
T-Cell Mediated Immunity in Merkel Cell Carcinoma


{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Tumor Microenvironment in MCC
3. Role of Tumor Infiltrating Lymphocytes in MCC
3.1. CD8+ and CD4+ T-Cells
3.2. Regulatory T-Cells
3.3. Other T-Cell Subsets
4. T-Cell Receptor Repertoires
5. Chemokines and Cytokines in T-Cell Recruitment
6. Formation of Tertiary Lymphoid Structures
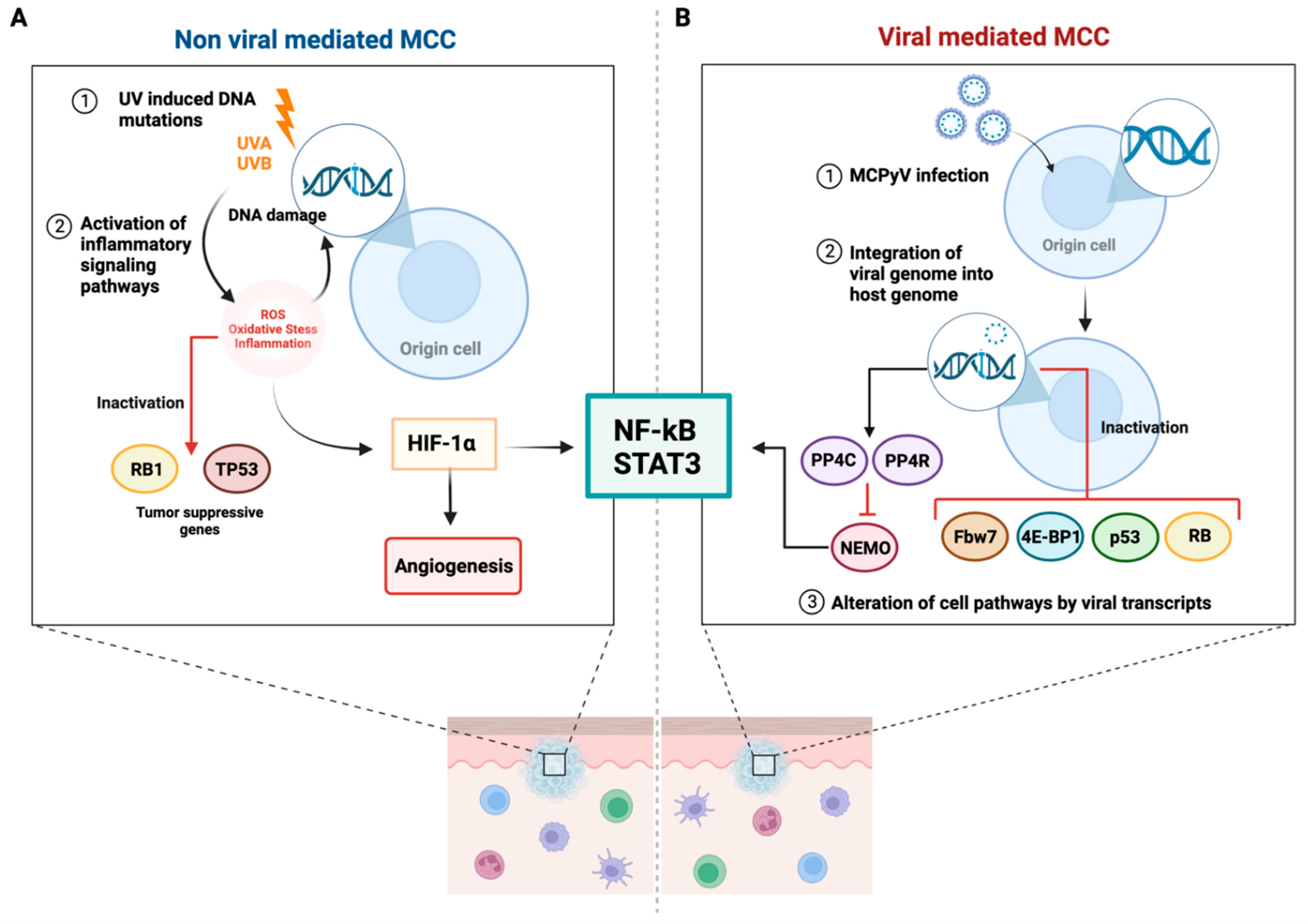
7. MCPyV-Positive and MCPyV-Negative MCC
7.1. T-Cell Responses to Oncogenic Merkel Cell Polyomavirus Proteins
7.2. T-Cell Responses in Virus-Negative MCC
8. Tumor Immune Escape and Targets for Immunotherapy
9. Potential Role of Extracellular Traps in MCC
9.1. Extracellular Traps: Strands in the Dark Web
9.2. Extracellular Traps: Keeping the Peace
10. Novel Methods for MCC Studies
11. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Cullison, C.R.; Zheng, D.X.; Levoska, M.A.; Scott, J.F.; Bordeaux, J.S. Tumor primary site as a prognostic factor for Merkel cell carcinoma disease-specific death. J. Am. Acad. Dermatol. 2021, 85, 1259–1266. [Google Scholar] [CrossRef] [PubMed]
- Schadendorf, D.; Lebbe, C.; Hausen, A.Z.; Avril, M.F.; Hariharan, S.; Bharmal, M.; Becker, J.C. Merkel Cell Carcinoma: Epidemiology, Prognosis, Therapy and Unmet Medical Needs. Eur. J. Cancer 2017, 71, 53–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McEvoy, A.M.; Lachance, K.; Hippe, D.S.; Cahill, K.; Moshiri, Y.; Lewis, C.W.; Singh, N.; Park, S.Y.; Thuesmunn, Z.; Cook, M.M.; et al. Recurrence and Mortality Risk of Merkel Cell Carcinoma by Cancer Stage and Time from Diagnosis. JAMA Dermatol. 2022, 158, 382. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Cao, D.; Zhao, J.; Zhu, B.; Xie, J. Clinical Features and Prognosis of Merkel Cell Carcinoma in Elderly Patients. Med. Sci. Monit. 2020, 26, e924570. [Google Scholar] [CrossRef]
- Paulson, K.; Park, S.Y.; Vandeven, N.A.; Lachance, K.; Thomas, H.; Chapuis, A.G.; Harms, K.L.; Thompson, J.A.; Bhatia, S.; Stang, A.; et al. Merkel cell carcinoma: Current US incidence and projected increases based on changing demographics. J. Am. Acad. Dermatol. 2018, 78, 457–463.e2. [Google Scholar] [CrossRef]
- Patel, P.; Hussain, K. Merkel Cell Carcinoma. Clin. Exp. Dermatol. 2021, 46, 814–819. [Google Scholar] [CrossRef]
- Ma, J.E.; Brewer, J.D. Merkel Cell Carcinoma in Immunosuppressed Patients. Cancers 2014, 6, 1328–1350. [Google Scholar] [CrossRef]
- Becker, J.C.; Stang, A.; DeCaprio, J.A.; Cerroni, L.; Lebbe, C.; Veness, M.; Nghiem, P. Merkel Cell Carcinoma. Nat. Rev. Dis. Prim. 2017, 3, 17077. [Google Scholar] [CrossRef]
- Feng, H.; Shuda, M.; Chang, Y.; Moore, P.S. Clonal Integration of a Polyomavirus in Human Merkel Cell Carcinoma. Science 2008, 319, 1096–1100. [Google Scholar] [CrossRef] [Green Version]
- Harms, P.; Vats, P.; Verhaegen, M.E.; Robinson, D.R.; Wu, Y.-M.; Dhanasekaran, S.M.; Palanisamy, N.; Siddiqui, J.; Cao, X.; Su, F.; et al. The Distinctive Mutational Spectra of Polyomavirus-Negative Merkel Cell Carcinoma. Cancer Res. 2015, 75, 3720–3727. [Google Scholar] [CrossRef]
- Goh, G.; Walradt, T.; Markarov, V.; Blom, A.; Riaz, N.; Doumani, R.; Stafstrom, K.; Moshiri, A.; Yelistratova, L.; Levinsohn, J.; et al. Mutational Landscape of Mcpyv-Positive and Mcpyv-Negative Merkel Cell Carcinomas with Implications for Immunotherapy. Oncotarget 2016, 7, 3403–3415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wendzicki, J.A.; Moore, P.S.; Chang, Y. Large T and small T antigens of Merkel cell polyomavirus. Curr. Opin. Virol. 2015, 11, 38–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spurgeon, M.E.; Cheng, J.; Ward-Shaw, E.; Dick, F.A.; DeCaprio, J.A.; Lambert, P.F. Merkel cell polyomavirus large T antigen binding to pRb promotes skin hyperplasia and tumor development. PLOS Pathog. 2022, 18, e1010551. [Google Scholar] [CrossRef]
- Shuda, M.; Guastafierro, A.; Geng, X.; Shuda, Y.; Ostrowski, S.M.; Lukianov, S.; Jenkins, F.J.; Honda, K.; Maricich, S.M.; Moore, P.; et al. Merkel Cell Polyomavirus Small T Antigen Induces Cancer and Embryonic Merkel Cell Proliferation in a Transgenic Mouse Model. PLoS ONE 2015, 10, e0142329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdul-Sada, H.; Muller, M.; Mehta, R.; Toth, R.; Arthur, J.S.C.; Whitehouse, A.; Macdonald, A. The Pp4r1 Sub-Unit of Protein Phosphatase Pp4 Is Essential for Inhibition of Nf-Kappab by Merkel Polyomavirus Small Tumour Antigen. Oncotarget 2017, 8, 25418–25432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nwogu, N.; Ortiz, L.E.; Kwun, H.J. Surface charge of Merkel cell polyomavirus small T antigen determines cell transformation through allosteric FBW7 WD40 domain targeting. Oncogenesis 2020, 9, 53. [Google Scholar] [CrossRef]
- Shuda, M.; Kwun, H.J.; Feng, H.; Chang, Y.; Moore, P.S. Human Merkel cell polyomavirus small T antigen is an oncoprotein targeting the 4E-BP1 translation regulator. J. Clin. Investig. 2011, 121, 3623–3634. [Google Scholar] [CrossRef]
- Laikova, K.V.; Oberemok, V.V.; Krasnodubets, A.M.; Gal’chinsky, N.V.; Useinov, R.Z.; Novikov, I.A.; Temirova, Z.Z.; Gorlov, M.V.; Shved, N.A.; Kumeiko, V.V.; et al. Advances in the Understanding of Skin Cancer: Ultraviolet Radiation, Mutations, and Antisense Oligonucleotides as Anticancer Drugs. Molecules 2019, 24, 1516. [Google Scholar] [CrossRef] [Green Version]
