

Viewing AML through a New Lens: Technological Advances in the Study of Epigenetic Regulation

{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Epigenetic Regulation in the Hematopoietic System
1.1. Histone Modifications and Their Functions in the Hematopoietic System
1.2. DNA Methylation and Its Function in the Hematopoietic System
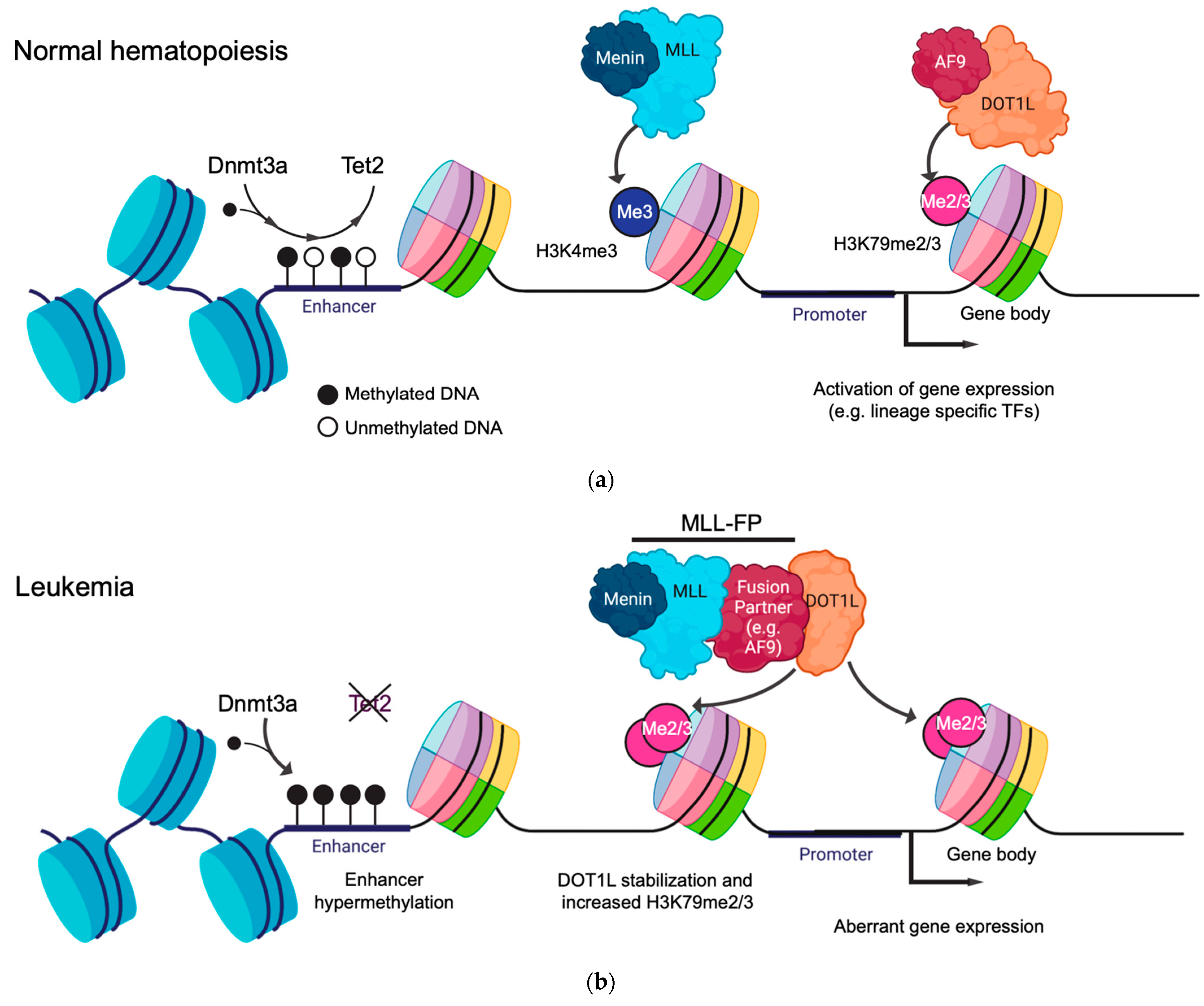
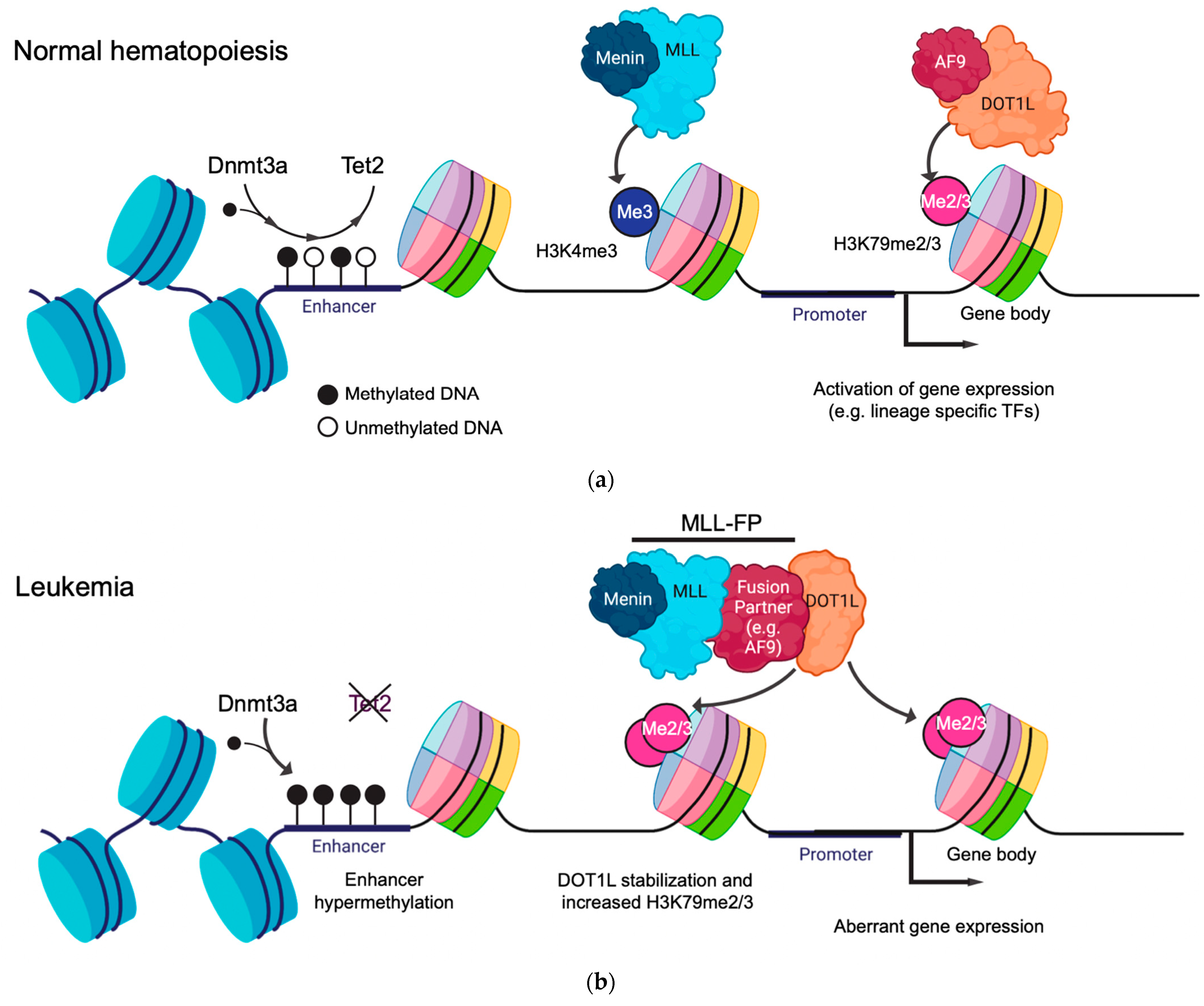
1.3. Gene Regulatory Function of Histone Modifications and DNA Methylation in Leukemia
2. New Models to Study the Role of Histone and DNA Methylation in AML
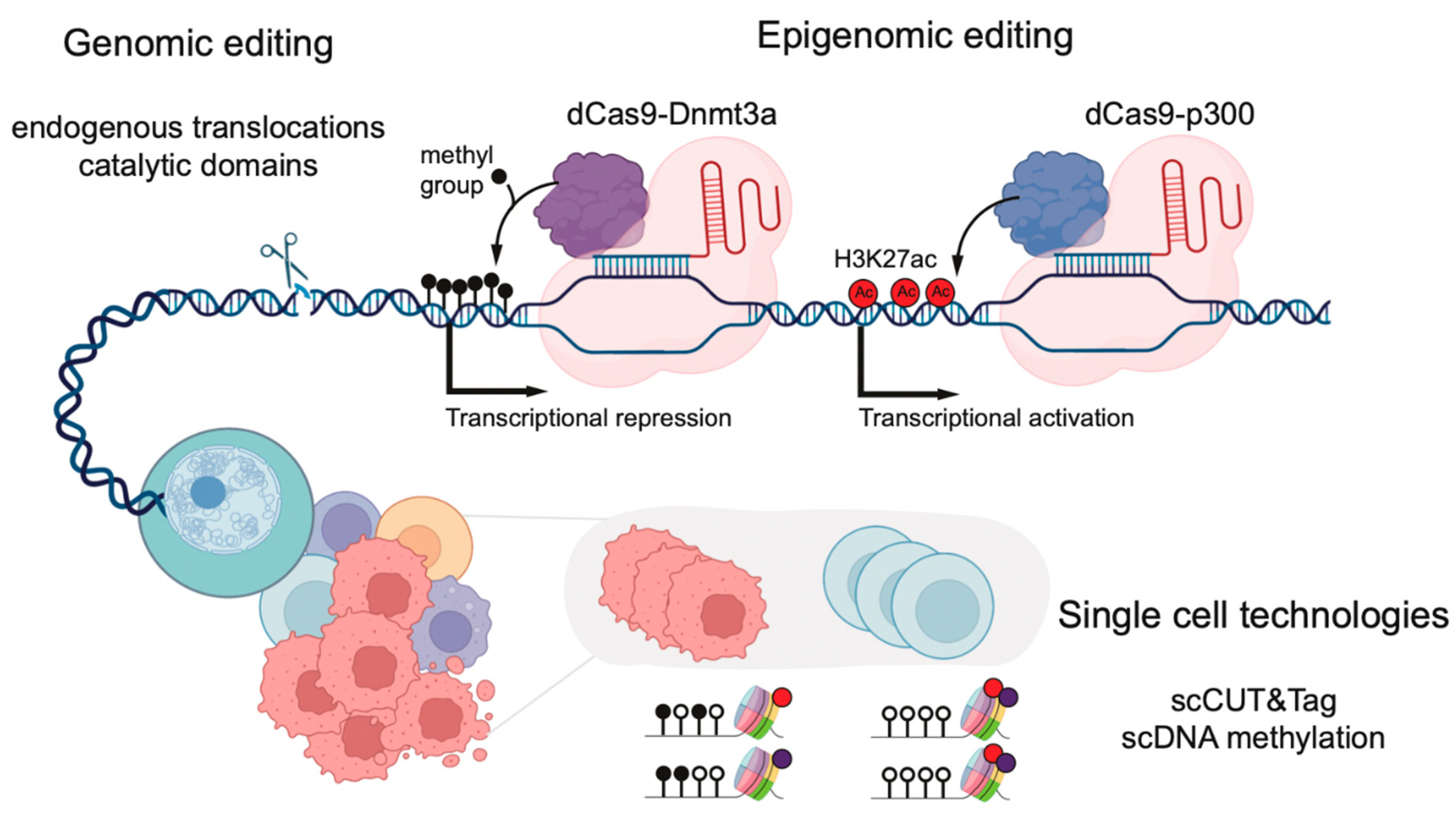
3. Epigenomic Editing
4. Single-Cell Epigenomic Technologies in Normal and Leukemic Hematopoiesis
5. Discussion
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 3D | Three-dimensional |
| 5mC | 5′-Methyl cytosine |
| AF10 | ALL1-fused gene from chromosome 10 |
| AF4 | ALL1-fused gene from chromosome 4 |
| AF9 | ALL1-fused gene from chromosome 9 |
| ALL | Acute lymphoid leukemia |
| AML | Acute myeloid leukemia |
| ASXL1 | Additional sex combs like-1 |
| ATAC-seq | Assay for transposase-accessible chromatin and sequencing |
| BER | Base excision repair |
| BS-seq | Bisulfite sequencing |
| CBP | Cyclic adenosine monophosphate response element binding protein |
| CD11B | Cluster of differentiation 11 B |
| CD34 | Cluster of differentiation 34 |
| CHIP | Clonal hematopoiesis of indeterminate potential |
| ChIP-seq | Chromatin immunoprecipitation and sequencing |
| Cre-LoxP | Cyclization recombinase, locus of x-over P1 |
| CRISPR | Clustered regularly interspaced short palindromic repeats |
| CRISPRa | CRISPR activation |
| CRISPRi | CRISPR inhibition |
| CTCF | CCCTC-binding factor |
| DNA | Deoxyribonucleic acid |
| DNMT1 | DNA methyltransferase 1 |
| DNMT3A | DNA methyltransferase 3A |
| DNMT3B | DNA methyltransferase 3B |
| DOT1L | Disrupter of telomeric silencing like-1 |
| ENL | Eleven-nineteen leukemia |
| EZH2 | Enhancer of zeste 2 (polycomb repressive complex 2 subunit) |
| GMP | Granulocyte and macrophage progenitors |
| H3K27ac | Histone 3 lysine 27 acetylation |
| H3K27M | Histone 3 lysine 27 mutant |
| H3K36me3 | Histone 3 lysine 36 trimethylation |
| H3K4me1 | Histone 3 lysine 4 monomethylation |
| H3K4me3 | Histone 3 lysine 4 trimethylation |
| H3K79me2/3 | Histone 3 lysine 79 di/trimethylation |
| HDAC | Histone deacetylase |
| HOXA | Homeobox A |
| HSC | Hematopoietic stem cell |
| IDH1 | Isocitrate dehydrogenase 1 |
| IDH2 | Isocitrate dehydrogenase 2 |
| IRX2 | Iroquois homeobox protein 2 |
| KLF1 | KLF transcription factor 1 |
| Lmo2 | LIM domain only 2 |
| LSD1 | Lysine-specific demethylase 1 |
| LSK | Lineage- Sca1+ kit+ |
| MEIS1 | Myeloid ecotropic viral integration site 1 |
| mESCs | mouse embryonic stem cells |
| MLL | Mixed-lineage leukemia gene |
| MLL::FP | Mixed-lineage leukemia fusion protein |
| MLL-r | Mixed-lineage leukemia gene rearranged |
| MNase | Micrococcal nuclease |
| Myc | Myelocytomatosis |
| Mx1 | MX dynamin-like GTPase 1 |
| p300 | E1A-associated protein |
| POU2AF1 | POU domain class 2-associating factor 1 |
| PRC | Polycomb repressive complex |
| PRMD9 | PR domain zinc finger protein 9 |
| PROTAC | Proteolysis targeting chimera |
| RNA | Ribonucleic acid |
| RNA-seq | Ribonucleic acid sequencing |
| RNPs | Ribonucleoproteins |
| RUNX1 | Runt-related transcription factor 1 |
| SEC | Super elongation complex |
| sgRNA | Single-guide RNA |
| Spi1 | Spleen focus forming virus proviral integration 1 |
| Suz12 | Suppressor of zeste 12 protein homolog |
| TAD | Topologically associated domains |
| TAPT1 | Transmembrane anterior posterior transformation 1 |
| TET | Ten-eleven translocation methylcytosine dioxygenase |
| Tn5 | Transposase 5 |
References
- Creyghton, M.P.; Cheng, A.W.; Welstead, G.G.; Kooistra, T.; Carey, B.W.; Steine, E.J.; Hanna, J.; Lodato, M.A.; Frampton, G.M.; Sharp, P.A.; et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 2010, 107, 21931–21936. [Google Scholar] [CrossRef] [Green Version]
- Heintzman, N.D.; Stuart, R.K.; Hon, G.; Fu, Y.; Ching, C.W.; Hawkins, R.D.; Barrera, L.O.; van Calcar, S.; Qu, C.; Ching, K.A.; et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 2007, 39, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Soares, L.M.; He, P.C.; Chun, Y.; Suh, H.; Kim, T.; Buratowski, S. Determinants of Histone H3K4 Methylation Patterns. Mol. Cell 2017, 68, 773–785.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milne, T.A.; Briggs, S.D.; Brock, H.W.; Martin, M.E.; Gibbs, D.; Allis, C.D.; Hess, J.L. MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol. Cell 2002, 10, 1107–1117. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, L.; Crump, N.T.; Thorne, R.; Lau, I.J.; Repapi, E.; Dimou, D.; Smith, A.L.; Harman, J.R.; Telenius, J.M.; Oudelaar, A.M.; et al. DOT1L inhibition reveals a distinct subset of enhancers dependent on H3K79 methylation. Nat. Commun. 2019, 10, 2803. [Google Scholar] [CrossRef] [Green Version]
