

The Cytotoxic Effects of Cannabidiol and Cannabigerol on Glioblastoma Stem Cells May Mostly Involve GPR55 and TRPV1 Signalling

 ,
,  ,
,  , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cannabinoids
2.2. Tissue Samples and Cell Lines
2.3. Gene Expression Analysis
2.4. Cell Viability Assay
2.5. Statistical Methods
3. Results
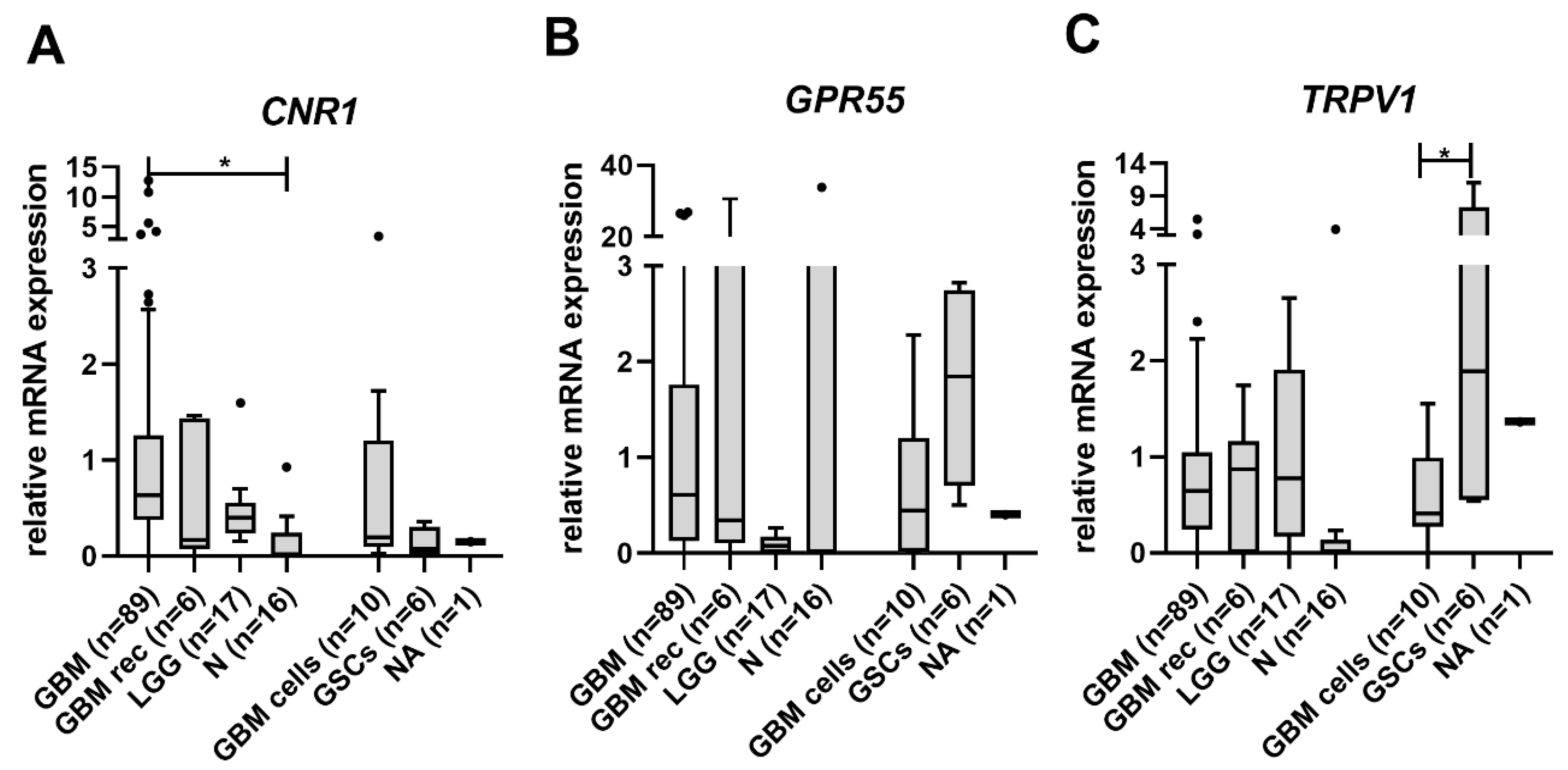
3.1. CNR1 mRNA Levels Are Increased in GBM Tissue Samples
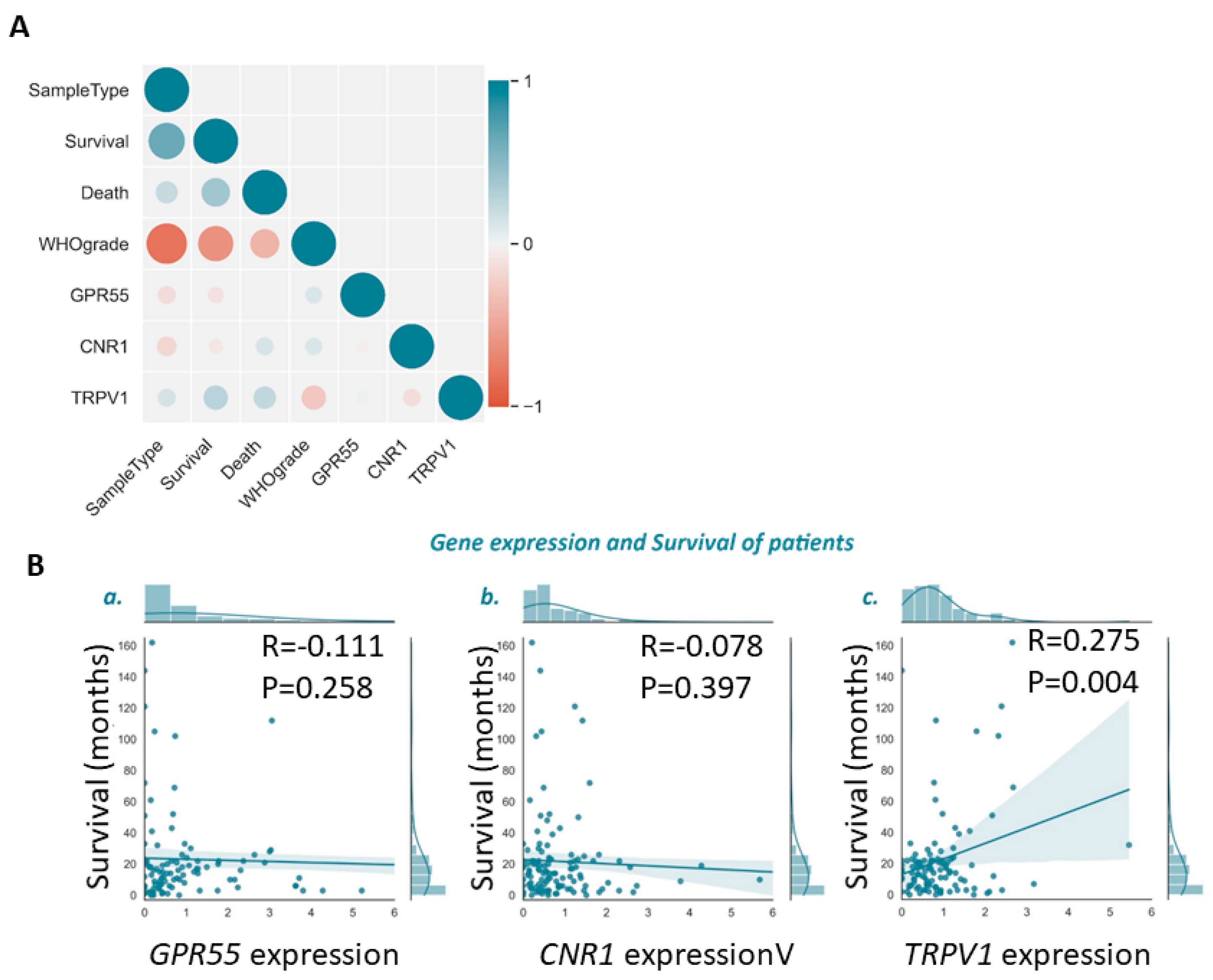
3.2. Lower TRPV1 mRNA Levels Indicate Shorter Glioma Patients’ Survival
3.3. Cannabinoid Receptors Expression and Association with Survival of GBM Patient Survival
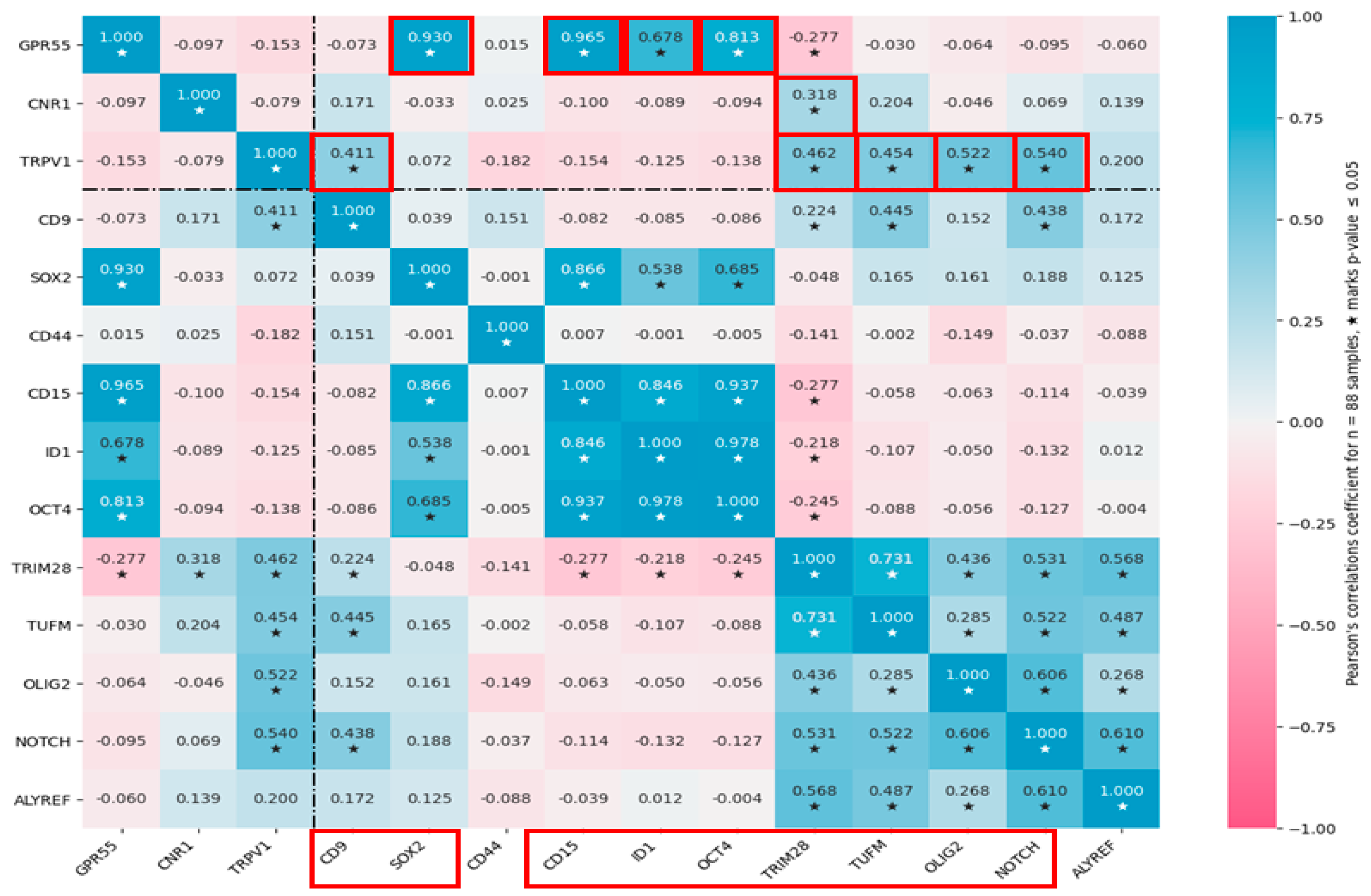
3.4. Cannabinoid Receptors Expression Correlate with GSC Biomarkers Expression in GBM Tissues
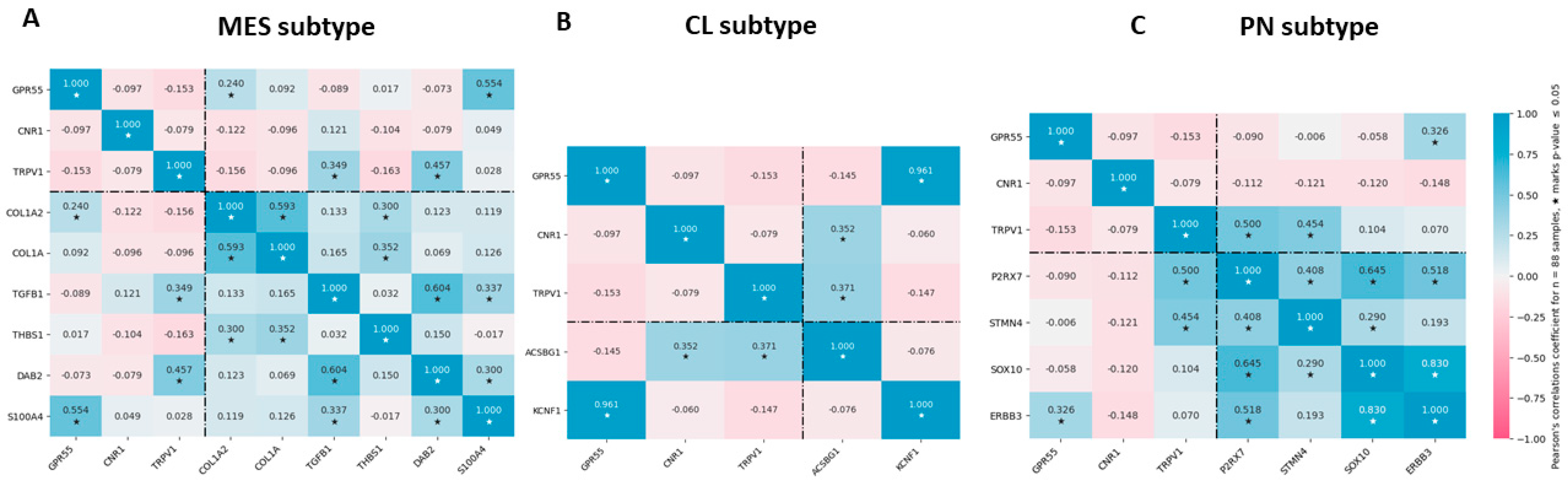
3.5. Association of Cannabinoid Receptor Expression with MES, PN, and CL GBM Subtype Markers
3.6. The Cannabinoids CBG and CBD Affect the Viability of Primary GBM Cells and GSCs
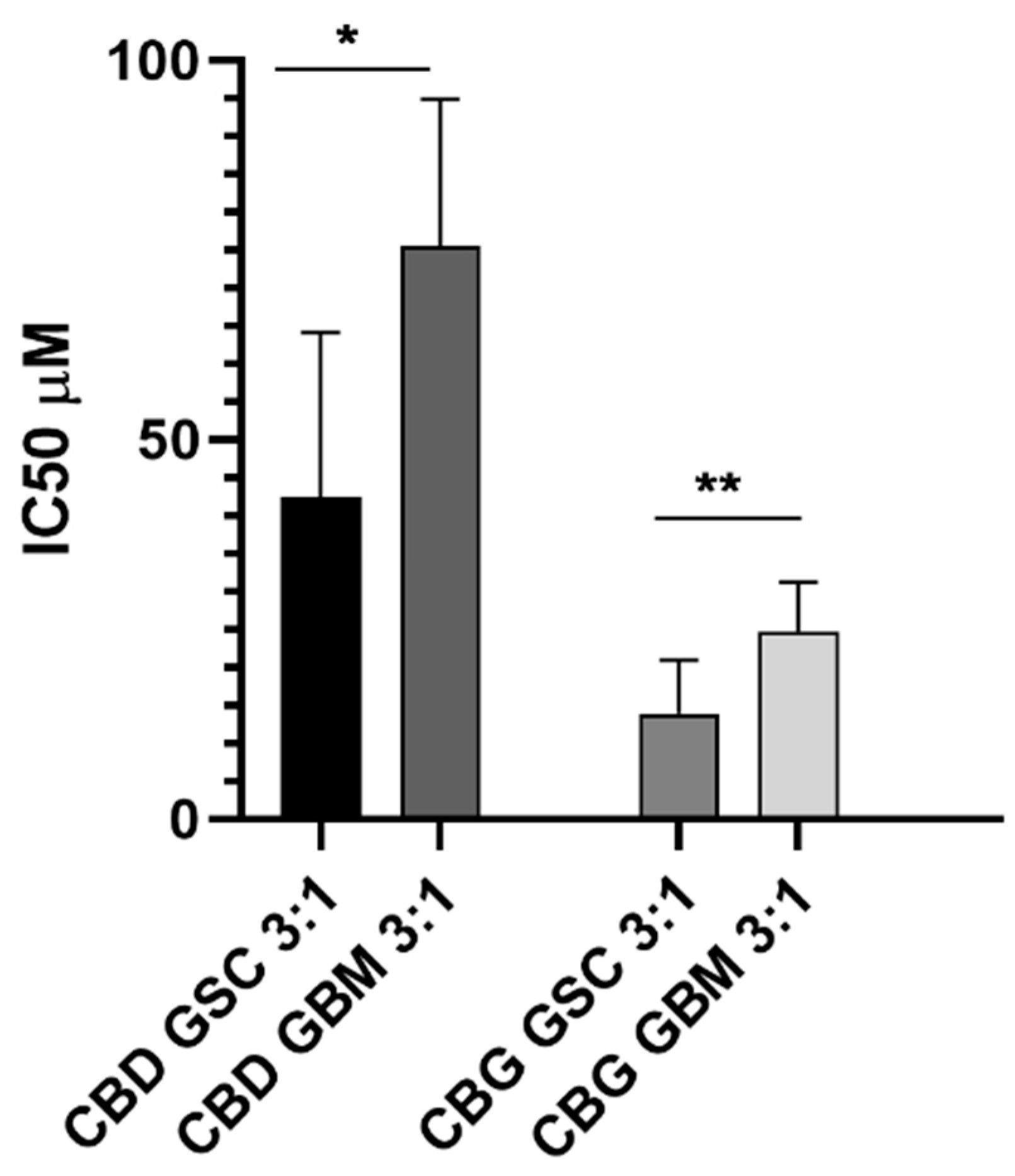
3.7. CBD and CBG Cytotoxic Effects Alone and at the Molar Ratio of 3:1 on GBM and GSC Cells
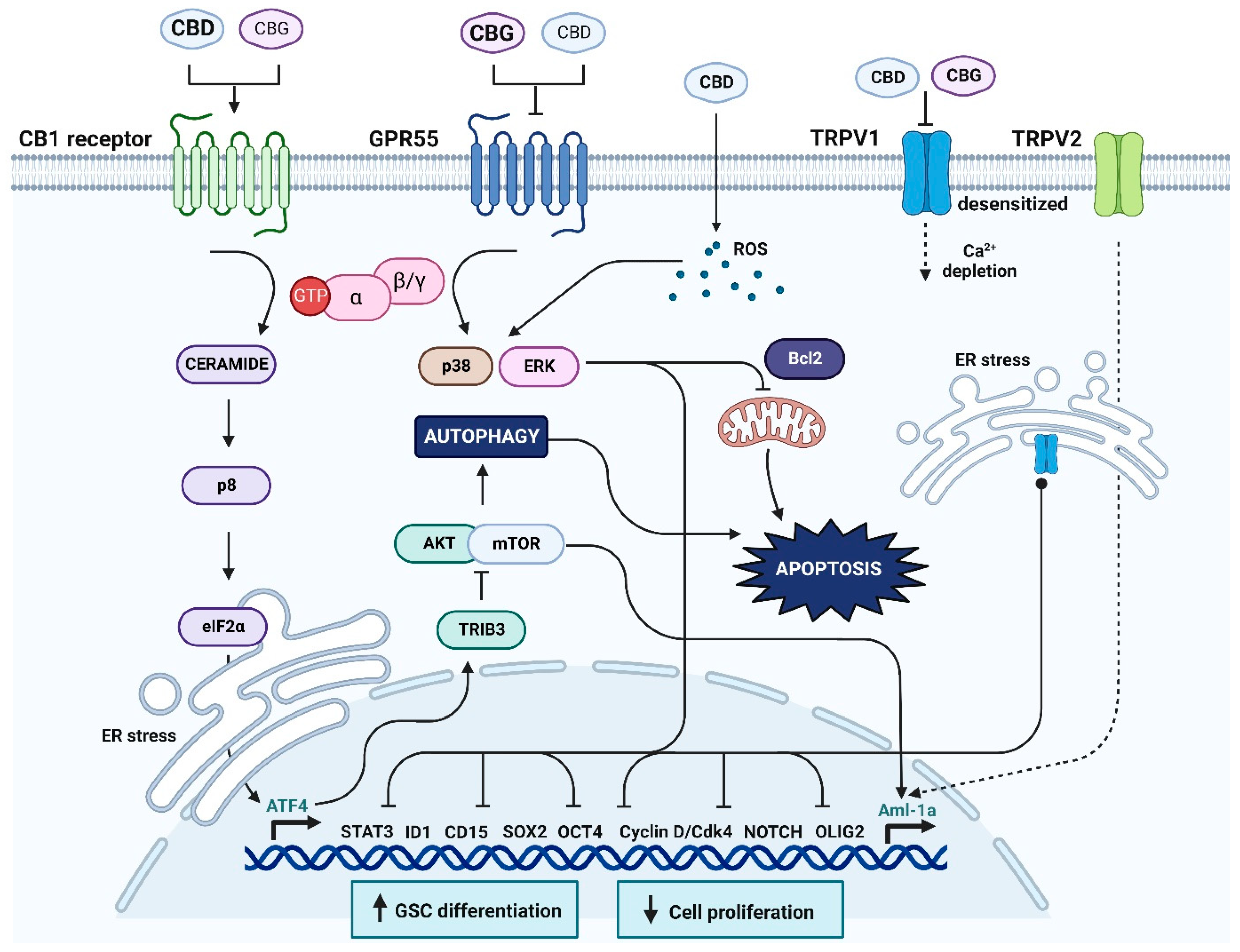
4. Discussion
4.1. Specific Cannabinoid Receptors in Glioma Progression
4.2. The Expression and Potential Role of GPR55 in Glioma
4.3. The Expression and Potential Role of Non-Selective TRPV1 in Glioma
4.4. Cannabinoids CBD and CBG Inhibit Viability of Glioblastoma Stem Cells
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACSBG1 | Acyl-CoA Synthetase Bubblegum Family Member 1 |
| AIF1 | Allograft Inflammatory Factor 1 |
| ALYREF | Aly/REF Export Factor |
| AML-1 | Acute Myeloid Leukemia 1 |
| ATCC | American Type Cell Collection |
| ATF3 | Activating Transcription Factor 3 |
| ATF4 | Activating Transcription Factor 4 |
| BCL2 | B-cell Lymphoma 2 |
| BE | Base Emulsion |
| bFGF | Beta Fibroblast Growth Factor |
| CAPSAICIN | Trans-8-methyl-N-vanillyl-6-nonenamide |
| CB1 | Cannabinoid Receptor Type 1 |
| CB2 | Cannabinoid Receptor Type 2 |
| CBD | Cannabidiol |
| CBG | Cannabigerol |
| CCA | Cholangiocarcinoma |
| CCL5 | Chemokine (C-C motif) ligand 5 |
| CCR3 | C-C Motif Chemokine Receptor 3 |
| CCR5 | C-C Motif Chemokine Receptor 5 |
| CD15 | Lewis X Antigen |
| CD44 | CD44 molecule |
| CD9 | CD9 Antigen |
| CDH1 | Cadherin 1 |
| CEBPA | CCAAT Enhancer Binding Protein Alpha |
| CHI3L1 | Chitinase-3-like protein 1; YKL-40 |
| CL | Classical |
| CLS | Cell Lines Service |
| CNR1 | Cannabinoid Receptor 1 gene |
| CNR2 | Cannabinoid Receptor 2 gene |
| CNRs | Cannabinoid Receptors |
| COL1A1 | Collagen Type I Alpha 1 Chain |
| COL1A2 | Collagen Type I Alpha 2 Chain |
| CSCs | Cancer Stem Cells |
| CST7 | Cystatin F gene |
| DAB2 | DAB Adaptor Protein 2 |
| DMEM | Dulbecco’s Modified Eagle Medium |
| DMSO | Dimethyl sulfoxide |