- Pfeifer, G.P.; Besaratinia, A. UV wavelength-dependent DNA damage and human non-melanoma and melanoma skin cancer. Photochem. Photobiol. Sci. 2011, 11, 90–97. [Google Scholar] [CrossRef] [Green Version]
- Ciążyńska, M.; Olejniczak-Staruch, I.; Sobolewska-Sztychny, D.; Narbutt, J.; Skibińska, M.; Lesiak, A. Ultraviolet Radiation and Chronic Inflammation—Molecules and Mechanisms Involved in Skin Carcinogenesis: A Narrative Review. Life 2021, 11, 326. [Google Scholar] [CrossRef]
- Horny, K.; Gerhardt, P.; Hebel-Cherouny, A.; Wülbeck, C.; Utikal, J.; Becker, J. Mutational Landscape of Virus- and UV-Associated Merkel Cell Carcinoma Cell Lines Is Comparable to Tumor Tissue. Cancers 2021, 13, 649. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.Q.; Waldeck, K.; Vergara, I.A.; Schröder, J.; Madore, J.; Wilmott, J.S.; Colebatch, A.J.; De Paoli-Iseppi, R.; Li, J.; Lupat, R.; et al. UV-Associated Mutations Underlie the Etiology of MCV-Negative Merkel Cell Carcinomas. Cancer Res. 2015, 75, 5228–5234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krump, N.A.; You, J. From Merkel Cell Polyomavirus Infection to Merkel Cell Carcinoma Oncogenesis. Front. Microbiol. 2021, 12, 739695. [Google Scholar] [CrossRef] [PubMed]
- Prezioso, C.; Carletti, R.; Obregon, F.; Piacentini, F.; Manicone, A.; Soda, G.; Moens, U.; Di Gioia, C.; Pietropaolo, V. Evaluation of Merkel Cell Polyomavirus DNA in Tissue Samples from Italian Patients with Diagnosis of MCC. Viruses 2021, 13, 61. [Google Scholar] [CrossRef] [PubMed]
- Iyer, J.G.; Afanasiev, O.K.; McClurkan, C.; Paulson, K.; Nagase, K.; Jing, L.; Marshak, J.O.; Dong, L.; Carter, J.; Lai, I.; et al. Merkel Cell Polyomavirus-Specific CD8+ and CD4+ T-cell Responses Identified in Merkel Cell Carcinomas and Blood. Clin. Cancer Res. 2011, 17, 6671–6680. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; He, W.; Wu, C.; Tan, Y.; He, Y.; Xu, B.; Chen, L.; Li, Q.; Jiang, J. Scoring System for Tumor-Infiltrating Lymphocytes and Its Prognostic Value for Gastric Cancer. Front. Immunol. 2019, 10, 71. [Google Scholar] [CrossRef] [Green Version]
- Roma-Rodrigues, C.; Mendes, R.; Baptista, P.V.; Fernandes, A.R. Targeting Tumor Microenvironment for Cancer Therapy. Int. J. Mol. Sci. 2019, 20, 840. [Google Scholar] [CrossRef] [Green Version]
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun. Signal. 2020, 18, 59. [Google Scholar] [CrossRef] [Green Version]
- Sundqvist, B.; Sihto, H.; von Willebrand, M.; Böhling, T.; Koljonen, V. LRIG1 is a positive prognostic marker in Merkel cell carcinoma and Merkel cell carcinoma expresses epithelial stem cell markers. Virchows Arch. 2021, 479, 1197–1207. [Google Scholar] [CrossRef]
- Albores-Saavedra, J.; Batich, K.; Chable-Montero, F.; Sagy, N.; Schwartz, A.M.; Henson, D.E. Merkel cell carcinoma demographics, morphology, and survival based on 3870 cases: A population based study. J. Cutan. Pathol. 2009, 37, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Fleming, K.E.; Ly, T.Y.; Pasternak, S.; Godlewski, M.; Doucette, S.; Walsh, N.M. Support for p63 expression as an adverse prognostic marker in Merkel cell carcinoma: Report on a Canadian cohort. Hum. Pathol. 2014, 45, 952–960. [Google Scholar] [CrossRef] [PubMed]
- Asioli, S.; Righi, A.; Volante, M.; Eusebi, V.; Bussolati, G. p63 expression as a new prognostic marker in Merkel cell carcinoma. Cancer 2007, 110, 640–647. [Google Scholar] [CrossRef] [PubMed]
- Feldmeyer, L.; Hudgens, C.W.; Ray-Lyons, G.; Nagarajan, P.; Aung, P.P.; Curry, J.L.; Torres-Cabala, C.A.; Mino, B.; Rodriguez-Canales, J.; Reuben, A.; et al. Density, Distribution, and Composition of Immune Infiltrates Correlate with Survival in Merkel Cell Carcinoma. Clin. Cancer Res. 2016, 22, 5553–5563. [Google Scholar] [CrossRef] [Green Version]
- Tarantola, T.I.; Vallow, L.A.; Halyard, M.Y.; Weenig, R.H.; Warschaw, K.E.; Grotz, T.E.; Jakub, J.W.; Roenigk, R.K.; Brewer, J.D.; Weaver, A.L.; et al. Prognostic factors in Merkel cell carcinoma: Analysis of 240 cases. J. Am. Acad. Dermatol. 2013, 68, 425–432. [Google Scholar] [CrossRef]
- Farah, M.; Reuben, A.; Spassova, I.; Yang, R.; Kubat, L.; Nagarajan, P.; Ning, J.; Li, W.; Aung, P.; Curry, J.; et al. T-cell Repertoire in Combination with T-cell Density Predicts Clinical Outcomes in Patients with Merkel Cell Carcinoma. J. Investig. Dermatol. 2020, 140, 2146–2456.e4. [Google Scholar] [CrossRef] [PubMed]
- Ricci, C.; Righi, A.; Ambrosi, F.; Gibertoni, D.; Maletta, F.; Uccella, S.; Sessa, F.; Asioli, S.; Pellilli, M.; Maragliano, R.; et al. Prognostic Impact of MCPyV and TIL Subtyping in Merkel Cell Carcinoma: Evidence from a Large European Cohort of 95 Patients. Endocr. Pathol. 2019, 31, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Dowlatshahi, M.; Huang, V.; Gehad, A.E.; Jiang, Y.; Calarese, A.; Teague, J.E.; Dorosario, A.A.; Cheng, J.; Nghiem, P.; Schanbacher, C.F.; et al. Tumor-Specific T Cells in Human Merkel Cell Carcinomas: A Possible Role for Tregs and T-Cell Exhaustion in Reducing T-Cell Responses. J. Investig. Dermatol. 2013, 133, 1879–1889. [Google Scholar] [CrossRef] [Green Version]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pagès, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, Density, and Location of Immune Cells Within Human Colorectal Tumors Predict Clinical Outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [Green Version]
- Kather, J.N.; Suarez-Carmona, M.; Charoentong, P.; Weis, C.-A.; Hirsch, D.; Bankhead, P.; Horning, M.; Ferber, D.; Kel, I.; Herpel, E.; et al. Topography of cancer-associated immune cells in human solid tumors. Elife 2018, 7, e36967. [Google Scholar] [CrossRef]
