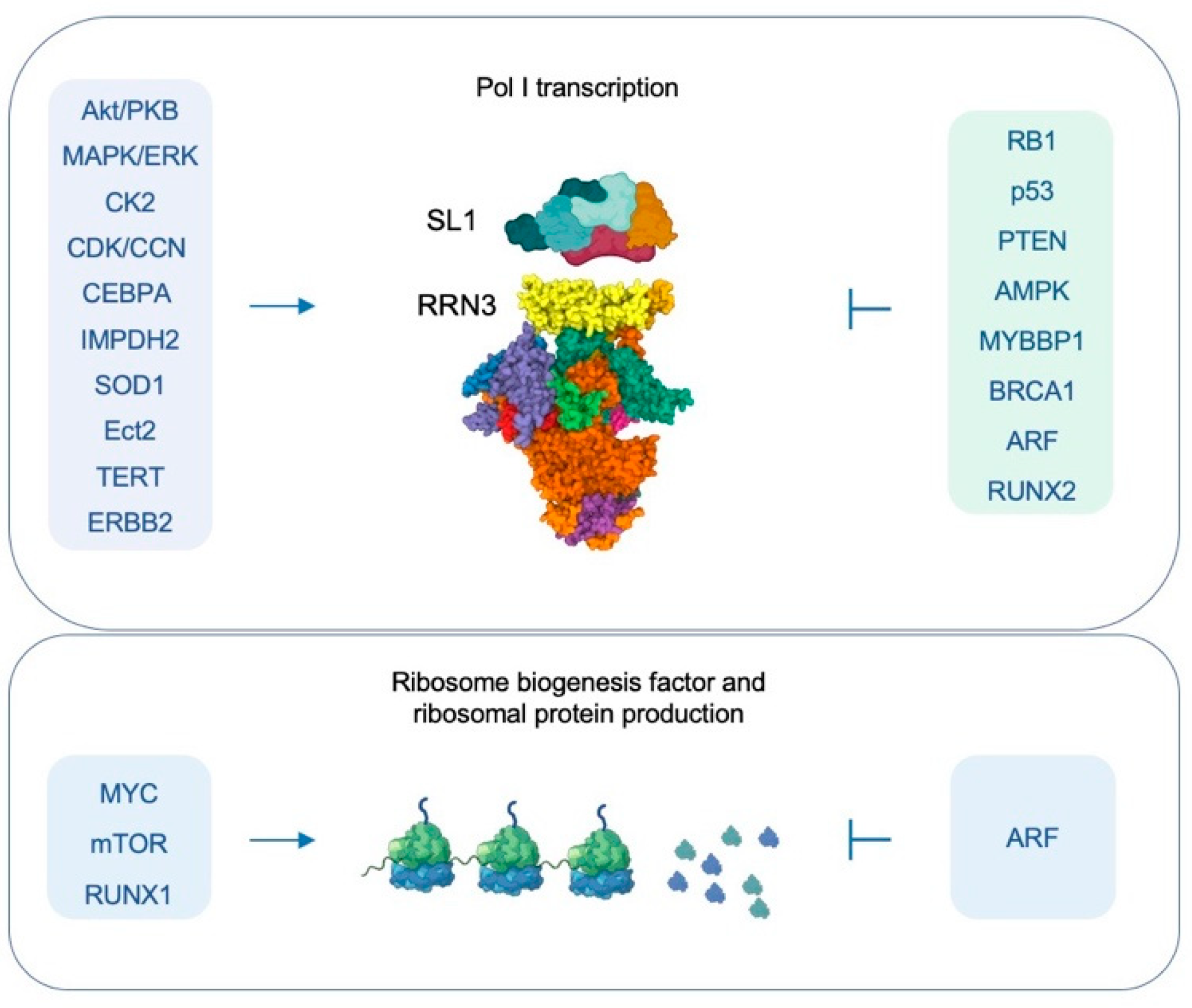
5. RNA Polymerase I and Cancer
5.1. Pol I Transcription Is Upregulated in Cancer
5.2. Cancer Drivers Promote Deregulated Pol I Transcription
5.3. Inactivation of Tumor Suppressors Leads to Uncontrolled Pol I Activity
5.4. Current Strategies to Target Pol I
6. Direct Regulation of the Pol I Enzyme to Treat Cancer: BMH-21, A First-in-Class Pol I Inhibitor
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AgNOR | argyrophilic nucleolar organizing region |
| DUB | Deubiquitinase |
| CDK | cyclin-dependent kinase |
| ETS | external transcribed spacer |
| ITS | internal transcribed spacer |
| IGS | intergenic spacer |
| NMP | nucleoside monophosphate |
| NOR | nucleolar organizing region |
| NoRC | nucleolar chromatin remodeling complex |
| NTP | nucleoside triphosphate |
| PIC | preinitiation complex |
| Pol | RNA polymerase |
| rDNA | ribosomal DNA |
| rRNA | ribosomal RNA |
| SL-1 | Selectivity factor-1 |
| TOP | Topoisomerase |
| UCE | upstream control element |
References
- Warner, J.R. The economics of ribosome biosynthesis in yeast. Trends Biochem. Sci. 1999, 24, 437–440. [Google Scholar] [CrossRef] [PubMed]
- Warner, J.R.; Vilardell, J.; Sohn, J.H. Economics of ribosome biosynthesis. Cold Spring Harb. Symp. Quant. Biol. 2001, 66, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Girbig, M.; Misiaszek, A.D.; Muller, C.W. Structural insights into nuclear transcription by eukaryotic DNA-dependent RNA polymerases. Nat. Rev. Mol. Cell Biol. 2022, 23, 603–622. [Google Scholar] [CrossRef]
- Bassler, J.; Hurt, E. Eukaryotic Ribosome Assembly. Annu. Rev. Biochem. 2019, 88, 281–306. [Google Scholar] [CrossRef]
- Klinge, S.; Woolford, J.L. Ribosome assembly coming into focus. Nat. Rev. Mol. Cell Biol. 2019, 20, 116–131. [Google Scholar] [CrossRef]
- Turowski, T.W.; Tollervey, D. Cotranscriptional events in eukaryotic ribosome synthesis. Wiley Interdiscip. Rev. RNA 2015, 6, 129–139. [Google Scholar] [CrossRef]
- Moss, T.; Langlois, F.; Gagnon-Kugler, T.; Stefanovsky, V. A housekeeper with power of attorney: The rRNA genes in ribosome biogenesis. Cell Mol. Life Sci. 2007, 64, 29–49. [Google Scholar] [CrossRef] [PubMed]
- Sharifi, S.; Bierhoff, H. Regulation of RNA Polymerase I Transcription in Development, Disease, and Aging. Annu. Rev. Biochem. 2018, 87, 51–73. [Google Scholar] [CrossRef] [PubMed]
- Grummt, I. Wisely chosen paths—regulation of rRNA synthesis: Delivered on 30 June 2010 at the 35th FEBS Congress in Gothenburg, Sweden. FEBS J. 2010, 277, 4626–4639. [Google Scholar] [CrossRef] [PubMed]
- Farley-Barnes, K.I.; Ogawa, L.M.; Baserga, S.J. Ribosomopathies: Old Concepts, New Controversies. Trends Genet. 2019, 35, 754–767. [Google Scholar] [CrossRef]
- Aspesi, A.; Ellis, S.R. Rare ribosomopathies: Insights into mechanisms of cancer. Nat. Rev. Cancer 2019, 19, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, J.; Thomas, G.; Volarevic, S. Ribosome biogenesis in cancer: New players and therapeutic avenues. Nat. Rev. Cancer 2018, 18, 51–63. [Google Scholar] [CrossRef]
- Roeder, R.G.; Rutter, W.J. Multiple forms of DNA-dependent RNA polymerase in eukaryotic organisms. Nature 1969, 224, 234–237. [Google Scholar] [CrossRef] [PubMed]
- Roeder, R.G. 50+ years of eukaryotic transcription: An expanding universe of factors and mechanisms. Nat. Struct. Mol. Biol. 2019, 26, 783–791. [Google Scholar] [CrossRef]
- White, R.J. RNA polymerases I and III, non-coding RNAs and cancer. Trends Genet. 2008, 24, 622–629. [Google Scholar] [CrossRef]
- Bywater, M.J.; Pearson, R.B.; McArthur, G.A.; Hannan, R.D. Dysregulation of the basal RNA polymerase transcription apparatus in cancer. Nat. Rev. Cancer 2013, 13, 299–314. [Google Scholar] [CrossRef]
- McStay, B. Nucleolar organizer regions: Genomic ‘dark matter’ requiring illumination. Genes. Dev. 2016, 30, 1598–1610. [Google Scholar] [CrossRef] [Green Version]
- Nurk, S.; Koren, S.; Rhie, A.; Rautiainen, M.; Bzikadze, A.V.; Mikheenko, A.; Vollger, M.R.; Altemose, N.; Uralsky, L.; Gershman, A.; et al. The complete sequence of a human genome. Science 2022, 376, 44–53. [Google Scholar] [CrossRef]
- Nelson, J.O.; Watase, G.J.; Warsinger-Pepe, N.; Yamashita, Y.M. Mechanisms of rDNA Copy Number Maintenance. Trends Genet. 2019, 35, 734–742. [Google Scholar] [CrossRef]
- Salim, D.; Gerton, J.L. Ribosomal DNA instability and genome adaptability. Chromosome Res. 2019, 27, 73–87. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Lemos, B. Ribosomal DNA copy number amplification and loss in human cancers is linked to tumor genetic context, nucleolus activity, and proliferation. PLoS Genet. 2017, 13, e1006994. [Google Scholar] [CrossRef]
- Fan, W.; Eklund, E.; Sherman, R.M.; Liu, H.; Pitts, S.; Ford, B.; Nv, R.; Laiho, M. Widespread genetic heterogeneity of human ribosomal RNA genes. RNA 2022, 28, 478–492. [Google Scholar] [CrossRef]
- Potapova, T.A.; Gerton, J.L. Ribosomal DNA and the nucleolus in the context of genome organization. Chromosome Res. 2019, 27, 109–127. [Google Scholar] [CrossRef]
- Moss, T.; Mars, J.C.; Tremblay, M.G.; Sabourin-Felix, M. The chromatin landscape of the ribosomal RNA genes in mouse and human. Chromosome Res. 2019, 27, 31–40. [Google Scholar] [CrossRef]
- De Winter, R.F.; Moss, T. Spacer promoters are essential for efficient enhancement of X. laevis ribosomal transcription. Cell 1986, 44, 313–318. [Google Scholar] [CrossRef]
- Reeder, R.H. Enhancers and ribosomal gene spacers. Cell 1984, 38, 349–351. [Google Scholar] [CrossRef]
- Osheim, Y.N.; Mougey, E.B.; Windle, J.; Anderson, M.; O’Reilly, M.; Miller, O.L., Jr.; Beyer, A.; Sollner-Webb, B. Metazoan rDNA enhancer acts by making more genes transcriptionally active. J. Cell Biol. 1996, 133, 943–954. [Google Scholar] [CrossRef] [Green Version]
- Mars, J.C.; Sabourin-Felix, M.; Tremblay, M.G.; Moss, T. A Deconvolution Protocol for ChIP-Seq Reveals Analogous Enhancer Structures on the Mouse and Human Ribosomal RNA Genes. G3 (Bethesda) 2018, 8, 303–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer, C.; Schmitz, K.M.; Li, J.; Grummt, I.; Santoro, R. Intergenic transcripts regulate the epigenetic state of rRNA genes. Mol. Cell 2006, 22, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Santoro, R.; Schmitz, K.M.; Sandoval, J.; Grummt, I. Intergenic transcripts originating from a subclass of ribosomal DNA repeats silence ribosomal RNA genes in trans. EMBO Rep. 2010, 11, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Russell, J.; Zomerdijk, J.C. RNA-polymerase-I-directed rDNA transcription, life and works. Trends Biochem. Sci. 2005, 30, 87–96. [Google Scholar] [CrossRef] [Green Version]
- Laferte, A.; Favry, E.; Sentenac, A.; Riva, M.; Carles, C.; Chedin, S. The transcriptional activity of RNA polymerase I is a key determinant for the level of all ribosome components. Genes Dev. 2006, 20, 2030–2040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullineux, S.T.; Lafontaine, D.L. Mapping the cleavage sites on mammalian pre-rRNAs: Where do we stand? Biochimie 2012, 94, 1521–1532. [Google Scholar] [CrossRef]