- Vakoc, C.R.; Sachdeva, M.M.; Wang, H.; Blobel, G.A. Profile of histone lysine methylation across transcribed mammalian chromatin. Mol. Cell Biol. 2006, 26, 9185–9195. [Google Scholar] [CrossRef] [Green Version]
- Bannister, A.J.; Schneider, R.; Myers, F.A.; Thorne, A.W.; Crane-Robinson, C.; Kouzarides, T. Spatial distribution of di-and tri-methyl lysine 36 of histone H3 at active genes. J. Biol. Chem. 2005, 280, 17732–17736. [Google Scholar] [CrossRef] [Green Version]
- Kouskouti, A.; Talianidis, I. Histone modifications defining active genes persist after transcriptional and mitotic inactivation. Embo J. 2005, 24, 347–357. [Google Scholar] [CrossRef] [Green Version]
- Bracken, A.P.; Dietrich, N.; Pasini, D.; Hansen, K.H.; Helin, K. Genome-wide mapping of Polycomb target genes unravels their roles in cell fate transitions. Genes Dev. 2006, 20, 1123–1136. [Google Scholar] [CrossRef] [Green Version]
- Bernt, K.M.; Zhu, N.; Sinha, A.U.; Vempati, S.; Faber, J.; Krivtsov, A.V.; Feng, Z.; Punt, N.; Daigle, A.; Bullinger, L.; et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell 2011, 20, 66–78. [Google Scholar] [CrossRef]
- Krivtsov, A.V.; Feng, Z.; Lemieux, M.E.; Faber, J.; Vempati, S.; Sinha, A.U.; Xia, X.; Jesneck, J.; Bracken, A.P.; Silverman, L.B.; et al. H3K79 methylation profiles define murine and human MLL-AF4 leukemias. Cancer Cell 2008, 14, 355–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Q.; Wang, H.; Ng, H.H.; Erdjument-Bromage, H.; Tempst, P.; Struhl, K.; Zhang, Y. Methylation of H3-lysine 79 is mediated by a new family of HMTases without a SET domain. Curr. Biol. 2002, 12, 1052–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, B.; Su, H.; Bhat, A.; Lei, H.; Bajko, J.; Hevi, S.; Baltus, G.A.; Kadam, S.; Zhai, H.; Valdez, R.; et al. The histone H3K79 methyltransferase Dot1L is essential for mammalian development and heterochromatin structure. PLoS Genet. 2008, 4, e1000190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.; Yang, Y.; Ortega, M.M.; Copeland, J.N.; Zhang, M.; Jacob, J.B.; Fields, T.A.; Vivian, J.L.; Fields, P.E. Early mammalian erythropoiesis requires the Dot1L methyltransferase. Blood 2010, 116, 4483–4491. [Google Scholar] [CrossRef] [Green Version]
- Steger, D.J.; Lefterova, M.I.; Ying, L.; Stonestrom, A.J.; Schupp, M.; Zhuo, D.; Vakoc, A.L.; Kim, J.E.; Chen, J.; Lazar, M.A.; et al. DOT1L/KMT4 recruitment and H3K79 methylation are ubiquitously coupled with gene transcription in mammalian cells. Mol. Cell Biol. 2008, 28, 2825–2839. [Google Scholar] [CrossRef] [Green Version]
- Mueller, D.; Bach, C.; Zeisig, D.; Garcia-Cuellar, M.P.; Monroe, S.; Sreekumar, A.; Zhou, R.; Nesvizhskii, A.; Chinnaiyan, A.; Hess, J.L.; et al. A role for the MLL fusion partner ENL in transcriptional elongation and chromatin modification. Blood 2007, 110, 4445–4454. [Google Scholar] [CrossRef]
- Leach, B.I.; Kuntimaddi, A.; Schmidt, C.R.; Cierpicki, T.; Johnson, S.A.; Bushweller, J.H. Leukemia fusion target AF9 is an intrinsically disordered transcriptional regulator that recruits multiple partners via coupled folding and binding. Structure 2013, 21, 176–183. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.; Garrett, A.S.; de Kumar, B.; Smith, E.R.; Gogol, M.; Seidel, C.; Krumlauf, R.; Shilatifard, A. Dynamic transcriptional events in embryonic stem cells mediated by the super elongation complex (SEC). Genes Dev. 2011, 25, 1486–1498. [Google Scholar] [CrossRef] [Green Version]
- Wan, L.; Wen, H.; Li, Y.; Lyu, J.; Xi, Y.; Hoshii, T.; Joseph, J.K.; Wang, X.; Loh, Y.E.; Erb, M.A.; et al. ENL links histone acetylation to oncogenic gene expression in acute myeloid leukaemia. Nature 2017, 543, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Godfrey, L.; Crump, N.T.; O’Byrne, S.; Lau, I.J.; Rice, S.; Harman, J.R.; Jackson, T.; Elliott, N.; Buck, G.; Connor, C.; et al. H3K79me2/3 controls enhancer–promoter interactions and activation of the pan-cancer stem cell marker PROM1/CD133 in MLL-AF4 leukemia cells. Leukemia 2021, 35, 90–106. [Google Scholar] [CrossRef]
