
Integrating Pharmacogenomics Data-Driven Computational Drug Prediction with Single-Cell RNAseq to Demonstrate the Efficacy of a NAMPT Inhibitor against Aggressive, Taxane-Resistant, and Stem-like Cells in Lethal Prostate Cancer

 , ,
, ,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Drugs and Reagents
2.2. Human Prostate Cancer Cell Lines
2.3. Patient Datasets
2.4. Identification of Secondary Drugs (secDrugs) in Lethal Prostate Cancer
2.5. In Vitro Cytotoxicity Assays and Drug Synergy Analysis
2.6. Caspase-3/7 Activity Assay
2.7. Annexin V and Propidium Iodide (PI) Staining
2.8. Assessment of Cellular and Nuclear Morphology
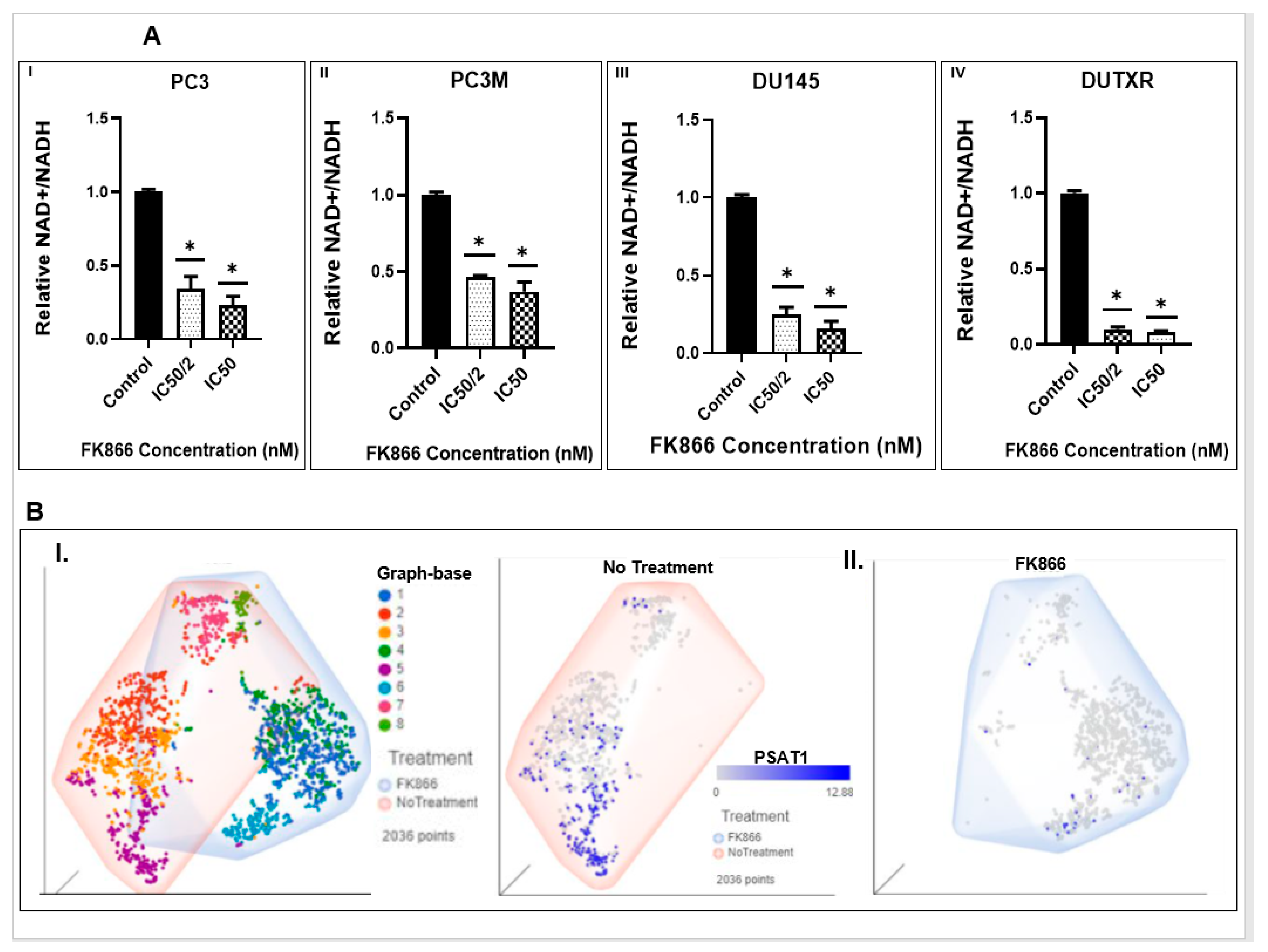