- Henras, A.K.; Plisson-Chastang, C.; O’Donohue, M.F.; Chakraborty, A.; Gleizes, P.E. An overview of pre-ribosomal RNA processing in eukaryotes. Wiley Interdiscip. Rev. RNA 2015, 6, 225–242. [Google Scholar] [CrossRef]
- Scull, C.E.; Schneider, D.A. Coordinated Control of rRNA Processing by RNA Polymerase, I. Trends Genet. 2019, 35, 724–733. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, A.; Perez-Fernandez, J.; Merkl, P.; Hamperl, S.; Gerber, J.; Griesenbeck, J.; Tschochner, H. RNA polymerase I termination: Where is the end? Biochim. Biophys. Acta 2013, 1829, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Manley, J.L. Beyond rRNA: Nucleolar transcription generates a complex network of RNAs with multiple roles in maintaining cellular homeostasis. Genes Dev. 2022, 36, 876–886. [Google Scholar] [CrossRef]
- Engel, C.; Sainsbury, S.; Cheung, A.C.; Kostrewa, D.; Cramer, P. RNA polymerase I structure and transcription regulation. Nature 2013, 502, 650–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Tornero, C.; Moreno-Morcillo, M.; Rashid, U.J.; Taylor, N.M.; Ruiz, F.M.; Gruene, T.; Legrand, P.; Steuerwald, U.; Muller, C.W. Crystal structure of the 14-subunit RNA polymerase I. Nature 2013, 502, 644–649. [Google Scholar] [CrossRef]
- Tafur, L.; Sadian, Y.; Hoffmann, N.A.; Jakobi, A.J.; Wetzel, R.; Hagen, W.J.H.; Sachse, C.; Muller, C.W. Molecular Structures of Transcribing RNA Polymerase I. Mol. Cell 2016, 64, 1135–1143. [Google Scholar] [CrossRef] [PubMed]
- Pilsl, M.; Crucifix, C.; Papai, G.; Krupp, F.; Steinbauer, R.; Griesenbeck, J.; Milkereit, P.; Tschochner, H.; Schultz, P. Structure of the initiation-competent RNA polymerase I and its implication for transcription. Nat. Commun. 2016, 7, 12126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engel, C.; Plitzko, J.; Cramer, P. RNA polymerase I-Rrn3 complex at 4.8 A resolution. Nat. Commun. 2016, 7, 12129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neyer, S.; Kunz, M.; Geiss, C.; Hantsche, M.; Hodirnau, V.V.; Seybert, A.; Engel, C.; Scheffer, M.P.; Cramer, P.; Frangakis, A.S. Structure of RNA polymerase I transcribing ribosomal DNA genes. Nature 2016, 540, 607–610. [Google Scholar] [CrossRef]
- Engel, C.; Gubbey, T.; Neyer, S.; Sainsbury, S.; Oberthuer, C.; Baejen, C.; Bernecky, C.; Cramer, P. Structural Basis of RNA Polymerase I Transcription Initiation. Cell 2017, 169, 120–131.e22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadian, Y.; Tafur, L.; Kosinski, J.; Jakobi, A.J.; Wetzel, R.; Buczak, K.; Hagen, W.J.; Beck, M.; Sachse, C.; Muller, C.W. Structural insights into transcription initiation by yeast RNA polymerase I. EMBO J. 2017, 36, 2698–2709. [Google Scholar] [CrossRef] [PubMed]
- Sadian, Y.; Baudin, F.; Tafur, L.; Murciano, B.; Wetzel, R.; Weis, F.; Muller, C.W. Molecular insight into RNA polymerase I promoter recognition and promoter melting. Nat. Commun. 2019, 10, 5543. [Google Scholar] [CrossRef] [Green Version]
- Pilsl, M.; Engel, C. Structural basis of RNA polymerase I pre-initiation complex formation and promoter melting. Nat. Commun. 2020, 11, 1206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geiger, S.R.; Lorenzen, K.; Schreieck, A.; Hanecker, P.; Kostrewa, D.; Heck, A.J.; Cramer, P. RNA polymerase I contains a TFIIF-related DNA-binding subcomplex. Mol. Cell 2010, 39, 583–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albert, B.; Leger-Silvestre, I.; Normand, C.; Ostermaier, M.K.; Perez-Fernandez, J.; Panov, K.I.; Zomerdijk, J.C.; Schultz, P.; Gadal, O. RNA polymerase I-specific subunits promote polymerase clustering to enhance the rRNA gene transcription cycle. J. Cell Biol. 2011, 192, 277–293. [Google Scholar] [CrossRef] [Green Version]
- Zhao, D.; Liu, W.; Chen, K.; Wu, Z.; Yang, H.; Xu, Y. Structure of the human RNA polymerase I elongation complex. Cell Discov. 2021, 7, 97. [Google Scholar] [CrossRef]
- Misiaszek, A.D.; Girbig, M.; Grotsch, H.; Baudin, F.; Murciano, B.; Lafita, A.; Muller, C.W. Cryo-EM structures of human RNA polymerase I. Nat. Struct. Mol. Biol. 2021, 28, 997–1008. [Google Scholar] [CrossRef] [PubMed]
- Khatter, H.; Vorlander, M.K.; Muller, C.W. RNA polymerase I and III: Similar yet unique. Curr. Opin. Struct. Biol. 2017, 47, 88–94. [Google Scholar] [CrossRef]
- Engel, C.; Neyer, S.; Cramer, P. Distinct Mechanisms of Transcription Initiation by RNA Polymerases I and II. Annu. Rev. Biophys. 2018, 47, 425–446. [Google Scholar] [CrossRef] [PubMed]
- Schneider, D.A. RNA polymerase I activity is regulated at multiple steps in the transcription cycle: Recent insights into factors that influence transcription elongation. Gene 2012, 493, 176–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- French, S.L.; Osheim, Y.N.; Cioci, F.; Nomura, M.; Beyer, A.L. In exponentially growing Saccharomyces cerevisiae cells, rRNA synthesis is determined by the summed RNA polymerase I loading rate rather than by the number of active genes. Mol. Cell Biol. 2003, 23, 1558–1568. [Google Scholar] [CrossRef] [Green Version]
- Herdman, C.; Mars, J.C.; Stefanovsky, V.Y.; Tremblay, M.G.; Sabourin-Felix, M.; Lindsay, H.; Robinson, M.D.; Moss, T. A unique enhancer boundary complex on the mouse ribosomal RNA genes persists after loss of Rrn3 or UBF and the inactivation of RNA polymerase I transcription. PLoS Genet. 2017, 13, e1006899. [Google Scholar] [CrossRef] [Green Version]
- Svetlov, V.; Nudler, E. Basic mechanism of transcription by RNA polymerase II. Biochim. Biophys. Acta 2013, 1829, 20–28. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, R.Q.; Ingram, Z.M.; Lucius, A.L.; Schneider, D.A. Defining the divergent enzymatic properties of RNA polymerases I and II. J. Biol. Chem. 2021, 296, 100051. [Google Scholar] [CrossRef]
- Clarke, A.M.; Engel, K.L.; Giles, K.E.; Petit, C.M.; Schneider, D.A. NETSeq reveals heterogeneous nucleotide incorporation by RNA polymerase I. Proc. Natl. Acad. Sci. USA 2018, 115, E11633–E11641. [Google Scholar] [CrossRef] [Green Version]
- Turowski, T.W.; Petfalski, E.; Goddard, B.D.; French, S.L.; Helwak, A.; Tollervey, D. Nascent Transcript Folding Plays a Major Role in Determining RNA Polymerase Elongation Rates. Mol. Cell 2020, 79, 488–503.e11. [Google Scholar] [CrossRef]
- Scull, C.E.; Clarke, A.M.; Lucius, A.L.; Schneider, D.A. Downstream sequence-dependent RNA cleavage and pausing by RNA polymerase I. J. Biol. Chem. 2020, 295, 1288–1299. [Google Scholar] [CrossRef] [PubMed]
- Merkl, P.E.; Pilsl, M.; Fremter, T.; Schwank, K.; Engel, C.; Langst, G.; Milkereit, P.; Griesenbeck, J.; Tschochner, H. RNA polymerase I (Pol I) passage through nucleosomes depends on Pol I subunits binding its lobe structure. J. Biol. Chem. 2020, 295, 4782–4795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lisica, A.; Engel, C.; Jahnel, M.; Roldan, E.; Galburt, E.A.; Cramer, P.; Grill, S.W. Mechanisms of backtrack recovery by RNA polymerases I and II. Proc. Natl. Acad. Sci. USA 2016, 113, 2946–2951. [Google Scholar] [CrossRef] [Green Version]