| DPYSL2 | Dihydropyrimidinase Like 2 |
| eCB | Endogenous Cannabinoid-like Ligands |
| ECS | Endocannabinoids System |
| EGF | Epidermal Growth Factor |
| EGFR | Epidermal growth factor receptor |
| eIF2α | Eukaryotic Initiation Factor 2 alpha |
| eIFKA3 | Eukaryotic Translation Initiation Factor 2-alpha kinase 3 |
| EMT | Epithelial-to-Mesenchymal Transition |
| ER | Endoplasmic Reticulum |
| ERBB3 | Erb-B2 Receptor Tyrosine Kinase 3 |
| EtOH | Ethanol |
| FBS | Foetal Bovine Serum |
| FREM2 | FRAS1 Related Extracellular Matrix 2 |
| FUT4 | Fucosyltransferase 4 |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
| GBM | Glioblastoma |
| GBM rec | Recurrent GBM |
| GFAP | Glial Fibrillary Acidic Protein |
| GPCRs | G protein-Coupled Receptors |
| GPR55 | G Protein-Coupled Receptor 55 |
| GSC | GBM stem cell |
| HLH | helix-loop-helix |
| HPRT1 | Hypoxanthine Phosphoribosyltransferase 1 |
| ID1 | Inhibitor of DNA Binding 1 |
| KCNF1 | Potassium Voltage-Gated Channel Modifier Subfamily F Member 1 |
| LGG | Low Grade Glioma |
| MAPK/ERK | Mitogen-activated protein kinases/extracellular signal-regulated kinases |
| MES | Mesenchymal |
| mRNA | Messenger RNA |
| MTS | 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium salt |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium-bromide |
| N | Non-malignant brain tissues |
| NA | Normal Astrocytes |
| NF-kB | Nuclear factor-κB |
| NFKB1 | Nuclear Factor Kappa B Subunit 1 gene |
| NIB | National Institute of Biology |
| NOTCH1 | Neurogenic locus notch homolog protein 1 |
| NSCs | Neural Stem Cells |
| OCT4 | Octamer-binding transcription factor 4 |
| OLIG2 | Oligodendrocyte Transcription Factor 2 |
| P2RX7 | Purinergic Receptor P2X 7 |
| PDGFR | Platelet-derived growth factor receptor |
| PI3K/Akt | Phosphoinositide-3-kinase/Protein kinase B |
| PN | Proneural |
| POU5F1B | POU Class 5 Homeobox 1B; OCT4-PG1 (OCT4 pseudogene) |
| PROM1 | prominin-1; CD133 antigen |
| ROS | Reactive Oxygen Species |
| S100A4 | S100 Calcium Binding Protein A4 |
| SNAI1 | Snail Family Transcriptional Repressor 1 |
| SOX10 | SRY-Box Transcription Factor 10 |
| SOX2 | SRY-Box Transcription Factor 2 |
| SPRY | Sprouty RTK Signaling Antagonist 1 |
| STAT3 | Signal Transducer And Activator Of Transcription 3 |
| STMN4 | Stathmin 4 |
| TCGA | The Cancer Genome Atlas |
| TGFB1 | Transforming growth factor beta 1 gene |
| TGFβ | Transforming growth factor beta |
| THBS1 | Thrombospondin 1 gene |
| THC | Δ9-Tetrahydrocannabinol |
| TMZ | Temozolomide |
| TRIB3 | Trible Homologue 3 |
| TRIM28 | Tripartite Motif Containing 28 |
| TRPs | Transient Receptor Potential Channels |
| TRPV1 | Transient Receptor Potential Cation Channel Subfamily V Member 1 |
| TRPV2 | Transient Receptor Potential Cation Channel Subfamily V Member 2 |
| TUBB3 | Tubulin Beta 3 Class III |