2.9. NAD/NADH Activity Assay
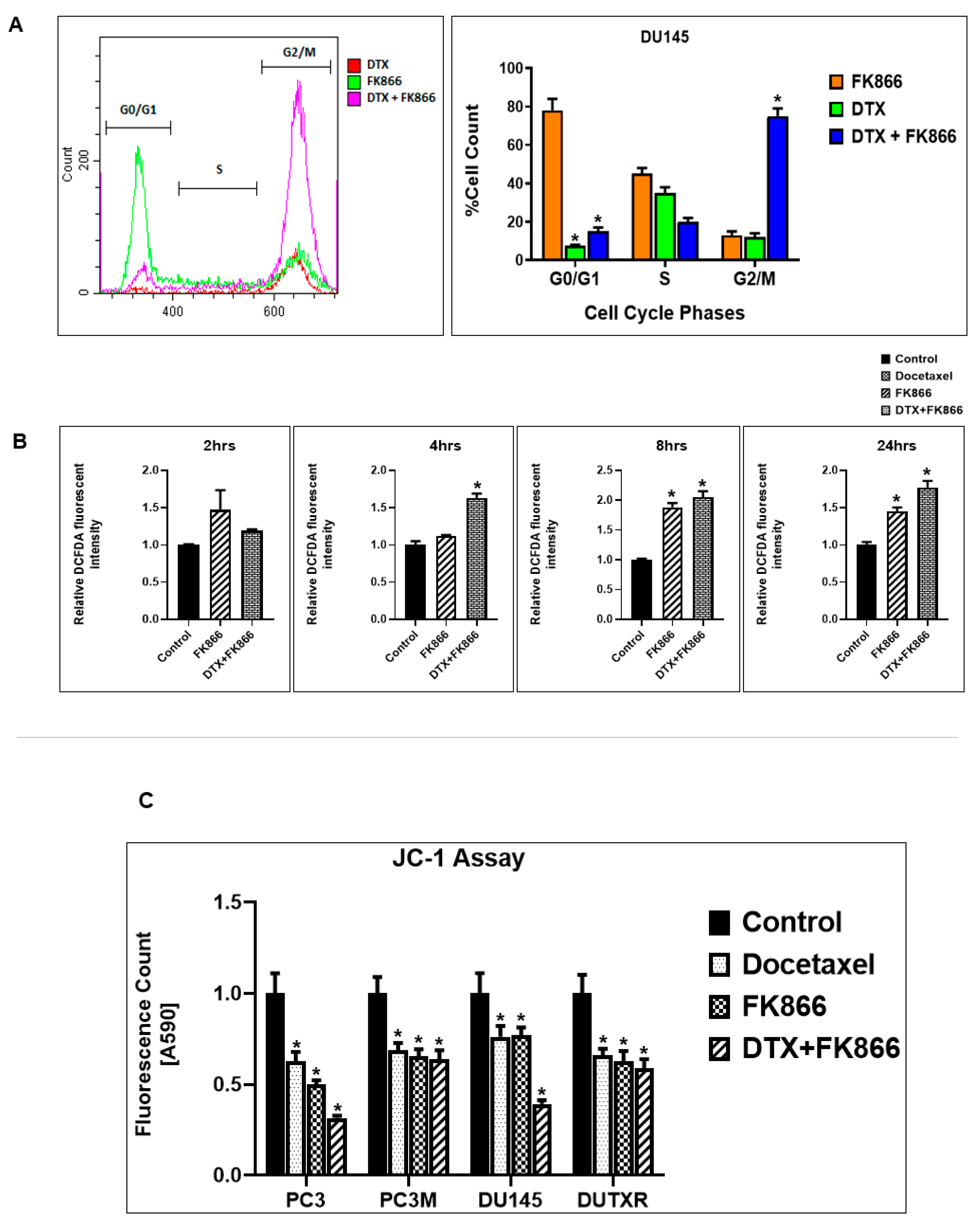
2.10. Assessment of Cell Cycle
2.11. Assessment of Intracellular ROS Levels (DCFDA Assay)
2.12. Assessment of Mitochondrial Membrane Potential
2.13. Assessment of Side Population
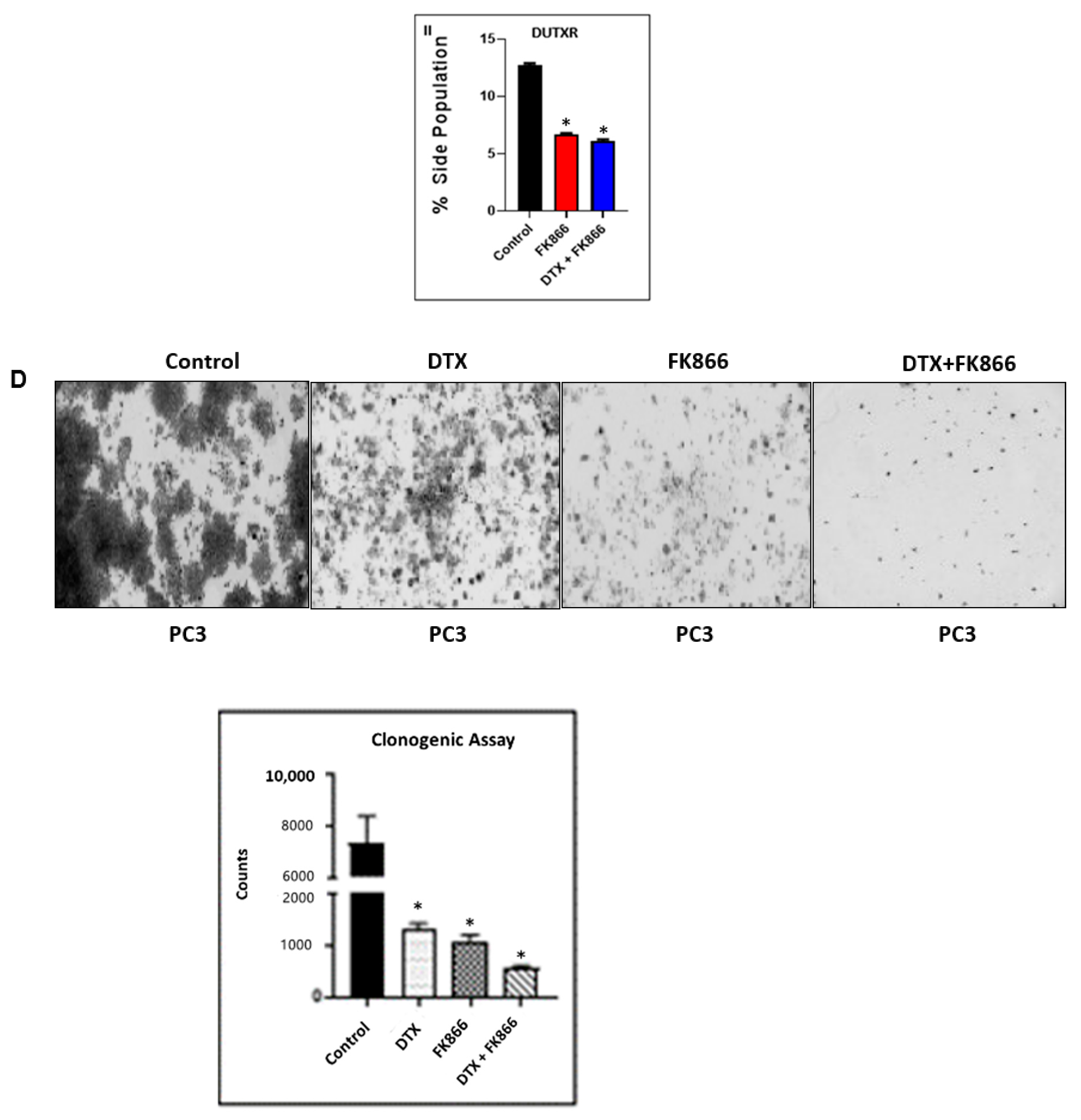
2.14. Colony Formation Assay
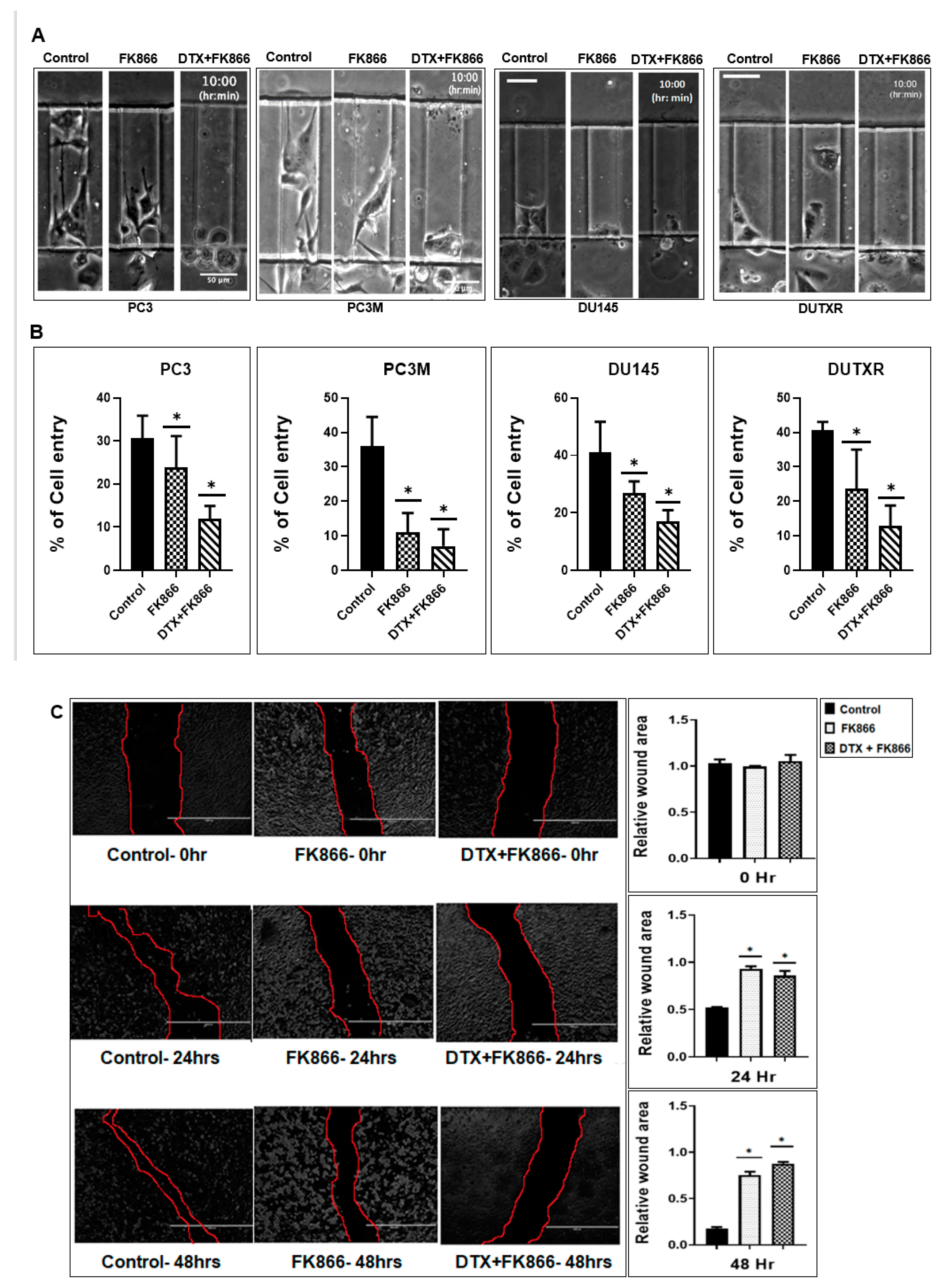
2.15. Microfluidic (μ)-Channel Cell Migration Assay
2.16. Cell Migration/Scratch Assay
2.17. Pre- and Post-Treatment Tumor mRNA Sequencing (RNAseq)
2.18. Pre- and Post-Treatment Single-Cell RNA Sequencing (scRNAseq)
2.19. Ingenuity Pathway Analysis (IPA)
2.20. Immunoblotting
2.21. Statistical Analysis
3. Results
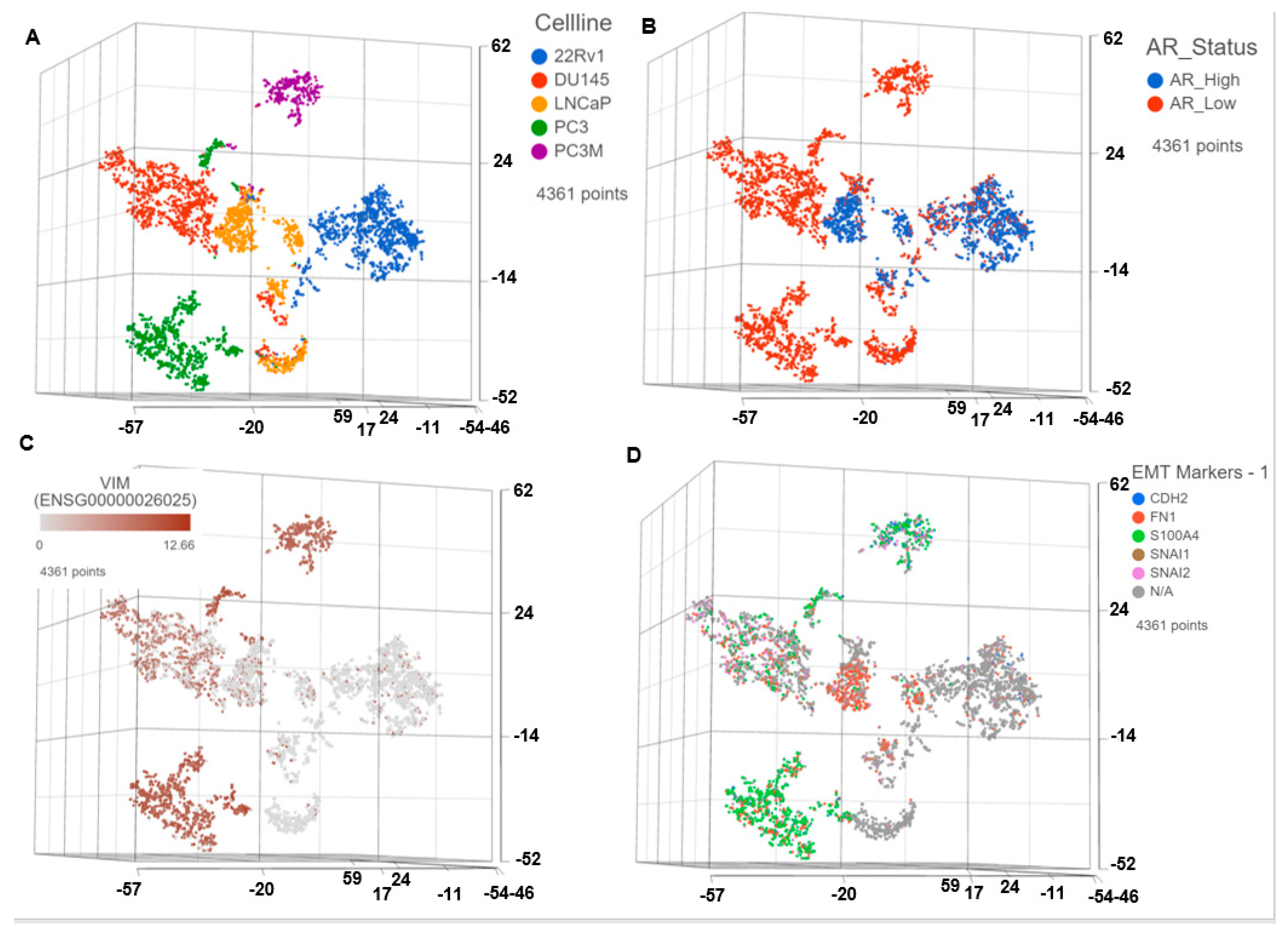