- Appling, F.D.; Scull, C.E.; Lucius, A.L.; Schneider, D.A. The A12.2 Subunit Is an Intrinsic Destabilizer of the RNA Polymerase I Elongation Complex. Biophys. J. 2018, 114, 2507–2515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwank, K.; Schmid, C.; Fremter, T.; Milkereit, P.; Griesenbeck, J.; Tschochner, H. RNA polymerase I (Pol I) lobe-binding subunit Rpa12.2 promotes RNA cleavage and proofreading. J. Biol. Chem. 2022, 298, 101862. [Google Scholar] [CrossRef]
- Gout, J.F.; Li, W.; Fritsch, C.; Li, A.; Haroon, S.; Singh, L.; Hua, D.; Fazelinia, H.; Smith, Z.; Seeholzer, S.; et al. The landscape of transcription errors in eukaryotic cells. Sci. Adv. 2017, 3, e1701484. [Google Scholar] [CrossRef] [Green Version]
- Nemeth, A.; Guibert, S.; Tiwari, V.K.; Ohlsson, R.; Langst, G. Epigenetic regulation of TTF-I-mediated promoter-terminator interactions of rRNA genes. EMBO J. 2008, 27, 1255–1265. [Google Scholar] [CrossRef]
- Denissov, S.; Lessard, F.; Mayer, C.; Stefanovsky, V.; van Driel, M.; Grummt, I.; Moss, T.; Stunnenberg, H.G. A model for the topology of active ribosomal RNA genes. EMBO Rep. 2011, 12, 231–237. [Google Scholar] [CrossRef] [Green Version]
- Schneider, D.A.; Nomura, M. RNA polymerase I remains intact without subunit exchange through multiple rounds of transcription in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 2004, 101, 15112–15117. [Google Scholar] [CrossRef] [Green Version]
- Wei, T.; Najmi, S.M.; Liu, H.; Peltonen, K.; Kucerova, A.; Schneider, D.A.; Laiho, M. Small-Molecule Targeting of RNA Polymerase I Activates a Conserved Transcription Elongation Checkpoint. Cell Rep. 2018, 23, 404–414. [Google Scholar] [CrossRef]
- Sanz-Murillo, M.; Xu, J.; Belogurov, G.A.; Calvo, O.; Gil-Carton, D.; Moreno-Morcillo, M.; Wang, D.; Fernandez-Tornero, C. Structural basis of RNA polymerase I stalling at UV light-induced DNA damage. Proc. Natl. Acad. Sci. USA 2018, 115, 8972–8977. [Google Scholar] [CrossRef] [Green Version]
- Knutson, B.A.; McNamar, R.; Rothblum, L.I. Dynamics of the RNA polymerase I TFIIF/TFIIE-like subcomplex: A mini-review. Biochem. Soc. Trans. 2020, 48, 1917–1927. [Google Scholar] [CrossRef] [PubMed]
- Scull, C.E.; Lucius, A.L.; Schneider, D.A. The N-terminal domain of the A12.2 subunit stimulates RNA polymerase I transcription elongation. Biophys. J. 2021, 120, 1883–1893. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Lee, C.Y.; Grzechnik, A.; Gonzales-Zubiate, F.; Vashisht, A.A.; Lee, A.; Wohlschlegel, J.; Chanfreau, G.F. RNA polymerase I stability couples cellular growth to metal availability. Mol. Cell 2013, 51, 105–115. [Google Scholar] [CrossRef] [Green Version]
- Richardson, L.A.; Reed, B.J.; Charette, J.M.; Freed, E.F.; Fredrickson, E.K.; Locke, M.N.; Baserga, S.J.; Gardner, R.G. A conserved deubiquitinating enzyme controls cell growth by regulating RNA polymerase I stability. Cell Rep. 2012, 2, 372–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, M.D.; Harreman, M.; Svejstrup, J.Q. Ubiquitylation and degradation of elongating RNA polymerase II: The last resort. Biochim. Biophys. Acta 2013, 1829, 151–157. [Google Scholar] [CrossRef]
- Son, K.; Scharer, O.D. Repair, Removal, and Shutdown: It All Hinges on RNA Polymerase II Ubiquitylation. Cell 2020, 180, 1039–1041. [Google Scholar] [CrossRef]
- Nakazawa, Y.; Hara, Y.; Oka, Y.; Komine, O.; van den Heuvel, D.; Guo, C.; Daigaku, Y.; Isono, M.; He, Y.; Shimada, M.; et al. Ubiquitination of DNA Damage-Stalled RNAPII Promotes Transcription-Coupled Repair. Cell 2020, 180, 1228–1244.e24. [Google Scholar] [CrossRef]
- Tufegdzic Vidakovic, A.; Mitter, R.; Kelly, G.P.; Neumann, M.; Harreman, M.; Rodriguez-Martinez, M.; Herlihy, A.; Weems, J.C.; Boeing, S.; Encheva, V.; et al. Regulation of the RNAPII Pool Is Integral to the DNA Damage Response. Cell 2020, 180, 1245–1261.e1221. [Google Scholar] [CrossRef]
- Lesniewska, E.; Ciesla, M.; Boguta, M. Repression of yeast RNA polymerase III by stress leads to ubiquitylation and proteasomal degradation of its largest subunit, C160. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 25–34. [Google Scholar] [CrossRef]
- Wang, Z.; Wu, C.; Aslanian, A.; Yates, J.R., III; Hunter, T. Defective RNA polymerase III is negatively regulated by the SUMO-Ubiquitin-Cdc48 pathway. Elife 2018, 7, e35447. [Google Scholar] [CrossRef] [PubMed]
- Klein, J.; Grummt, I. Cell cycle-dependent regulation of RNA polymerase I transcription: The nucleolar transcription factor UBF is inactive in mitosis and early G1. Proc. Natl. Acad. Sci. USA 1999, 96, 6096–6101. [Google Scholar] [CrossRef] [Green Version]
- McStay, B.; Grummt, I. The epigenetics of rRNA genes: From molecular to chromosome biology. Annu. Rev. Cell Dev. Biol. 2008, 24, 131–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefanovsky, V.Y.; Pelletier, G.; Hannan, R.; Gagnon-Kugler, T.; Rothblum, L.I.; Moss, T. An immediate response of ribosomal transcription to growth factor stimulation in mammals is mediated by ERK phosphorylation of UBF. Mol. Cell 2001, 8, 1063–1073. [Google Scholar] [CrossRef]
- Grummt, I. Life on a planet of its own: Regulation of RNA polymerase I transcription in the nucleolus. Genes Dev. 2003, 17, 1691–1702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Yuan, X.; Frodin, M.; Grummt, I. ERK-dependent phosphorylation of the transcription initiation factor TIF-IA is required for RNA polymerase I transcription and cell growth. Mol. Cell 2003, 11, 405–413. [Google Scholar] [CrossRef]
- Bierhoff, H.; Dundr, M.; Michels, A.A.; Grummt, I. Phosphorylation by casein kinase 2 facilitates rRNA gene transcription by promoting dissociation of TIF-IA from elongating RNA polymerase I. Mol. Cell Biol. 2008, 28, 4988–4998. [Google Scholar] [CrossRef] [Green Version]
- Mayer, C.; Zhao, J.; Yuan, X.; Grummt, I. mTOR-dependent activation of the transcription factor TIF-IA links rRNA synthesis to nutrient availability. Genes Dev. 2004, 18, 423–434. [Google Scholar] [CrossRef] [Green Version]
- Muth, V.; Nadaud, S.; Grummt, I.; Voit, R. Acetylation of TAF(I)68, a subunit of TIF-IB/SL1, activates RNA polymerase I transcription. EMBO J. 2001, 20, 1353–1362. [Google Scholar] [CrossRef] [Green Version]
- Mayer, C.; Bierhoff, H.; Grummt, I. The nucleolus as a stress sensor: JNK2 inactivates the transcription factor TIF-IA and down-regulates rRNA synthesis. Genes Dev. 2005, 19, 933–941. [Google Scholar] [CrossRef]
- Hoppe, S.; Bierhoff, H.; Cado, I.; Weber, A.; Tiebe, M.; Grummt, I.; Voit, R. AMP-activated protein kinase adapts rRNA synthesis to cellular energy supply. Proc. Natl. Acad. Sci. USA 2009, 106, 17781–17786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, Y.; Yano, H.; Ogasawara, S.; Yoshioka, S.; Imamura, H.; Okamoto, K.; Tsuneoka, M. Mild Glucose Starvation Induces KDM2A-Mediated H3K36me2 Demethylation through AMPK To Reduce rRNA Transcription and Cell Proliferation. Mol. Cell Biol. 2015, 35, 4170–4184. [Google Scholar] [CrossRef] [Green Version]