- Ji, H.; Ehrlich, L.I.R.; Seita, J.; Murakami, P.; Doi, A.; Lindau, P.; Lee, H.; Aryee, M.J.; Irizarry, R.A.; Kim, K.; et al. Comprehensive methylome map of lineage commitment from haematopoietic progenitors. Nature 2010, 467, 338–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonkers, I.; Lis, J.T. Getting up to speed with transcription elongation by RNA polymerase II. Nat. Rev. Mol. Cell Biol. 2015, 16, 167–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borgel, J.; Guibert, S.; Li, Y.; Chiba, H.; Schübeler, D.; Sasaki, H.; Forné, T.; Weber, M. Targets and dynamics of promoter DNA methylation during early mouse development. Nat. Genet. 2010, 42, 1093–1100. [Google Scholar] [CrossRef] [PubMed]
- Flavahan, W.A.; Drier, Y.; Liau, B.B.; Gillespie, S.M.; Venteicher, A.S.; Stemmer-Rachamimov, A.O.; Suvà, M.L.; Bernstein, B.E. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 2016, 529, 110–114. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Song, F.; Lyu, H.; Kobayashi, M.; Zhang, B.; Zhao, Z.; Hou, Y.; Wang, X.; Luan, Y.; Jia, B.; et al. Subtype-specific 3D genome alteration in acute myeloid leukaemia. Nature 2022, 611, 387–398. [Google Scholar] [CrossRef]
- Fu, A.Q.; Genereux, D.P.; Stöger, R.; Burden, A.F.; Laird, C.D.; Stephens, M. Statistical inference of in vivo properties of human DNA methyltransferases from double-stranded methylation patterns. PLoS ONE 2012, 7, e32225. [Google Scholar] [CrossRef]
- Zhou, W.; Dinh, H.Q.; Ramjan, Z.; Weisenberger, D.J.; Nicolet, C.M.; Shen, H.; Laird, P.W.; Berman, B.P. DNA methylation loss in late-replicating domains is linked to mitotic cell division. Nat. Genet. 2018, 50, 591–602. [Google Scholar] [CrossRef] [Green Version]
- Hermann, A.; Goyal, R.; Jeltsch, A. The Dnmt1 DNA-(cytosine-C5)-methyltransferase methylates DNA processively with high preference for hemimethylated target sites. J. Biol. Chem. 2004, 279, 48350–48359. [Google Scholar] [CrossRef] [Green Version]
- Beerman, I.; Bock, C.; Garrison, B.S.; Smith, Z.D.; Gu, H.; Meissner, A.; Rossi, D.J. Proliferation-dependent alterations of the DNA methylation landscape underlie hematopoietic stem cell aging. Cell Stem Cell 2013, 12, 413–425. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Liu, Y.; Rappaport, A.R.; Kitzing, T.; Schultz, N.; Zhao, Z.; Shroff, A.S.; Dickins, R.A.; Vakoc, C.R.; Bradner, J.E.; et al. MLL3 is a haploinsufficient 7q tumor suppressor in acute myeloid leukemia. Cancer Cell 2014, 25, 652–665. [Google Scholar] [CrossRef]
- Nikoloski, G.; Langemeijer, S.M.; Kuiper, R.P.; Knops, R.; Massop, M.; Tönnissen, E.R.; van der Heijden, A.; Scheele, T.N.; Vandenberghe, P.; de Witte, T.; et al. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat. Genet. 2010, 42, 665–667. [Google Scholar] [CrossRef]
- Ernst, T.; Chase, A.J.; Score, J.; Hidalgo-Curtis, C.E.; Bryant, C.; Jones, A.V.; Waghorn, K.; Zoi, K.; Ross, F.M.; Reiter, A.; et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat. Genet. 2010, 42, 722–726. [Google Scholar] [CrossRef] [PubMed]
- Chou, W.C.; Huang, H.H.; Hou, H.A.; Chen, C.Y.; Tang, J.L.; Yao, M.; Tsay, W.; Ko, B.S.; Wu, S.J.; Huang, S.Y.; et al. Distinct clinical and biological features of de novo acute myeloid leukemia with additional sex comb-like 1 (ASXL1) mutations. Blood 2010, 116, 4086–4094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, W.J.; Huang, X.; Lynch, J.T.; Spencer, G.J.; Hitchin, J.R.; Li, Y.; Ciceri, F.; Blaser, J.G.; Greystoke, B.F.; Jordan, A.M.; et al. The histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cells. Cancer Cell 2012, 21, 473–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boileau, M.; Shirinian, M.; Gayden, T.; Harutyunyan, A.S.; Chen, C.C.L.; Mikael, L.G.; Duncan, H.M.; Neumann, A.L.; Arreba-Tutusaus, P.; de Jay, N.; et al. Mutant H3 histones drive human pre-leukemic hematopoietic stem cell expansion and promote leukemic aggressiveness. Nat. Commun. 2019, 10, 2891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehnertz, B.; Zhang, Y.W.; Boivin, I.; Mayotte, N.; Tomellini, E.; Chagraoui, J.; Lavallée, V.P.; Hébert, J.; Sauvageau, G. H3(K27M/I) mutations promote context-dependent transformation in acute myeloid leukemia with RUNX1 alterations. Blood 2017, 130, 2204–2214. [Google Scholar] [CrossRef] [PubMed]
- Issa, G.C.; Zarka, J.; Sasaki, K.; Qiao, W.; Pak, D.; Ning, J.; Short, N.J.; Haddad, F.; Tang, Z.; Patel, K.P.; et al. Predictors of outcomes in adults with acute myeloid leukemia and KMT2A rearrangements. Blood Cancer J. 2021, 11, 162. [Google Scholar] [CrossRef] [PubMed]