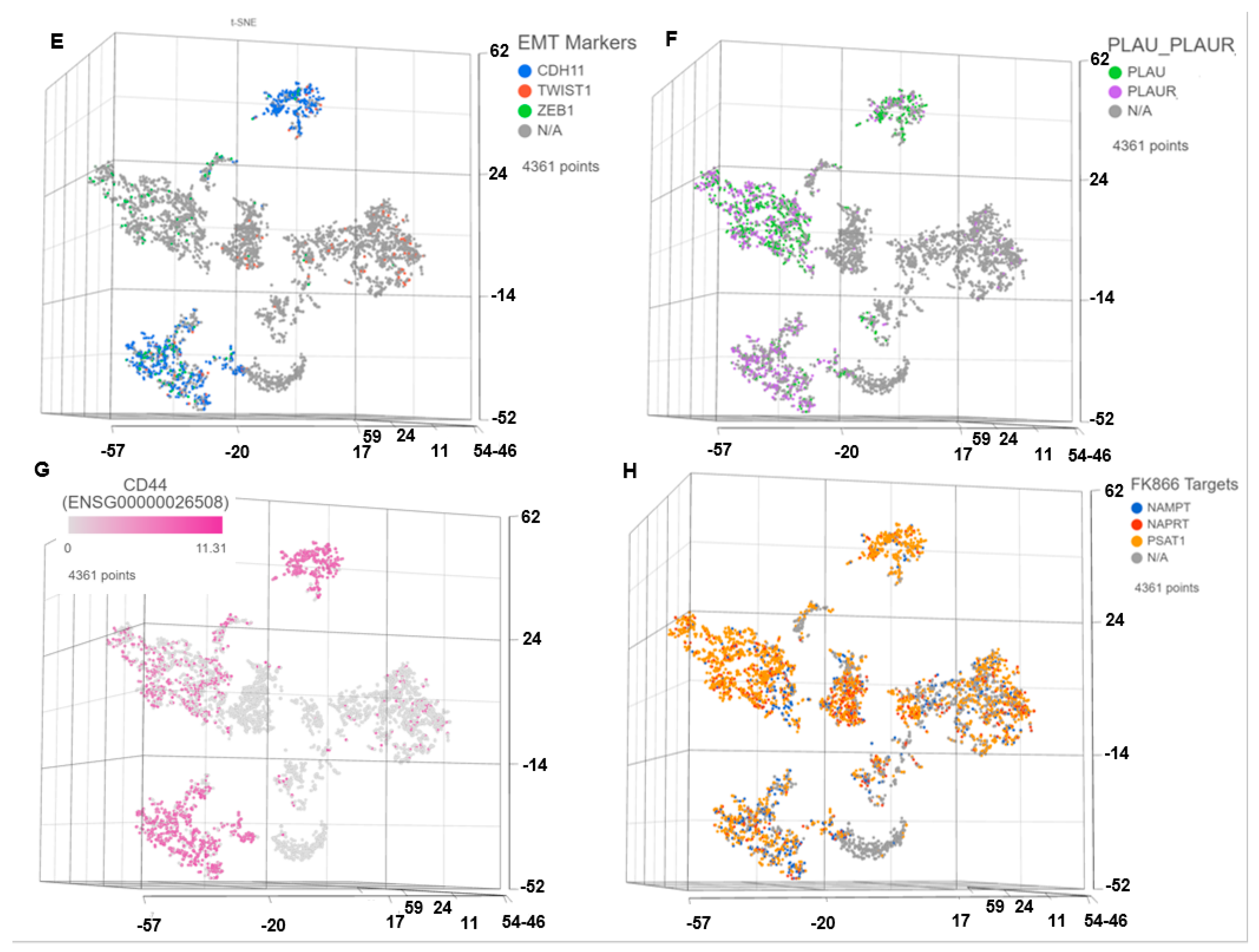
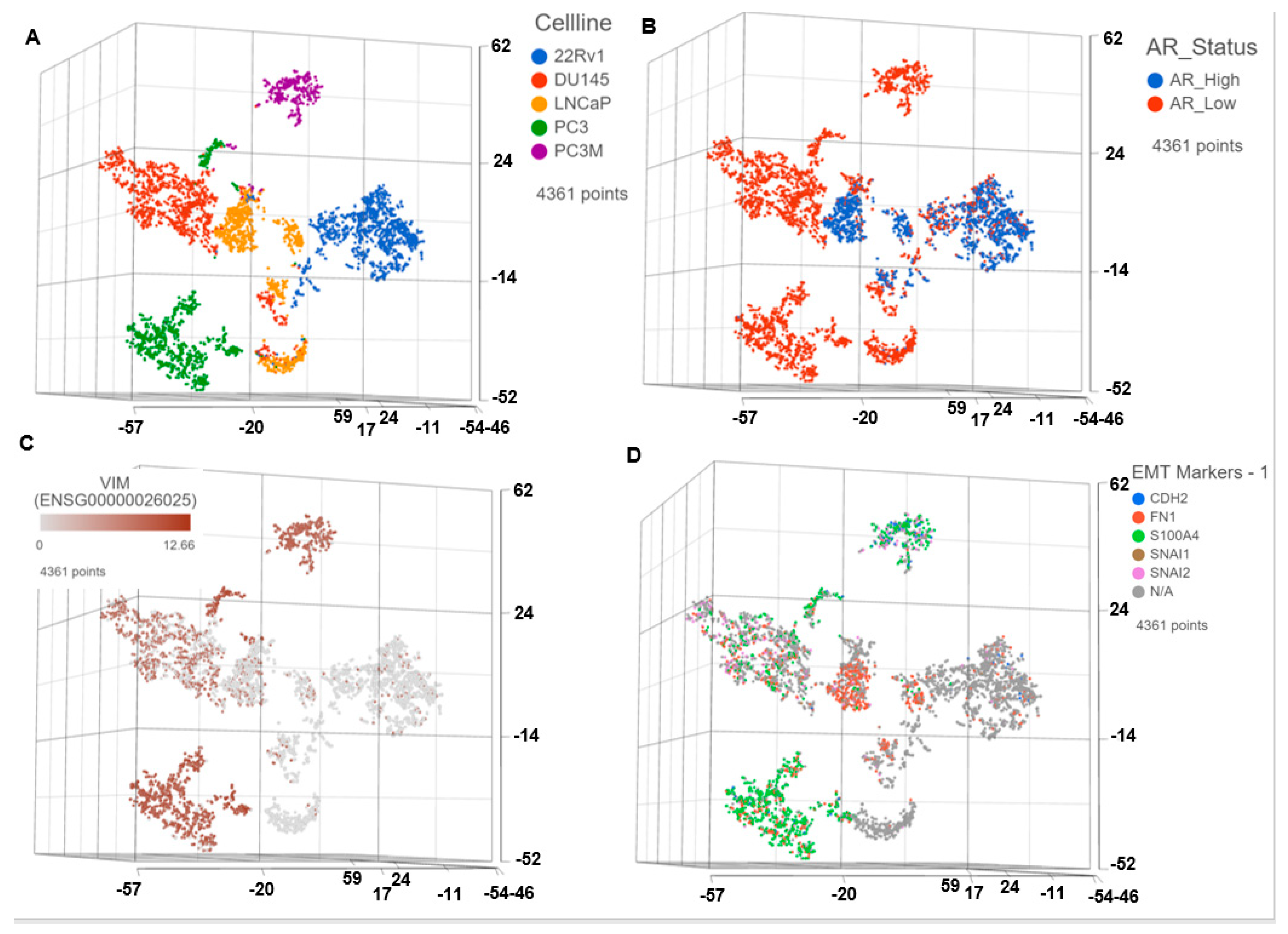
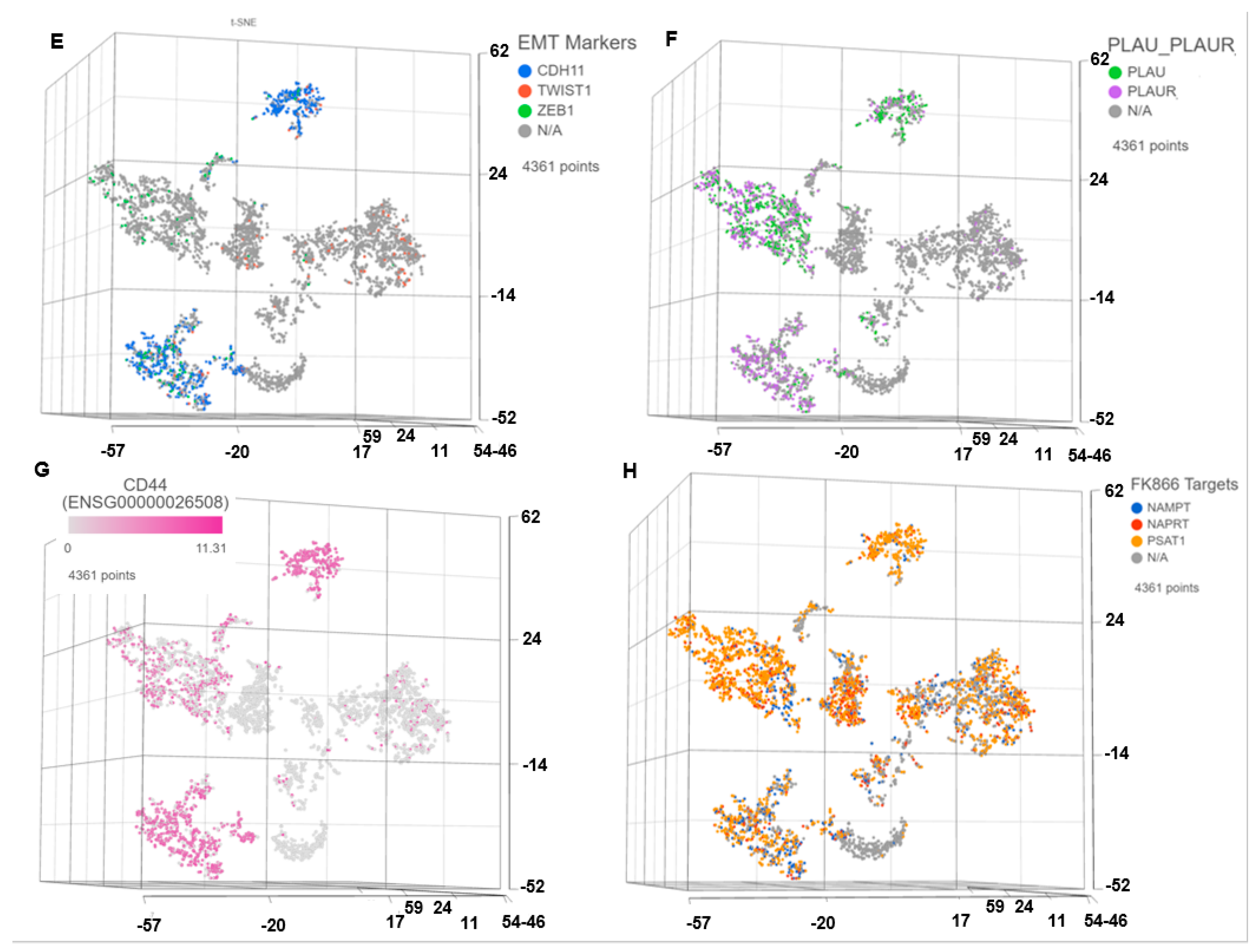
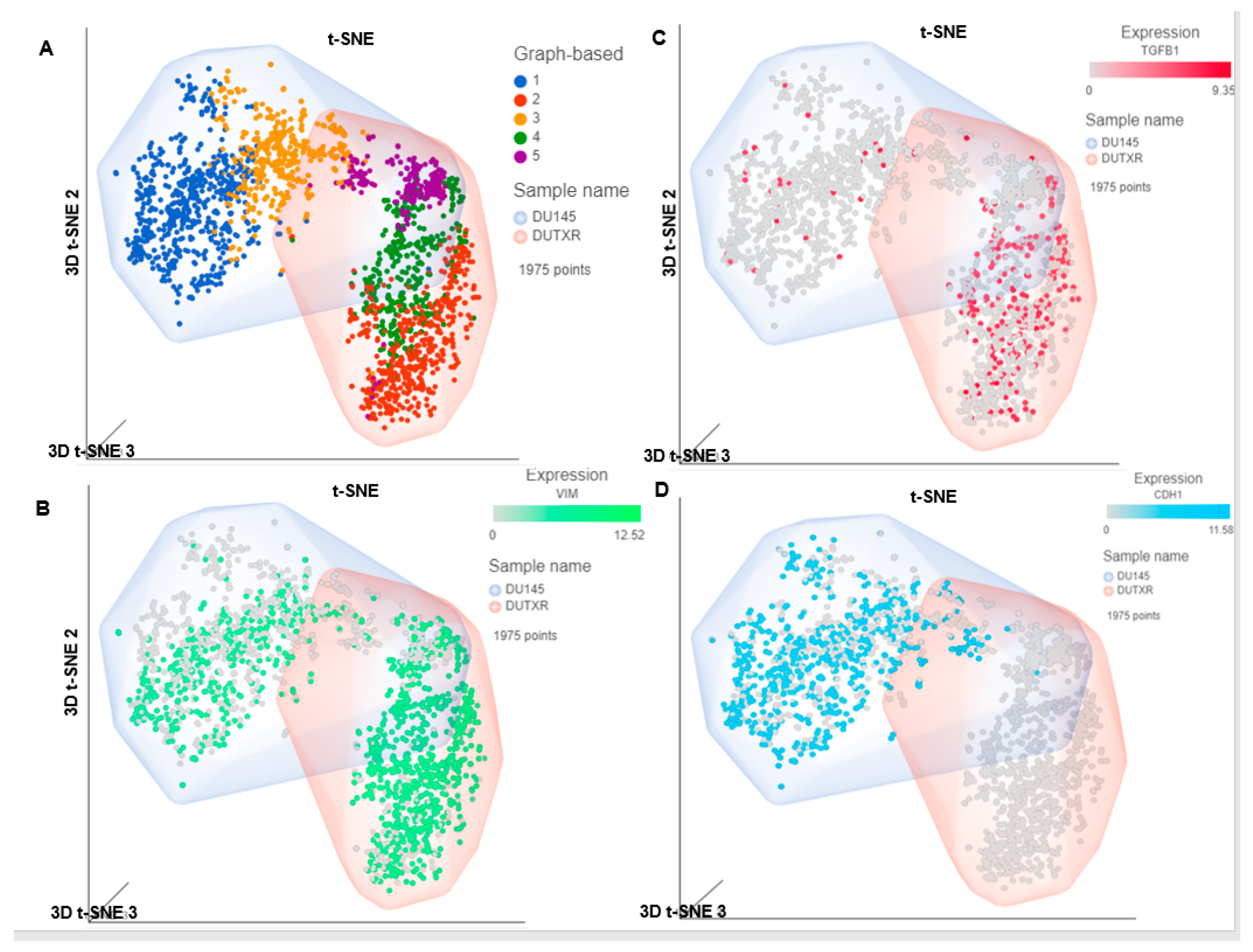
3.1. Single Cell Transcriptomics (scRNAseq) Showed ARlow PCa Cells with Signatures of Epithelial-Mesenchymal Transition (EMT) and Cancer ‘Stemness’
3.2. Pharmacogenomics Data-Driven Algorithm and scRNAseq-Based ‘Double-Hit’ Screening Predicted FK866 as a Top secDrug Potentially Effective against Lethal PCa
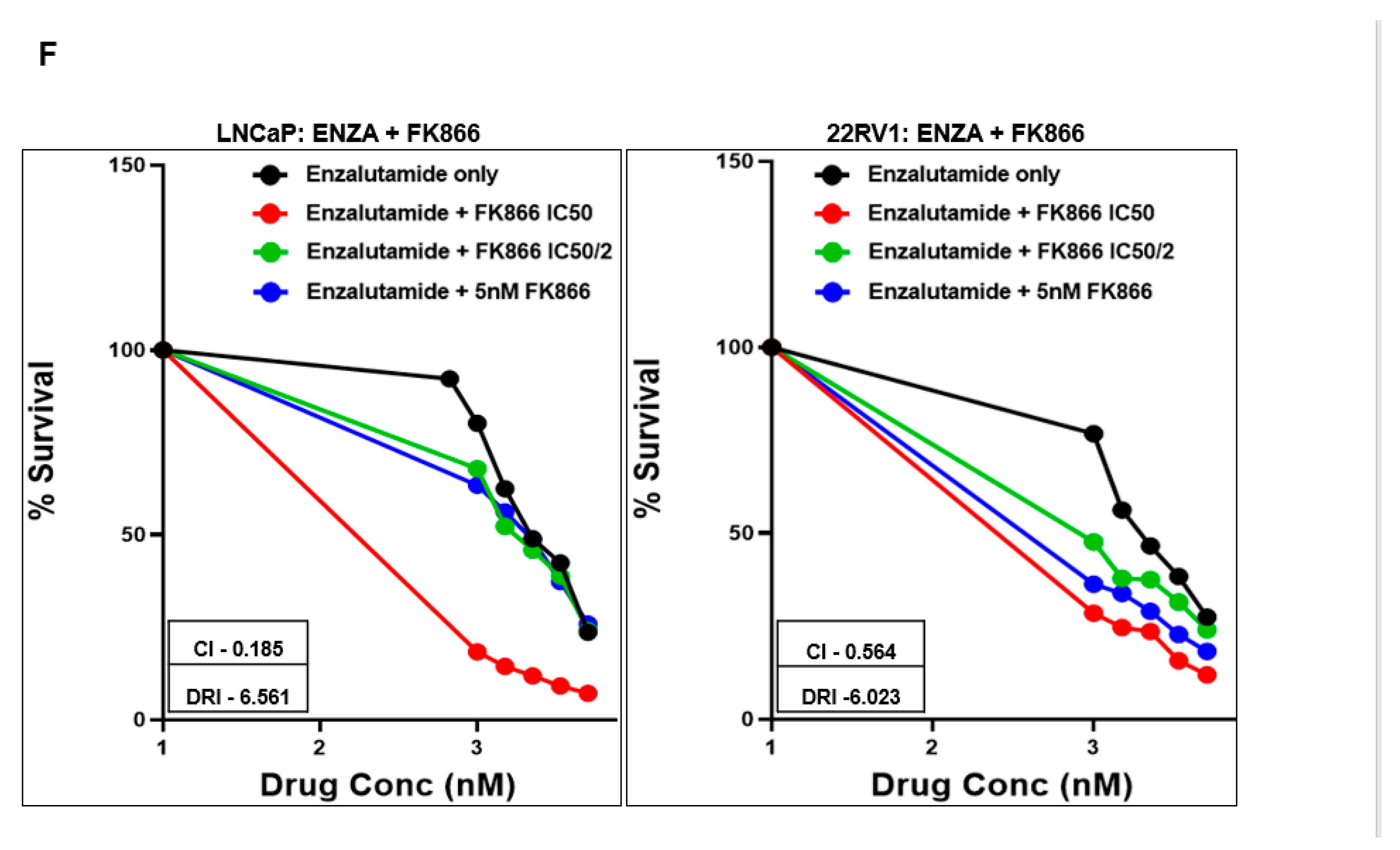
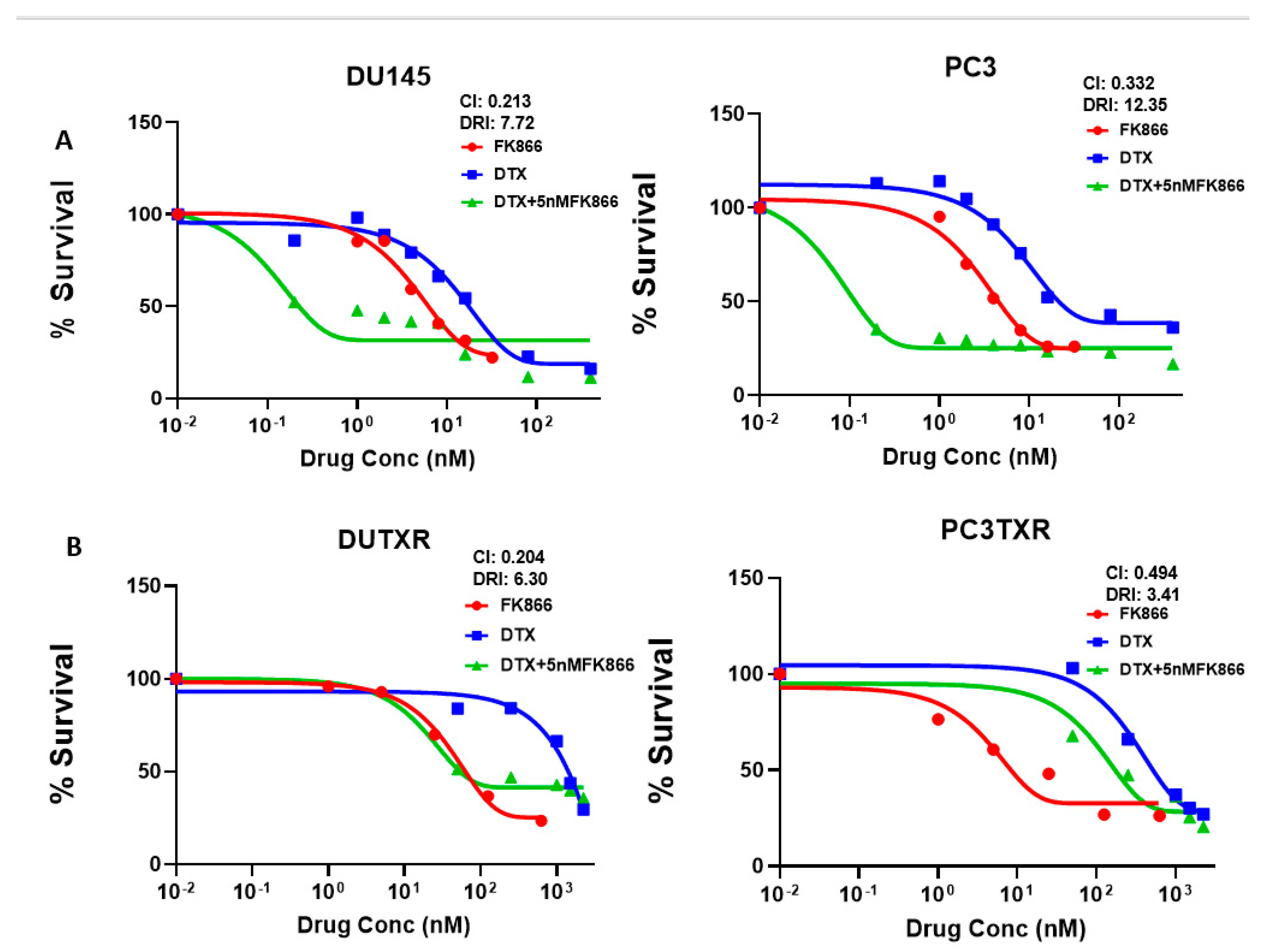
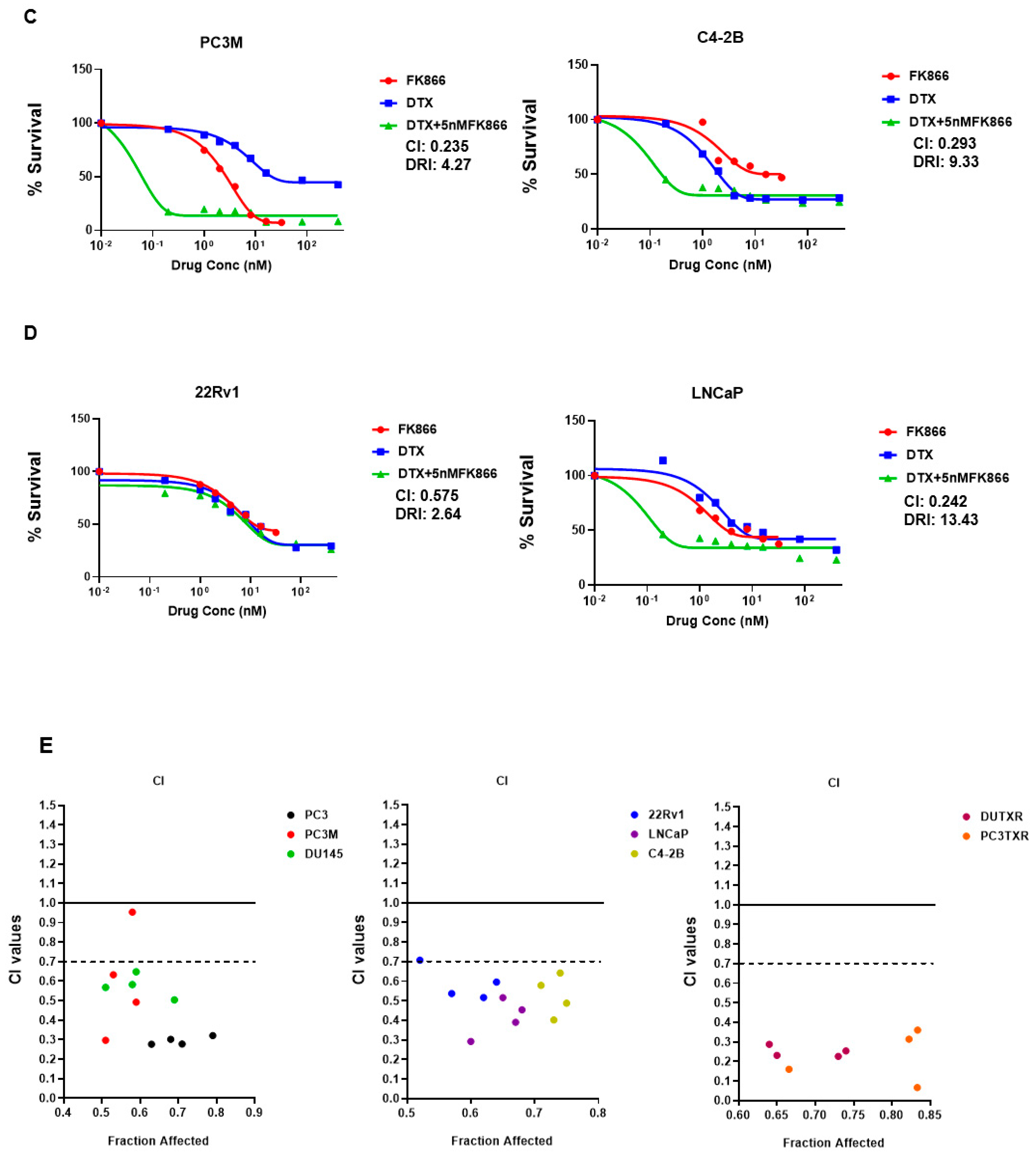
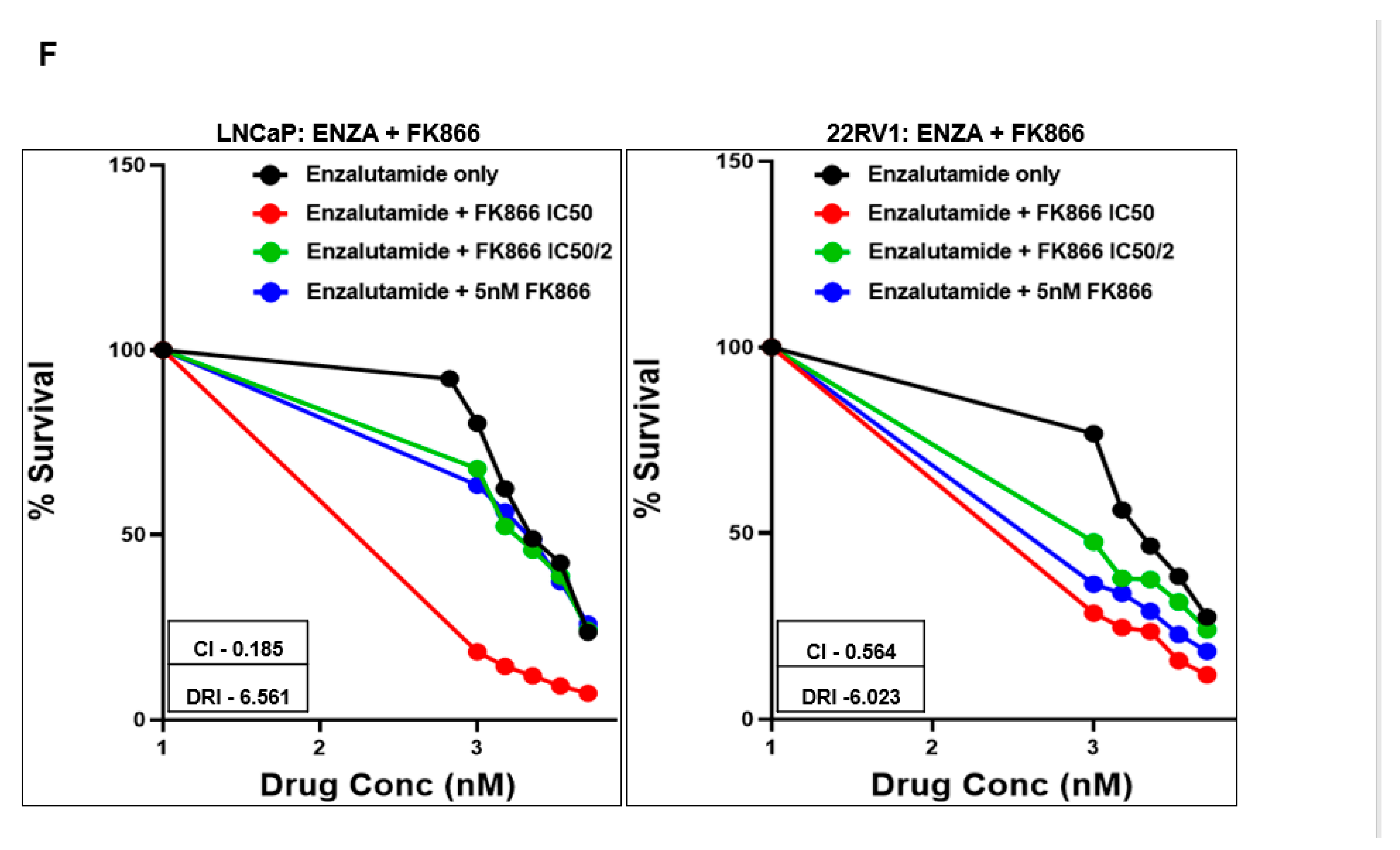