- Zheng, B.; Ma, J.; Tian, L.; Dong, L.; Song, G.; Pan, J.; Liu, Y.; Yang, S.; Wang, X.; Zhang, X.; et al. The distribution of immune cells within combined hepatocellular carcinoma and cholangiocarcinoma predicts clinical outcome. Clin. Transl. Med. 2020, 10, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Afanasiev, O.K.; Yelistratova, L.; Miller, N.; Nagase, K.; Paulson, K.; Iyer, J.G.; Ibrani, D.; Koelle, D.M.; Nghiem, P. Merkel Polyomavirus-Specific T Cells Fluctuate with Merkel Cell Carcinoma Burden and Express Therapeutically Targetable PD-1 and Tim-3 Exhaustion Markers. Clin. Cancer Res. 2013, 19, 5351–5360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buisseret, L.; Garaud, S.; de Wind, A.; van den Eynden, G.; Boisson, A.; Solinas, C.; Gu-Trantien, C.; Naveaux, C.; Lodewyckx, J.N.; Duvillier, H.; et al. Tumor-Infiltrating Lymphocyte Composition, Organization and Pd-1/ Pd-L1 Expression Are Linked in Breast Cancer. Oncoimmunology 2017, 6, e1257452. [Google Scholar] [CrossRef]
- Koch, M.; Beckhove, P.; den Winkel, J.O.; Autenrieth, D.; Wagner, P.; Nummer, D.; Specht, S.; Antolovic, D.; Galindo, L.; Schmitz-Winnenthal, F.H.; et al. Tumor Infiltrating T Lymphocytes in Colorectal Cancer: Tumor-Selective Activation and Cytotoxic Activity in Situ. Ann. Surg. 2006, 244, 986–992. [Google Scholar] [CrossRef] [PubMed]
- Stanton, S.E.; Disis, M.L. Clinical significance of tumor-infiltrating lymphocytes in breast cancer. J. Immunother. Cancer 2016, 4, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, S.A.; Hanniford, D.; Hernando, E.; Osman, I. Revisiting determinants of prognosis in cutaneous melanoma. Cancer 2015, 121, 4108–4123. [Google Scholar] [CrossRef] [Green Version]
- Brambilla, E.; Le Teuff, G.; Marguet, S.; Lantuejoul, S.; Dunant, A.; Graziano, S.; Pirker, R.; Douillard, J.-Y.; Le Chevalier, T.; Filipits, M.; et al. Prognostic Effect of Tumor Lymphocytic Infiltration in Resectable Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2016, 34, 1223–1230. [Google Scholar] [CrossRef] [Green Version]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Landskron, G.; De La Fuente, M.; Thuwajit, P.; Thuwajit, C.; Hermoso, M.A. Chronic Inflammation and Cytokines in the Tumor Microenvironment. J. Immunol. Res. 2014, 2014, 149185. [Google Scholar] [CrossRef] [Green Version]
- Lima, L.G.; Ham, S.; Shin, H.; Chai, E.P.Z.; Lek, E.S.H.; Lobb, R.J.; Müller, A.F.; Mathivanan, S.; Yeo, B.; Choi, Y.; et al. Tumor microenvironmental cytokines bound to cancer exosomes determine uptake by cytokine receptor-expressing cells and biodistribution. Nat. Commun. 2021, 12, 3543. [Google Scholar] [CrossRef]
- Maibach, F.; Sadozai, H.; Jafari, S.M.S.; Hunger, R.E.; Schenk, M. Tumor-Infiltrating Lymphocytes and Their Prognostic Value in Cutaneous Melanoma. Front. Immunol. 2020, 11, 2105. [Google Scholar] [CrossRef] [PubMed]
- Gherardin, N.A.; Waldeck, K.; Caneborg, A.; Martelotto, L.G.; Balachander, S.; Zethoven, M.; Petrone, P.M.; Pattison, A.; Wilmott, J.S.; Quinones-Parra, S.M.; et al. Gammadelta T Cells in Merkel Cell Carcinomas Have a Proinflammatory Profile Prognostic of Patient Survival. Cancer Immunol. Res. 2021, 9, 612–623. [Google Scholar] [CrossRef] [PubMed]
- Copeland, A.R.; Shindorf, M.L.; McDonald, J.D.; Hernandez, J.M.; Davis, J.L.; Margolin, K.A.; Blakely, A.M. Tumor-infiltrating lymphocytes are associated with improved survival in node-positive Merkel cell carcinoma: A national cohort analysis. J. Am. Acad. Dermatol. 2021, 86, 1172–1174. [Google Scholar] [CrossRef] [PubMed]
- Butala, A.A.; Jain, V.; Reddy, V.K.; Sebro, R.A.; Song, Y.; Karakousis, G.; Mitchell, T.C.; Lukens, J.N.; Shabason, J.E. Impact of Tumor-Infiltrating Lymphocytes on Overall Survival in Merkel Cell Carcinoma. Oncologist 2020, 26, 63–69. [Google Scholar] [CrossRef]
- Paulson, K.G.; Iyer, J.G.; Tegeder, A.R.; Thibodeau, R.; Schelter, J.; Koba, S.; Schrama, D.; Simonson, W.T.; Lemos, B.D.; Byrd, D.R.; et al. Transcriptome-Wide Studies of Merkel Cell Carcinoma and Validation of Intratumoral CD8+ Lymphocyte Invasion as an Independent Predictor of Survival. J. Clin. Oncol. 2011, 29, 1539–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulson, K.G.; Iyer, J.G.; Simonson, W.T.; Blom, A.; Thibodeau, R.M.; Schmidt, M.; Pietromonaco, S.; Sokil, M.; Warton, E.M.; Asgari, M.M.; et al. Cd8+ Lymphocyte Intratumoral Infiltration as a Stage-Independent Predictor of Merkel Cell Carcinoma Survival: A Population-Based Study. Am. J. Clin. Pathol. 2014, 142, 452–458. [Google Scholar] [CrossRef] [Green Version]
- Shuda, M.; Feng, H.; Kwun, H.J.; Rosen, S.T.; Gjoerup, O.; Moore, P.S.; Chang, Y. T antigen mutations are a human tumor-specific signature for Merkel cell polyomavirus. Proc. Natl. Acad. Sci. USA 2008, 105, 16272–16277. [Google Scholar] [CrossRef] [Green Version]
- Samimi, M.; Benlalam, H.; Aumond, P.; Gaboriaud, P.; Fradin, D.; Kervarrec, T.; Florenceau, L.; Vignard, V.; Blom, A.; Touzé, A.; et al. Viral and tumor antigen-specific CD8 T-cell responses in Merkel cell carcinoma. Cell. Immunol. 2019, 344, 103961. [Google Scholar] [CrossRef]
- Bacher, P.; Scheffold, A. Flow-cytometric analysis of rare antigen-specific T cells. Cytom. Part A 2013, 83, 692–701. [Google Scholar] [CrossRef]