- Young, D.W.; Hassan, M.Q.; Pratap, J.; Galindo, M.; Zaidi, S.K.; Lee, S.H.; Yang, X.; Xie, R.; Javed, A.; Underwood, J.M.; et al. Mitotic occupancy and lineage-specific transcriptional control of rRNA genes by Runx2. Nature 2007, 445, 442–446. [Google Scholar] [CrossRef] [PubMed]
- Jouffe, C.; Cretenet, G.; Symul, L.; Martin, E.; Atger, F.; Naef, F.; Gachon, F. The circadian clock coordinates ribosome biogenesis. PLoS Biol. 2013, 11, e1001455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Mauvoisin, D.; Martin, E.; Atger, F.; Galindo, A.N.; Dayon, L.; Sizzano, F.; Palini, A.; Kussmann, M.; Waridel, P.; et al. Nuclear Proteomics Uncovers Diurnal Regulatory Landscapes in Mouse Liver. Cell Metab. 2017, 25, 102–117. [Google Scholar] [CrossRef] [Green Version]
- Sinturel, F.; Gerber, A.; Mauvoisin, D.; Wang, J.; Gatfield, D.; Stubblefield, J.J.; Green, C.B.; Gachon, F.; Schibler, U. Diurnal Oscillations in Liver Mass and Cell Size Accompany Ribosome Assembly Cycles. Cell 2017, 169, 651–663.e614. [Google Scholar] [CrossRef] [Green Version]
- Kermekchiev, M.; Workman, J.L.; Pikaard, C.S. Nucleosome binding by the polymerase I transactivator upstream binding factor displaces linker histone H1. Mol. Cell Biol. 1997, 17, 5833–5842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamdane, N.; Stefanovsky, V.Y.; Tremblay, M.G.; Nemeth, A.; Paquet, E.; Lessard, F.; Sanij, E.; Hannan, R.; Moss, T. Conditional inactivation of Upstream Binding Factor reveals its epigenetic functions and the existence of a somatic nucleolar precursor body. PLoS Genet. 2014, 10, e1004505. [Google Scholar] [CrossRef] [Green Version]
- Salifou, K.; Ray, S.; Verrier, L.; Aguirrebengoa, M.; Trouche, D.; Panov, K.I.; Vandromme, M. The histone demethylase JMJD2A/KDM4A links ribosomal RNA transcription to nutrients and growth factors availability. Nat. Commun. 2016, 7, 10174. [Google Scholar] [CrossRef] [Green Version]
- Stefanovsky, V.; Langlois, F.; Gagnon-Kugler, T.; Rothblum, L.I.; Moss, T. Growth factor signaling regulates elongation of RNA polymerase I transcription in mammals via UBF phosphorylation and r-chromatin remodeling. Mol. Cell 2006, 21, 629–639. [Google Scholar] [CrossRef]
- Strohner, R.; Nemeth, A.; Jansa, P.; Hofmann-Rohrer, U.; Santoro, R.; Langst, G.; Grummt, I. NoRC--a novel member of mammalian ISWI-containing chromatin remodeling machines. EMBO J. 2001, 20, 4892–4900. [Google Scholar] [CrossRef] [Green Version]
- Santoro, R.; Li, J.; Grummt, I. The nucleolar remodeling complex NoRC mediates heterochromatin formation and silencing of ribosomal gene transcription. Nat. Genet. 2002, 32, 393–396. [Google Scholar] [CrossRef]
- Guetg, C.; Lienemann, P.; Sirri, V.; Grummt, I.; Hernandez-Verdun, D.; Hottiger, M.O.; Fussenegger, M.; Santoro, R. The NoRC complex mediates the heterochromatin formation and stability of silent rRNA genes and centromeric repeats. EMBO J. 2010, 29, 2135–2146. [Google Scholar] [CrossRef]
- Moore, H.M.; Bai, B.; Boisvert, F.M.; Latonen, L.; Rantanen, V.; Simpson, J.C.; Pepperkok, R.; Lamond, A.I.; Laiho, M. Quantitative proteomics and dynamic imaging of the nucleolus reveal distinct responses to UV and ionizing radiation. Mol. Cell Proteom. 2011, 10, M111.009241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lafontaine, D.L.J.; Riback, J.A.; Bascetin, R.; Brangwynne, C.P. The nucleolus as a multiphase liquid condensate. Nat. Rev. Mol. Cell Biol. 2021, 22, 165–182. [Google Scholar] [CrossRef] [PubMed]
- Boulon, S.; Westman, B.J.; Hutten, S.; Boisvert, F.M.; Lamond, A.I. The nucleolus under stress. Mol. Cell 2010, 40, 216–227. [Google Scholar] [CrossRef] [PubMed]
- van Sluis, M.; McStay, B. Nucleolar reorganization in response to rDNA damage. Curr. Opin. Cell Biol. 2017, 46, 81–86. [Google Scholar] [CrossRef]
- Lohrum, M.A.; Ludwig, R.L.; Kubbutat, M.H.; Hanlon, M.; Vousden, K.H. Regulation of HDM2 activity by the ribosomal protein L11. Cancer Cell 2003, 3, 577–587. [Google Scholar] [CrossRef] [Green Version]
- Macias, E.; Jin, A.; Deisenroth, C.; Bhat, K.; Mao, H.; Lindstrom, M.S.; Zhang, Y. An ARF-independent c-MYC-activated tumor suppression pathway mediated by ribosomal protein-Mdm2 Interaction. Cancer Cell 2010, 18, 231–243. [Google Scholar] [CrossRef] [Green Version]
- Bursac, S.; Brdovcak, M.C.; Pfannkuchen, M.; Orsolic, I.; Golomb, L.; Zhu, Y.; Katz, C.; Daftuar, L.; Grabusic, K.; Vukelic, I.; et al. Mutual protection of ribosomal proteins L5 and L11 from degradation is essential for p53 activation upon ribosomal biogenesis stress. Proc. Natl. Acad. Sci. USA 2012, 109, 20467–20472. [Google Scholar] [CrossRef]
- Deisenroth, C.; Franklin, D.A.; Zhang, Y. The Evolution of the Ribosomal Protein-MDM2-p53 Pathway. Cold Spring Harb. Perspect. Med. 2016, 6, a026138. [Google Scholar] [CrossRef] [Green Version]
- Zisi, A.; Bartek, J.; Lindstrom, M.S. Targeting Ribosome Biogenesis in Cancer: Lessons Learned and Way Forward. Cancers 2022, 14, 2126. [Google Scholar] [CrossRef]
- Aoi, Y.; Takahashi, Y.H.; Shah, A.P.; Iwanaszko, M.; Rendleman, E.J.; Khan, N.H.; Cho, B.K.; Goo, Y.A.; Ganesan, S.; Kelleher, N.L.; et al. SPT5 stabilization of promoter-proximal RNA polymerase II. Mol. Cell 2021, 81, 4413–4424.e5. [Google Scholar] [CrossRef] [PubMed]
- Caron, P.; Pankotai, T.; Wiegant, W.W.; Tollenaere, M.A.X.; Furst, A.; Bonhomme, C.; Helfricht, A.; de Groot, A.; Pastink, A.; Vertegaal, A.C.O.; et al. WWP2 ubiquitylates RNA polymerase II for DNA-PK-dependent transcription arrest and repair at DNA breaks. Genes Dev. 2019, 33, 684–704. [Google Scholar] [CrossRef] [Green Version]
- Harreman, M.; Taschner, M.; Sigurdsson, S.; Anindya, R.; Reid, J.; Somesh, B.; Kong, S.E.; Banks, C.A.; Conaway, R.C.; Conaway, J.W.; et al. Distinct ubiquitin ligases act sequentially for RNA polymerase II polyubiquitylation. Proc. Natl. Acad. Sci. USA 2009, 106, 20705–20710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huibregtse, J.M.; Yang, J.C.; Beaudenon, S.L. The large subunit of RNA polymerase II is a substrate of the Rsp5 ubiquitin-protein ligase. Proc. Natl. Acad. Sci. USA 1997, 94, 3656–3661. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Wolgast, M.; Beebe, L.M.; Reese, J.C. Ccr4-Not maintains genomic integrity by controlling the ubiquitylation and degradation of arrested RNAPII. Genes Dev. 2019, 33, 705–717. [Google Scholar] [CrossRef] [PubMed]
- Kleiman, F.E.; Wu-Baer, F.; Fonseca, D.; Kaneko, S.; Baer, R.; Manley, J.L. BRCA1/BARD1 inhibition of mRNA 3’ processing involves targeted degradation of RNA polymerase II. Genes Dev. 2005, 19, 1227–1237. [Google Scholar] [CrossRef] [Green Version]
- Kuznetsova, A.V.; Meller, J.; Schnell, P.O.; Nash, J.A.; Ignacak, M.L.; Sanchez, Y.; Conaway, J.W.; Conaway, R.C.; Czyzyk-Krzeska, M.F. von Hippel-Lindau protein binds hyperphosphorylated large subunit of RNA polymerase II through a proline hydroxylation motif and targets it for ubiquitination. Proc. Natl. Acad. Sci. USA 2003, 100, 2706–2711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Zhang, Z.; Wang, B.; Zhang, J.; Zhao, Y.; Jin, Y. Wwp2-mediated ubiquitination of the RNA polymerase II large subunit in mouse embryonic pluripotent stem cells. Mol. Cell Biol. 2007, 27, 5296–5305. [Google Scholar] [CrossRef] [PubMed]