3.3. FK866 Induced Loss of Viability in PCa Cell Lines as Single-Agent Treatment and Showed Synergy with Taxanes and AR Inhibitors
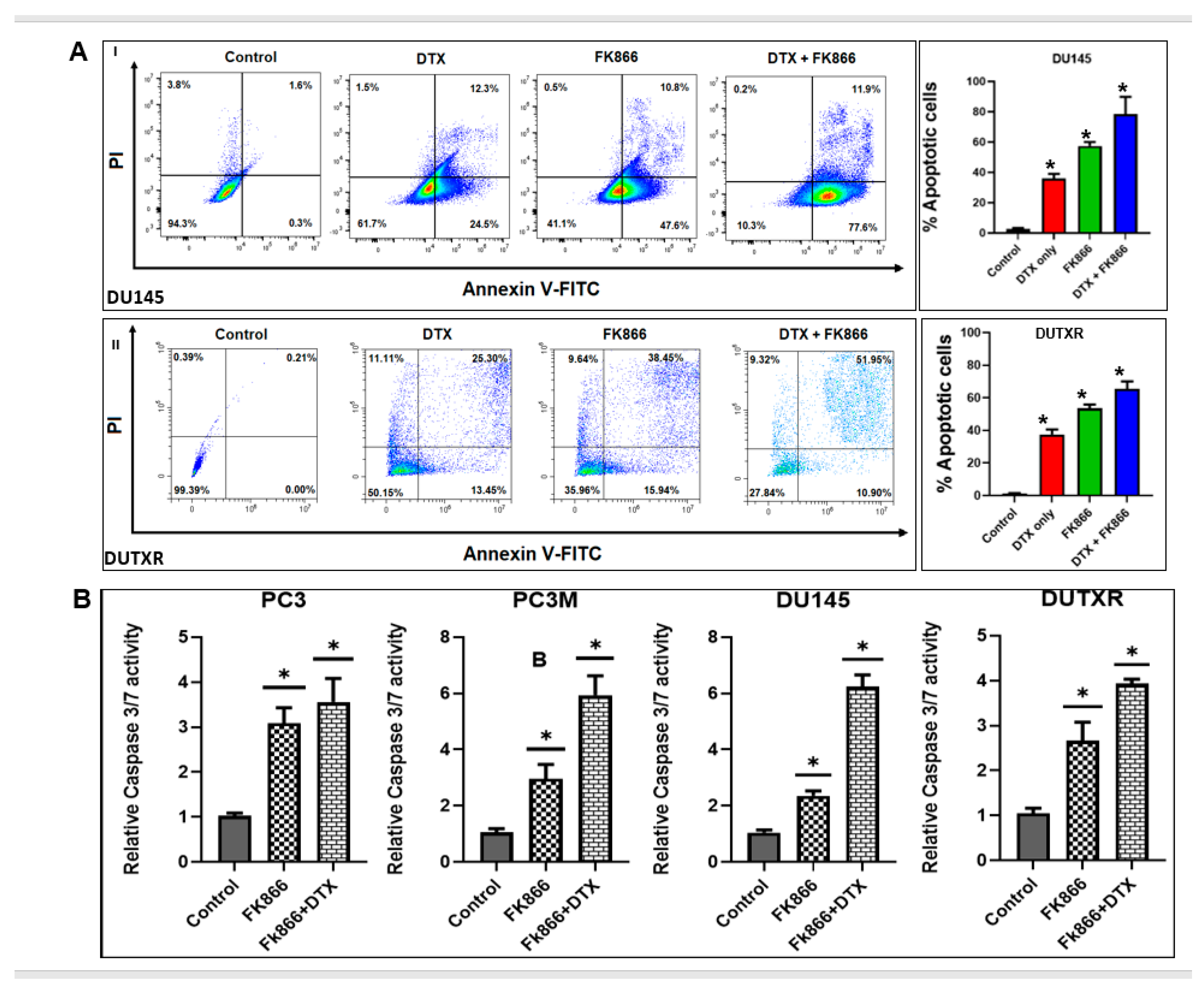
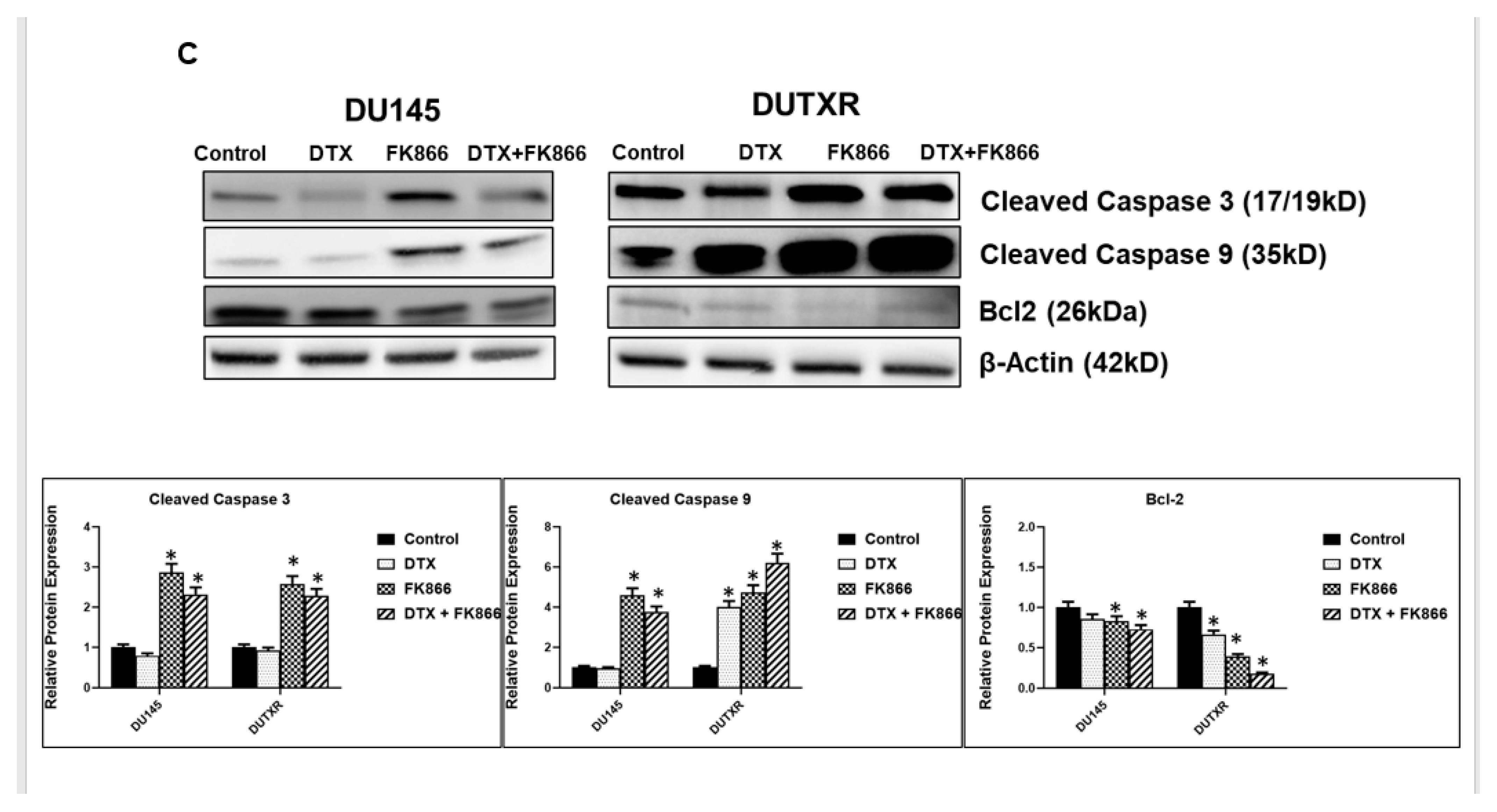
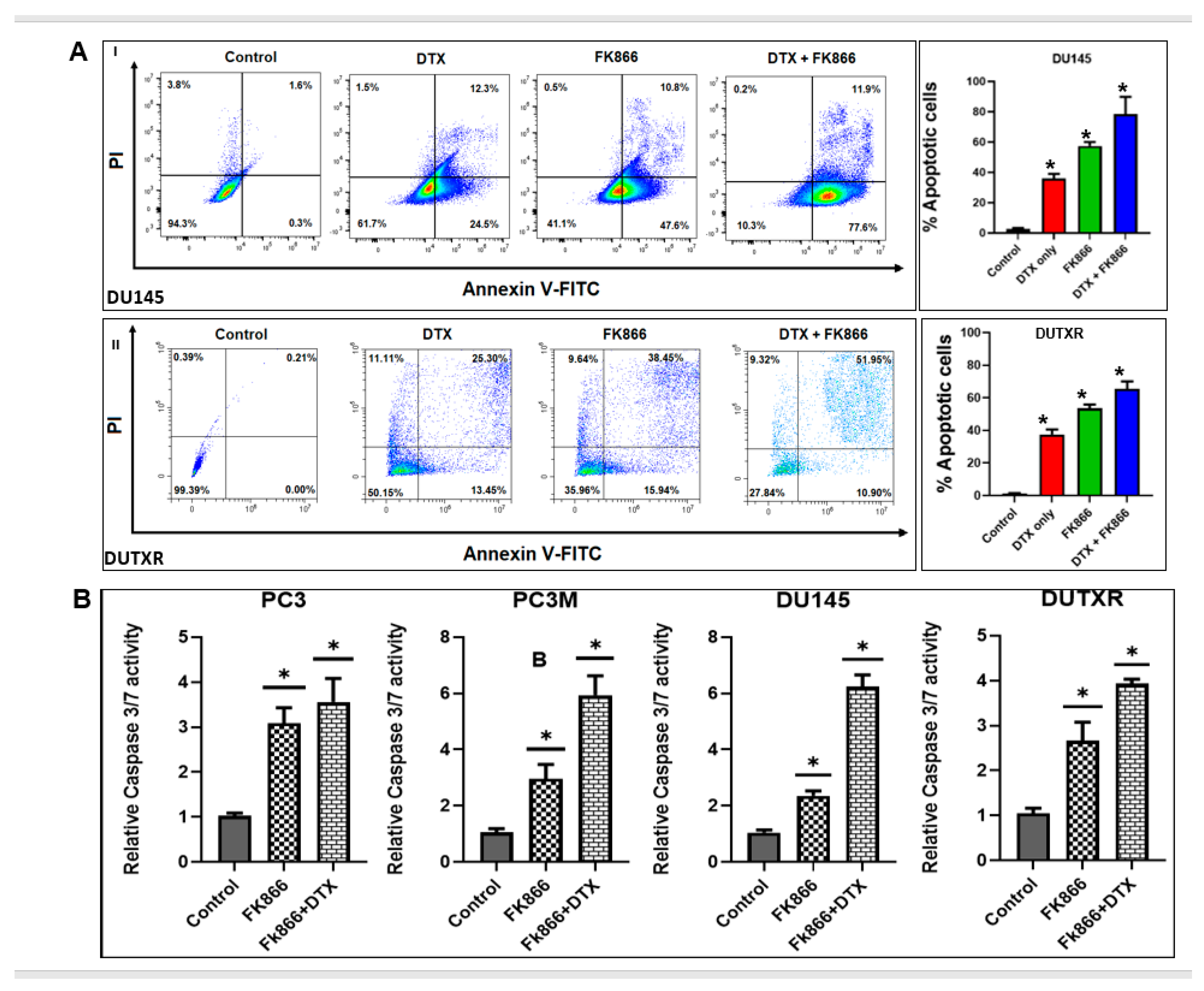
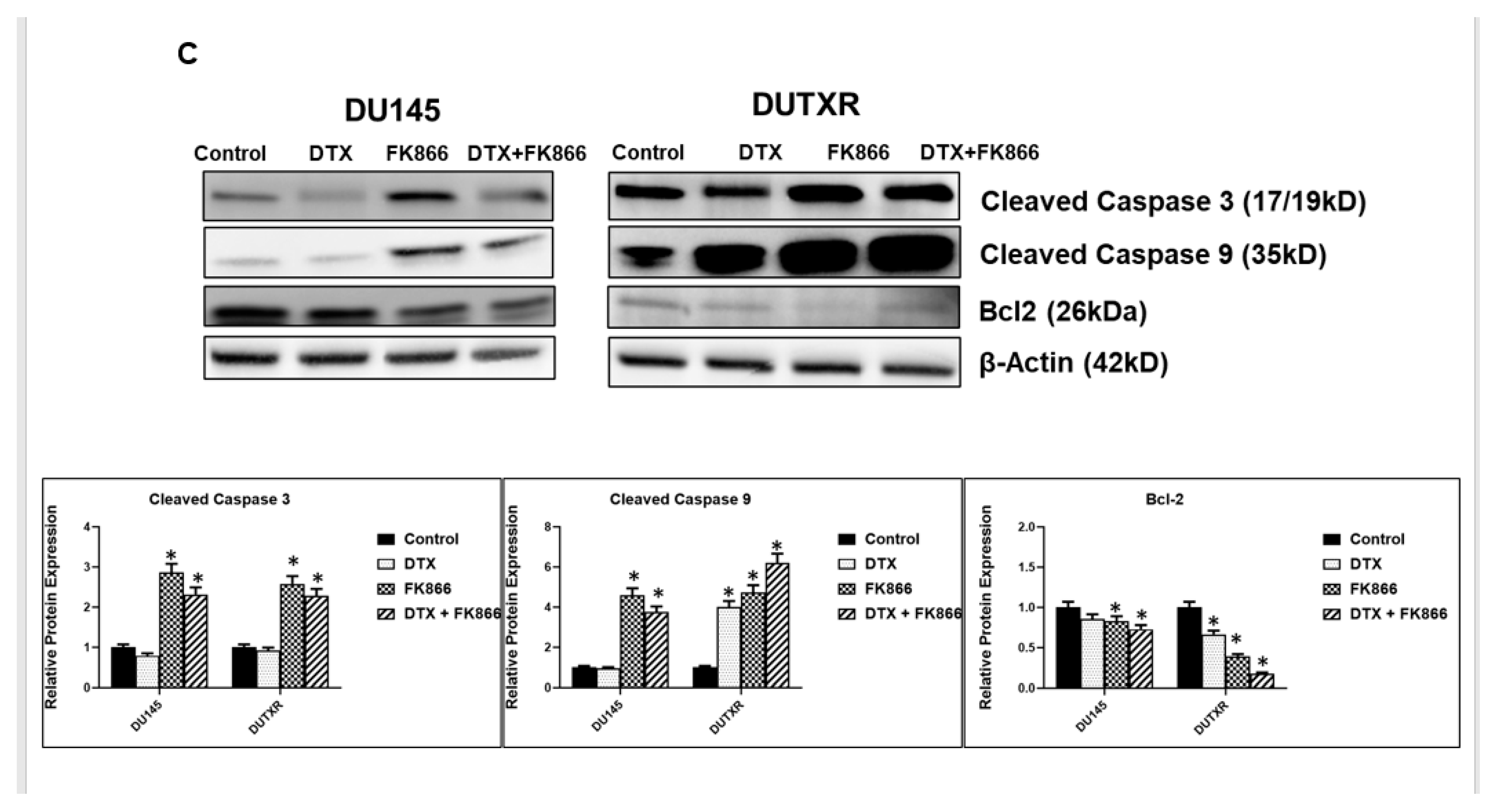
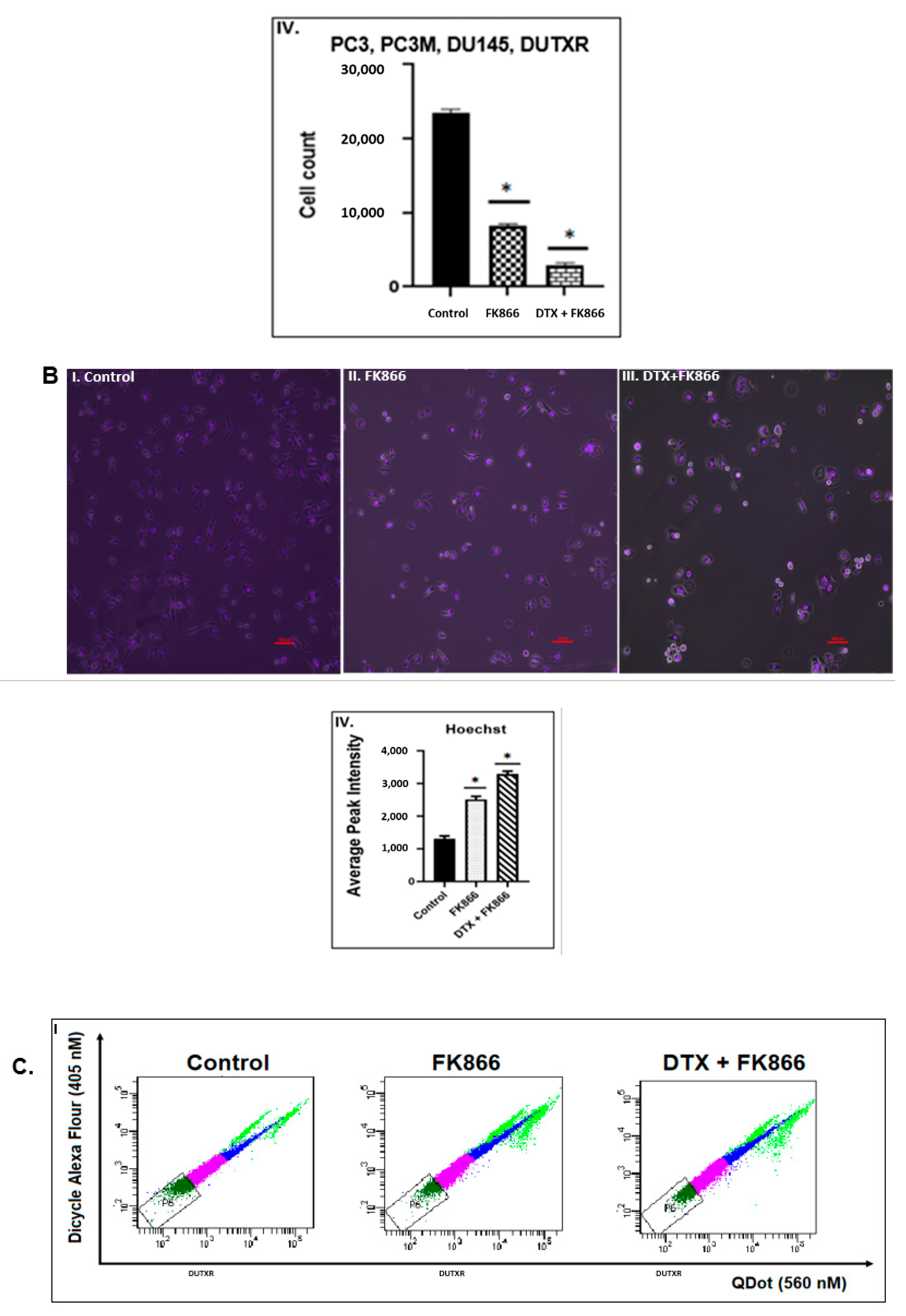
3.4. FK866 Induced Apoptosis in PCa Cell Lines
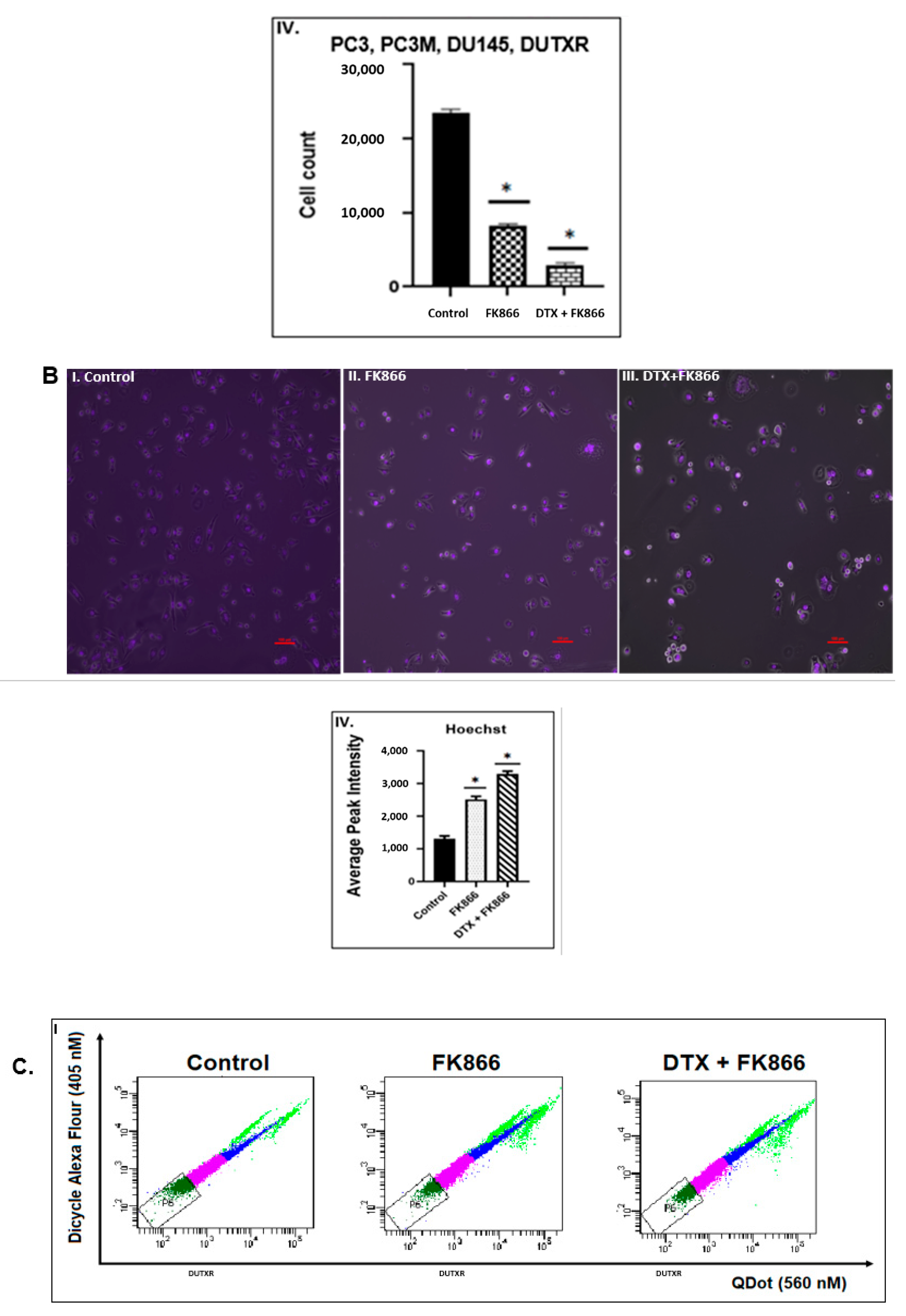
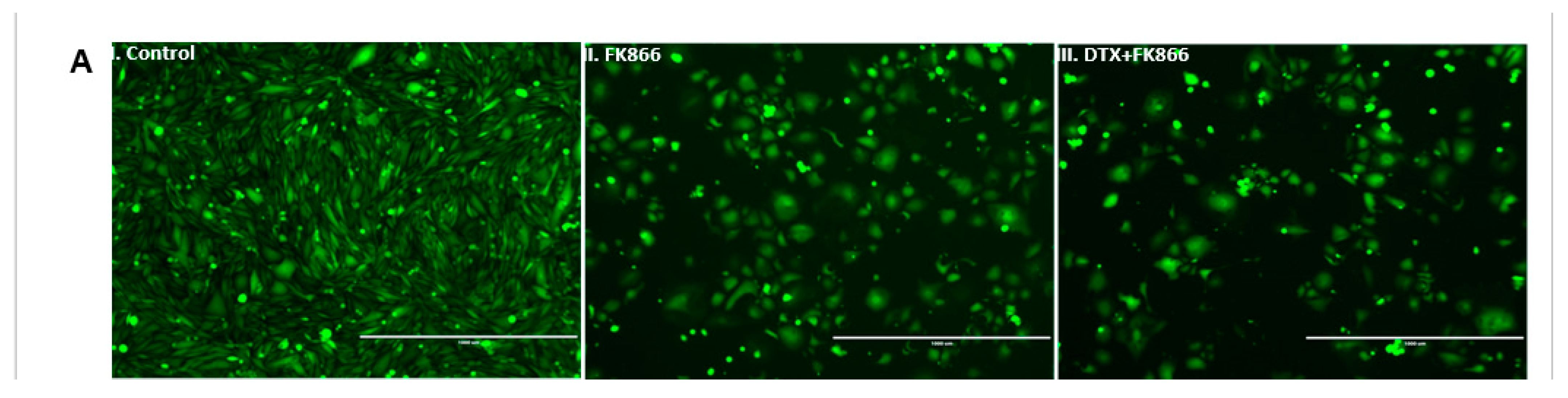
3.5. FK866 Treatment Diminished Cell Density and Altered Nuclear Morphology of PCa Cell Lines
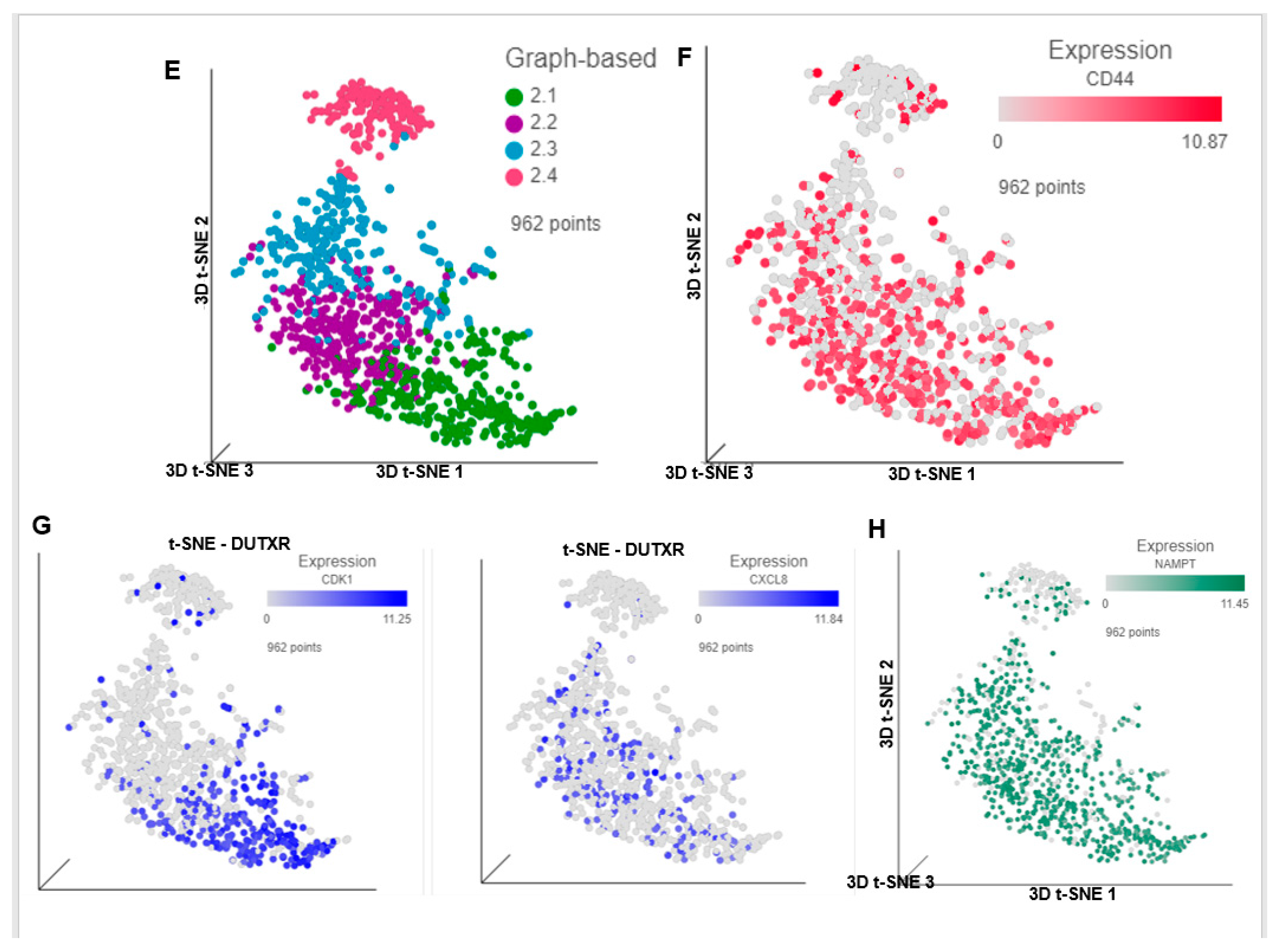
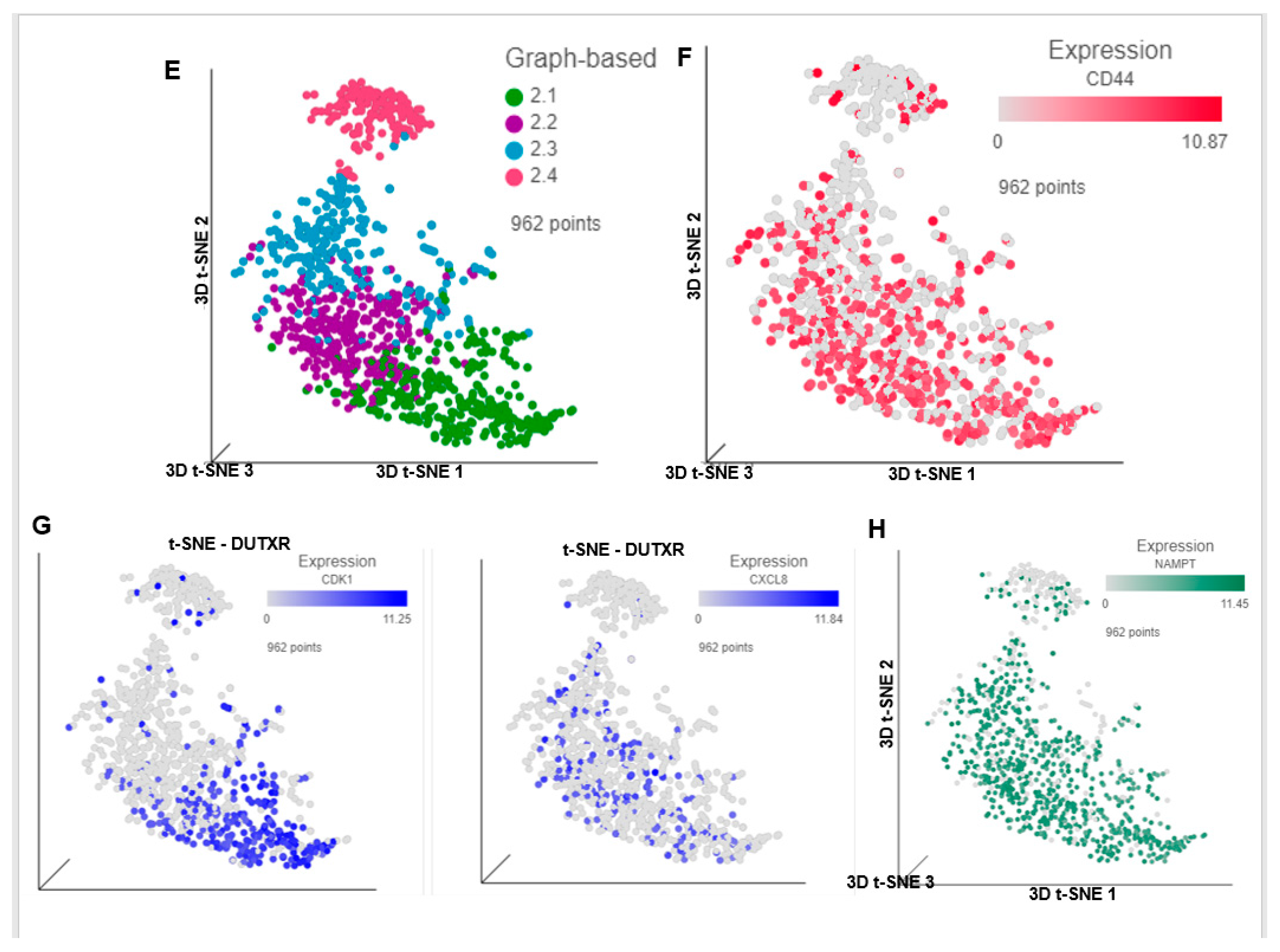
3.6. FK866 Potentially Decreased Stem Cell Load in Lethal PCa
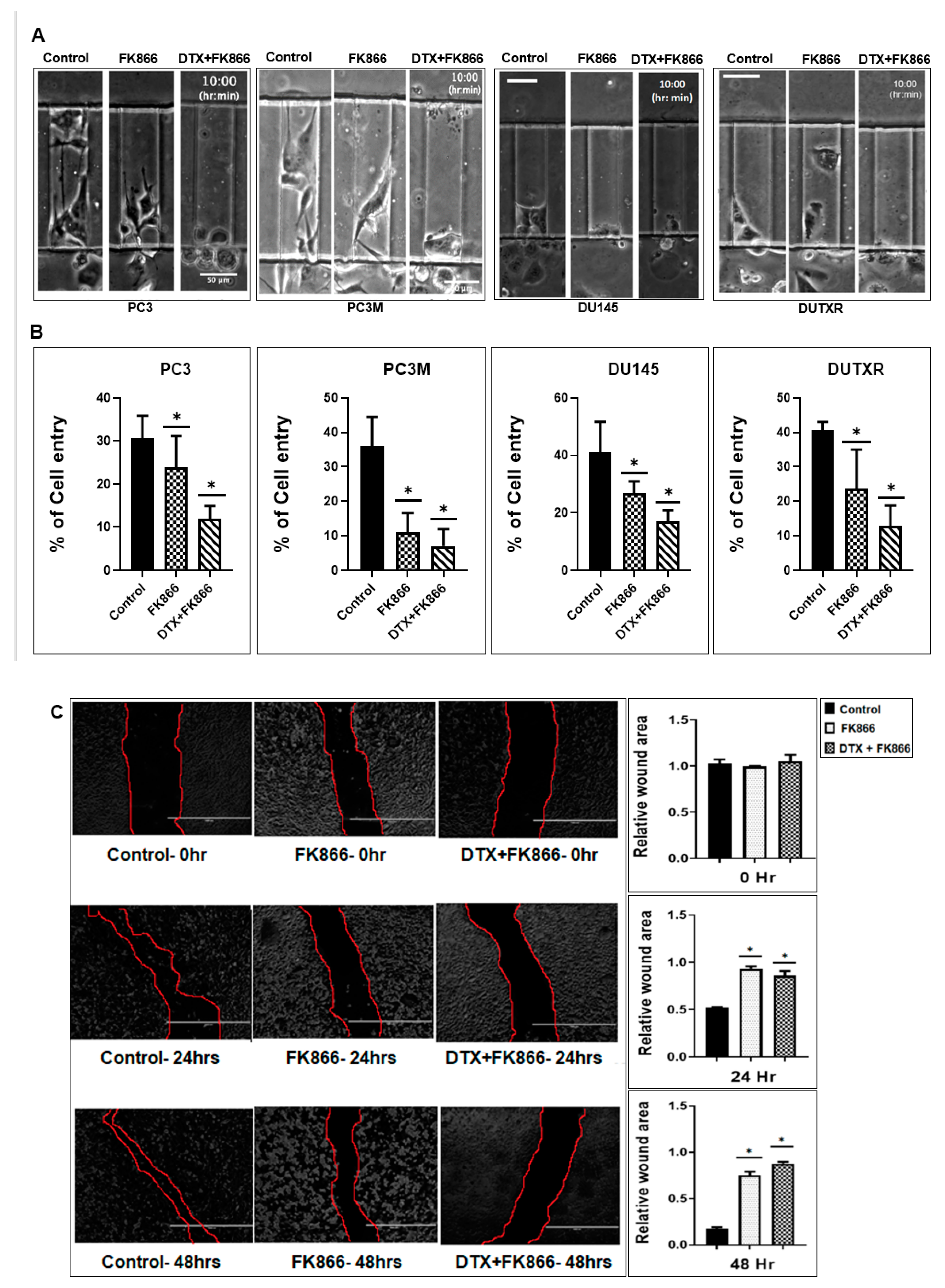