| TUFM | Tu Translation Elongation Factor, Mitochondrial |
| VIM | Vimentin |
References
- IARC: Home. Available online: https://www.iarc.who.int/ (accessed on 25 September 2022).
- World Cancer Report—IARC. Available online: https://www.iarc.who.int/world-cancer-report-content-overview/ (accessed on 25 September 2022).
- Philips, A.; Henshaw, D.L.; Lamburn, G.; O’Carroll, M.J. Brain tumours: Rise in glioblastoma multiforme incidence in England 1995-2015 Suggests an Adverse Environmental or Lifestyle Factor. J. Environ. Public Health 2018, 2018, 7910754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; van den Bent, M.; Preusser, M.; le Rhun, E.; Tonn, J.C.; Minniti, G.; Bendszus, M.; Balana, C.; Chinot, O.; Dirven, L.; et al. EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nat. Rev. Clin. Oncol. 2021, 18, 170–186. [Google Scholar] [CrossRef]
- Park, J.; Shim, J.K.; Yoon, S.J.; Kim, S.H.; Chang, J.H.; Kang, S.G. Transcriptome profiling-based identification of prognostic subtypes and multi-omics signatures of glioblastoma. Sci. Rep. 2019, 9, 10555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.M.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavare, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gimple, R.C.; Yang, K.; Halbert, M.E.; Agnihotri, S.; Rich, J.N. Brain cancer stem cells: Resilience through adaptive plasticity and hierarchical heterogeneity. Nat. Rev. Cancer 2022, 22, 497–514. [Google Scholar] [CrossRef] [PubMed]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef] [Green Version]
- Biserova, K.; Jakovlevs, A.; Uljanovs, R.; Strumfa, I. Cancer Stem Cells: Significance in Origin, Pathogenesis and Treatment of Glioblastoma. Cells 2021, 10, 621. [Google Scholar] [CrossRef]
- Alves, T.R.; Lima, F.R.S.; Kahn, S.A.; Lobo, D.; Dubois, L.G.F.; Soletti, R.; Borges, H.; Neto, V.M. Glioblastoma cells: A heterogeneous and fatal tumor interacting with the parenchyma. Life Sci. 2011, 89, 532–539. [Google Scholar] [CrossRef] [Green Version]
- Aguado, T.; Carracedo, A.; Julien, B.; Velasco, G.; Milman, G.; Mechoulamluis, R.; Alvarez, L.; Guzmán, M.; Galve-Roperh, I. Cannabinoids induce glioma stem-like cell differentiation and inhibit gliomagenesis. J. Biol. Chem. 2007, 282, 6854–6862. [Google Scholar] [CrossRef]
- Costas-Insua, C.; Guzmán, M. Endocannabinoid signaling in glioma. Glia 2022, 71, 127–138. [Google Scholar] [CrossRef]
- Nabissi, M.; Morelli, M.B.; Amantini, C.; Liberati, S.; Santoni, M.; Ricci-Vitiani, L.; Pallini, R.; Santoni, G. Cannabidiol stimulates AML-1a-dependent glial differentiation and inhibits glioma stem-like cells proliferation by inducing autophagy in a TRPV2-dependent manner. Int. J. Cancer 2015, 137, 1855–1869. [Google Scholar] [CrossRef] [Green Version]