- Starita, L.M.; Horwitz, A.A.; Keogh, M.C.; Ishioka, C.; Parvin, J.D.; Chiba, N. BRCA1/BARD1 ubiquitinate phosphorylated RNA polymerase II. J. Biol. Chem. 2005, 280, 24498–24505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Weegen, Y.; Golan-Berman, H.; Mevissen, T.E.T.; Apelt, K.; Gonzalez-Prieto, R.; Goedhart, J.; Heilbrun, E.E.; Vertegaal, A.C.O.; van den Heuvel, D.; Walter, J.C.; et al. The cooperative action of CSB, CSA, and UVSSA target TFIIH to DNA damage-stalled RNA polymerase II. Nat. Commun. 2020, 11, 2104. [Google Scholar] [CrossRef] [PubMed]
- Verma, R.; Oania, R.; Fang, R.; Smith, G.T.; Deshaies, R.J. Cdc48/p97 mediates UV-dependent turnover of RNA Pol II. Mol. Cell 2011, 41, 82–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, M.D.; Harreman, M.; Taschner, M.; Reid, J.; Walker, J.; Erdjument-Bromage, H.; Tempst, P.; Svejstrup, J.Q. Proteasome-mediated processing of Def1, a critical step in the cellular response to transcription stress. Cell 2013, 154, 983–995. [Google Scholar] [CrossRef] [Green Version]
- Yasukawa, T.; Kamura, T.; Kitajima, S.; Conaway, R.C.; Conaway, J.W.; Aso, T. Mammalian Elongin A complex mediates DNA-damage-induced ubiquitylation and degradation of Rpb1. EMBO J. 2008, 27, 3256–3266. [Google Scholar] [CrossRef] [Green Version]
- Pianese, G. Beitrag zur Histologie und Aetiologie des Carcinoms: Histologische und experimentelle Untersuchungen; Gustav Fischer: Jena, Germany, 1896. [Google Scholar]
- Montanaro, L.; Trere, D.; Derenzini, M. Nucleolus, ribosomes, and cancer. Am. J. Pathol. 2008, 173, 301–310. [Google Scholar] [CrossRef] [Green Version]
- Derenzini, M.; Trere, D.; Pession, A.; Montanaro, L.; Sirri, V.; Ochs, R.L. Nucleolar function and size in cancer cells. Am. J. Pathol. 1998, 152, 1291–1297. [Google Scholar]
- Derenzini, M.; Montanaro, L.; Trere, D. What the nucleolus says to a tumour pathologist. Histopathology 2009, 54, 753–762. [Google Scholar] [CrossRef]
- Aydin, H.; Zhou, M.; Herawi, M.; Epstein, J.I. Number and location of nucleoli and presence of apoptotic bodies in diagnostically challenging cases of prostate adenocarcinoma on needle biopsy. Hum. Pathol. 2005, 36, 1172–1177. [Google Scholar] [CrossRef]
- Pich, A.; Chiusa, L.; Margaria, E. Prognostic relevance of AgNORs in tumor pathology. Micron 2000, 31, 133–141. [Google Scholar] [CrossRef]
- Guner, G.; Sirajuddin, P.; Zheng, Q.; Bai, B.; Brodie, A.; Liu, H.; Af Hallstrom, T.; Kulac, I.; Laiho, M.; De Marzo, A.M. Novel Assay to Detect RNA Polymerase I Activity In Vivo. Mol. Cancer Res. 2017, 15, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Tsoi, H.; Lam, K.C.; Dong, Y.; Zhang, X.; Lee, C.K.; Zhang, J.; Ng, S.C.; Ng, S.S.M.; Zheng, S.; Chen, Y.; et al. Pre-45s rRNA promotes colon cancer and is associated with poor survival of CRC patients. Oncogene 2017, 36, 6109–6118. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Wang, Y.; Lv, Q.; Zhang, J.; Wang, Q.; Gao, F.; Hou, H.; Zhang, H.; Zhang, W.; Li, L. Overexpression of Ribosomal RNA in the Development of Human Cervical Cancer Is Associated with rDNA Promoter Hypomethylation. PLoS ONE 2016, 11, e0163340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uemura, M.; Zheng, Q.; Koh, C.M.; Nelson, W.G.; Yegnasubramanian, S.; De Marzo, A.M. Overexpression of ribosomal RNA in prostate cancer is common but not linked to rDNA promoter hypomethylation. Oncogene 2012, 31, 1254–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, C.M.; Gurel, B.; Sutcliffe, S.; Aryee, M.J.; Schultz, D.; Iwata, T.; Uemura, M.; Zeller, K.I.; Anele, U.; Zheng, Q.; et al. Alterations in nucleolar structure and gene expression programs in prostatic neoplasia are driven by the MYC oncogene. Am. J. Pathol. 2011, 178, 1824–1834. [Google Scholar] [CrossRef] [PubMed]
- Marcel, V.; Ghayad, S.E.; Belin, S.; Therizols, G.; Morel, A.P.; Solano-Gonzalez, E.; Vendrell, J.A.; Hacot, S.; Mertani, H.C.; Albaret, M.A.; et al. p53 acts as a safeguard of translational control by regulating fibrillarin and rRNA methylation in cancer. Cancer Cell 2013, 24, 318–330. [Google Scholar] [CrossRef] [Green Version]
- Ueda, M.; Iguchi, T.; Nambara, S.; Saito, T.; Komatsu, H.; Sakimura, S.; Hirata, H.; Uchi, R.; Takano, Y.; Shinden, Y.; et al. Overexpression of Transcription Termination Factor 1 is Associated with a Poor Prognosis in Patients with Colorectal Cancer. Ann. Surg. Oncol. 2015, 22, 1490–1498. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, H.; Iguchi, T.; Ueda, M.; Nambara, S.; Saito, T.; Hirata, H.; Sakimura, S.; Takano, Y.; Uchi, R.; Shinden, Y.; et al. Clinical and biological significance of transcription termination factor, RNA polymerase I in human liver hepatocellular carcinoma. Oncol. Rep. 2016, 35, 2073–2080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolezal, J.M.; Dash, A.P.; Prochownik, E.V. Diagnostic and prognostic implications of ribosomal protein transcript expression patterns in human cancers. BMC Cancer 2018, 18, 275. [Google Scholar] [CrossRef] [Green Version]
- Ebright, R.Y.; Lee, S.; Wittner, B.S.; Niederhoffer, K.L.; Nicholson, B.T.; Bardia, A.; Truesdell, S.; Wiley, D.F.; Wesley, B.; Li, S.; et al. Deregulation of ribosomal protein expression and translation promotes breast cancer metastasis. Science 2020, 367, 1468–1473. [Google Scholar] [CrossRef] [PubMed]
- Hannan, K.M.; Sanij, E.; Rothblum, L.I.; Hannan, R.D.; Pearson, R.B. Dysregulation of RNA polymerase I transcription during disease. Biochim. Biophys. Acta 2013, 1829, 342–360. [Google Scholar] [CrossRef] [PubMed]
- Ruggero, D. Revisiting the nucleolus: From marker to dynamic integrator of cancer signaling. Sci. Signal 2012, 5, pe38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Riggelen, J.; Yetil, A.; Felsher, D.W. MYC as a regulator of ribosome biogenesis and protein synthesis. Nat. Rev. Cancer 2010, 10, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V. MYC on the path to cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef] [Green Version]
- Boon, K.; Caron, H.N.; van Asperen, R.; Valentijn, L.; Hermus, M.C.; van Sluis, P.; Roobeek, I.; Weis, I.; Voute, P.A.; Schwab, M.; et al. N-myc enhances the expression of a large set of genes functioning in ribosome biogenesis and protein synthesis. EMBO J. 2001, 20, 1383–1393. [Google Scholar] [CrossRef]