- Winters, A.C.; Bernt, K.M. MLL-Rearranged Leukemias-An Update on Science and Clinical Approaches. Front. Pediatr 2017, 5, 4. [Google Scholar] [CrossRef] [Green Version]
- Kerry, J.; Godfrey, L.; Repapi, E.; Tapia, M.; Blackledge, N.P.; Ma, H.; Ballabio, E.; O’Byrne, S.; Ponthan, F.; Heidenreich, O.; et al. MLL-AF4 Spreading Identifies Binding Sites that Are Distinct from Super-Enhancers and that Govern Sensitivity to DOT1L Inhibition in Leukemia. Cell Rep. 2017, 18, 482–495. [Google Scholar] [CrossRef] [Green Version]
- Daigle, S.R.; Olhava, E.J.; Therkelsen, C.A.; Basavapathruni, A.; Jin, L.; Boriack-Sjodin, P.A.; Allain, C.J.; Klaus, C.R.; Raimondi, A.; Scott, M.P.; et al. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood 2013, 122, 1017–1025. [Google Scholar] [CrossRef]
- Daigle, S.R.; Olhava, E.J.; Therkelsen, C.A.; Majer, C.R.; Sneeringer, C.J.; Song, J.; Johnston, L.D.; Scott, M.P.; Smith, J.J.; Xiao, Y.; et al. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell 2011, 20, 53–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsen, S.N.; Godfrey, L.; Healy, J.P.; Choi, Y.A.; Kai, Y.; Hatton, C.; Perner, F.; Haarer, E.L.; Nabet, B.; Yuan, G.C.; et al. MLL::AF9 degradation induces rapid changes in transcriptional elongation and subsequent loss of an active chromatin landscape. Mol. Cell 2022, 82, 1140–1155.e11. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.; Hofmann, J.; Burmeister, T.; Gröger, D.; Park, T.S.; Emerenciano, M.; de Oliveira, M.P.; Renneville, A.; Villarese, P.; Macintyre, E.; et al. The MLL recombinome of acute leukemias in 2013. Leukemia 2013, 27, 2165–2176. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.; Burmeister, T.; Gröger, D.; Tsaur, G.; Fechina, L.; Renneville, A.; Sutton, R.; Venn, N.C.; Emerenciano, M.; Pombo-de-Oliveira, M.S.; et al. The MLL recombinome of acute leukemias in 2017. Leukemia 2018, 32, 273–284. [Google Scholar] [CrossRef]
- Pui, C.H.; Ribeiro, R.C.; Hancock, M.L.; Rivera, G.K.; Evans, W.E.; Raimondi, S.C.; Head, D.R.; Behm, F.G.; Mahmoud, M.H.; Sandlund, J.T.; et al. Acute myeloid leukemia in children treated with epipodophyllotoxins for acute lymphoblastic leukemia. N. Engl. J. Med. 1991, 325, 1682–1687. [Google Scholar] [CrossRef]
- Collins, E.C.; Pannell, R.; Simpson, E.M.; Forster, A.; Rabbitts, T.H. Inter-chromosomal recombination of Mll and Af9 genes mediated by cre-loxP in mouse development. EMBO Rep. 2000, 1, 127–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krivtsov, A.V.; Twomey, D.; Feng, Z.; Stubbs, M.C.; Wang, Y.; Faber, J.; Levine, J.E.; Wang, J.; Hahn, W.C.; Gilliland, D.G.; et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature 2006, 442, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Somervaille, T.C.; Cleary, M.L. Identification and characterization of leukemia stem cells in murine MLL-AF9 acute myeloid leukemia. Cancer Cell 2006, 10, 257–268. [Google Scholar] [CrossRef]
- Krivtsov, A.V.; Figueroa, M.E.; Sinha, A.U.; Stubbs, M.C.; Feng, Z.; Valk, P.J.; Delwel, R.; Döhner, K.; Bullinger, L.; Kung, A.L.; et al. Cell of origin determines clinically relevant subtypes of MLL-rearranged AML. Leukemia 2013, 27, 852–860. [Google Scholar] [CrossRef] [Green Version]
- Stavropoulou, V.; Kaspar, S.; Brault, L.; Sanders, M.A.; Juge, S.; Morettini, S.; Tzankov, A.; Iacovino, M.; Lau, I.J.; Milne, T.A.; et al. MLL-AF9 Expression in Hematopoietic Stem Cells Drives a Highly Invasive AML Expressing EMT-Related Genes Linked to Poor Outcome. Cancer Cell 2016, 30, 43–58. [Google Scholar] [CrossRef]
- Mitchell, E.; Chapman, M.S.; Williams, N.; Dawson, K.J.; Mende, N.; Calderbank, E.F.; Jung, H.; Mitchell, T.; Coorens, T.H.H.; Spencer, D.H.; et al. Clonal dynamics of haematopoiesis across the human lifespan. Nature 2022, 606, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Fabre, M.A.; de Almeida, J.G.; Fiorillo, E.; Mitchell, E.; Damaskou, A.; Rak, J.; Orrù, V.; Marongiu, M.; Chapman, M.S.; Vijayabaskar, M.S.; et al. The longitudinal dynamics and natural history of clonal haematopoiesis. Nature 2022, 606, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [Green Version]
- Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, A.; Hoadley, K.; Triche, T.J., Jr.; Laird, P.W.; Baty, J.D.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [PubMed] [Green Version]