3.7. A Microfluidic Screen Followed by ‘Scratch Assay’ Showed FK866 Is Potentially Effective against EMT Transdifferentiation and Metastasis in Treatment-Refractory Aggressive Subclones
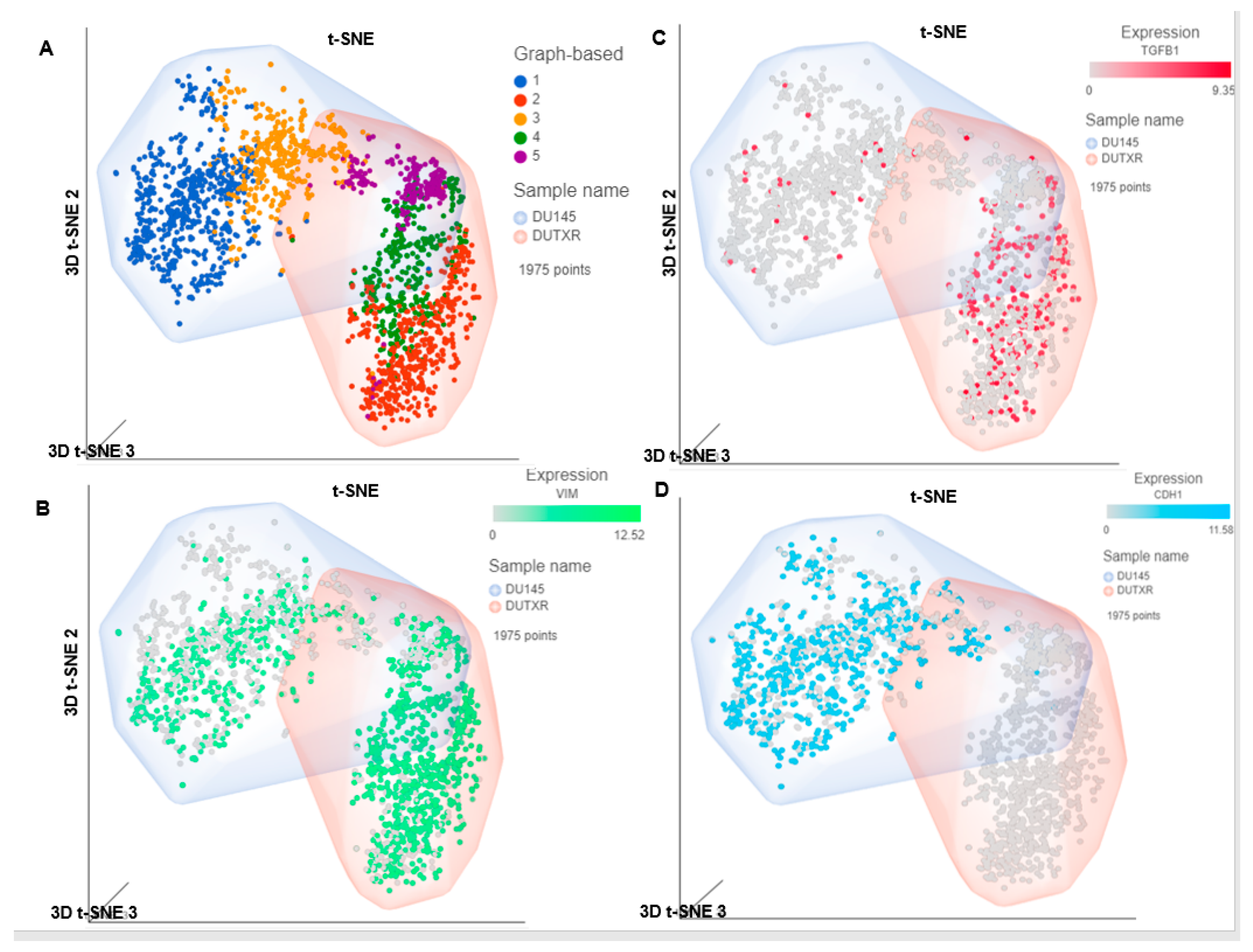
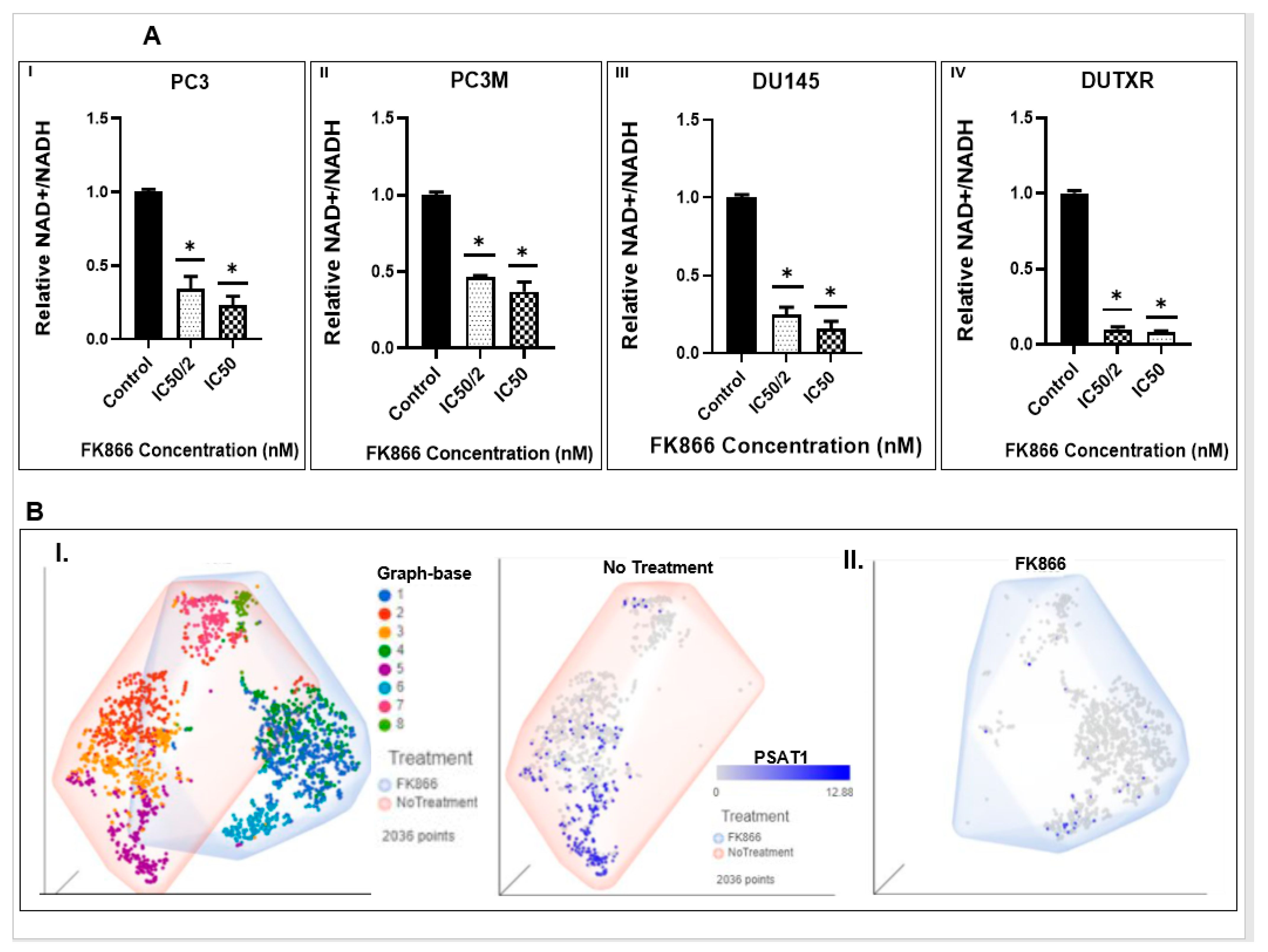
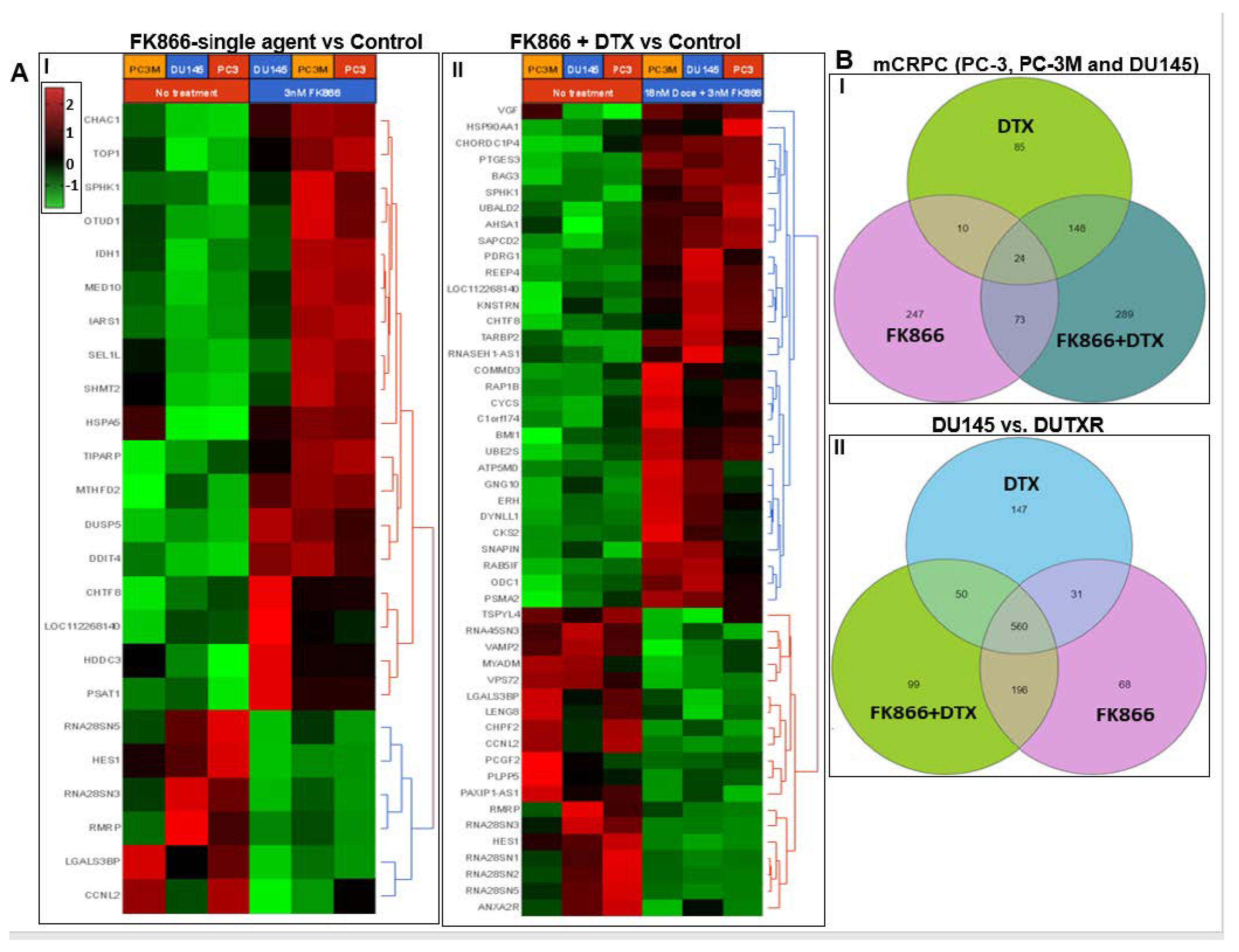
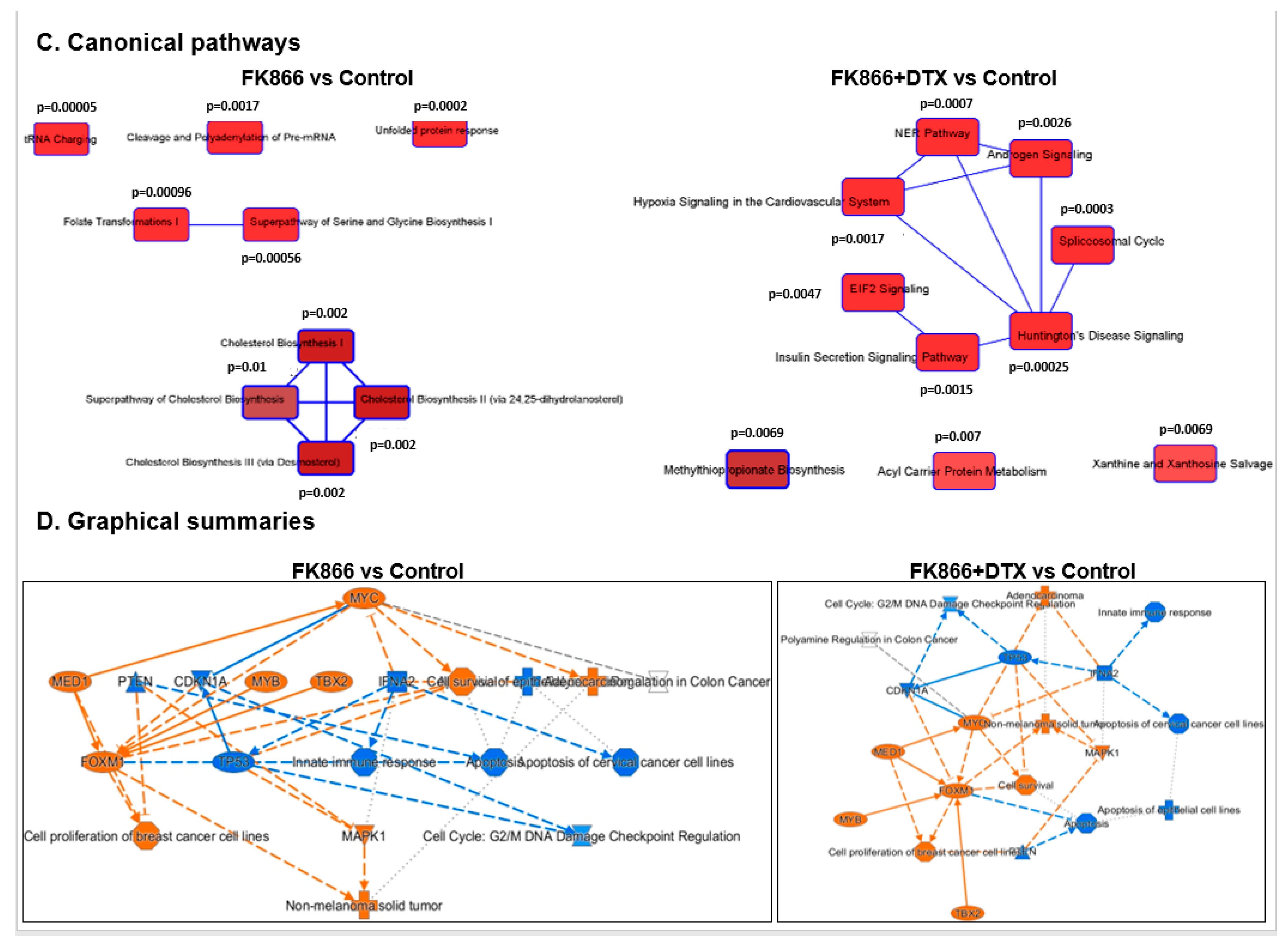
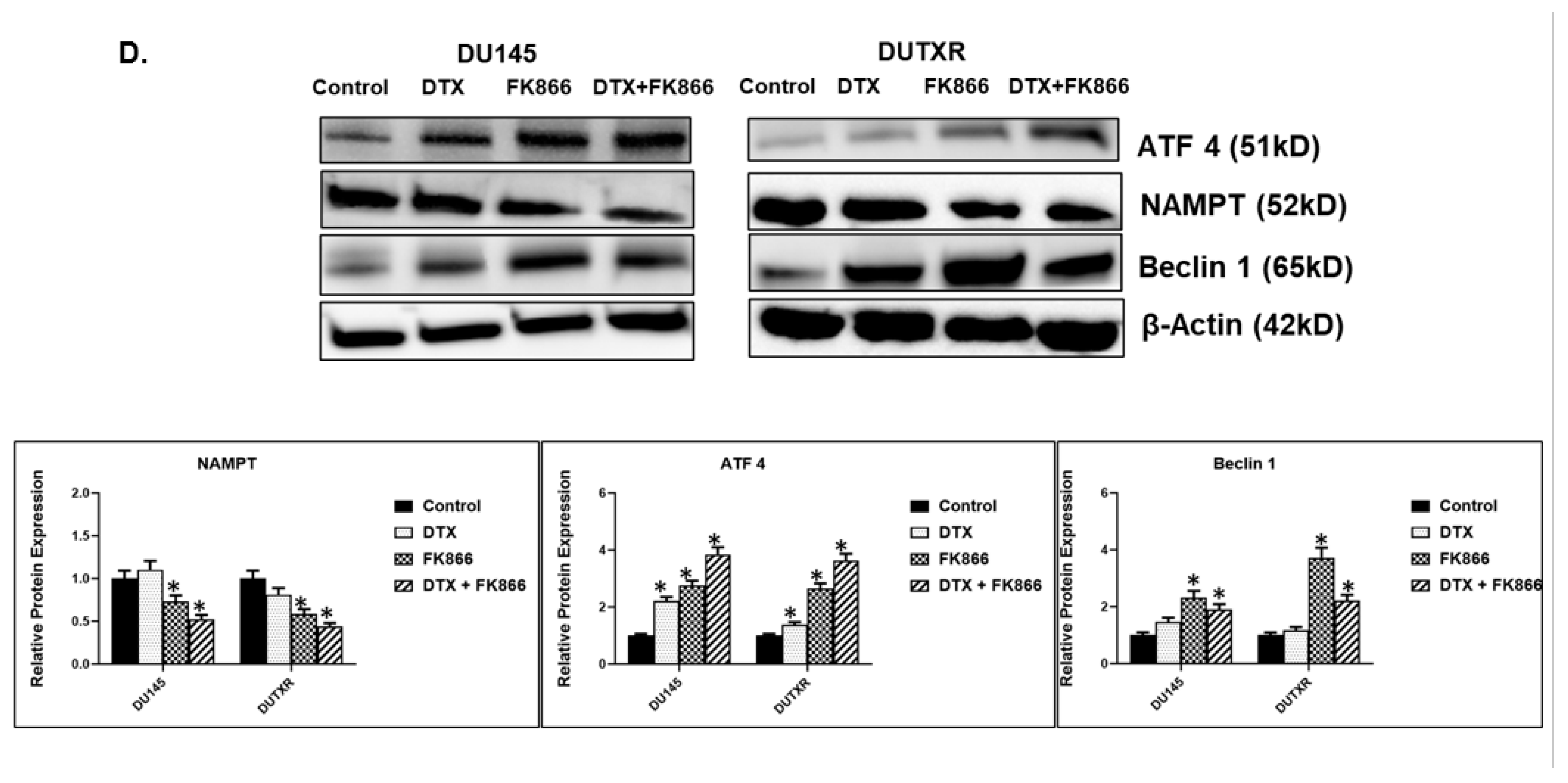
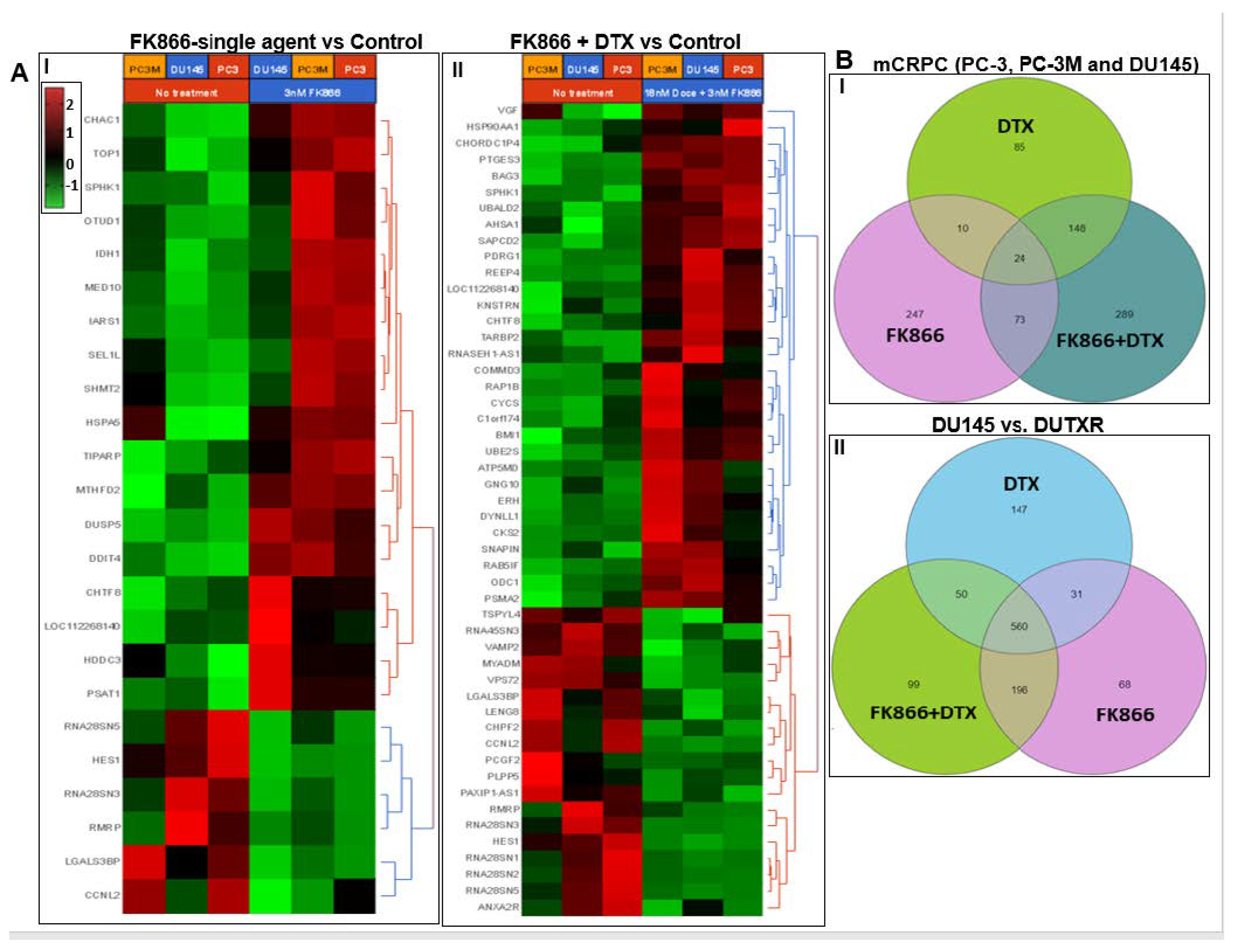
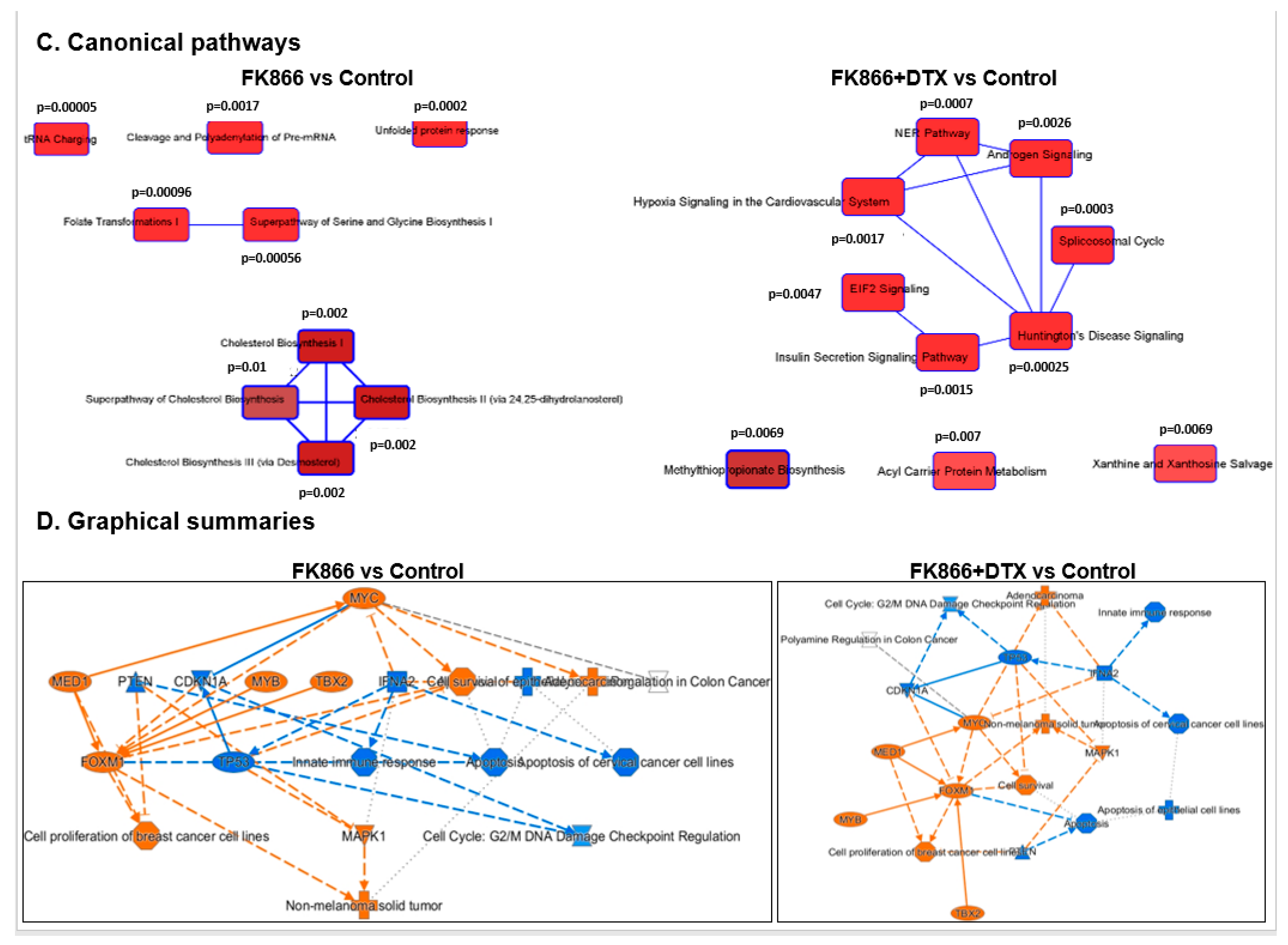
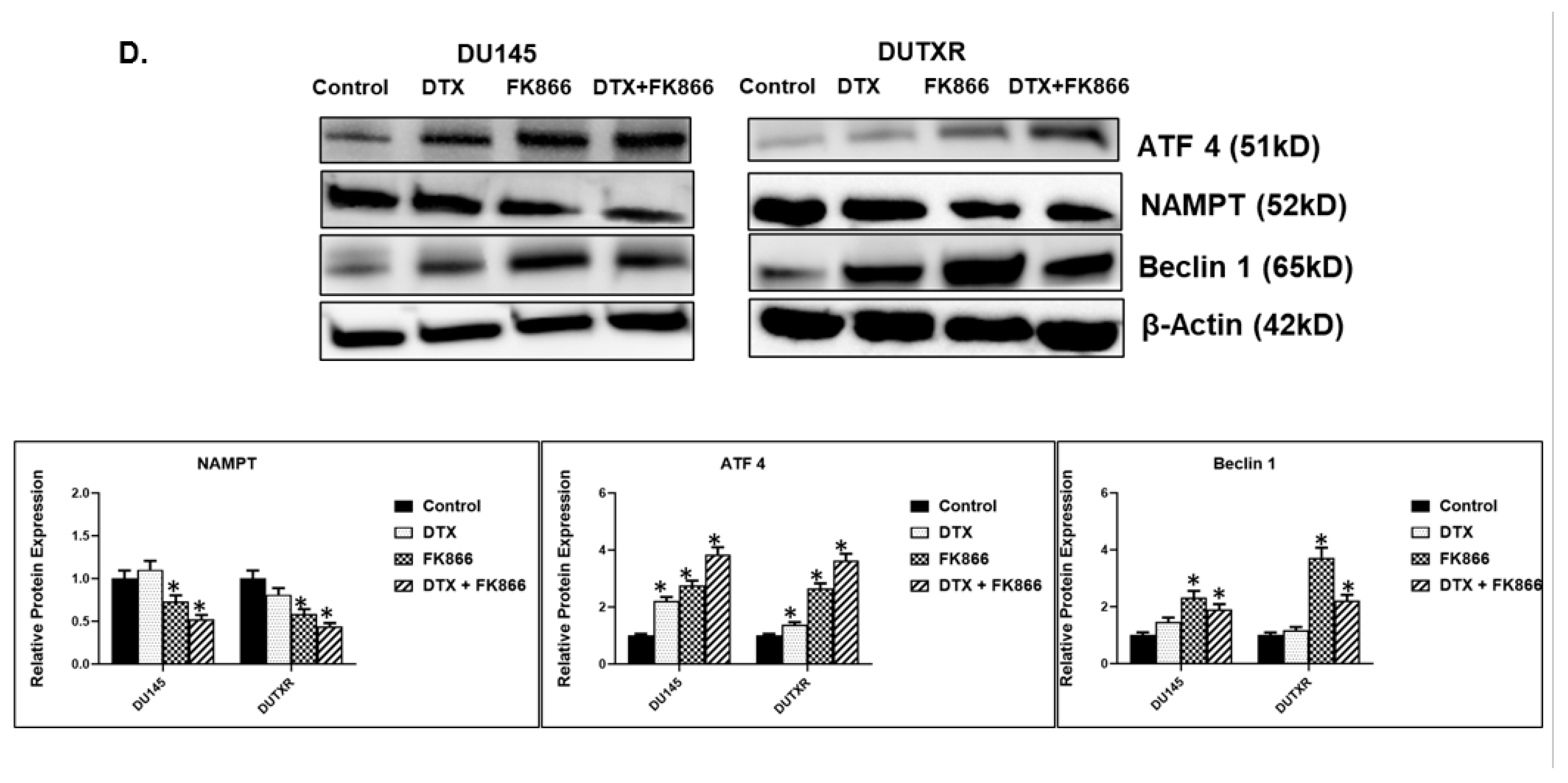
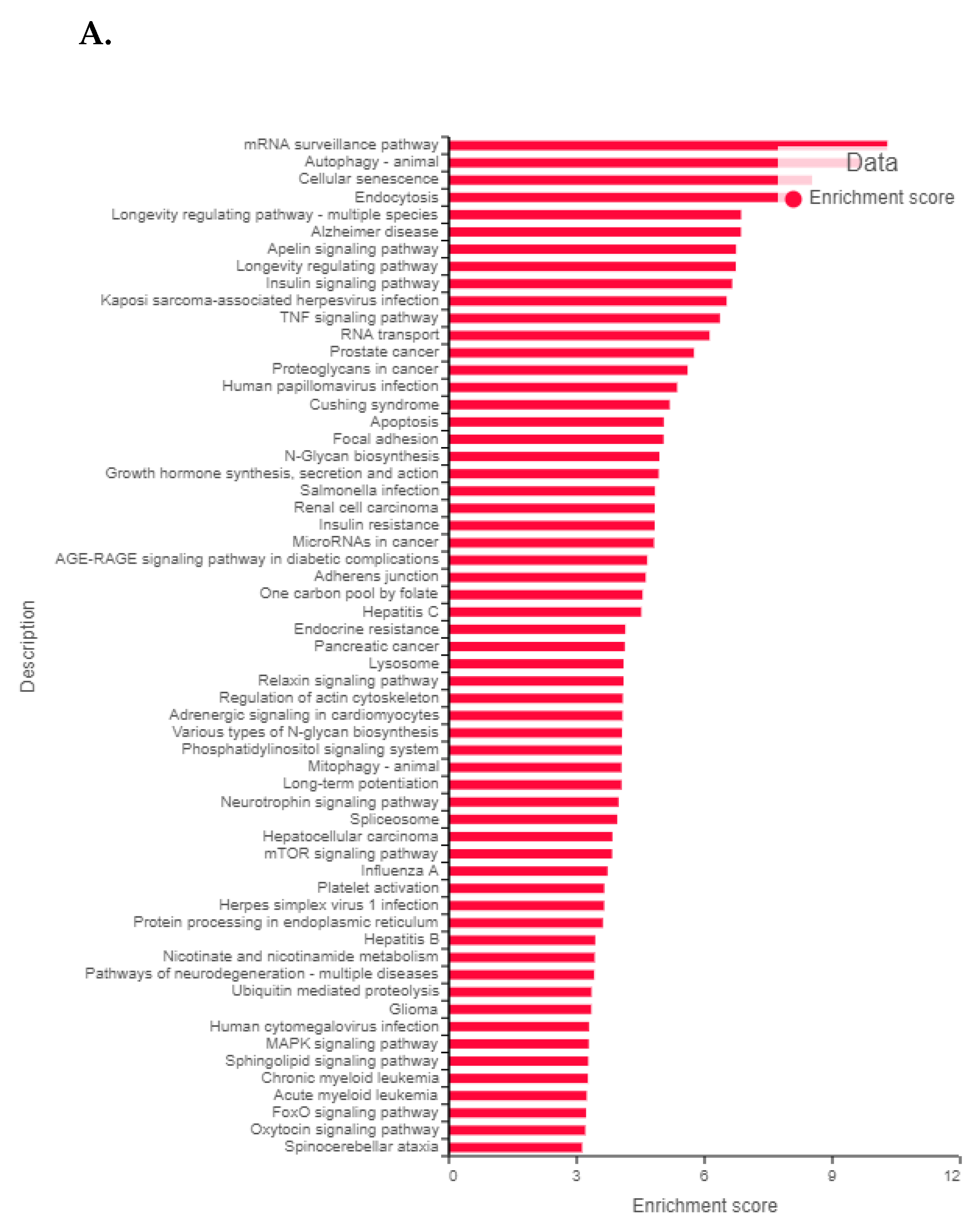
3.8. FK866 Showed Selective on-Target Inhibition of NAMPT Activity and a Distinct Impact on Gene Expression Signature