- Arabi, A.; Wu, S.; Ridderstrale, K.; Bierhoff, H.; Shiue, C.; Fatyol, K.; Fahlen, S.; Hydbring, P.; Soderberg, O.; Grummt, I.; et al. c-Myc associates with ribosomal DNA and activates RNA polymerase I transcription. Nat. Cell Biol. 2005, 7, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Grandori, C.; Gomez-Roman, N.; Felton-Edkins, Z.A.; Ngouenet, C.; Galloway, D.A.; Eisenman, R.N.; White, R.J. c-Myc binds to human ribosomal DNA and stimulates transcription of rRNA genes by RNA polymerase I. Nat. Cell Biol. 2005, 7, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.W.; Wu, N.; Kim, Y.C.; Cheng, P.F.; Basom, R.; Kim, D.; Dunn, C.T.; Lee, A.Y.; Kim, K.; Lee, C.S.; et al. Genetic requirement for Mycl and efficacy of RNA Pol I inhibition in mouse models of small cell lung cancer. Genes Dev. 2016, 30, 1289–1299. [Google Scholar] [CrossRef] [Green Version]
- Bonetti, P.; Davoli, T.; Sironi, C.; Amati, B.; Pelicci, P.G.; Colombo, E. Nucleophosmin and its AML-associated mutant regulate c-Myc turnover through Fbw7 gamma. J. Cell Biol 2008, 182, 19–26. [Google Scholar] [CrossRef] [Green Version]
- Muller, C.; Bremer, A.; Schreiber, S.; Eichwald, S.; Calkhoven, C.F. Nucleolar retention of a translational C/EBPalpha isoform stimulates rDNA transcription and cell size. EMBO J. 2010, 29, 897–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thoreen, C.C.; Chantranupong, L.; Keys, H.R.; Wang, T.; Gray, N.S.; Sabatini, D.M. A unifying model for mTORC1-mediated regulation of mRNA translation. Nature 2012, 485, 109–113. [Google Scholar] [CrossRef]
- Iadevaia, V.; Liu, R.; Proud, C.G. mTORC1 signaling controls multiple steps in ribosome biogenesis. Semin. Cell Dev. Biol. 2014, 36, 113–120. [Google Scholar] [CrossRef]
- Philippi, A.; Steinbauer, R.; Reiter, A.; Fath, S.; Leger-Silvestre, I.; Milkereit, P.; Griesenbeck, J.; Tschochner, H. TOR-dependent reduction in the expression level of Rrn3p lowers the activity of the yeast RNA Pol I machinery, but does not account for the strong inhibition of rRNA production. Nucleic Acids Res. 2010, 38, 5315–5326. [Google Scholar] [CrossRef] [Green Version]
- Hannan, K.M.; Brandenburger, Y.; Jenkins, A.; Sharkey, K.; Cavanaugh, A.; Rothblum, L.; Moss, T.; Poortinga, G.; McArthur, G.A.; Pearson, R.B.; et al. mTOR-dependent regulation of ribosomal gene transcription requires S6K1 and is mediated by phosphorylation of the carboxy-terminal activation domain of the nucleolar transcription factor UBF. Mol. Cell Biol. 2003, 23, 8862–8877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Claypool, J.A.; French, S.L.; Johzuka, K.; Eliason, K.; Vu, L.; Dodd, J.A.; Beyer, A.L.; Nomura, M. Tor pathway regulates Rrn3p-dependent recruitment of yeast RNA polymerase I to the promoter but does not participate in alteration of the number of active genes. Mol. Biol. Cell 2004, 15, 946–956. [Google Scholar] [CrossRef]
- Li, H.; Tsang, C.K.; Watkins, M.; Bertram, P.G.; Zheng, X.F. Nutrient regulates Tor1 nuclear localization and association with rDNA promoter. Nature 2006, 442, 1058–1061. [Google Scholar] [CrossRef] [PubMed]
- Iadevaia, V.; Zhang, Z.; Jan, E.; Proud, C.G. mTOR signaling regulates the processing of pre-rRNA in human cells. Nucleic Acids Res. 2012, 40, 2527–2539. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Gao, L.; Teng, L.; Ge, J.; Oo, Z.M.; Kumar, A.R.; Gilliland, D.G.; Mason, P.J.; Tan, K.; Speck, N.A. Runx1 Deficiency Decreases Ribosome Biogenesis and Confers Stress Resistance to Hematopoietic Stem and Progenitor Cells. Cell Stem Cell 2015, 17, 165–177. [Google Scholar] [CrossRef] [Green Version]
- Prakash, V.; Carson, B.B.; Feenstra, J.M.; Dass, R.A.; Sekyrova, P.; Hoshino, A.; Petersen, J.; Guo, Y.; Parks, M.M.; Kurylo, C.M.; et al. Ribosome biogenesis during cell cycle arrest fuels EMT in development and disease. Nat. Commun. 2019, 10, 2110. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zhang, H.; Sapio, R.; Yang, J.; Wong, J.; Zhang, X.; Guo, J.Y.; Pine, S.; Van Remmen, H.; Li, H.; et al. SOD1 regulates ribosome biogenesis in KRAS mutant non-small cell lung cancer. Nat. Commun. 2021, 12, 2259. [Google Scholar] [CrossRef]
- Justilien, V.; Ali, S.A.; Jamieson, L.; Yin, N.; Cox, A.D.; Der, C.J.; Murray, N.R.; Fields, A.P. Ect2-Dependent rRNA Synthesis Is Required for KRAS-TRP53-Driven Lung Adenocarcinoma. Cancer Cell 2017, 31, 256–269. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, O.G.; Assfalg, R.; Koch, S.; Schelling, A.; Meena, J.K.; Kraus, J.; Lechel, A.; Katz, S.F.; Benes, V.; Scharffetter-Kochanek, K.; et al. Telomerase stimulates ribosomal DNA transcription under hyperproliferative conditions. Nat. Commun. 2014, 5, 4599. [Google Scholar] [CrossRef] [Green Version]
- Li, L.Y.; Chen, H.; Hsieh, Y.H.; Wang, Y.N.; Chu, H.J.; Chen, Y.H.; Chen, H.Y.; Chien, P.J.; Ma, H.T.; Tsai, H.C.; et al. Nuclear ErbB2 enhances translation and cell growth by activating transcription of ribosomal RNA genes. Cancer Res. 2011, 71, 4269–4279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, J.C.; Hannan, K.M.; Riddell, K.; Ng, P.Y.; Peck, A.; Lee, R.S.; Hung, S.; Astle, M.V.; Bywater, M.; Wall, M.; et al. AKT promotes rRNA synthesis and cooperates with c-MYC to stimulate ribosome biogenesis in cancer. Sci. Signal 2011, 4, ra56. [Google Scholar] [CrossRef] [PubMed]
- Ghoshal, K.; Majumder, S.; Datta, J.; Motiwala, T.; Bai, S.; Sharma, S.M.; Frankel, W.; Jacob, S.T. Role of human ribosomal RNA (rRNA) promoter methylation and of methyl-CpG-binding protein MBD2 in the suppression of rRNA gene expression. J. Biol. Chem. 2004, 279, 6783–6793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powell, M.A.; Mutch, D.G.; Rader, J.S.; Herzog, T.J.; Huang, T.H.; Goodfellow, P.J. Ribosomal DNA methylation in patients with endometrial carcinoma: An independent prognostic marker. Cancer 2002, 94, 2941–2952. [Google Scholar] [CrossRef] [PubMed]
- Voit, R.; Schafer, K.; Grummt, I. Mechanism of repression of RNA polymerase I transcription by the retinoblastoma protein. Mol. Cell Biol. 1997, 17, 4230–4237. [Google Scholar] [CrossRef] [Green Version]
- Zhai, W.; Comai, L. Repression of RNA polymerase I transcription by the tumor suppressor p53. Mol. Cell Biol. 2000, 20, 5930–5938. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Comai, L.; Johnson, D.L. PTEN represses RNA Polymerase I transcription by disrupting the SL1 complex. Mol. Cell Biol. 2005, 25, 6899–6911. [Google Scholar] [CrossRef] [Green Version]
- Johnston, R.; D’Costa, Z.; Ray, S.; Gorski, J.; Harkin, D.P.; Mullan, P.; Panov, K.I. The identification of a novel role for BRCA1 in regulating RNA polymerase I transcription. Oncotarget 2016, 7, 68097–68110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayrault, O.; Andrique, L.; Fauvin, D.; Eymin, B.; Gazzeri, S.; Seite, P. Human tumor suppressor p14ARF negatively regulates rRNA transcription and inhibits UBF1 transcription factor phosphorylation. Oncogene 2006, 25, 7577–7586. [Google Scholar] [CrossRef]