- Moran-Crusio, K.; Reavie, L.; Shih, A.; Abdel-Wahab, O.; Ndiaye-Lobry, D.; Lobry, C.; Figueroa, M.E.; Vasanthakumar, A.; Patel, J.; Zhao, X.; et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell 2011, 20, 11–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Challen, G.A.; Sun, D.; Jeong, M.; Luo, M.; Jelinek, J.; Berg, J.S.; Bock, C.; Vasanthakumar, A.; Gu, H.; Xi, Y.; et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat. Genet. 2011, 44, 23–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trowbridge, J.J.; Snow, J.W.; Kim, J.; Orkin, S.H. DNA methyltransferase 1 is essential for and uniquely regulates hematopoietic stem and progenitor cells. Cell Stem Cell 2009, 5, 442–449. [Google Scholar] [CrossRef] [Green Version]
- Broske, A.M.; Vockentanz, L.; Kharazi, S.; Huska, M.R.; Mancini, E.; Scheller, M.; Kuhl, C.; Enns, A.; Prinz, M.; Jaenisch, R.; et al. DNA methylation protects hematopoietic stem cell multipotency from myeloerythroid restriction. Nat. Genet. 2009, 41, 1207–1215. [Google Scholar] [CrossRef]
- Trowbridge, J.J.; Sinha, A.U.; Zhu, N.; Li, M.; Armstrong, S.A.; Orkin, S.H. Haploinsufficiency of Dnmt1 impairs leukemia stem cell function through derepression of bivalent chromatin domains. Genes Dev. 2012, 26, 344–349. [Google Scholar] [CrossRef] [Green Version]
- Ziller, M.J.; Gu, H.; Müller, F.; Donaghey, J.; Tsai, L.T.; Kohlbacher, O.; de Jager, P.L.; Rosen, E.D.; Bennett, D.A.; Bernstein, B.E.; et al. Charting a dynamic DNA methylation landscape of the human genome. Nature 2013, 500, 477–481. [Google Scholar] [CrossRef]
- Wilkinson, A.C.; Ishida, R.; Kikuchi, M.; Sudo, K.; Morita, M.; Crisostomo, R.V.; Yamamoto, R.; Loh, K.M.; Nakamura, Y.; Watanabe, M.; et al. Long-term ex vivo haematopoietic-stem-cell expansion allows nonconditioned transplantation. Nature 2019, 571, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, A.C.; Ishida, R.; Nakauchi, H.; Yamazaki, S. Long-term ex vivo expansion of mouse hematopoietic stem cells. Nat. Protoc. 2020, 15, 628–648. [Google Scholar] [CrossRef] [PubMed]
- Fares, I.; Chagraoui, J.; Gareau, Y.; Gingras, S.; Ruel, R.; Mayotte, N.; Csaszar, E.; Knapp, D.J.; Miller, P.; Ngom, M.; et al. Cord blood expansion. Pyrimidoindole derivatives are agonists of human hematopoietic stem cell self-renewal. Science 2014, 345, 1509–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkinson, A.C.; Dever, D.P.; Baik, R.; Camarena, J.; Hsu, I.; Charlesworth, C.T.; Morita, C.; Nakauchi, H.; Porteus, M.H. Cas9-AAV6 gene correction of beta-globin in autologous HSCs improves sickle cell disease erythropoiesis in mice. Nat. Commun. 2021, 12, 686. [Google Scholar] [CrossRef]
- Christen, F.; Hablesreiter, R.; Hoyer, K.; Hennch, C.; Maluck-Bottcher, A.; Segler, A.; Madadi, A.; Frick, M.; Bullinger, L.; Briest, F.; et al. Modeling clonal hematopoiesis in umbilical cord blood cells by CRISPR/Cas9. Leukemia 2022, 36, 1102–1110. [Google Scholar] [CrossRef]
- Nakauchi, Y.; Azizi, A.; Thomas, D.; Corces, M.R.; Reinisch, A.; Sharma, R.; Hernandez, D.C.; Köhnke, T.; Karigane, D.; Fan, A.; et al. The Cell Type-Specific 5hmC Landscape and Dynamics of Healthy Human Hematopoiesis and TET2-Mutant Preleukemia. Blood Cancer Discov. 2022, 3, 346–367. [Google Scholar] [CrossRef]
- Jeong, J.; Jager, A.; Domizi, P.; Pavel-Dinu, M.; Gojenola, L.; Iwasaki, M.; Wei, M.C.; Pan, F.; Zehnder, J.L.; Porteus, M.H.; et al. High-efficiency CRISPR induction of t(9;11) chromosomal translocations and acute leukemias in human blood stem cells. Blood Adv. 2019, 3, 2825–2835. [Google Scholar] [CrossRef] [Green Version]
- Reimer, J.; Knöß, S.; Labuhn, M.; Charpentier, E.M.; Göhring, G.; Schlegelberger, B.; Klusmann, J.H.; Heckl, D. CRISPR-Cas9-induced t(11;19)/MLL-ENL translocations initiate leukemia in human hematopoietic progenitor cells in vivo. Haematologica 2017, 102, 1558–1566. [Google Scholar] [CrossRef] [Green Version]
- Rice, S.; Jackson, T.; Crump, N.T.; Fordham, N.; Elliott, N.; O’Byrne, S.; Fanego, M.; Addy, D.; Crabb, T.; Dryden, C.; et al. A human fetal liver-derived infant MLL-AF4 acute lymphoblastic leukemia model reveals a distinct fetal gene expression program. Nat. Commun. 2021, 12, 6905. [Google Scholar] [CrossRef]
- Secker, K.A.; Bruns, L.; Keppeler, H.; Jeong, J.; Hentrich, T.; Schulze-Hentrich, J.M.; Mankel, B.; Fend, F.; Schneidawind, D.; Schneidawind, C. Only Hematopoietic Stem and Progenitor Cells from Cord Blood Are Susceptible to Malignant Transformation by MLL-AF4 Translocations. Cancers 2020, 12, 1487. [Google Scholar] [CrossRef]
- Sarrou, E.; Richmond, L.; Carmody, R.J.; Gibson, B.; Keeshan, K. CRISPR Gene Editing of Murine Blood Stem and Progenitor Cells Induces MLL-AF9 Chromosomal Translocation and MLL-AF9 Leukaemogenesis. Int. J. Mol. Sci. 2020, 21, 4266. [Google Scholar] [CrossRef] [PubMed]