- Dumitru, C.A.; Sandalcioglu, I.E.; Karsak, M. Cannabinoids in glioblastoma therapy: New applications for old drugs. Front. Mol. Neurosci. 2018, 11, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- dos Reis Rosa Franco, G.; Smid, S.; Viegas, C. Phytocannabinoids: General Aspects and Pharmacological Potential in Neurodegenerative Diseases. Curr. Neuropharmacol. 2020, 19, 449–464. [Google Scholar] [CrossRef]
- Deiana, S. Potential Medical Uses of Cannabigerol: A Brief Overview. In Handbook of Cannabis and Related Pathologies: Biology, Pharmacology, Diagnosis, and Treatment; Elsevier Inc.: Amsterdam, The Netherlands, 2017. [Google Scholar] [CrossRef]
- Ligresti, A.; de Petrocellis, L.; di Marzo, V. From Phytocannabinoids to Cannabinoid Receptors and Endocannabinoids: Pleiotropic Physiological and Pathological Roles Through Complex Pharmacology. Physiol. Rev. 2016, 96, 1593–1659. [Google Scholar] [CrossRef] [Green Version]
- Moreno, E.; Cavic, M.; Krivokuca, A.; Canela, E.I. The Interplay between Cancer Biology and the Endocannabinoid System-Significance for Cancer Risk, Prognosis and Response to Treatment. Cancers 2020, 12, 3275. [Google Scholar] [CrossRef]
- O’Reilly, E.M.; Cosgrave, J.M.; Gallagher, W.M.; Perry, A.S. Plant-derived cannabinoids as anticancer agents. Trends Cancer 2022, 8, 350–357. [Google Scholar] [CrossRef]
- Held-Feindt, J.; Dörner, L.; Sahan, G.; Mehdorn, H.M.; Mentlein, R. Cannabinoid receptors in human astroglial tumors. J. Neurochem. 2006, 98, 886–893. [Google Scholar] [CrossRef]
- Nahler, G. Cannabidiol and Other Phytocannabinoids as Cancer Therapeutics. Pharm. Med. 2022, 36, 99–129. [Google Scholar] [CrossRef]
- Kolbe, M.R.; Hohmann, T.; Hohmann, U.; Ghadban, C.; Mackie, K.; Zöller, C.; Prell, J.; Illert, J.; Strauss, C.; Dehghani, F. THC Reduces Ki67-Immunoreactive Cells Derived from Human Primary Glioblastoma in a GPR55-Dependent Manner. Cancers 2021, 13, 1064. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Lyu, Y.; Wang, H. TRP Channels as Molecular Targets to Relieve Endocrine-Related Diseases. Front. Mol. Biosci. 2022, 9, 391. [Google Scholar] [CrossRef] [PubMed]
- De Petrocellis, L.; Nabissi, M.; Santoni, G.; Ligresti, A. Actions and Regulation of Ionotropic Cannabinoid Receptors. Adv. Pharmacol. 2017, 80, 249–289. [Google Scholar] [CrossRef] [PubMed]
- Zhai, K.; Liskova, A.; Kubatka, P.; Büsselberg, D. Calcium Entry through TRPV1: A Potential Target for the Regulation of Proliferation and Apoptosis in Cancerous and Healthy Cells. Int. J. Mol. Sci. 2020, 21, 4177. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Chen, C.; Xiang, Q.; Fan, S.; Xiao, T.; Chen, Y.; Zheng, D. Transient Receptor Potential Cation Channel Subfamily V Member 1 Expression Promotes Chemoresistance in Non-Small-Cell Lung Cancer. Front. Oncol. 2022, 12, 773654. [Google Scholar] [CrossRef]
- de Almeida, A.S.; de Barros Bernardes, L.; Trevisan, G. TRP channels in cancer pain. Eur. J. Pharmacol. 2021, 904, 174185. [Google Scholar] [CrossRef]