- Sugimoto, M.; Kuo, M.L.; Roussel, M.F.; Sherr, C.J. Nucleolar Arf tumor suppressor inhibits ribosomal RNA processing. Mol. Cell 2003, 11, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Maggi, L.B., Jr.; Winkeler, C.L.; Miceli, A.P.; Apicelli, A.J.; Brady, S.N.; Kuchenreuther, M.J.; Weber, J.D. ARF tumor suppression in the nucleolus. Biochim. Biophys. Acta 2014, 1842, 831–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hochstatter, J.; Holzel, M.; Rohrmoser, M.; Schermelleh, L.; Leonhardt, H.; Keough, R.; Gonda, T.J.; Imhof, A.; Eick, D.; Langst, G.; et al. Myb-binding protein 1a (Mybbp1a) regulates levels and processing of pre-ribosomal RNA. J. Biol. Chem. 2012, 287, 24365–24377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, S.A.; Dobson, J.R.; Lian, J.B.; Stein, J.L.; van Wijnen, A.J.; Zaidi, S.K.; Stein, G.S. A RUNX2-HDAC1 co-repressor complex regulates rRNA gene expression by modulating UBF acetylation. J. Cell Sci. 2012, 125, 2732–2739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roche, B.; Arcangioli, B.; Martienssen, R.A. RNA interference is essential for cellular quiescence. Science 2016, 354, aah5651. [Google Scholar] [CrossRef] [Green Version]
- Roche, B.; Arcangioli, B.; Martienssen, R. New roles for Dicer in the nucleolus and its relevance to cancer. Cell Cycle 2017, 16, 1643–1653. [Google Scholar] [CrossRef] [Green Version]
- Burger, K.; Muhl, B.; Harasim, T.; Rohrmoser, M.; Malamoussi, A.; Orban, M.; Kellner, M.; Gruber-Eber, A.; Kremmer, E.; Holzel, M.; et al. Chemotherapeutic drugs inhibit ribosome biogenesis at various levels. J. Biol. Chem. 2010, 285, 12416–12425. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, M.; Kastan, M.B. The DNA damage response: Implications for tumor responses to radiation and chemotherapy. Annu. Rev. Med. 2015, 66, 129–143. [Google Scholar] [CrossRef] [Green Version]
- Zhai, X.; Beckmann, H.; Jantzen, H.M.; Essigmann, J.M. Cisplatin-DNA adducts inhibit ribosomal RNA synthesis by hijacking the transcription factor human upstream binding factor. Biochemistry 1998, 37, 16307–16315. [Google Scholar] [CrossRef]
- Hamdane, N.; Herdman, C.; Mars, J.C.; Stefanovsky, V.; Tremblay, M.G.; Moss, T. Depletion of the cisplatin targeted HMGB-box factor UBF selectively induces p53-independent apoptotic death in transformed cells. Oncotarget 2015, 6, 27519–27536. [Google Scholar] [CrossRef] [PubMed]
- Bruno, P.M.; Liu, Y.; Park, G.Y.; Murai, J.; Koch, C.E.; Eisen, T.J.; Pritchard, J.R.; Pommier, Y.; Lippard, S.J.; Hemann, M.T. A subset of platinum-containing chemotherapeutic agents kills cells by inducing ribosome biogenesis stress. Nat. Med. 2017, 23, 461–471. [Google Scholar] [CrossRef] [Green Version]
- Perry, R.P.; Kelley, D.E. Inhibition of RNA synthesis by actinomycin D: Characteristic dose-response of different RNA species. J. Cell Physiol. 1970, 76, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Sobell, H.M. Actinomycin and DNA transcription. Proc. Natl. Acad. Sci. USA 1985, 82, 5328–5331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mischo, H.E.; Hemmerich, P.; Grosse, F.; Zhang, S. Actinomycin D induces histone gamma-H2AX foci and complex formation of gamma-H2AX with Ku70 and nuclear DNA helicase II. J. Biol. Chem. 2005, 280, 9586–9594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nitiss, J.L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. [Google Scholar] [CrossRef] [Green Version]
- Pommier, Y.; Sun, Y.; Huang, S.N.; Nitiss, J.L. Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat. Rev. Mol. Cell Biol. 2016, 17, 703–721. [Google Scholar] [CrossRef]
- Stiborova, M.; Frei, E. Ellipticines as DNA-targeted chemotherapeutics. Curr. Med. Chem. 2014, 21, 575–591. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Panova, T.; Miller, G.; Volkov, A.; Porter, A.C.; Russell, J.; Panov, K.I.; Zomerdijk, J.C. Topoisomerase IIalpha promotes activation of RNA polymerase I transcription by facilitating pre-initiation complex formation. Nat. Commun. 2013, 4, 1598. [Google Scholar] [CrossRef] [Green Version]
- Andrews, W.J.; Panova, T.; Normand, C.; Gadal, O.; Tikhonova, I.G.; Panov, K.I. Old drug, new target: Ellipticines selectively inhibit RNA polymerase I transcription. J. Biol. Chem. 2013, 288, 4567–4582. [Google Scholar] [CrossRef] [Green Version]
- Kosiol, N.; Juranek, S.; Brossart, P.; Heine, A.; Paeschke, K. G-quadruplexes: A promising target for cancer therapy. Mol. Cancer 2021, 20, 40. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Hurley, L.H. A first-in-class clinical G-quadruplex-targeting drug. The bench-to-bedside translation of the fluoroquinolone QQ58 to CX-5461 (Pidnarulex). Bioorg. Med. Chem. Lett. 2022, 77, 129016. [Google Scholar] [CrossRef]
- Sanchez-Martin, V.; Schneider, D.A.; Ortiz-Gonzalez, M.; Soriano-Lerma, A.; Linde-Rodriguez, A.; Perez-Carrasco, V.; Gutierrez-Fernandez, J.; Cuadros, M.; Gonzalez, C.; Soriano, M.; et al. Targeting ribosomal G-quadruplexes with naphthalene-diimides as RNA polymerase I inhibitors for colorectal cancer treatment. Cell Chem. Biol. 2021, 28, 1590–1601.e1594. [Google Scholar] [CrossRef]
- Drygin, D.; Siddiqui-Jain, A.; O’Brien, S.; Schwaebe, M.; Lin, A.; Bliesath, J.; Ho, C.B.; Proffitt, C.; Trent, K.; Whitten, J.P.; et al. Anticancer activity of CX-3543: A direct inhibitor of rRNA biogenesis. Cancer Res. 2009, 69, 7653–7661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drygin, D.; Lin, A.; Bliesath, J.; Ho, C.B.; O’Brien, S.E.; Proffitt, C.; Omori, M.; Haddach, M.; Schwaebe, M.K.; Siddiqui-Jain, A.; et al. Targeting RNA polymerase I with an oral small molecule CX-5461 inhibits ribosomal RNA synthesis and solid tumor growth. Cancer Res. 2011, 71, 1418–1430. [Google Scholar] [CrossRef] [Green Version]
- Hilton, J.; Gelmon, K.; Bedard, P.L.; Tu, D.; Xu, H.; Tinker, A.V.; Goodwin, R.; Laurie, S.A.; Jonker, D.; Hansen, A.R.; et al. Results of the phase I CCTG IND.231 trial of CX-5461 in patients with advanced solid tumors enriched for DNA-repair deficiencies. Nat. Commun. 2022, 13, 3607. [Google Scholar] [CrossRef] [PubMed]
- Mars, J.C.; Tremblay, M.G.; Valere, M.; Sibai, D.S.; Sabourin-Felix, M.; Lessard, F.; Moss, T. The chemotherapeutic agent CX-5461 irreversibly blocks RNA polymerase I initiation and promoter release to cause nucleolar disruption, DNA damage and cell inviability. NAR Cancer 2020, 2, zcaa032. [Google Scholar] [CrossRef]
- Xu, H.; Di Antonio, M.; McKinney, S.; Mathew, V.; Ho, B.; O’Neil, N.J.; Santos, N.D.; Silvester, J.; Wei, V.; Garcia, J.; et al. CX-5461 is a DNA G-quadruplex stabilizer with selective lethality in BRCA1/2 deficient tumours. Nat. Commun. 2017, 8, 14432. [Google Scholar] [CrossRef] [Green Version]
- Bossaert, M.; Pipier, A.; Riou, J.F.; Noirot, C.; Nguyen, L.T.; Serre, R.F.; Bouchez, O.; Defrancq, E.; Calsou, P.; Britton, S.; et al. Transcription-associated topoisomerase 2alpha (TOP2A) activity is a major effector of cytotoxicity induced by G-quadruplex ligands. Elife 2021, 10, e65184. [Google Scholar] [CrossRef] [PubMed]
- Lehman, S.L.; Schwartz, K.R.; Maheshwari, S.; Camphausen, K.; Tofilon, P.J. CX-5461 induces radiosensitization through modification of the DNA damage response and not inhibition of RNA polymerase I. Sci. Rep. 2022, 12, 4059. [Google Scholar] [CrossRef]
- Bruno, P.M.; Lu, M.; Dennis, K.A.; Inam, H.; Moore, C.J.; Sheehe, J.; Elledge, S.J.; Hemann, M.T.; Pritchard, J.R. The primary mechanism of cytotoxicity of the chemotherapeutic agent CX-5461 is topoisomerase II poisoning. Proc. Natl. Acad. Sci. USA 2020, 117, 4053–4060. [Google Scholar] [CrossRef]