- Van den Boom, V.; Maat, H.; Geugien, M.; López, A.R.; Sotoca, A.M.; Jaques, J.; Brouwers-Vos, A.Z.; Fusetti, F.; Groen, R.W.J.; Yuan, H.; et al. Non-canonical PRC1.1 Targets Active Genes Independent of H3K27me3 and Is Essential for Leukemogenesis. Cell Rep. 2016, 14, 332–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, J.; Chen, S.A.; Local, A.; Liu, T.; Qiu, Y.; Dorighi, K.M.; Preissl, S.; Rivera, C.M.; Wang, C.; Ye, Z.; et al. Histone H3 lysine 4 monomethylation modulates long-range chromatin interactions at enhancers. Cell Res. 2018, 28, 204–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorighi, K.M.; Swigut, T.; Henriques, T.; Bhanu, N.V.; Scruggs, B.S.; Nady, N.; Still, C.D., 2nd; Garcia, B.A.; Adelman, K.; Wysocka, J. Mll3 and Mll4 Facilitate Enhancer RNA Synthesis and Transcription from Promoters Independently of H3K4 Monomethylation. Mol. Cell 2017, 66, 568–576.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sankar, A.; Mohammad, F.; Sundaramurthy, A.K.; Wang, H.; Lerdrup, M.; Tatar, T.; Helin, K. Histone editing elucidates the functional roles of H3K27 methylation and acetylation in mammals. Nat. Genet. 2022, 54, 754–760. [Google Scholar] [CrossRef]
- Nakamura, M.; Gao, Y.; Dominguez, A.A.; Qi, L.S. CRISPR technologies for precise epigenome editing. Nat. Cell Biol. 2021, 23, 11–22. [Google Scholar] [CrossRef]
- Hilton, I.B.; D’Ippolito, A.M.; Vockley, C.M.; Thakore, P.I.; Crawford, G.E.; Reddy, T.E.; Gersbach, C.A. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat. Biotechnol. 2015, 33, 510–517. [Google Scholar] [CrossRef] [Green Version]
- Cano-Rodriguez, D.; Gjaltema, R.A.; Jilderda, L.J.; Jellema, P.; Dokter-Fokkens, J.; Ruiters, M.H.; Rots, M.G. Writing of H3K4Me3 overcomes epigenetic silencing in a sustained but context-dependent manner. Nat. Commun. 2016, 7, 12284. [Google Scholar] [CrossRef] [Green Version]
- Kwon, D.Y.; Zhao, Y.T.; Lamonica, J.M.; Zhou, Z. Locus-specific histone deacetylation using a synthetic CRISPR-Cas9-based HDAC. Nat. Commun. 2017, 8, 15315. [Google Scholar] [CrossRef] [Green Version]
- Bester, A.C.; Lee, J.D.; Chavez, A.; Lee, Y.R.; Nachmani, D.; Vora, S.; Victor, J.; Sauvageau, M.; Monteleone, E.; Rinn, J.L.; et al. An Integrated Genome-wide CRISPRa Approach to Functionalize lncRNAs in Drug Resistance. Cell 2018, 173, 649–664.e20. [Google Scholar] [CrossRef]
- Gilbert, L.A.; Horlbeck, M.A.; Adamson, B.; Villalta, J.E.; Chen, Y.; Whitehead, E.H.; Guimaraes, C.; Panning, B.; Ploegh, H.L.; Bassik, M.C.; et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 2014, 159, 647–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, Y.; Zhang, X.; Su, J.; Jeong, M.; Gundry, M.C.; Huang, Y.-H.; Zhou, Y.; Li, W.; Goodell, M.A. Targeted DNA methylation in vivo using an engineered dCas9-MQ1 fusion protein. Nat. Commun. 2017, 8, 16026. [Google Scholar] [CrossRef] [Green Version]
- Amabile, A.; Migliara, A.; Capasso, P.; Biffi, M.; Cittaro, D.; Naldini, L.; Lombardo, A. Inheritable Silencing of Endogenous Genes by Hit-and-Run Targeted Epigenetic Editing. Cell 2016, 167, 219–232.e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.-H.; Su, J.; Lei, Y.; Brunetti, L.; Gundry, M.C.; Zhang, X.; Jeong, M.; Li, W.; Goodell, M.A. DNA epigenome editing using CRISPR-Cas SunTag-directed DNMT3A. Genome Biol. 2017, 18, 176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, M.; Kanamori, M.; Someya, K.; Nakatsukasa, H.; Yoshimura, A. Stabilization of Foxp3 expression by CRISPR-dCas9-based epigenome editing in mouse primary T cells. Epigenetics Chromatin 2017, 10, 24. [Google Scholar] [CrossRef] [Green Version]
- Gemberling, M.P.; Siklenka, K.; Rodriguez, E.; Tonn-Eisinger, K.R.; Barrera, A.; Liu, F.; Kantor, A.; Li, L.; Cigliola, V.; Hazlett, M.F.; et al. Transgenic mice for in vivo epigenome editing with CRISPR-based systems. Nat. Methods 2021, 18, 965–974. [Google Scholar] [CrossRef] [PubMed]
- Buenrostro, J.D.; Giresi, P.G.; Zaba, L.C.; Chang, H.Y.; Greenleaf, W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position, Nature Methods. Nat. Methods 2013, 10, 1213–1218. [Google Scholar] [CrossRef]
- Buenrostro, J.D.; Wu, B.; Litzenburger, U.M.; Ruff, D.; Gonzales, M.L.; Snyder, M.P.; Chang, H.Y.; Greenleaf, W.J. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 2015, 523, 486–490. [Google Scholar] [CrossRef] [Green Version]