- Touzé, A.; Gaitan, J.; Arnold, F.; Cazal, R.; Fleury, M.J.; Combelas, N.; Sizaret, P.-Y.; Guyetant, S.; Maruani, A.; Baay, M.; et al. Generation of Merkel Cell Polyomavirus (MCV)-Like Particles and Their Application to Detection of MCV Antibodies. J. Clin. Microbiol. 2010, 48, 1767–1770. [Google Scholar] [CrossRef]
- Tornesello, A.L.; Tagliamonte, M.; Buonaguro, F.M.; Tornesello, M.L.; Buonaguro, L. Virus-like Particles as Preventive and Therapeutic Cancer Vaccines. Vaccines 2022, 10, 227. [Google Scholar] [CrossRef] [PubMed]
- Longino, N.; Yang, J.; Iyer, J.; Ibrani, D.; Chow, I.; Laing, K.; Campbell, V.; Paulson, K.; Kulikauskas, R.; Church, C.; et al. Human CD4+ T cells specific for Merkel cell polyomavirus localize to Merkel cell carcinomas and target a required oncogenic domain. Cancer Immunol. Res. 2019, 7, 1727–1739. [Google Scholar] [CrossRef] [PubMed]
- Togashi, Y.; Shitara, K.; Nishikawa, H. Regulatory T cells in cancer immunosuppression—Implications for anticancer therapy. Nat. Rev. Clin. Oncol. 2019, 16, 356–371. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Kuniyasu, Y.; Toda, M.; Sakaguchi, N.; Itoh, M.; Iwata, M.; Shimizu, J. Immunologic self-tolerance maintained by CD25+CD4+ naturally anergic and suppressive T cells: Induction of autoimmune disease by breaking their anergic/suppressive state. Int. Immunol. 1998, 10, 1969–1980. [Google Scholar] [CrossRef] [Green Version]
- Collison, L.W.; Workman, C.J.; Kuo, T.T.; Boyd, K.; Wang, Y.; Vignali, K.M.; Cross, R.; Sehy, D.; Blumberg, R.S.; Vignali, D.A.A. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature 2007, 450, 566–569. [Google Scholar] [CrossRef]
- Yu, X.; Harden, K.; Gonzalez, L.C.; Francesco, M.; Chiang, E.; Irving, B.A.; Tom, I.; Ivelja, S.; Refino, C.J.; Clark, H.; et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat. Immunol. 2008, 10, 48–57. [Google Scholar] [CrossRef]
- Fourcade, J.; Sun, Z.; Chauvin, J.-M.; Ka, M.; Davar, D.; Pagliano, O.; Wang, H.; Saada, S.; Menna, C.; Amin, R.; et al. CD226 opposes TIGIT to disrupt Tregs in melanoma. JCI Insight 2018, 3, e121157. [Google Scholar] [CrossRef] [Green Version]
- Chauvin, J.-M.; Zarour, H.M. TIGIT in cancer immunotherapy. J. Immunother. Cancer 2020, 8, e000957. [Google Scholar] [CrossRef]
- Joller, N.; Lozano, E.; Burkett, P.R.; Patel, B.; Xiao, S.; Zhu, C.; Xia, J.; Tan, T.G.; Sefik, E.; Yajnik, V.; et al. Treg Cells Expressing the Coinhibitory Molecule TIGIT Selectively Inhibit Proinflammatory Th1 and Th17 Cell Responses. Immunity 2014, 40, 569–581. [Google Scholar] [CrossRef] [Green Version]
- Wei, S.; Kryczek, I.; Zou, L.; Daniel, B.; Cheng, P.; Mottram, P.; Curiel, T.; Lange, A.; Zou, W. Plasmacytoid Dendritic Cells Induce CD8+ Regulatory T Cells in Human Ovarian Carcinoma. Cancer Res. 2005, 65, 5020–5026. [Google Scholar] [CrossRef]
- Yano, H.; Andrews, L.P.; Workman, C.J.; Vignali, D.A.A. Intratumoral Regulatory T Cells: Markers, Subsets and Their Impact on Anti-Tumor Immunity. Immunology 2019, 157, 232–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morath, A.; Schamel, W.W. Alphabeta and Gammadelta T Cell Receptors: Similar but Different. J. Leukoc. Biol. 2020, 107, 1045–1055. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, C. The Role of Human Gammadelta T Cells in Anti-Tumor Immunity and Their Potential for Cancer Immunotherapy. Cells 2020, 9, 1206. [Google Scholar] [CrossRef]
- Wo, J.; Zhang, F.; Li, Z.; Sun, C.; Zhang, W.; Sun, G. The Role of Gamma-Delta T Cells in Diseases of the Central Nervous System. Front. Immunol. 2020, 11, 580304. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, A.M.; Michels, A.W. T cell receptor sequencing in autoimmunity. JoLS J. Life Sci. 2020, 2, 38–58. [Google Scholar] [CrossRef]
- Minervina, A.A.; Komech, E.A.; Titov, A.; Koraichi, M.B.; Rosati, E.; Mamedov, I.Z.; Franke, A.; Efimov, G.A.; Chudakov, D.M.; Mora, T.; et al. Longitudinal high-throughput TCR repertoire profiling reveals the dynamics of T-cell memory formation after mild COVID-19 infection. eLife 2021, 10, e63502. [Google Scholar] [CrossRef]
- Valpione, S.; Mundra, P.A.; Galvani, E.; Campana, L.G.; Lorigan, P.; de Rosa, F.; Gupta, A.; Weightman, J.; Mills, S.; Dhomen, N.; et al. The T Cell Receptor Repertoire of Tumor Infiltrating T Cells Is Predictive and Prognostic for Cancer Survival. Nat. Commun. 2021, 12, 4098. [Google Scholar] [CrossRef]
- Spassova, I.; Ugurel, S.; Terheyden, P.; Sucker, A.; Hassel, J.C.; Ritter, C.; Kubat, L.; Habermann, D.; Farahpour, F.; Saeedghalati, M.; et al. Predominance of Central Memory T Cells with High T-Cell Receptor Repertoire Diversity Is Associated with Response to Pd-1/Pd-L1 Inhibition in Merkel Cell Carcinoma. Clin. Cancer Res. 2020, 26, 2257–2267. [Google Scholar] [CrossRef]
- Crusz, S.M.; Balkwill, F. Inflammation and cancer: Advances and new agents. Nat. Rev. Clin. Oncol. 2015, 12, 584–596. [Google Scholar] [CrossRef]
- Lan, T.; Chen, L.; Wei, X. Inflammatory Cytokines in Cancer: Comprehensive Understanding and Clinical Progress in Gene Therapy. Cells 2021, 10, 100. [Google Scholar] [CrossRef]
- Bridge, J.A.; Lee, J.C.; Daud, A.; Wells, J.W.; Bluestone, J.A. Cytokines, Chemokines, and Other Biomarkers of Response for Checkpoint Inhibitor Therapy in Skin Cancer. Front. Med. 2018, 5, 351. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Guo, Y.; Liu, S.; Wang, H.; Zhu, J.; Ou, L.; Xu, X. Targeting regulatory T cells for immunotherapy in melanoma. Mol. Biomed. 2021, 2, 11. [Google Scholar] [CrossRef] [PubMed]