- Stock, K.; Kumar, J.; Synowitz, M.; Petrosino, S.; Imperatore, R.; Smith, E.S.J.; Wend, P.; Purfürst, B.; Nuber, U.A.; Gurok, U.; et al. Neural precursor cells induce cell death of high-grade astrocytomas through stimulation of TRPV1. Nat. Med. 2012, 18, 1232. [Google Scholar] [CrossRef] [Green Version]
- Valeri, A.; Mazzon, E. Cannabinoids and Neurogenesis: The Promised Solution for Neurodegeneration? Molecules 2021, 26, 6313. [Google Scholar] [CrossRef]
- Pagano, C.; Navarra, G.; Coppola, L.; Bifulco, M.; Laezza, C. Molecular Mechanism of Cannabinoids in Cancer Progression. Int. J. Mol. Sci. 2021, 22, 3680. [Google Scholar] [CrossRef]
- Walsh, K.B.; McKinney, A.E.; Holmes, A.E. Minor Cannabinoids: Biosynthesis, Molecular Pharmacology and Potential Therapeutic Uses. Front. Pharmacol. 2021, 12, 3366. [Google Scholar] [CrossRef] [PubMed]
- Lah, T.T.; Novak, M.; Pena Almidon, M.A.; Marinelli, O.; Žvar Baškovič, B.; Majc, B.; Mlinar, M.; Bošnjak, R.; Breznik, B.; Zomer, R.; et al. Cannabigerol Is a Potential Therapeutic Agent in a Novel Combined Therapy for Glioblastoma. Cells 2021, 10, 340. [Google Scholar] [CrossRef] [PubMed]
- Novak, M.; Krajnc, M.K.; Hrastar, B.; Breznik, B.; Majc, B.; Mlinar, M.; Rotter, A.; Porčnik, A.; Mlakar, J.; Stare, K.; et al. CCR5-mediated signaling is involved in invasion of glioblastoma cells in its microenvironment. Int. J. Mol. Sci. 2020, 21, 4199. [Google Scholar] [CrossRef] [PubMed]
- Peeri, H.; Koltai, H. Cannabis Biomolecule Effects on Cancer Cells and Cancer Stem Cells: Cytotoxic, Anti-Proliferative, and Anti-Migratory Activities. Biomolecules 2022, 12, 491. [Google Scholar] [CrossRef] [PubMed]
- Ramšak, Ž.; Coll, A.; Stare, T.; Tzfadia, O.; Baebler, Š.; Van de Peer, Y.; Gruden, K. Network Modeling Unravels Mechanisms of Crosstalk between Ethylene and Salicylate Signaling in Potato. Plant Physiol. 2018, 178, 488–499. [Google Scholar] [CrossRef] [Green Version]
- Behnan, J.; Stangeland, B.; Hosainey, S.A.M.; Joel, M.; Olsen, T.K.; Micci, F.; Glover, J.C.; Isakson, P.; Brinchmann, J.E. Differential propagation of stroma and cancer stem cells dictates tumorigenesis and multipotency. Oncogene 2016, 36, 570–584. [Google Scholar] [CrossRef] [Green Version]
- Iozzo, M.; Sgrignani, G.; Comito, G.; Chiarugi, P.; Giannoni, E. Endocannabinoid System and Tumour Microenvironment: New Intertwined Connections for Anticancer Approaches. Cells 2021, 10, 3396. [Google Scholar] [CrossRef]
- Peeri, H.; Shalev, N.; Vinayaka, A.C.; Nizar, R.; Kazimirsky, G.; Namdar, D.; Anil, S.M.; Belausov, E.; Brodie, C.; Koltai, H. Specific Compositions of Cannabis sativa Compounds Have Cytotoxic Activity and Inhibit Motility and Colony Formation of Human Glioblastoma Cells In Vitro. Cancers 2021, 13, 1720. [Google Scholar] [CrossRef]