- Corces, M.R.; Buenrostro, J.D.; Wu, B.; Greenside, P.G.; Chan, S.M.; Koenig, J.L.; Snyder, M.P.; Pritchard, J.K.; Kundaje, A.; Greenleaf, W.J.; et al. Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat. Genet. 2016, 48, 1193–1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granja, J.M.; Klemm, S.; McGinnis, L.M.; Kathiria, A.S.; Mezger, A.; Corces, M.R.; Parks, B.; Gars, E.; Liedtke, M.; Zheng, G.X.Y.; et al. Single-cell multiomic analysis identifies regulatory programs in mixed-phenotype acute leukemia. Nat. Biotechnol. 2019, 37, 1458–1465. [Google Scholar] [CrossRef] [PubMed]
- Grosselin, K.; Durand, A.; Marsolier, J.; Poitou, A.; Marangoni, E.; Nemati, F.; Dahmani, A.; Lameiras, S.; Reyal, F.; Frenoy, O.; et al. High-throughput single-cell ChIP-seq identifies heterogeneity of chromatin states in breast cancer. Nat. Genet. 2019, 51, 1060–1066. [Google Scholar] [CrossRef] [PubMed]
- Rotem, A.; Ram, O.; Shoresh, N.; Sperling, R.A.; Goren, A.; Weitz, D.A.; Bernstein, B.E. Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nat. Biotechnol. 2015, 33, 1165–1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Galen, P.; Viny, A.D.; Ram, O.; Ryan, R.J.H.; Cotton, M.J.; Donohue, L.; Sievers, C.; Drier, Y.; Liau, B.B.; Gillespie, S.M.; et al. A Multiplexed System for Quantitative Comparisons of Chromatin Landscapes. Mol. Cell 2016, 61, 170–180. [Google Scholar] [CrossRef] [Green Version]
- Kaya-Okur, H.S.; Wu, S.J.; Codomo, C.A.; Pledger, E.S.; Bryson, T.D.; Henikoff, J.G.; Ahmad, K.; Henikoff, S. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun. 2019, 10, 1930. [Google Scholar]
- Skene, P.J.; Henikoff, S. An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. eLife 2017, 6, e21856. [Google Scholar] [CrossRef]
- Hainer, S.J.; Bošković, A.; McCannell, K.N.; Rando, O.J.; Fazzio, T.G. Profiling of Pluripotency Factors in Single Cells and Early Embryos. Cell 2019, 177, 1319–1329.e11. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.J.; Furlan, S.N.; Mihalas, A.B.; Kaya-Okur, H.S.; Feroze, A.H.; Emerson, S.N.; Zheng, Y.; Carson, K.; Cimino, P.J.; Keene, C.D.; et al. Single-cell CUT&Tag analysis of chromatin modifications in differentiation and tumor progression. Nat. Biotechnol. 2021, 39, 819–824. [Google Scholar]
- Janssens, D.H.; Meers, M.P.; Wu, S.J.; Babaeva, E.; Meshinchi, S.; Sarthy, J.F.; Ahmad, K.; Henikoff, S. Automated CUT&Tag profiling of chromatin heterogeneity in mixed-lineage leukemia. Nat. Genet. 2021, 53, 1586–1596. [Google Scholar]
- Yang, L.; Chan, A.K.N.; Miyashita, K.; Delaney, C.D.; Wang, X.; Li, H.; Pokharel, S.P.; Li, S.; Li, M.; Xu, X.; et al. High-resolution characterization of gene function using single-cell CRISPR tiling screen. Nat. Commun. 2021, 12, 4063. [Google Scholar] [CrossRef]
- Karemaker, I.D.; Vermeulen, M. Single-Cell DNA Methylation Profiling: Technologies and Biological Applications. Trends Biotechnol. 2018, 36, 952–965. [Google Scholar] [CrossRef]
- Nam, A.S.; Dusaj, N.; Izzo, F.; Murali, R.; Myers, R.M.; Mouhieddine, T.; Sotelo, J.; Benbarche, S.; Waarts, M.; Gaiti, F.; et al. Single-cell multi-omics of human clonal hematopoiesis reveals that DNMT3A R882 mutations perturb early progenitor states through selective hypomethylation. Nat. Genet. 2022, 54, 1514–1526. Available online: https://www.nature.com/articles/s41588-022-01179-9 (accessed on 31 October 2022). [CrossRef] [PubMed]
- Gaiti, F.; Chaligne, R.; Gu, H.; Brand, R.M.; Kothen-Hill, S.; Schulman, R.C.; Grigorev, K.; Risso, D.; Kim, K.-T.; Pastore, A.; et al. Epigenetic evolution and lineage histories of chronic lymphocytic leukaemia. Nature 2019, 569, 576–580. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.; Kowarz, E.; Hofmann, J.; Renneville, A.; Zuna, J.; Trka, J.; Abdelali, R.B.; Macintyre, E.; de Braekeleer, E.; de Braekeleer, M.; et al. New insights to the MLL recombinome of acute leukemias. Leukemia 2009, 23, 1490–1499. [Google Scholar] [CrossRef] [PubMed]
- Alonso, C.N.; Longo, P.L.; Gallego, M.S.; Medina, A.; Felice, M.S. A novel AF9 breakpoint in MLL-AF9-positive acute monoblastic leukemia. Pediatr. Blood Cancer 2008, 50, 869–871. [Google Scholar] [CrossRef]
- Lyu, J.; Liu, Y.; Gong, L.; Chen, M.; Madanat, Y.F.; Zhang, Y.; Cai, F.; Gu, Z.; Cao, H.; Kaphle, P.; et al. Disabling Uncompetitive Inhibition of Oncogenic IDH Mutations Drives Acquired Resistance. Cancer Discov. 2022. [Google Scholar] [CrossRef]
- Galonska, C.; Charlton, J.; Mattei, A.L.; Donaghey, J.; Clement, K.; Gu, H.; Mohammad, A.W.; Stamenova, E.K.; Cacchiarelli, D.; Klages, S.; et al. Genome-wide tracking of dCas9-methyltransferase footprints. Nat. Commun. 2018, 9, 597. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, K.; Das, V.; Vyas, P.; Hajduch, M. Nucleosidic DNA demethylating epigenetic drugs—A comprehensive review from discovery to clinic. Pharmacol. Ther. 2018, 188, 45–79. [Google Scholar] [CrossRef]