- Monteagudo, C.; Ramos, D.; Pellín-Carcelén, A.; Gil, R.; Callaghan, R.C.; Martín, J.M.; Alonso, V.; Murgui, A.; Navarro, L.; Calabuig, S.; et al. CCL27–CCR10 and CXCL12–CXCR4 chemokine ligand-receptor mRNA expression ratio: New predictive factors of tumor progression in cutaneous malignant melanoma. Clin. Exp. Metastasis 2012, 29, 625–637. [Google Scholar] [CrossRef] [PubMed]
- Kuhnelt-Leddihn, L.; Muller, H.; Eisendle, K.; Zelger, B.; Weinlich, G. Overexpression of the Chemokine Receptors Cxcr4, Ccr7, Ccr9, and Ccr10 in Human Primary Cutaneous Melanoma: A Potential Prognostic Value for Ccr7 and Ccr10? Arch. Dermatol. Res. 2012, 304, 185–193. [Google Scholar] [CrossRef]
- Martin-Garcia, D.; Silva-Vilches, C.; Will, R.; Enk, A.H.; Lonsdorf, A.S. Tumor-derived CCL20 affects B16 melanoma growth in mice. J. Dermatol. Sci. 2019, 97, 57–65. [Google Scholar] [CrossRef]
- Shields, J.D.; Kourtis, I.C.; Tomei, A.A.; Roberts, J.M.; Swartz, M.A. Induction of Lymphoidlike Stroma and Immune Escape by Tumors That Express the Chemokine CCL21. Science 2010, 328, 749–752. [Google Scholar] [CrossRef] [Green Version]
- Riboni, L.; Hadi, L.A.; Navone, S.E.; Guarnaccia, L.; Campanella, R.; Marfia, G. Sphingosine-1-Phosphate in the Tumor Microenvironment: A Signaling Hub Regulating Cancer Hallmarks. Cells 2020, 9, 337. [Google Scholar] [CrossRef] [Green Version]
- Rasheed, K.; Abdulsalam, I.; Fismen, S.; Grimstad, O.; Sveinbjornsson, B.; Moens, U. Ccl17/Tarc and Ccr4 Expression in Merkel Cell Carcinoma. Oncotarget 2018, 9, 31432–31447. [Google Scholar] [CrossRef] [Green Version]
- Kubo, T.; Ichimiya, S.; Tonooka, A.; Nagashima, T.; Kikuchi, T.; Sato, N. p63 Induces CD4+ T-Cell Chemoattractant TARC/CCL17 in Human Epithelial Cells. J. Interf. Cytokine Res. 2008, 28, 725–732. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Larsen, K.M.; Minaya, M.K.; Vaish, V.; Pena, M.M.O. The Role of Il-33/St2 Pathway in Tumorigenesis. Int. J. Mol. Sci. 2018, 19, 2676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griesenauer, B.; Paczesny, S. The St2/Il-33 Axis in Immune Cells During Inflammatory Diseases. Front. Immunol. 2017, 8, 475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasheed, K.; Moens, U.; Policastro, B.; Johnsen, J.I.; Koljonen, V.; Sihto, H.; Lui, W.O.; Sveinbjornsson, B. The Merkel Cell Polyomavirus T-Antigens and Il-33/St2-Il1racp Axis: Possible Role in Merkel Cell Carcinoma. Int. J. Mol. Sci. 2022, 23, 3702. [Google Scholar] [CrossRef]
- Jovanovic, I.P.; Pejnovic, N.N.; Radosavljevic, G.D.; Pantic, J.M.; Milovanovic, M.Z.; Arsenijevic, N.N.; Lukic, M.L. Interleukin-33/St2 Axis Promotes Breast Cancer Growth and Metastases by Facilitating Intratumoral Accumulation of Immunosuppressive and Innate Lymphoid Cells. Int. J. Cancer 2014, 134, 1669–1682. [Google Scholar] [CrossRef]
- Chen, Y.-L.; Gutowska-Owsiak, D.; Hardman, C.S.; Westmoreland, M.; MacKenzie, T.; Cifuentes, L.; Waithe, D.; Lloyd-Lavery, A.; Marquette, A.; Londei, M.; et al. Proof-of-concept clinical trial of etokimab shows a key role for IL-33 in atopic dermatitis pathogenesis. Sci. Transl. Med. 2019, 11, eaax2945. [Google Scholar] [CrossRef]
- Akimoto, M.; Hayashi, J.-I.; Nakae, S.; Saito, H.; Takenaga, K. Interleukin-33 enhances programmed oncosis of ST2L-positive low-metastatic cells in the tumour microenvironment of lung cancer. Cell Death Dis. 2016, 7, e2057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Jeught, K.; Sun, Y.; Fang, Y.; Zhou, Z.; Jiang, H.; Yu, T.; Yang, J.; Kamocka, M.M.; So, K.M.; Li, Y.; et al. ST2 as checkpoint target for colorectal cancer immunotherapy. J. Clin. Investig. 2020, 5, 136073. [Google Scholar] [CrossRef]
- Pastille, E.; Wasmer, M.H.; Adamczyk, A.; Vu, V.P.; Mager, L.F.; Phuong, N.N.T.; Palmieri, V.; Simillion, C.; Hansen, W.; Kasper, S.; et al. The Il-33/St2 Pathway Shapes the Regulatory T Cell Phenotype to Promote Intestinal Cancer. Mucosal Immunol. 2019, 12, 990–1003. [Google Scholar] [CrossRef] [Green Version]
- Sautès-Fridman, C.; Petitprez, F.; Calderaro, J.; Fridman, W.H. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat. Rev. Cancer 2019, 19, 307–325. [Google Scholar] [CrossRef]
- Goc, J.; Germain, C.; Vo-Bourgais, T.K.D.; Lupo, A.; Klein, C.; Knockaert, S.; de Chaisemartin, L.; Ouakrim, H.; Becht, E.; Alifano, M.; et al. Dendritic Cells in Tumor-Associated Tertiary Lymphoid Structures Signal a Th1 Cytotoxic Immune Contexture and License the Positive Prognostic Value of Infiltrating CD8+ T Cells. Cancer Res 2014, 74, 705–715. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Thommen, D.S. Tertiary lymphoid structures in cancer. Science 2022, 375, abf9419. [Google Scholar] [CrossRef] [PubMed]
- Behr, D.S.; Peitsch, W.K.; Hametner, C.; Lasitschka, F.; Houben, R.; Schönhaar, K.; Michel, J.; Dollt, C.; Goebeler, M.; Marx, A.; et al. Prognostic value of immune cell infiltration, tertiary lymphoid structures and PD-L1 expression in Merkel cell carcinomas. Int. J. Clin. Exp. Pathol. 2014, 7, 7610–7621. [Google Scholar] [PubMed]
- Nakamura, M.; Magara, T.; Kano, S.; Matsubara, A.; Kato, H.; Morita, A. Tertiary Lymphoid Structures and Chemokine Landscape in Virus-Positive and Virus-Negative Merkel Cell Carcinoma. Front. Oncol. 2022, 12, 811586. [Google Scholar] [CrossRef] [PubMed]
- Nghiem, P.T.; Bhatia, S.; Lipson, E.J.; Kudchadkar, R.R.; Miller, N.J.; Annamalai, L.; Berry, S.; Chartash, E.K.; Daud, A.; Fling, S.P.; et al. PD-1 Blockade with Pembrolizumab in Advanced Merkel-Cell Carcinoma. N. Engl. J. Med. 2016, 374, 2542–2552. [Google Scholar] [CrossRef]