- Brown, A.J. Novel cannabinoid receptors. Br. J. Pharmacol. 2007, 152, 567–575. [Google Scholar] [CrossRef] [Green Version]
- Andradas, C.; Caffarel, M.M.; Pérez-Gómez, E.; Salazar, M.; Lorente, M.; Velasco, G.; Guzmán, M.; Sánchez, C. The orphan G protein-coupled receptor GPR55 promotes cancer cell proliferation via ERK. Oncogene 2011, 30, 245–252. [Google Scholar] [CrossRef]
- Navarro, G.; Varani, K.; Lillo, A.; Vincenzi, F.; Rivas-Santisteban, R.; Raïch, I.; Reyes-Resina, I.; Ferreiro-Vera, C.; Borea, P.A.; Sánchez de Medina, V.; et al. Pharmacological data of cannabidiol- and cannabigerol-type phytocannabinoids acting on cannabinoid CB1, CB2 and CB1/CB2 heteromer receptors. Pharmacol. Res. 2020, 159, 104940. [Google Scholar] [CrossRef]
- Navarro, G.; Cordomí, A.; Brugarolas, M.; Moreno, E.; Aguinaga, D.; Pérez-Benito, L.; Ferre, S.; Cortés, A.; Casadó, V.; Mallol, J.; et al. Cross-communication between Gi and Gs in a G-protein-coupled receptor heterotetramer guided by a receptor C-terminal domain. BMC Biol. 2018, 16, 24. [Google Scholar] [CrossRef] [PubMed]
- Singer, E.; Judkins, J.; Salomonis, N.; Matlaf, L.; Soteropoulos, P.; McAllister, S.; Soroceanu, L. Reactive oxygen species-mediated therapeutic response and resistance in glioblastoma. Cell Death Dis. 2015, 6, e1601. [Google Scholar] [CrossRef] [Green Version]
- Viereckl, M.J.; Krutsinger, K.; Apawu, A.; Gu, J.; Cardona, B.; Barratt, D.; Han, Y. Cannabidiol and Cannabigerol Inhibit Cholangiocarcinoma Growth In Vitro via Divergent Cell Death Pathways. Biomolecules 2022, 12, 854. [Google Scholar] [CrossRef]
- López-Valero, I.; Saiz-Ladera, C.; Torres, S.; Hernández-Tiedra, S.; García-Taboada, E.; Rodríguez-Fornés, F.; Barba, M.; Dávila, D.; Salvador-Tormo, N.; Guzmán, M.; et al. Targeting Glioma Initiating Cells with A combined therapy of cannabinoids and temozolomide. Biochem. Pharmacol. 2018, 157, 266–274. [Google Scholar] [CrossRef]
- Cui, C.; Merritt, R.; Fu, L.; Pan, Z. Targeting calcium signaling in cancer therapy. Acta Pharm. Sin. B 2017, 7, 3–17. [Google Scholar] [CrossRef]
- Bode, A.M.; Cho, Y.Y.; Zheng, D.; Zhu, F.; Ericson, M.E.; Ma, W.Y.; Yao, K.; Dong, Z. Transient receptor potential type vanilloid 1 suppresses skin carcinogenesis. Cancer Res. 2009, 69, 905–913. [Google Scholar] [CrossRef] [Green Version]
- De Jong, P.R.; Takahashi, N.; Harris, A.R.; Lee, J.; Bertin, S.; Jeffries, J.; Jung, M.; Duong, J.; Triano, A.I.; Lee, J.; et al. Ion channel TRPV1-dependent activation of PTP1B suppresses EGFR-associated intestinal tumorigenesis. J. Clin. Investig. 2014, 124, 3793–3806. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Guo, W.; Ma, J.; Xu, P.; Zhang, W.; Guo, S.; Liu, L.; Ma, J.; Shi, Q.; Jian, Z.; et al. Downregulated TRPV1 Expression Contributes to Melanoma Growth via the Calcineurin-ATF3-p53 Pathway. J. Investig. Dermatol. 2018, 138, 2205–2215. [Google Scholar] [CrossRef] [Green Version]