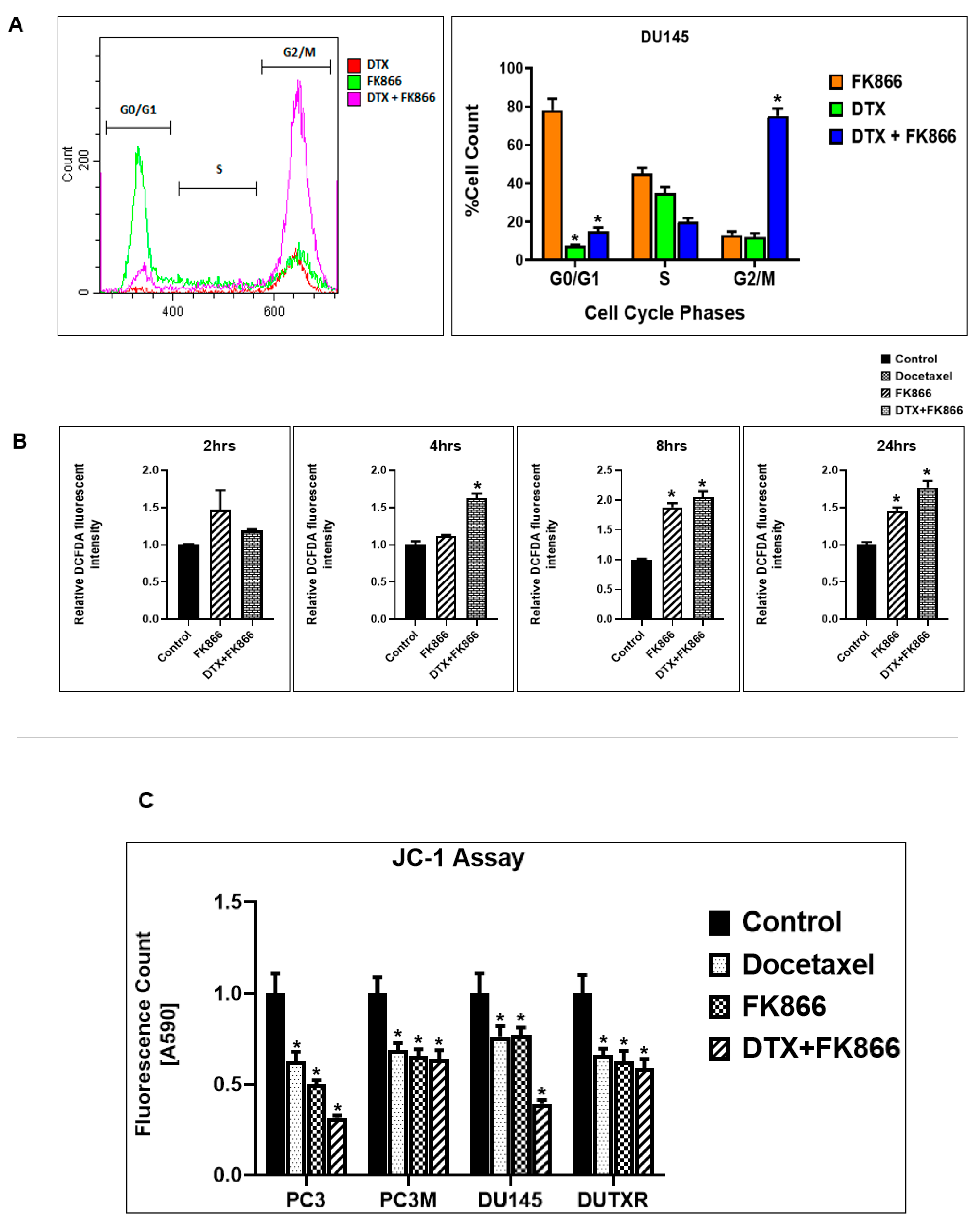
3.9. FK866 Treatment Results in Differential Regulation of Cell Cycle and Checkpoint Regulation Genes
3.10. FK866 Treatment Promotes Oxidative Stress and Mitochondrial-Mediated Pathway Gene Dysregulation
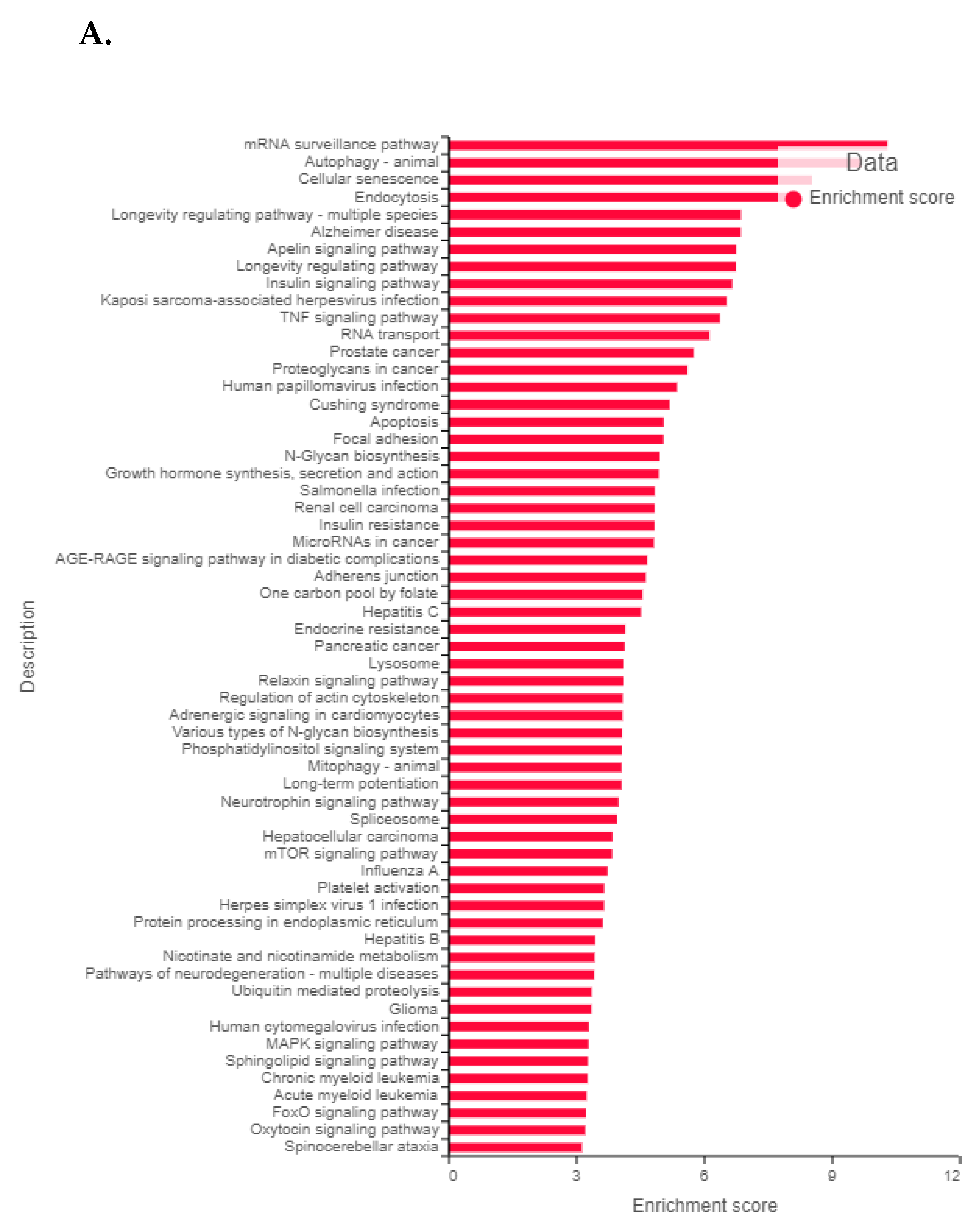
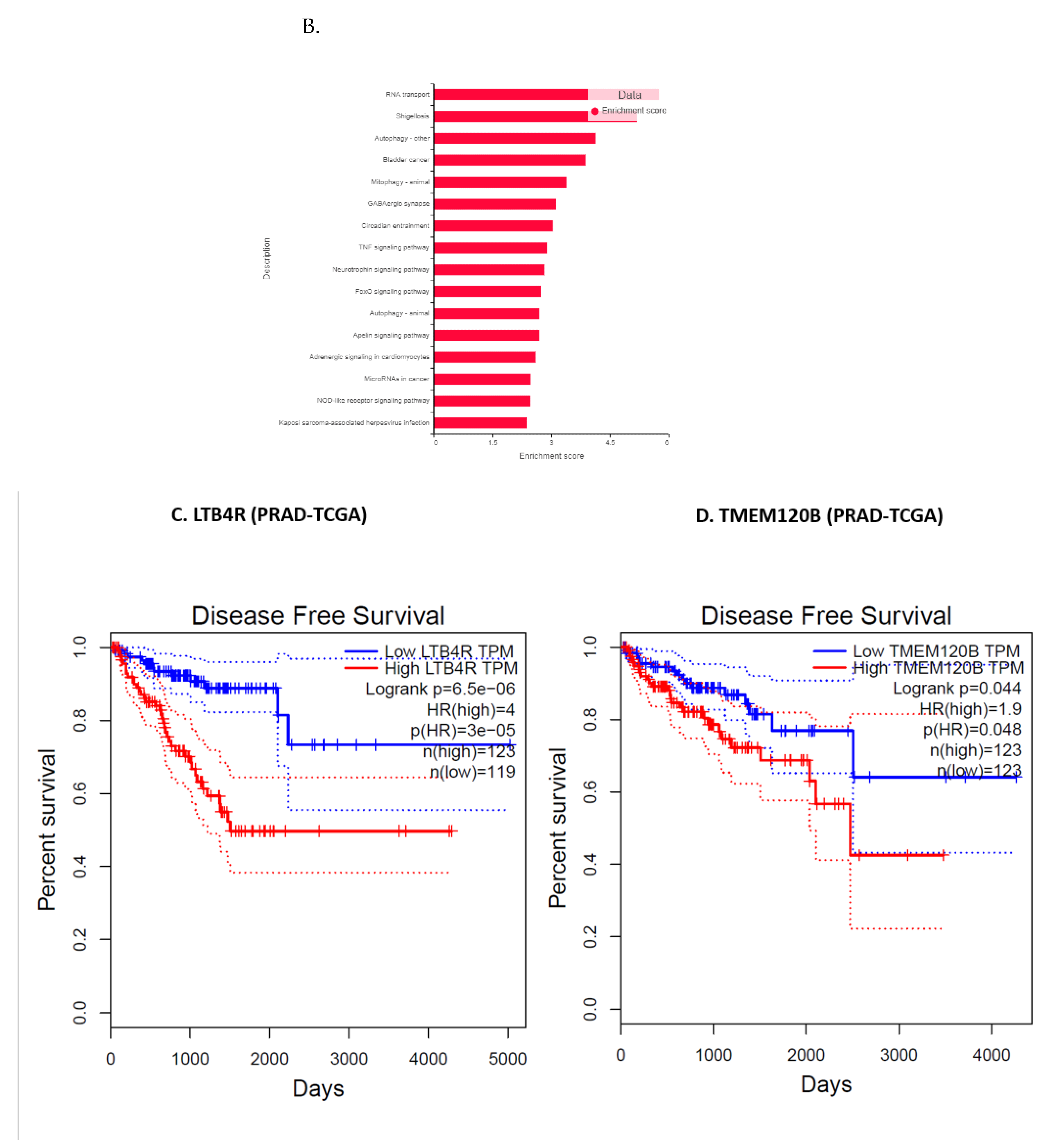
3.11. Validation of FK866 Treatment-Induced Gene Signatures Using Patient Datasets
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- American Cancer Society (2016). Cancer Facts and Figures 2016. American Cancer Society, Atlanta. Available online: http://www.cancer.org/acs/groups/content/@research/documents/document/acspc-047079.pdf (accessed on 31 August 2022).