- Starrett, G.; Thakuria, M.; Chen, T.; Marcelus, C.; Cheng, J.; Nomburg, J.; Thorner, A.R.; Slevin, M.K.; Powers, W.; Burns, R.T.; et al. Clinical and molecular characterization of virus-positive and virus-negative Merkel cell carcinoma. Genome Med. 2020, 12, 30. [Google Scholar] [CrossRef] [Green Version]
- Miller, N.; Church, C.; Dong, L.; Crispin, D.; FitzGibbon, M.; Lachance, K.; Jing, L.; Shinohara, M.; Gavvovidis, I.; Willimsky, G.; et al. Tumor-infiltrating Merkel cell polyomavirus-specific T cells are diverse and associated with improved patient survival. Cancer Immunol. Res. 2018, 5, 137–147. [Google Scholar] [CrossRef] [Green Version]
- Lyngaa, R.; Pedersen, N.W.; Schrama, D.; Thrue, C.A.; Ibrani, D.; Met, O.; Straten, P.T.; Nghiem, P.; Becker, J.C.; Hadrup, S.R. T-cell Responses to Oncogenic Merkel Cell Polyomavirus Proteins Distinguish Patients with Merkel Cell Carcinoma from Healthy Donors. Clin. Cancer Res. 2014, 20, 1768–1778. [Google Scholar] [CrossRef] [Green Version]
- Sihto, H.; Böhling, T.; Kavola, H.; Koljonen, V.; Salmi, M.; Jalkanen, S.; Joensuu, H. Tumor Infiltrating Immune Cells and Outcome of Merkel Cell Carcinoma: A Population-Based Study. Clin. Cancer Res. 2012, 18, 2872–2881. [Google Scholar] [CrossRef] [Green Version]
- Stachyra, K.; Dudzisz-Śledź, M.; Bylina, E.; Szumera-Ciećkiewicz, A.; Spałek, M.; Bartnik, E.; Rutkowski, P.; Czarnecka, A. Merkel Cell Carcinoma from Molecular Pathology to Novel Therapies. Int. J. Mol. Sci. 2021, 22, 6305. [Google Scholar] [CrossRef]
- Bichakjian, C.K.; Olencki, T.; Aasi, S.Z.; Alam, M.; Andersen, J.S.; Blitzblau, R.; Bowen, G.M.; Contreras, C.M.; Daniels, G.A.; Decker, R.; et al. Merkel Cell Carcinoma, Version 1.2018, Nccn Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2018, 16, 742–774. [Google Scholar] [CrossRef]
- Miller, N.J.; Church, C.D.; Fling, S.P.; Kulikauskas, R.; Ramchurren, N.; Shinohara, M.M.; Kluger, H.M.; Bhatia, S.; Lundgren, L.; Cheever, M.A.; et al. Merkel cell polyomavirus-specific immune responses in patients with Merkel cell carcinoma receiving anti-PD-1 therapy. J. Immunother. Cancer 2018, 6, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritter, C.; Fan, K.; Paulson, K.G.; Nghiem, P.; Schrama, D.; Becker, J.C. Reversal of epigenetic silencing of MHC class I chain-related protein A and B improves immune recognition of Merkel cell carcinoma. Sci. Rep. 2016, 6, 21678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prendergast, G.C. Immune escape as a fundamental trait of cancer: Focus on IDO. Oncogene 2008, 27, 3889–3900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatia, S.; Afanasiev, O.; Nghiem, P. Immunobiology of Merkel Cell Carcinoma: Implications for Immunotherapy of a Polyomavirus-Associated Cancer. Curr. Oncol. Rep. 2011, 13, 488–497. [Google Scholar] [CrossRef] [Green Version]
- Paulson, K.G.; Tegeder, A.; Willmes, C.; Iyer, J.G.; Afanasiev, O.K.; Schrama, D.; Koba, S.; Thibodeau, R.; Nagase, K.; Simonson, W.T.; et al. Downregulation of MHC-I Expression Is Prevalent but Reversible in Merkel Cell Carcinoma. Cancer Immunol. Res. 2014, 2, 1071–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houben, R.; Dreher, C.; Angermeyer, S.; Borst, A.; Utikal, J.; Haferkamp, S.; Peitsch, W.K.; Schrama, D.; Hesbacher, S. Mechanisms of p53 Restriction in Merkel Cell Carcinoma Cells Are Independent of the Merkel Cell Polyoma Virus T Antigens. J. Investig. Dermatol. 2013, 133, 2453–2460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houben, R.; Adam, C.; Baeurle, A.; Hesbacher, S.; Grimm, J.; Angermeyer, S.; Henzel, K.; Hauser, S.; Elling, R.; Bröcker, E.-B.; et al. An intact retinoblastoma protein-binding site in Merkel cell polyomavirus large T antigen is required for promoting growth of Merkel cell carcinoma cells. Int. J. Cancer 2011, 130, 847–856. [Google Scholar] [CrossRef]
- Raulet, D.H.; Gasser, S.; Gowen, B.G.; Deng, W.; Jung, H. Regulation of Ligands for the NKG2D Activating Receptor. Annu. Rev. Immunol. 2013, 31, 413–441. [Google Scholar] [CrossRef] [Green Version]
- Mazziotta, C.; Lanzillotti, C.; Gafà, R.; Touzé, A.; Durand, M.-A.; Martini, F.; Rotondo, J.C. The Role of Histone Post-Translational Modifications in Merkel Cell Carcinoma. Front. Oncol. 2022, 12, 832047. [Google Scholar] [CrossRef]
- Ritter, C.; Fan, K.; Paschen, A.; Hadrup, S.R.; Ferrone, S.; Nghiem, P.; Ugurel, S.; Schrama, D.; Becker, J.C. Epigenetic priming restores the HLA class-I antigen processing machinery expression in Merkel cell carcinoma. Sci. Rep. 2017, 7, 2290. [Google Scholar] [CrossRef]
- Chan, I.; Bhatia, S.; Kaufman, H.L.; Lipson, E.J. Immunotherapy for Merkel cell carcinoma: A turning point in patient care. J. Immunother. Cancer 2018, 6, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.-P.; Lampert, J.C.; Chen, X.; Leitão, C.; Popović, J.; Muller, W.; Blankenstein, T. Transgenic mice with a diverse human T cell antigen receptor repertoire. Nat. Med. 2010, 16, 1029–1034. [Google Scholar] [CrossRef] [PubMed]
- Gavvovidis, I.; Leisegang, M.; Willimsky, G.; Miller, N.J.; Nghiem, P.; Blankenstein, T. Targeting Merkel Cell Carcinoma by Engineered T Cells Specific to T-Antigens of Merkel Cell Polyomavirus. Clin. Cancer Res. 2018, 24, 3644–3655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyake, K.; Karasuyama, H. The Role of Trogocytosis in the Modulation of Immune Cell Functions. Cells 2021, 10, 1255. [Google Scholar] [CrossRef]