- Starkus, J.; Jansen, C.; Shimoda, L.M.N.; Stokes, A.J.; Small-Howard, A.L.; Turner, H. Diverse TRPV1 responses to cannabinoids. Channels 2019, 13, 172. [Google Scholar] [CrossRef]
- Podergajs, N.; Motaln, H.; Rajčević, U.; Verbovšek, U.; Koršič, M.; Obad, N.; Espedal, H.; Vittori, M.; Herold-mende, C.; Miletic, H.; et al. Transmembrane protein CD9 is glioblastoma biomarker, relevant for maintenance of glioblastoma stem cells. Oncotarget 2015, 7, 593–609. [Google Scholar] [CrossRef] [Green Version]
- Porčnik, A.; Novak, M.; Breznik, B.; Majc, B.; Hrastar, B.; Šamec, N.; Zottel, A.; Jovčevska, I.; Vittori, M.; Rotter, A.; et al. TRIM28 Selective Nanobody Reduces Glioblastoma Stem Cell Invasion. Molecules 2021, 26, 5141. [Google Scholar] [CrossRef] [PubMed]
- Jovčevska, I.; Zupanec, N.; Kočevar, N.; Cesselli, D.; Podergajs, N.; Stokin, C.L.; Myers, M.P.; Muyldermans, S.; Ghassabeh, G.H.; Motaln, H.; et al. TRIM28 and β-actin identified via nanobody-based reverse proteomics approach as possible human glioblastoma biomarkers. PLoS ONE 2014, 9, e113688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majc, B.; Sever, T.; Zarić, M.; Breznik, B.; Turk, B.; Lah, T.T. Epithelial-to-mesenchymal transition as the driver of changing carcinoma and glioblastoma microenvironment. Biochim. Biophys. Acta-Mol. Cell Res. 2020, 1867, 118782. [Google Scholar] [CrossRef] [PubMed]
- Aguado, T.; Palazuelos, J.; Monory, K.; Stella, N.; Cravatt, B.; Lutz, B.; Marsicano, G.; Kokaia, Z.; Guzmán, M.; Galve-Roperh, I. The endocannabinoid system promotes astroglial differentiation by acting on neural progenitor cells. J. Neurosci. 2006, 26, 1551–1561. [Google Scholar] [CrossRef] [Green Version]
- Stock, K.; Garthe, A.; de Almeida Sassi, F.; Glass, R.; Wolf, S.A.; Kettenmann, H. The capsaicin receptor TRPV1 as a novel modulator of neural precursor cell proliferation. Stem Cells 2014, 32, 3183–3195. [Google Scholar] [CrossRef]
- Afrin, F.; Chi, M.; Eamens, A.L.; Duchatel, R.J.; Douglas, A.M.; Schneider, J.; Gedye, C.; Woldu, A.S.; Dun, M.D. Can Hemp Help? Low-THC Cannabis and Non-THC Cannabinoids for the Treatment of Cancer. Cancers 2020, 12, 1033. [Google Scholar] [CrossRef] [Green Version]
- McAllister, S.D.; Chan, C.; Taft, R.J.; Luu, T.; Abood, M.E.; Moore, D.H.; Aldape, K.; Yount, G. Cannabinoids selectively inhibit proliferation and induce death of cultured human glioblastoma multiforme cells. J. Neuro-Oncol. 2005, 74, 31–40. [Google Scholar] [CrossRef]
- Guzmán, M.; Duarte, M.J.; Blázquez, C.; Ravina, J.; Rosa, M.C.; Galve-Roperh, I.; Sánchez, C.; Velasco, G.; González-Feria, L. A pilot clinical study of Δ9-tetrahydrocannabinol in patients with recurrent glioblastoma multiforme. Br. J. Cancer 2006, 95, 197–203. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| p-Value | |||||||