- Pan, M.; Wright, W.C.; Chapple, R.H.; Zubair, A.; Sandhu, M.; Batchelder, J.E.; Huddle, B.C.; Low, J.; Blankenship, K.B.; Wang, Y.; et al. The chemotherapeutic CX-5461 primarily targets TOP2B and exhibits selective activity in high-risk neuroblastoma. Nat. Commun. 2021, 12, 6468. [Google Scholar] [CrossRef]
- Espinoza, J.A.; Zisi, A.; Kanellis, D.C.; Carreras-Puigvert, J.; Henriksson, M.; Huhn, D.; Watanabe, K.; Helleday, T.; Lindstrom, M.S.; Bartek, J. The antimalarial drug amodiaquine stabilizes p53 through ribosome biogenesis stress, independently of its autophagy-inhibitory activity. Cell Death Differ. 2020, 27, 773–789. [Google Scholar] [CrossRef] [Green Version]
- Anikin, L.; Pestov, D.G. 9-Aminoacridine Inhibits Ribosome Biogenesis by Targeting Both Transcription and Processing of Ribosomal RNA. Int. J. Mol. Sci. 2022, 23, 1260. [Google Scholar] [CrossRef]
- Peltonen, K.; Colis, L.; Liu, H.; Trivedi, R.; Moubarek, M.S.; Moore, H.M.; Bai, B.; Rudek, M.A.; Bieberich, C.J.; Laiho, M. A targeting modality for destruction of RNA polymerase I that possesses anticancer activity. Cancer Cell 2014, 25, 77–90. [Google Scholar] [CrossRef] [Green Version]
- Peltonen, K.; Colis, L.; Liu, H.; Jaamaa, S.; Zhang, Z.; Af Hallstrom, T.; Moore, H.M.; Sirajuddin, P.; Laiho, M. Small molecule BMH-compounds that inhibit RNA polymerase I and cause nucleolar stress. Mol. Cancer Ther. 2014, 13, 2537–2546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.T.; Chen, J.J.; Wang, H.T. Targeting RNA Polymerase I with Hernandonine Inhibits Ribosomal RNA Synthesis and Tumor Cell Growth. Mol. Cancer Res. 2019, 17, 2294–2305. [Google Scholar] [CrossRef]
- Caggiano, C.; Guida, E.; Todaro, F.; Bielli, P.; Mori, M.; Ghirga, F.; Quaglio, D.; Botta, B.; Moretti, F.; Grimaldi, P.; et al. Sempervirine inhibits RNA polymerase I transcription independently from p53 in tumor cells. Cell Death Discov. 2020, 6, 111. [Google Scholar] [CrossRef] [PubMed]
- Peltonen, K.; Colis, L.; Liu, H.; Jaamaa, S.; Moore, H.M.; Enback, J.; Laakkonen, P.; Vaahtokari, A.; Jones, R.J.; af Hallstrom, T.M.; et al. Identification of novel p53 pathway activating small-molecule compounds reveals unexpected similarities with known therapeutic agents. PLoS ONE 2010, 5, e12996. [Google Scholar] [CrossRef] [PubMed]
- Colis, L.; Peltonen, K.; Sirajuddin, P.; Liu, H.; Sanders, S.; Ernst, G.; Barrow, J.C.; Laiho, M. DNA intercalator BMH-21 inhibits RNA polymerase I independent of DNA damage response. Oncotarget 2014, 5, 4361–4369. [Google Scholar] [CrossRef] [Green Version]
- Pitts, S.; Liu, H.; Ibrahim, A.; Garg, A.; Felgueira, C.M.; Begum, A.; Fan, W.; Teh, S.; Low, J.Y.; Ford, B.; et al. Identification of an E3 ligase that targets the catalytic subunit of RNA polymerase I upon transcription stress. J. Biol. Chem. 2022, 102690. [Google Scholar] [CrossRef]
- Low, J.Y.; Sirajuddin, P.; Moubarek, M.; Agarwal, S.; Rege, A.; Guner, G.; Liu, H.; Yang, Z.; De Marzo, A.M.; Bieberich, C.; et al. Effective targeting of RNA polymerase I in treatment-resistant prostate cancer. Prostate 2019, 79, 1837–1851. [Google Scholar] [CrossRef]
- Colis, L.; Ernst, G.; Sanders, S.; Liu, H.; Sirajuddin, P.; Peltonen, K.; DePasquale, M.; Barrow, J.C.; Laiho, M. Design, synthesis, and structure-activity relationships of pyridoquinazolinecarboxamides as RNA polymerase I inhibitors. J. Med. Chem. 2014, 57, 4950–4961. [Google Scholar] [CrossRef] [Green Version]
- Dorado, T.E.; de Leon, P.; Begum, A.; Liu, H.; Chen, D.; Rajeshkumar, N.V.; Rey-Rodriguez, R.; Hoareau-Aveilla, C.; Alcouffe, C.; Laiho, M.; et al. Discovery and Evaluation of Novel Angular Fused Pyridoquinazolinonecarboxamides as RNA Polymerase I Inhibitors. ACS Med. Chem. Lett. 2022, 13, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Morgado-Palacin, L.; Llanos, S.; Urbano-Cuadrado, M.; Blanco-Aparicio, C.; Megias, D.; Pastor, J.; Serrano, M. Non-genotoxic activation of p53 through the RPL11-dependent ribosomal stress pathway. Carcinogenesis 2014, 35, 2822–2830. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.L.; Zhao, C.L.; Chen, Q.; Xu, K.; Qiao, X.; Xu, J.Y. Targeting RNA polymerase I transcription machinery in cancer cells by a novel monofunctional platinum-based agent. Eur. J. Med. Chem. 2018, 155, 434–444. [Google Scholar] [CrossRef]
- Jacobs, R.Q.; Huffines, A.K.; Laiho, M.; Schneider, D.A. The small-molecule BMH-21 directly inhibits transcription elongation and DNA occupancy of RNA polymerase I in vivo and in vitro. J. Biol. Chem. 2022, 298, 101450. [Google Scholar] [CrossRef]
- Jacobs, R.Q.; Fuller, K.B.; Cooper, S.L.; Carter, Z.I.; Laiho, M.; Lucius, A.L.; Schneider, D.A. RNA polymerase I is uniquely vulnerable to the small-molecule inhibitor BMH-21. Cancers 2022, 14, 5544. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Agent | Drug class | DNA Damage | Clinical/Preclinical | Effect on Pol I Stability | Ref. |
|---|---|---|---|---|---|
| Actinomycin D | DNA intercalator | Yes | Clinical | No effect | [179] |
| Amodiaquine | DNA intercalator | No | Clinical | Destabilization | [204] |
| 9-aminoacridine | DNA intercalator | No | Clinical (topical) | NA * | [205] |
| BMH-21 | DNA intercalator | No | Preclinical | Destabilization | [206] |
| BMH-9, -22, -23 | DNA intercalator | No | Preclinical | Destabilization | [207] |
| Cisplatin | DNA crosslinker | Yes | Clinical | No effect | [179] |
| CX-5461 | TOP2 inhibitor/G4-stabilizer | Yes | Clinical trials (I/II) | No effect | [199,200,202,203] |
| CX-3543 | G4-stabilizer | NA | Clinical trials (I/II) | No effect | [195] |
| T5 | G4-stabilizer | NA | Preclinical | Destabilization | [194] |
| Doxorubicin | TOP2 inhibitor | Yes | Clinical | No effect | [179] |
| Ellipticine | TOP1/2 inhibitor | Yes | Preclinical | NA | [191] |
| Hernandonine | Alkaloid | No | Preclinical | Destabilization | [208] |
| Mitoxantrone | TOP2 inhibitor | Yes | Clinical | No effect | [179] |
| Mitomycin C | DNA intercalator | Yes | Clinical | NA | [179] |
| Oxaliplatin | DNA crosslinker | Yes | Clinical | No effect | [183] |
| Sempervirine | Nucleic acid binding | No | Preclinical | Destabilization | [209] |
| Topotecan | Top1 inhibitor | Yes | Clinical | No effect | [179] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pitts, S.; Laiho, M. Regulation of RNA Polymerase I Stability and Function. Cancers 2022, 14, 5776. https://doi.org/10.3390/cancers14235776
Pitts S, Laiho M. Regulation of RNA Polymerase I Stability and Function. Cancers. 2022; 14(23):5776. https://doi.org/10.3390/cancers14235776
Chicago/Turabian StylePitts, Stephanie, and Marikki Laiho. 2022. "Regulation of RNA Polymerase I Stability and Function" Cancers 14, no. 23: 5776. https://doi.org/10.3390/cancers14235776
APA StylePitts, S., & Laiho, M. (2022). Regulation of RNA Polymerase I Stability and Function. Cancers, 14(23), 5776. https://doi.org/10.3390/cancers14235776