- Lu, Z.; McBrearty, N.; Chen, J.; Tomar, V.S.; Zhang, H.; De Rosa, G.; Tan, A.; Weljie, A.M.; Beiting, D.P.; Miao, Z.; et al. ATF3 and CH25H regulate effector trogocytosis and anti-tumor activities of endogenous and immunotherapeutic cytotoxic T lymphocytes. Cell Metab. 2022, 34, 1342–1358.e7. [Google Scholar] [CrossRef]
- Takei, H.; Araki, A.; Watanabe, H.; Ichinose, A.; Sendo, F. Rapid killing of human neutrophils by the potent activator phorbol 12-myristate 13-acetate (PMA) accompanied by changes different from typical apoptosis or necrosis. J. Leukoc. Biol. 1996, 59, 229–240. [Google Scholar] [CrossRef]
- Goldmann, O.; Medina, E. The expanding world of extracellular traps: Not only neutrophils but much more. Front. Immunol. 2013, 3, 420. [Google Scholar] [CrossRef] [Green Version]
- Agak, G.W.; Mouton, A.; Teles, R.M.; Weston, T.A.; Morselli, M.; Andrade, P.R.; Pellegrini, M.; Modlin, R.L. Extracellular traps released by antimicrobial TH17 cells contribute to host defense. J. Clin. Investig. 2021, 131, e141594. [Google Scholar] [CrossRef]
- Liu, P.; Wu, X.; Liao, C.; Liu, X.; Du, J.; Shi, H.; Wang, X.; Bai, X.; Peng, P.; Yu, L.; et al. Escherichia coli and Candida albicans Induced Macrophage Extracellular Trap-Like Structures with Limited Microbicidal Activity. PLoS ONE 2014, 9, e90042. [Google Scholar] [CrossRef] [Green Version]
- Loureiro, A.; Pais, C.; Sampaio, P. Relevance of Macrophage Extracellular Traps in C. Albicans Killing. Front. Immunol. 2019, 10, 2767. [Google Scholar] [CrossRef]
- Mohanan, S.; Horibata, S.; McElwee, J.L.; Dannenberg, A.J.; Coonrod, S.A. Identification of Macrophage Extracellular Trap-Like Structures in Mammary Gland Adipose Tissue: A Preliminary Study. Front. Immunol. 2013, 4, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz-Caro, T.; Silva, L.M.R.; Ritter, C.; Taubert, A.; Hermosilla, C. Besnoitia besnoiti tachyzoites induce monocyte extracellular trap formation. Parasitol. Res. 2014, 113, 4189–4197. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, M.; Lacy, P.; Ueki, S. Eosinophil Extracellular Traps and Inflammatory Pathologies-Untangling the Web! Front. Immunol. 2018, 9, 2763. [Google Scholar] [CrossRef]
- Yousefi, S.; Morshed, M.; Amini, P.; Stojkov, D.; Simon, D.; von Gunten, S.; Kaufmann, T.; Simon, H.U. Basophils Exhibit Antibacterial Activity through Extracellular Trap Formation. Allergy 2015, 70, 1184–1188. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.W.; Jacobs, W.R., Jr. Mycobacterium Tuberculosis Exploits Human Interferon Gamma to Stimulate Macrophage Extracellular Trap Formation and Necrosis. J. Infect. Dis. 2013, 208, 109–119. [Google Scholar] [CrossRef] [Green Version]
- Guimarães-Costa, A.B.; Nascimento, M.T.C.; Froment, G.S.; Soares, R.P.P.; Morgado, F.N.; Conceição-Silva, F.; Saraiva, E.M. Leishmania amazonensis promastigotes induce and are killed by neutrophil extracellular traps. Proc. Natl. Acad. Sci. USA 2009, 106, 6748–6753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campillo-Navarro, M.; Leyva-Paredes, K.; Donis-Maturano, L.; González-Jiménez, M.; Paredes-Vivas, Y.; Cerbulo-Vázquez, A.; Serafín-López, J.; García-Pérez, B.; Ullrich, S.E.; Flores-Romo, L.; et al. Listeria monocytogenes induces mast cell extracellular traps. Immunobiology 2017, 222, 432–439. [Google Scholar] [CrossRef]
- Ouyang, K.; Oparaugo, N.; Nelson, A.M.; Agak, G.W. T Cell Extracellular Traps: Tipping the Balance Between Skin Health and Disease. Front. Immunol. 2022, 13, 900634. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, L. The pro-tumor effect and the anti-tumor effect of neutrophils extracellular traps. Biosci. Trends 2019, 13, 469–475. [Google Scholar] [CrossRef] [Green Version]
- Triner, D.; Devenport, S.N.; Ramakrishnan, S.K.; Ma, X.; Frieler, R.A.; Greenson, J.K.; Inohara, N.; Nunez, G.; Colacino, J.A.; Mortensen, R.M.; et al. Neutrophils Restrict Tumor-Associated Microbiota to Reduce Growth and Invasion of Colon Tumors in Mice. Gastroenterology 2019, 156, 1467–1482. [Google Scholar] [CrossRef]
- Chen, Q.; Zhang, L.; Li, X.; Zhuo, W. Neutrophil Extracellular Traps in Tumor Metastasis: Pathological Functions and Clinical Applications. Cancers 2021, 13, 2832. [Google Scholar] [CrossRef] [PubMed]
- Shao, B.-Z.; Yao, Y.; Li, J.-P.; Chai, N.-L.; Linghu, E.-Q. The Role of Neutrophil Extracellular Traps in Cancer. Front. Oncol. 2021, 11, 714357. [Google Scholar] [CrossRef] [PubMed]
- Tohme, S.; Yazdani, H.O.; Al-Khafaji, A.B.; Chidi, A.P.; Loughran, P.; Mowen, A.K.; Wang, Y.; Simmons, R.L.; Huang, H.; Tsung, A. Neutrophil extracellular traps promote the development and progression of liver metastases after surgical stress. Cancer Res. 2016, 76, 1367–1380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cools-Lartigue, J.; Spicer, J.; McDonald, B.; Gowing, S.; Chow, S.; Giannias, B.; Bourdeau, F.; Kubes, P.; Ferri, L. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J. Clin. Investig. 2013, 123, 3446–3458. [Google Scholar] [CrossRef] [PubMed]
- Cedervall, J.; Zhang, Y.; Olsson, A.-K. Tumor-Induced NETosis as a Risk Factor for Metastasis and Organ Failure. Cancer Res 2016, 76, 4311–4315. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Liu, Q.; Zhang, X.; Liu, X.; Zhou, B.; Chen, J.; Huang, D.; Li, J.; Li, H.; Chen, F.; et al. DNA of neutrophil extracellular traps promotes cancer metastasis via CCDC25. Nature 2020, 583, 133–138. [Google Scholar] [CrossRef]