- Cronin, K.A.; Lake, A.J.; Scott, S.; Sherman, R.L.; Noone, A.-M.; Howlader, N.; Henley, S.J.; Anderson, R.N.; Firth, A.U.; Ma, J.; et al. Annual Report to the Nation on the Status of Cancer, Part I: National Cancer Statistics. Cancer 2018, 124, 2785–2800. [Google Scholar] [CrossRef] [Green Version]
- Huggins, C.; Hodges, C.V. Studies on Prostatic Cancer i. the Effect of Castration, of Estrogen and of Androgen Injection on Serum Phosphatases in Metastatic Carcinoma of the Prostate. Cancer Res. 1941, 1, 293–297. [Google Scholar] [CrossRef]
- Nishiyama, T.; Hashimoto, Y.; Takahashi, K. The Influence of Androgen Deprivation Therapy on Dihydrotestosterone Levels in the Prostatic Tissue of Patients with Prostate Cancer. Clin. Cancer Res. 2004, 10, 7121–7126. [Google Scholar] [CrossRef] [Green Version]
- D’Amico, A.V.; Manola, J.; Loffredo, M.; Renshaw, A.A.; DellaCroce, A.; Kantoff, P.W. 6-Month Androgen Suppression plus Radiation Therapy vs Radiation Therapy Alone for Patients with Clinically Localized Prostate Cancer: A Randomized Controlled Trial. J. Am. Med. Assoc. 2004, 292, 821–827. [Google Scholar] [CrossRef] [Green Version]
- Pilepich, M.V.; Winter, K.; Lawton, C.A.; Krisch, R.E.; Wolkov, H.B.; Movsas, B.; Hug, E.B.; Asbell, S.O.; Grignon, D. Androgen Suppression Adjuvant to Definitive Radiotherapy in Prostate Carcinoma—Long-Term Results of Phase III RTOG 85–31. Int. J. Radiat. Oncol. Biol. Phys. 2005, 61, 1285–1290. [Google Scholar] [CrossRef]
- Scher, H.I.; Halabi, S.; Tannock, I.; Morris, M.; Sternberg, C.N.; Michael, A.; Eisenberger, M.A.; Higano, C.; Bubley, G.J.; Dreicer, R.; et al. NIH Public Access Progressive Prostate Cancer and Castrate Levels of Trials Working Group. J. Clin. Oncol. 2014, 26, 1148–1159. [Google Scholar] [CrossRef]
- Karantanos, T.; Corn, P.G.; Thompson, T.C. Prostate Cancer Progression after Androgen Deprivation Therapy: Mechanisms of Castrate-Resistance and Novel Therapeutic Approaches Theodoros. Curr. Opin. Support. Palliat. Care 2013, 7, 258–264. [Google Scholar] [CrossRef]
- Kapoor, A.; Wu, C.; Shayegan, B.; Rybak, A.P. Contemporary Agents in the Management of Metastatic Castration-Resistant Prostate Cancer. J. Can. Urol. Assoc. 2016, 10, E414–E423. [Google Scholar] [CrossRef] [Green Version]
- Wadosky, K.M.; Koochekpour, S. Molecular Mechanisms Underlying Resistance to Androgen Deprivation Therapy in Prostate Cancer. Oncotarget 2016, 7, 64447–64470. [Google Scholar] [CrossRef] [Green Version]
- Scher, H.I.; Solo, K.; Valant, J.; Todd, M.B.; Mehra, M. Prevalence of Prostate Cancer Clinical States and Mortality in the United States: Estimates Using a Dynamic Progression Model. PLoS ONE 2015, 10, e0139440. [Google Scholar] [CrossRef]
- American Cancer Society (2022). Cancer Facts & Figures 2022. American Cancer Society, Atlanta. Available online: https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2022/2022-cancer-facts-and-figures.pdf (accessed on 31 August 2022).
- Petrylak, D.P.; Tangen, C.M.; Hussain, M.H.A.; Lara, P.N.; Jones, J.A.; Taplin, M.E.; Burch, P.A.; Berry, D.; Moinpour, C.; Kohli, M.; et al. Docetaxel and Estramustine Compared with Mitoxantrone and Prednisone for Advanced Refractory Prostate Cancer. N. Engl. J. Med. 2004, 351, 1513–1520. [Google Scholar] [CrossRef] [Green Version]
- Sartor, A.O. Progression of Metastatic Castrate-Resistant Prostate Cancer: Impact of Therapeutic Intervention in the Post-Docetaxel Space. J. Hematol. Oncol. 2011, 4, 18. [Google Scholar] [CrossRef] [Green Version]
- Tannock, I.F.; de Wit, R.; Berry, W.R.; Horti, J.; Pluzanska, A.; Chi, K.N.; Oudard, S.; Théodore, C.; James, N.D.; Turesson, I.; et al. Docetaxel plus Prednisone or Mitoxantrone plus Prednisone for Advanced Prostate Cancer. N. Engl. J. Med. 2004, 351, 1502–1512. [Google Scholar] [CrossRef] [Green Version]
- Saad, F.; Miller, K. Treatment Options in Castration-Resistant Prostate Cancer: Current Therapies and Emerging Docetaxel-Based Regimens. Urol. Oncol. Semin. Orig. Investig. 2014, 32, 70–79. [Google Scholar] [CrossRef]
- De Bono, J.S.; Oudard, S.; Ozguroglu, M.; Hansen, S.; MacHiels, J.P.; Kocak, I.; Gravis, G.; Bodrogi, I.; MacKenzie, M.J.; Shen, L.; et al. Prednisone plus Cabazitaxel or Mitoxantrone for Metastatic Castration-Resistant Prostate Cancer Progressing after Docetaxel Treatment: A Randomised Open-Label Trial. Lancet 2010, 376, 1147–1154. [Google Scholar] [CrossRef]
- Cornford, P.; Bellmunt, J.; Bolla, M.; Briers, E.; De Santis, M.; Gross, T.; Henry, A.M.; Joniau, S.; Lam, T.B.; Mason, M.D.; et al. EAU-ESTRO-SIOG Guidelines on Prostate Cancer. Part II: Treatment of Relapsing, Metastatic, and Castration-Resistant Prostate Cancer. Eur. Urol. 2017, 71, 630–642. [Google Scholar] [CrossRef]
- Scher, H.I.; Fizazi, K.; Saad, F.; Taplin, M.-E.; Sternberg, C.N.; Miller, K.; de Wit, R.; Mulders, P.; Chi, K.N.; Shore, N.D.; et al. Increased Survival with Enzalutamide in Prostate Cancer after Chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. [Google Scholar] [CrossRef] [Green Version]
- Galletti, G.; Leach, B.I.; Lam, L.; Tagawa, S.T. Mechanisms of Resistance to Systemic Therapy in Metastatic Castration-Resistant Prostate Cancer. Cancer Treat. Rev. 2017, 57, 16–27. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, J.; Wu, Z.; Ding, W.; Gao, S.; Gao, Y.; Xu, C. Mechanisms of Enzalutamide Resistance in Castration-Resistant Prostate Cancer and Therapeutic Strategies to Overcome It. Br. J. Pharmacol. 2021, 178, 239–261. [Google Scholar] [CrossRef]
- Conteduca, V.; Oromendia, C.; Eng, K.W.; Bareja, R.; Sigouros, M.; Molina, A.; Faltas, B.M.; Sboner, A.; Mosquera, J.M.; Elemento, O.; et al. Clinical Features of Neuroendocrine Prostate Cancer. Eur. J. Cancer 2019, 121, 7–18. [Google Scholar] [CrossRef]
- Vashchenko, N.; Abrahamsson, P.-A. Neuroendocrine Differentiation in Prostate Cancer: Implications for New Treatment Modalities. Eur. Urol. 2005, 47, 147–155. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, H.; Cannon, V.; Wolcott, K.M.; Song, H.; Yates, C. Side Population Rather than CD133+ Cells Distinguishes Enriched Tumorigenicity in HTERT-Immortalized Primary Prostate Cancer Cells. Mol. Cancer 2011, 10, 112. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Yang, R.; Gao, W.-Q. Contributions of Epithelial-Mesenchymal Transition and Cancer Stem Cells to the Development of Castration Resistance of Prostate Cancer. Mol. Cancer 2014, 13, 55. [Google Scholar] [CrossRef] [Green Version]
- Contreras, H.R.; López-Moncada, F.; Castellón, E.A. Cancer Stem Cell and Mesenchymal Cell Cooperative Actions in Metastasis Progression and Hormone Resistance in Prostate Cancer: Potential Role of Androgen and Gonadotropin-releasing Hormone Receptors (Review). Int. J. Oncol. 2020, 56, 1075–1082. [Google Scholar] [CrossRef]
- Kumar, H.; Mazumder, S.; Chakravarti, S.; Sharma, N.; Mukherjee, U.K.; Kumar, S.; Baughn, L.B.; Van Ness, B.G.; Mitra, A.K. SecDrug: A Pipeline to Discover Novel Drug Combinations to Kill Drug-Resistant Multiple Myeloma Cells Using a Greedy Set Cover Algorithm and Single-Cell Multi-Omics. Blood Cancer J. 2022, 12, 39. [Google Scholar] [CrossRef] [PubMed]
- Sauer, H.; Kampmann, H.; Khosravi, F.; Sharifpanah, F.; Wartenberg, M. The Nicotinamide Phosphoribosyltransferase Antagonist FK866 Inhibits Growth of Prostate Tumour Spheroids and Increases Doxorubicin Retention without Changes in Drug Transporter and Cancer Stem Cell Protein Expression. Clin. Exp. Pharmacol. Physiol. 2021, 48, 422–434. [Google Scholar] [CrossRef] [PubMed]
- Bowlby, S.C.; Thomas, M.J.; D’Agostino, R.B.; Kridel, S.J. Nicotinamide Phosphoribosyl Transferase (Nampt) Is Required for de Novo Lipogenesis in Tumor Cells. PLoS ONE 2012, 7, e40195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, M.; Mizokami, A.; Mamiya, K.; Li, Y.Q.; Zhang, J.; Keller, E.T.; Namiki, M. The Establishment of Two Paclitaxel-Resistant Prostate Cancer Cell Lines and the Mechanisms of Paclitaxel Resistance with Two Cell Lines. Prostate 2007, 67, 955–967. [Google Scholar] [CrossRef] [Green Version]
- Long, Q.; Xu, J.; Osunkoya, A.O.; Sannigrahi, S.; Johnson, B.A.; Zhou, W.; Gillespie, T.; Park, J.Y.; Nam, R.K.; Sugar, L.; et al. Global Transcriptome Analysis of Formalin-Fixed Prostate Cancer Specimens Identifies Biomarkers of Disease Recurrence. Cancer Res. 2014, 74, 3228–3237. [Google Scholar] [CrossRef] [Green Version]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.V.S.K.; Varambally, S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Zhang, G.; Zhang, L.; Wang, Q.; Li, H.; Han, Y.; Xie, L.; Yan, Z.; Li, Y.; An, Y.; et al. Comprehensive Review of Web Servers and Bioinformatics Tools for Cancer Prognosis Analysis. Front. Oncol. 2020, 10, 68. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A Resource for Therapeutic Biomarker Discovery in Cancer Cells. Nucleic Acids Res. 2013, 41, D955-61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, T.C. The Mass-Action Law Based Algorithm for Cost-Effective Approach for Cancer Drug Discovery and Development. Am. J. Cancer Res. 2011, 1, 925–954. [Google Scholar]
- Wisniewski, E.O.; Mistriotis, P.; Bera, K.; Law, R.A.; Zhang, J.; Nikolic, M.; Weiger, M.; Parlani, M.; Tuntithavornwat, S.; Afthinos, A.; et al. Dorsoventral Polarity Directs Cell Responses to Migration Track Geometries. Sci. Adv. 2020, 6, eaba6505. [Google Scholar] [CrossRef] [PubMed]
- Krämer, A.; Green, J.; Pollard, J.; Tugendreich, S. Causal Analysis Approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef]
- Deng, L.; Gu, X.; Zeng, T.; Xu, F.; Dong, Z.; Liu, C.; Chao, H. Identification and Characterization of Biomarkers and Their Functions for Docetaxel-resistant Prostate Cancer Cells. Oncol. Lett. 2019, 18, 3236–3248. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Zhao, J.; Luo, Y.; Wang, Y.; Wei, N.; Jiang, Y. Isolation and Identification of Cancer Stem-like Cells from Side Population of Human Prostate Cancer Cells. J. Huazhong Univ. Sci. Technolog. Med. Sci. 2012, 32, 697–703. [Google Scholar] [CrossRef] [PubMed]
- Weigelin, B.; Bakker, G.-J.; Friedl, P. Intravital Third Harmonic Generation Microscopy of Collective Melanoma Cell Invasion: Principles of Interface Guidance and Microvesicle Dynamics. Intravital 2012, 1, 32–43. [Google Scholar] [CrossRef]
- Paul, C.D.; Mistriotis, P.; Konstantopoulos, K. Cancer Cell Motility: Lessons from Migration in Confined Spaces. Nat. Rev. Cancer 2017, 17, 131–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mistriotis, P.; Wisniewski, E.O.; Bera, K.; Keys, J.; Li, Y.; Tuntithavornwat, S.; Law, R.A.; Perez-Gonzalez, N.A.; Erdogmus, E.; Zhang, Y.; et al. Confinement Hinders Motility by Inducing RhoA-Mediated Nuclear Influx, Volume Expansion, and Blebbing. J. Cell Biol. 2019, 218, 4093–4111. [Google Scholar] [CrossRef]
- Wong, B.S.; Shea, D.J.; Mistriotis, P.; Tuntithavornwat, S.; Law, R.A.; Bieber, J.M.; Zheng, L.; Konstantopoulos, K. A Direct Podocalyxin-Dynamin-2 Interaction Regulates Cytoskeletal Dynamics to Promote Migration and Metastasis in Pancreatic Cancer Cells. Cancer Res. 2019, 79, 2878–2891. [Google Scholar] [CrossRef] [PubMed]
- Galli, U.; Colombo, G.; Travelli, C.; Tron, G.C.; Genazzani, A.A.; Grolla, A.A. Recent Advances in NAMPT Inhibitors: A Novel Immunotherapic Strategy. Front. Pharmacol. 2020, 11, 656. [Google Scholar] [CrossRef] [PubMed]
- Kanayama, M.; Luo, J. CD38-Induced Apoptosis and Mitochondrial Damage Is Restored by Nicotinamide in Prostate Cancer. Front. Mol. Biosci. 2022, 9, 890402. [Google Scholar] [CrossRef] [PubMed]
- Heske, C.M. Beyond Energy Metabolism: Exploiting the Additional Roles of NAMPT for Cancer Therapy. Front. Oncol. 2019, 9, 1514. [Google Scholar] [CrossRef] [Green Version]
- Audrito, V. The Dual Face of NAMPT: Intracellular/Extracellular Protein and Diagnostic/Therapeutic Target in Cancer. EBioMedicine 2020, 62, 103109. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.; Young, D.A.; Lu, Z.-H.; Wang, T.; Meier, T.I.; Shepard, R.L.; Roth, K.; Zhai, Y.; Huss, K.; Kuo, M.-S.; et al. Pharmacological Inhibition of Nicotinamide Phosphoribosyltransferase (NAMPT), an Enzyme Essential for NAD+ Biosynthesis, in Human Cancer Cells: Metabolic Basis and Potential Clinical Implications. J. Biol. Chem. 2013, 288, 3500–3511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Hasan, M.K.; Alvarado, E.; Yuan, H.; Wu, H.; Chen, W.Y. NAMPT Overexpression in Prostate Cancer and Its Contribution to Tumor Cell Survival and Stress Response. Oncogene 2011, 30, 907–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, P.; Zhang, L.; Shi, Q.-J.; Lu, Y.-B.; Wu, M.; Wei, E.-Q.; Zhang, W.-P. Nicotinamide Phosphoribosyltransferase Inhibitor APO866 Induces C6 Glioblastoma Cell Death via Autophagy. Pharmazie 2015, 70, 650–655. [Google Scholar] [PubMed]
- Nacarelli, T.; Fukumoto, T.; Zundell, J.A.; Fatkhutdinov, N.; Jean, S.; Cadungog, M.G.; Borowsky, M.E.; Zhang, R. NAMPT Inhibition Suppresses Cancer Stem-like Cells Associated with Therapy-Induced Senescence in Ovarian Cancer. Cancer Res. 2020, 80, 890–900. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Chen, L.; Fan, Y.; Hong, Y.; Yang, X.; Li, Y.; Lu, J.; Lv, J.; Pan, X.; Qu, F.; et al. IFITM3 Promotes Bone Metastasis of Prostate Cancer Cells by Mediating Activation of the TGF-β Signaling Pathway. Cell Death Dis. 2019, 10, 517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajapaksa, U.S.; Jin, C.; Dong, T. Malignancy and IFITM3: Friend or Foe? Front. Oncol. 2020, 10, 593245. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Gao, H.; Chen, P.; Jia, J.; Wu, S. Knockdown of Interferon-Induced Transmembrane Protein 3 Expression Suppresses Breast Cancer Cell Growth and Colony Formation and Affects the Cell Cycle. Oncol. Rep. 2013, 30, 171–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, B.; Wang, H.; Zong, G.; Li, P. The Role of IFITM3 in the Growth and Migration of Human Glioma Cells. BMC Neurol. 2013, 13, 210. [Google Scholar] [CrossRef] [Green Version]
- Howlader, N.; Noone, A.M.; Krapcho, M.; Miller, D.; Bishop, K.; Altekruse, S.F.; Kosary, C.L.; Yu, M.; Ruhl, J.; Tatalovich, Z. SEER Cancer Statistics Review, 1975–2013; National Cancer Institute: Bethesda, MD, USA, 2015. Available online: http//seer.cancer.gov/csr/1975_2013/ (accessed on 31 August 2022).