|---|---|---|---|---|---|---|---|
| Sample Type | 0 | ||||||
| Survival | <0.001 | 0 | |||||
| Death | 0.012 | <0.001 | 0 | ||||
| WHO grade | <0.001 | <0.001 | <0.001 | 0 | |||
| GPR55 | 0.144 | 0.258 | 0.903 | 0.219 | 0 | ||
| CNR1 | 0.054 | 0.397 | 0.128 | 0.187 | 0.701 | 0 | |
| TRPV1 | 0.132 | 0.004 | 0.012 | 0.004 | 0.712 | 0.164 | 0 |
| Sample Type | Survival | Death | WHO grade | GPR55 | CNR1 | TRPV1 | |
| GBM Cell Lines | IC50 Values (μM) | CBG | CBD:CBG = 3:1 |
|---|---|---|---|
| U373 1 (GB) | 99 | 61 | 68:22 |
| Primary patients derived at NIB | |||
| NIB140 (GB) | 114 | 175 | 79:27 |
| NIB142 (GB) | 71 | 53 | 80:26 |
| NIB138 (GB) | 61 | 54 | 46:15 |
| NIB180 (GB) | 83 | 127 | 98:32 |
| NIB185 (GB) | 104 | 144 | 103:34 |
| NIB182 (GB) | 47 | 156 | 95:31 |
| NIB167 (GB) | 99 | 107 | 71:23 |
| NIB258 (GB) | 68 | 83 | 65:21 |
| NIB255 (GB) | 43 | 42 | 50:16 |
| Means | 78.9 ± 7.8 | 100 ± 15.3 | 76 ± 6.6:25 ± 2.1 |
| GSC lines | |||
| NCH644 2 (GSC) | 34 | 100 | 34:11 |
| Primary patients derived at NIB | |||
| K26 (GSC) | 73 | 138 | 54:18 |
| NIB216 (GSC) | 24 | 90 | 30:10 |
| NIB237 (GSC) | 50 | 132 | 27:9 |
| NIB225 (GSC) | 50 | 34 | 28:9 |
| NIB220 (GSC) | 84 | 98 | 38:12 |
| NIB249 (GSC) | 52 | 61 | 46:15 |
| NIB253 (GSC) | 35 | 18 | 30:10 |
| Means | 50 ± 7.1 | 84 ± 15.3 | 42 ± 3.4:16 ± 1.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lah, T.T.; Majc, B.; Novak, M.; Sušnik, A.; Breznik, B.; Porčnik, A.; Bošnjak, R.; Sadikov, A.; Malavolta, M.; Halilčević, S.; et al. The Cytotoxic Effects of Cannabidiol and Cannabigerol on Glioblastoma Stem Cells May Mostly Involve GPR55 and TRPV1 Signalling. Cancers 2022, 14, 5918. https://doi.org/10.3390/cancers14235918
Lah TT, Majc B, Novak M, Sušnik A, Breznik B, Porčnik A, Bošnjak R, Sadikov A, Malavolta M, Halilčević S, et al. The Cytotoxic Effects of Cannabidiol and Cannabigerol on Glioblastoma Stem Cells May Mostly Involve GPR55 and TRPV1 Signalling. Cancers. 2022; 14(23):5918. https://doi.org/10.3390/cancers14235918
Chicago/Turabian StyleLah, Tamara T., Bernarda Majc, Metka Novak, Ajda Sušnik, Barbara Breznik, Andrej Porčnik, Roman Bošnjak, Aleksander Sadikov, Marta Malavolta, Selma Halilčević, and et al. 2022. "The Cytotoxic Effects of Cannabidiol and Cannabigerol on Glioblastoma Stem Cells May Mostly Involve GPR55 and TRPV1 Signalling" Cancers 14, no. 23: 5918. https://doi.org/10.3390/cancers14235918
APA StyleLah, T. T., Majc, B., Novak, M., Sušnik, A., Breznik, B., Porčnik, A., Bošnjak, R., Sadikov, A., Malavolta, M., Halilčević, S., Mlakar, J., & Zomer, R. (2022). The Cytotoxic Effects of Cannabidiol and Cannabigerol on Glioblastoma Stem Cells May Mostly Involve GPR55 and TRPV1 Signalling. Cancers, 14(23), 5918. https://doi.org/10.3390/cancers14235918