- Heath, M.; Jaimes, N.; Lemos, B.; Mostaghimi, A.; Wang, L.C.; Peñas, P.F.; Nghiem, P. Clinical characteristics of Merkel cell carcinoma at diagnosis in 195 patients: The AEIOU features. J. Am. Acad. Dermatol. 2008, 58, 375–381. [Google Scholar] [CrossRef] [Green Version]
- Sihto, H.; Joensuu, H. Tumor-Infiltrating Lymphocytes and Outcome in Merkel Cell Carcinoma, a Virus-Associated Cancer. Oncoimmunology 2012, 1, 1420–1421. [Google Scholar] [CrossRef] [Green Version]
- Yousefi, S.; Mihalache, C.; Kozlowski, E.; Schmid, I.; Simon, H.U. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ. 2009, 16, 1438–1444. [Google Scholar] [CrossRef]
- Zhang, Q.; Itagaki, K.; Hauser, C.J. Mitochondrial DNA Is Released by Shock and Activates Neutrophils Via P38 Map Kinase. Shock 2010, 34, 55–59. [Google Scholar] [CrossRef]
- Simmons, J.D.; Lee, Y.-L.; Mulekar, S.; Kuck, J.L.; Brevard, S.B.; Gonzalez, R.P.; Gillespie, M.N.; Richards, W.O. Elevated Levels of Plasma Mitochondrial DNA DAMPs Are Linked to Clinical Outcome in Severely Injured Human Subjects. Ann. Surg. 2013, 258, 591–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McIlroy, D.J.; Jarnicki, A.G.; Au, G.G.; Lott, N.; Smith, D.W.; Hansbro, P.M.; Balogh, Z.J. Mitochondrial DNA neutrophil extracellular traps are formed after trauma and subsequent surgery. J. Crit. Care 2014, 29, 1133.e1–1133.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, H.E.; Raucci, A. Alarmin(G) News About Danger: Workshop on Innate Danger Signals and Hmgb1. EMBO Rep. 2006, 7, 774–778. [Google Scholar] [CrossRef] [Green Version]
- Balogh, Z.J.; McIlroy, D.J.; Smith, D.W.; Hansbro, P.M. The origin and the role of mitochondrial DNA in postinjury inflammation. J. Crit. Care 2013, 28, 1099–1100. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Kim, G.B.; Krump, N.A.; Zhou, Y.; Riley, J.L.; You, J. Selective reactivation of STING signaling to target Merkel cell carcinoma. Proc. Natl. Acad. Sci. USA 2020, 117, 13730–13739. [Google Scholar] [CrossRef]
- Barber, G.N. Sting: Infection, Inflammation and Cancer. Nat. Rev. Immunol. 2015, 15, 760–770. [Google Scholar] [CrossRef] [Green Version]
- Fu, J.; Kanne, D.B.; Leong, M.; Glickman, L.H.; McWhirter, S.M.; Lemmens, E.; Mechette, K.; Leong, J.J.; Lauer, P.; Liu, W.; et al. Sting Agonist Formulated Cancer Vaccines Can Cure Established Tumors Resistant to Pd-1 Blockade. Sci. Transl. Med. 2015, 7, 283ra52. [Google Scholar] [CrossRef] [Green Version]
- Xia, T.; Konno, H.; Ahn, J.; Barber, G.N. Deregulation of STING Signaling in Colorectal Carcinoma Constrains DNA Damage Responses and Correlates with Tumorigenesis. Cell Rep. 2015, 14, 282–297. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, H.; Barber, G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef] [Green Version]
- Weiss, J.M.; Guérin, M.V.; Regnier, F.; Renault, G.; Galy-Fauroux, I.; Vimeux, L.; Feuillet, V.; Peranzoni, E.; Thoreau, M.; Trautmann, A.; et al. The STING agonist DMXAA triggers a cooperation between T lymphocytes and myeloid cells that leads to tumor regression. OncoImmunology 2017, 6, e1346765. [Google Scholar] [CrossRef]
- Woo, S.-R.; Fuertes, M.B.; Corrales, L.; Spranger, S.; Furdyna, M.J.; Leung, M.Y.K.; Duggan, R.; Wang, Y.; Barber, G.N.; Fitzgerald, K.A.; et al. STING-Dependent Cytosolic DNA Sensing Mediates Innate Immune Recognition of Immunogenic Tumors. Immunity 2014, 41, 830–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greene, E.; Finak, G.; D’Amico, L.A.; Bhardwaj, N.; Church, C.D.; Morishima, C.; Ramchurren, N.; Taube, J.M.; Nghiem, P.T.; Cheever, M.A.; et al. New interpretable machine-learning method for single-cell data reveals correlates of clinical response to cancer immunotherapy. Patterns 2021, 2, 100372. [Google Scholar] [CrossRef] [PubMed]
- Verhaegen, M.E.; Harms, P.W.; Van Goor, J.J.; Arche, J.; Patrick, M.T.; Wilbert, D.; Zabawa, H.; Grachtchouk, M.; Liu, C.-J.; Hu, K.; et al. Direct cellular reprogramming enables development of viral T antigen–driven Merkel cell carcinoma in mice. J. Clin. Investig. 2022, 132, e152069. [Google Scholar] [CrossRef] [PubMed]
- Toker, C. Trabecular Carcinoma of the Skin. Arch. Dermatol. 1972, 105, 107. [Google Scholar] [CrossRef]
- Triozzi, P.L.; Fernandez, A.P. The Role of the Immune Response in Merkel Cell Carcinoma. Cancers 2013, 5, 234–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarnicki, A.G.; Lysaght, J.; Todryk, S.; Mills, K.H. Suppression of Antitumor Immunity by Il-10 and Tgf-Beta-Producing T Cells Infiltrating the Growing Tumor: Influence of Tumor Environment on the Induction of Cd4+ and Cd8+ Regulatory T Cells. J. Immunol. 2006, 177, 896–904. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ouyang, K.; Zheng, D.X.; Agak, G.W. T-Cell Mediated Immunity in Merkel Cell Carcinoma. Cancers 2022, 14, 6058. https://doi.org/10.3390/cancers14246058
Ouyang K, Zheng DX, Agak GW. T-Cell Mediated Immunity in Merkel Cell Carcinoma. Cancers. 2022; 14(24):6058. https://doi.org/10.3390/cancers14246058
Chicago/Turabian StyleOuyang, Kelsey, David X. Zheng, and George W. Agak. 2022. "T-Cell Mediated Immunity in Merkel Cell Carcinoma" Cancers 14, no. 24: 6058. https://doi.org/10.3390/cancers14246058
APA StyleOuyang, K., Zheng, D. X., & Agak, G. W. (2022). T-Cell Mediated Immunity in Merkel Cell Carcinoma. Cancers, 14(24), 6058. https://doi.org/10.3390/cancers14246058