- Pappalardi, M.B.; Keenan, K.; Cockerill, M.; Kellner, W.A.; Stowell, A.; Sherk, C.; Wong, K.; Pathuri, S.; Briand, J.; Steidel, M.; et al. Discovery of a first-in-class reversible DNMT1-selective inhibitor with improved tolerability and efficacy in acute myeloid leukemia. Nat. Cancer 2021, 2, 1002–1017. [Google Scholar] [CrossRef]
- Krivtsov, A.V.; Evans, K.; Gadrey, J.Y.; Eschle, B.K.; Hatton, C.; Uckelmann, H.J.; Ross, K.N.; Perner, F.; Olsen, S.N.; Pritchard, T.; et al. A Menin-MLL Inhibitor Induces Specific Chromatin Changes and Eradicates Disease in Models of MLL-Rearranged Leukemia. Cancer Cell 2019, 36, 660–673.e11. [Google Scholar] [CrossRef]
- Swaminathan, M.; Bourgeois, W.; Armstrong, S.A.; Wang, E.S. Menin Inhibitors in Acute Myeloid Leukemia—What Does the Future Hold? Cancer J. 2022, 28, 62–66. [Google Scholar] [CrossRef]
- Stein, E.M.; Garcia-Manero, G.; Rizzieri, D.A.; Tibes, R.; Berdeja, J.G.; Savona, M.R.; Jongen-Lavrenic, M.; Altman, J.K.; Thomson, B.; Blakemore, S.J.; et al. The DOT1L inhibitor pinometostat reduces H3K79 methylation and has modest clinical activity in adult acute leukemia. Blood 2018, 131, 2661–2669. [Google Scholar] [CrossRef] [PubMed]
- Campbell, C.T.; Haladyna, J.N.; Drubin, D.A.; Thomson, T.M.; Maria, M.J.; Yamauchi, T.; Waters, N.J.; Olhava, E.J.; Pollock, R.M.; Smith, J.J.; et al. Mechanisms of Pinometostat (EPZ-5676) Treatment-Emergent Resistance in MLL-Rearranged Leukemia. Mol. Cancer 2017, 16, 1669–1679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, E.M.; Aldoss, I.; DiPersio, J.F.; Stone, R.M.; Arellano, M.L.; Rosen, G.; Meyers, M.L.; Huang, Y.; Smith, S.; Bagley, R.G.; et al. Safety and Efficacy of Menin Inhibition in Patients (Pts) with MLL-Rearranged and NPM1 Mutant Acute Leukemia: A Phase (Ph) 1, First-in-Human Study of SNDX-5613 (AUGMENT 101. Blood 2021, 138, 699. [Google Scholar] [CrossRef]
- Wang, E.S.; Altman, J.K.; Pettit, K.; de Botton, S.; Walter, R.P.; Fenaux, P.; Burrows, F.; Tomkinson, B.E.; Martell, B.; Fathi, A.T. Preliminary Data on a Phase 1/2A First in Human Study of the Menin-KMT2A (MLL) Inhibitor KO-539 in Patients with Relapsed or Refractory Acute Myeloid Leukemia. Blood 2020, 136 (Suppl. 1), 7–8. [Google Scholar] [CrossRef]
- van Gils, N.; Verhagen, H.J.M.P.; Broux, M.; Martiáñez, T.; Denkers, F.; Vermue, E.; Rutten, A.; Csikós, T.; Demeyer, S.; Çil, M.; et al. Targeting histone methylation to reprogram the transcriptional state that drives survival of drug-tolerant myeloid leukemia persisters. iScience 2022, 25, 105013. [Google Scholar] [CrossRef] [PubMed]
- Scalea, S.; Maresca, C.; Catalanotto, C.; Marino, R.; Cogoni, C.; Reale, A.; Zampieri, M.; Zardo, G. Modifications of H3K4 methylation levels are associated with DNA hypermethylation in acute myeloid leukemia. FEBS J. 2020, 287, 1155–1175. [Google Scholar] [CrossRef]
- Rau, R.E.; Rodriguez, B.A.; Luo, M.; Jeong, M.; Rosen, A.; Rogers, J.H.; Campbell, C.T.; Daigle, S.R.; Deng, L.; Song, Y.; et al. DOT1L as a therapeutic target for the treatment of DNMT3A-mutant acute myeloid leukemia. Blood 2016, 128, 971–981. [Google Scholar] [CrossRef] [Green Version]
- Lu, R.; Wang, P.; Parton, T.; Zhou, Y.; Chrysovergis, K.; Rockowitz, S.; Chen, W.-Y.; Abdel-Wahab, O.; Wade, P.; Zheng, D.; et al. Epigenetic Perturbations by Arg882-Mutated DNMT3A Potentiate Aberrant Stem Cell Gene-Expression Program and Acute Leukemia Development. Cancer Cell 2016, 30, 92–107. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Godfrey, L.C.; Rodriguez-Meira, A. Viewing AML through a New Lens: Technological Advances in the Study of Epigenetic Regulation. Cancers 2022, 14, 5989. https://doi.org/10.3390/cancers14235989
Godfrey LC, Rodriguez-Meira A. Viewing AML through a New Lens: Technological Advances in the Study of Epigenetic Regulation. Cancers. 2022; 14(23):5989. https://doi.org/10.3390/cancers14235989
Chicago/Turabian StyleGodfrey, Laura C., and Alba Rodriguez-Meira. 2022. "Viewing AML through a New Lens: Technological Advances in the Study of Epigenetic Regulation" Cancers 14, no. 23: 5989. https://doi.org/10.3390/cancers14235989
APA StyleGodfrey, L. C., & Rodriguez-Meira, A. (2022). Viewing AML through a New Lens: Technological Advances in the Study of Epigenetic Regulation. Cancers, 14(23), 5989. https://doi.org/10.3390/cancers14235989