- Larré, S.; Tran, N.; Fan, C.; Hamadeh, H.; Champigneulles, J.; Azzouzi, R.; Cussenot, O.; Mangin, P.; Olivier, J.L. PGE2 and LTB4 Tissue Levels in Benign and Cancerous Prostates. Prostaglandins Other Lipid Mediat. 2008, 87, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Magrini, E.; Garlanda, C. Noncanonical Functions of C1s Complement Its Canonical Functions in Renal Cancer. Cancer Immunol. Res. 2021, 9, 855. [Google Scholar] [CrossRef]
- Cao, Z.; Xu, Y.; Guo, F.; Chen, X.; Ji, J.; Xu, H.; He, J.; Yu, Y.; Sun, Y.; Lu, X.; et al. FASN Protein Overexpression Indicates Poor Biochemical Recurrence-Free Survival in Prostate Cancer. Dis. Markers 2020, 2020, 3904947. [Google Scholar] [CrossRef]
- Gutierrez, L.S.; Noria, F.; Finol, H.; Sun, L.; Castellino, F.; Pollard, M. Fas Ligand Expression and Its Correlation with Apoptosis and Proliferation in Lobund-Wistar Prostate Carcinomas. Pathobiology 2005, 72, 260–268. [Google Scholar] [CrossRef]
- Gao, S.; Lee, P.; Wang, H.; Gerald, W.; Adler, M.; Zhang, L.; Wang, Y.-F.; Wang, Z. The Androgen Receptor Directly Targets the Cellular Fas/FasL-Associated Death Domain Protein-like Inhibitory Protein Gene to Promote the Androgen-Independent Growth of Prostate Cancer Cells. Mol. Endocrinol. 2005, 19, 1792–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Y.; Wang, M.; Tong, H.; Tan, Y.; Hu, X.; Wang, K.; Wan, X. Plasma Exosomes from Endometrial Cancer Patients Contain LGALS3BP to Promote Endometrial Cancer Progression. Oncogene 2021, 40, 633–646. [Google Scholar] [CrossRef]
- Maldonado, L.Y.; Arsene, D.; Mato, J.M.; Lu, S.C. Methionine Adenosyltransferases in Cancers: Mechanisms of Dysregulation and Implications for Therapy. Exp. Biol. Med. (Maywood) 2018, 243, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Munkley, J.; Maia, T.M.; Ibarluzea, N.; Livermore, K.E.; Vodak, D.; Ehrmann, I.; James, K.; Rajan, P.; Barbosa-Morais, N.L.; Elliott, D.J. Androgen-Dependent Alternative MRNA Isoform Expression in Prostate Cancer Cells. F1000Research 2018, 7, 1189. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Ci, M.; Yoshioka, M.; Mayumi, Y.; Boivin, A.; André, B.; Belleau, P.; Pascal, B.; Gan, L.; Lin, G.; et al. Prostate-Specific Genes and Their Regulation by Dihydrotestosterone. Prostate 2008, 68, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Toraih, E.A.; Fawzy, M.S.; El-Falouji, A.I.; Hamed, E.O.; Nemr, N.A.; Hussein, M.H.; Abd El Fadeal, N.M. Stemness-Related Transcriptional Factors and Homing Gene Expression Profiles in Hepatic Differentiation and Cancer. Mol. Med. 2016, 22, 653–663. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, X.; Jessen, W.J.; Al-Ahmadie, H.; Serio, A.M.; Lin, Y.; Shih, W.-J.; Reuter, V.E.; Scardino, P.T.; Shen, M.M.; Aronow, B.J.; et al. Activator Protein-1 Transcription Factors Are Associated with Progression and Recurrence of Prostate Cancer. Cancer Res. 2008, 68, 2132–2144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riedel, M.; Cai, H.; Stoltze, I.C.; Vendelbo, M.H.; Wagner, E.F.; Bakiri, L.; Thomsen, M.K. Targeting AP-1 Transcription Factors by CRISPR in the Prostate. Oncotarget 2021, 12, 1956–1961. [Google Scholar] [CrossRef] [PubMed]
- Shankar, E.; Song, K.; Corum, S.L.; Bane, K.L.; Wang, H.; Kao, H.-Y.; Danielpour, D. A Signaling Network Controlling Androgenic Repression of C-Fos Protein in Prostate Adenocarcinoma Cells. J. Biol. Chem. 2016, 291, 5512–5526. [Google Scholar] [CrossRef] [Green Version]
- Yao, S.; Bee, A.; Brewer, D.; Dodson, A.; Beesley, C.; Ke, Y.; Ambroisine, L.; Fisher, G.; Møller, H.; Dickinson, T.; et al. PRKC-ζ Expression Promotes the Aggressive Phenotype of Human Prostate Cancer Cells and Is a Novel Target for Therapeutic Intervention. Genes Cancer 2010, 1, 444–464. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Watson, J.T.; Marengo, S.R.; Decker, K.S.; Coleman, I.; Nelson, P.S.; Sikes, R.A. Gene Expression in the LNCaP Human Prostate Cancer Progression Model: Progression Associated Expression in Vitro Corresponds to Expression Changes Associated with Prostate Cancer Progression in Vivo. Cancer Lett. 2006, 244, 274–288. [Google Scholar] [CrossRef]
- Cheng, Y.; Li, L.; Qin, Z.; Li, X.; Qi, F. Identification of Castration-Resistant Prostate Cancer-Related Hub Genes Using Weighted Gene Co-Expression Network Analysis. J. Cell. Mol. Med. 2020, 24, 8006–8017. [Google Scholar] [CrossRef]
- Su, W.; Li, S.; Chen, X.; Yin, L.; Ma, P.; Ma, Y.; Su, B. GABARAPL1 Suppresses Metastasis by Counteracting PI3K/Akt Pathway in Prostate Cancer. Oncotarget 2017, 8, 4449–4459. [Google Scholar] [CrossRef] [Green Version]
- Xie, C.-W.; Zhou, Y.; Liu, S.-L.; Fang, Z.-Y.; Su, B.; Zhang, W. Gabarapl1 Mediates Androgen-Regulated Autophagy in Prostate Cancer. Tumour Biol. 2015, 36, 8727–8733. [Google Scholar] [CrossRef]
- Deng, J.; Hou, G.; Fang, Z.; Liu, J.; Lv, X.-D. Distinct Expression and Prognostic Value of OTU Domain-Containing Proteins in Non-Small-Cell Lung Cancer. Oncol. Lett. 2019, 18, 5417–5427. [Google Scholar] [CrossRef] [Green Version]
- He, S.; Zhang, M.; Ye, Y.; Zhuang, J.; Ma, X.; Song, Y.; Xia, W. ChaC Glutathione Specific γ-Glutamylcyclotransferase 1 Inhibits Cell Viability and Increases the Sensitivity of Prostate Cancer Cells to Docetaxel by Inducing Endoplasmic Reticulum Stress and Ferroptosis. Exp. Ther. Med. 2021, 22, 997. [Google Scholar] [CrossRef] [PubMed]
- Saavedra-García, P.; Roman-Trufero, M.; Al-Sadah, H.A.; Blighe, K.; López-Jiménez, E.; Christoforou, M.; Penfold, L.; Capece, D.; Xiong, X.; Miao, Y.; et al. Systems Level Profiling of Chemotherapy-Induced Stress Resolution in Cancer Cells Reveals Druggable Trade-Offs. Proc. Natl. Acad. Sci. USA 2021, 118, e2018229118. [Google Scholar] [CrossRef]
- Crawford, R.R.; Prescott, E.T.; Sylvester, C.F.; Higdon, A.N.; Shan, J.; Kilberg, M.S.; Mungrue, I.N. Human CHAC1 Protein Degrades Glutathione, and MRNA Induction Is Regulated by the Transcription Factors ATF4 and ATF3 and a Bipartite ATF/CRE Regulatory Element. J. Biol. Chem. 2015, 290, 15878–15891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez-Álvarez, M.; Strippoli, R.; Donadelli, M.; Bazhin, A.V.; Cordani, M. Sestrins as a Therapeutic Bridge between ROS and Autophagy in Cancer. Cancers 2019, 11, 1415. [Google Scholar] [CrossRef] [Green Version]
- Cackowski, F.C.; Eber, M.R.; Rhee, J.; Decker, A.M.; Yumoto, K.; Berry, J.E.; Lee, E.; Shiozawa, Y.; Jung, Y.; Aguirre-Ghiso, J.A.; et al. Mer Tyrosine Kinase Regulates Disseminated Prostate Cancer Cellular Dormancy. J. Cell. Biochem. 2017, 118, 891–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, C.; Marquardt, S.; Li, F.; Spitschak, A.; Murr, N.; Edelhäuser, B.A.H.; Iliakis, G.; Pützer, B.M.; Logotheti, S. Rewiring E2F1 with Classical NHEJ via APLF Suppression Promotes Bladder Cancer Invasiveness. J. Exp. Clin. Cancer Res. 2019, 38, 292. [Google Scholar] [CrossRef] [PubMed]
- Majumder, A.; Syed, K.M.; Mukherjee, A.; Lankadasari, M.B.; Azeez, J.M.; Sreeja, S.; Harikumar, K.B.; Pillai, M.R.; Dutta, D. Enhanced Expression of Histone Chaperone APLF Associate with Breast Cancer. Mol. Cancer 2018, 17, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Y.; Qin, F.; Liu, A.; Li, H. Recurrent Fusion RNA DUS4L-BCAP29 in Non-Cancer Human Tissues and Cells. Oncotarget 2017, 8, 31415–31423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.-P.; Cho, G.-A.; Han, S.-W.; Shin, J.-Y.; Jeong, E.-G.; Song, S.-H.; Lee, W.-C.; Lee, K.-H.; Bang, D.; Seo, J.-S.; et al. Novel Fusion Transcripts in Human Gastric Cancer Revealed by Transcriptome Analysis. Oncogene 2014, 33, 5434–5441. [Google Scholar] [CrossRef]
- Wu, H.; Li, X.; Li, H. Gene Fusions and Chimeric RNAs, and Their Implications in Cancer. Genes Dis. 2019, 6, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-M.; Wu, H.-L.; Yu, X.; Tang, K.; Wang, S.-G.; Ye, Z.-Q.; Hu, J. The Putative Tumour Suppressor MiR-1-3p Modulates Prostate Cancer Cell Aggressiveness by Repressing E2F5 and PFTK1. J. Exp. Clin. Cancer Res. 2018, 37, 219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kojima, S.; Chiyomaru, T.; Kawakami, K.; Yoshino, H.; Enokida, H.; Nohata, N.; Fuse, M.; Ichikawa, T.; Naya, Y.; Nakagawa, M.; et al. Tumour Suppressors MiR-1 and MiR-133a Target the Oncogenic Function of Purine Nucleoside Phosphorylase (PNP) in Prostate Cancer. Br. J. Cancer 2012, 106, 405–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudson, R.S.; Yi, M.; Esposito, D.; Watkins, S.K.; Hurwitz, A.A.; Yfantis, H.G.; Lee, D.H.; Borin, J.F.; Naslund, M.J.; Alexander, R.B.; et al. MicroRNA-1 Is a Candidate Tumor Suppressor and Prognostic Marker in Human Prostate Cancer. Nucleic Acids Res. 2012, 40, 3689–3703. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.-C.; Huang, J.-C.; Zhang, L.-J.; Gan, B.-L.; Wen, D.-Y.; Chen, G.; Li, S.-H.; Yan, H.-B. Exploration of the Diagnostic Value and Molecular Mechanism of MiR-1 in Prostate Cancer: A Study Based on Meta-analyses and Bioinformatics. Mol. Med. Rep. 2018, 18, 5630–5646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Dalrymple, S.L.; Coleman, I.; Zheng, S.L.; Xu, J.; Hooper, J.E.; Antonarakis, E.S.; De Marzo, A.M.; Meeker, A.K.; Nelson, P.S.; et al. Role of Androgen Receptor Splice Variant-7 (AR-V7) in Prostate Cancer Resistance to 2nd-Generation Androgen Receptor Signaling Inhibitors. Oncogene 2020, 39, 6935–6949. [Google Scholar] [CrossRef]
- Liu, Z.-H.; Dai, X.-M.; Du, B. Hes1: A Key Role in Stemness, Metastasis and Multidrug Resistance. Cancer Biol. Ther. 2015, 16, 353–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soylu, H.; Kırca, M.; Avcı, S.; Ozpolat, B.; Ustunel, I. Antiandrogen Abiraterone and Docetaxel Treatments Affect Notch1, Jagged1 and Hes1 Expressions in Metastatic Prostate Cancer Cells. Exp. Mol. Pathol. 2021, 119, 104607. [Google Scholar] [CrossRef]
- Soylu, H.; Acar, N.; Ozbey, O.; Unal, B.; Koksal, I.T.; Bassorgun, I.; Ciftcioglu, A.; Ustunel, I. Characterization of Notch Signalling Pathway Members in Normal Prostate, Prostatic Intraepithelial Neoplasia (PIN) and Prostatic Adenocarcinoma. Pathol. Oncol. Res. 2016, 22, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, F.L.F.; Marchionni, L.; Gupta, A.; Kummangal, B.A.; Schaeffer, E.M.; Ross, A.E.; Berman, D.M. HES6 Promotes Prostate Cancer Aggressiveness Independently of Notch Signalling. J. Cell. Mol. Med. 2015, 19, 1624–1636. [Google Scholar] [CrossRef] [PubMed]
- Welch, D.R.; Hurst, D.R. Defining the Hallmarks of Metastasis. Cancer Res. 2019, 79, 3011–3027. [Google Scholar] [CrossRef] [PubMed]
- Van Zijl, F.; Krupitza, G.; Mikulits, W. Initial Steps of Metastasis: Cell Invasion and Endothelial Transmigration. Mutat. Res. 2011, 728, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.H.; Yang, J. Epithelial-Mesenchymal Plasticity in Carcinoma Metastasis. Genes Dev. 2013, 27, 2192–2206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Lee, S.J.; Park, S.; Konstantopoulos, K.; Glunde, K.; Chen, Y.; Barman, I. Cancer Cells Display Increased Migration and Deformability in Pace with Metastatic Progression. FASEB J. 2020, 34, 9307–9315. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.-H.V.; Middleton, K.; You, L.; Sun, Y. A Review of Microfluidic Approaches for Investigating Cancer Extravasation during Metastasis. Microsyst. Nanoeng. 2018, 4, 17104. [Google Scholar] [CrossRef] [Green Version]
- Thomas, E.; Thankan, R.S.; Purushottamachar, P.; Huang, W.; Kane, M.A.; Zhang, Y.; Ambulos, N.; Weber, D.J.; Njar, V.C.O. Transcriptome Profiling Reveals That VNPP433-3β, the Lead next-Generation Galeterone Analog Inhibits Prostate Cancer Stem Cells by Downregulating Epithelial-Mesenchymal Transition and Stem Cell Markers. Mol. Carcinog. 2022, 61, 643–654. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Name | Target | Target Pathway | |
|---|---|---|---|
| 1 | Afatinib | ERBB2, EGFR | EGFR signaling |
| 2 | AKT inhibitor VIII | AKT1, AKT2, AKT3 | PI3K/AKT pathway |
| 3 | AMG-706 (Motesanib) | VEGFR, RET, KIT, PDGFR | RTK signaling |
| 4 | AZD6482 | PI3Kβ | PI3K/MTOR signaling |
| 5 | Cetuximab | EGFR | EGFR signaling |
| 6 | CP724714 | ERBB2 | RTK signaling |
| 7 | FH535 | PPARγ, PPARδ | Wnt/β-catenin signaling |
| 8 | FK866 | NAMPT | NAD+ salvage pathway |
| 9 | GSK2126458 (Omipalisib) | PI3K (class 1), MTORC1, MTORC2 | PI3K/MTOR signaling |
| 10 | GW441756 | NTRK1 | RTK signaling |
| 11 | KIN001-260 | IKKB | NF-κB pathway |
| 12 | LY317615 | PKCB | Other, kinases |
| 13 | MK-2206 | AKT1, AKT2 | PI3K/MTOR signaling |
| 14 | Navitoclax | BCL2, BCL-XL, BCL-W | Apoptosis regulation |
| 15 | NSC-87877 | SHP-1 (PTPN6), SHP-2 (PTPN11) | Other |
| 16 | PD-0325901 | MEK1, MEK2 | ERK MAPK signaling |
| 17 | PD-173074 | FGFR1, FGFR2, FGFR3 | RTK signaling |
| 18 | PI-103 | PI3Kα, DAPK3, CLK4, PIM3, HIPK2 | Other, kinases |
| 19 | RDEA119 | MEK1, MEK2 | ERK MAPK signaling |
| 20 | SNX-2112 | HSP90 | Protein stability and degradation |
| 21 | TAK-715 | p38α, p38β | JNK and p38 signaling |
| 22 | TL-2-105 | CRAF | ERK MAPK signaling |
| 23 | WZ3105 | SRC, ROCK2, NTRK2, FLT3, IRAK1 | Other |
| 24 | XAV939 | TNKS1, TNKS2 | WNT signaling |
| 25 | YM155 | BIRC5 | Apoptosis regulation |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mazumder, S.; Mitra Ghosh, T.; Mukherjee, U.K.; Chakravarti, S.; Amiri, F.; Waliagha, R.S.; Hemmati, F.; Mistriotis, P.; Ahmed, S.; Elhussin, I.; et al. Integrating Pharmacogenomics Data-Driven Computational Drug Prediction with Single-Cell RNAseq to Demonstrate the Efficacy of a NAMPT Inhibitor against Aggressive, Taxane-Resistant, and Stem-like Cells in Lethal Prostate Cancer. Cancers 2022, 14, 6009. https://doi.org/10.3390/cancers14236009
Mazumder S, Mitra Ghosh T, Mukherjee UK, Chakravarti S, Amiri F, Waliagha RS, Hemmati F, Mistriotis P, Ahmed S, Elhussin I, et al. Integrating Pharmacogenomics Data-Driven Computational Drug Prediction with Single-Cell RNAseq to Demonstrate the Efficacy of a NAMPT Inhibitor against Aggressive, Taxane-Resistant, and Stem-like Cells in Lethal Prostate Cancer. Cancers. 2022; 14(23):6009. https://doi.org/10.3390/cancers14236009
Chicago/Turabian StyleMazumder, Suman, Taraswi Mitra Ghosh, Ujjal K. Mukherjee, Sayak Chakravarti, Farshad Amiri, Razan S. Waliagha, Farnaz Hemmati, Panagiotis Mistriotis, Salsabil Ahmed, Isra Elhussin, and et al. 2022. "Integrating Pharmacogenomics Data-Driven Computational Drug Prediction with Single-Cell RNAseq to Demonstrate the Efficacy of a NAMPT Inhibitor against Aggressive, Taxane-Resistant, and Stem-like Cells in Lethal Prostate Cancer" Cancers 14, no. 23: 6009. https://doi.org/10.3390/cancers14236009
APA StyleMazumder, S., Mitra Ghosh, T., Mukherjee, U. K., Chakravarti, S., Amiri, F., Waliagha, R. S., Hemmati, F., Mistriotis, P., Ahmed, S., Elhussin, I., Salam, A. -B., Dean-Colomb, W., Yates, C., Arnold, R. D., & Mitra, A. K. (2022). Integrating Pharmacogenomics Data-Driven Computational Drug Prediction with Single-Cell RNAseq to Demonstrate the Efficacy of a NAMPT Inhibitor against Aggressive, Taxane-Resistant, and Stem-like Cells in Lethal Prostate Cancer. Cancers, 14(23), 6009. https://doi.org/10.3390/cancers14236009