
Phosphorylation-Mediated Activation of β-Catenin-TCF4-CEGRs/ALCDs Pathway Is an Essential Event in Development of Aggressive Hepatoblastoma
 , ,
, ,
Abstract
:Simple Summary
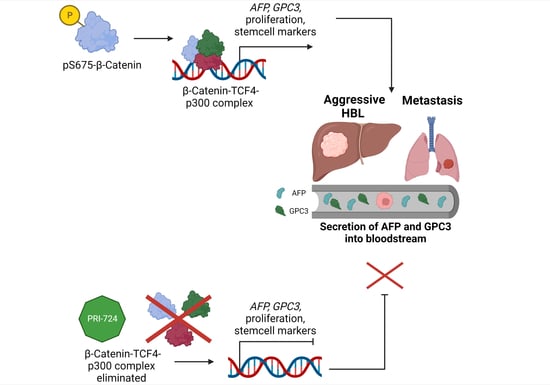
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Pediatric HBL Patients
2.2. List of Antibodies Used in This Study
2.3. Immunohistochemistry
2.4. List of TaqMan Probes Used in This Study
2.5. Real-Time Quantitative Reverse-Transcriptase PCR
2.6. Protein Isolation, Western Blotting, Co-Immunoprecipitation
2.7. Pull-Down Assay
2.8. Chromatin Immunoprecipitation (ChIP) Assay
2.9. Proliferation Assay
2.10. Locations of CEGRs/ALCDs within Corresponding Genes
2.11. Statistical Analysis
3. Results
3.1. Patients with Aggressive HBL Have Increased Levels of β-Catenin, Phosphorylated at S675
3.2. β-Catenin-TCF4-p300 Complexes Increase the Expression of CEGRs/ALCDs-Containing Oncogenes in Patients with Aggressive HBL
3.3. Expression of Glypican 3, a Target of β-Catenin, Is Strongly Elevated in Patients with Active β-Catenin-TCF4-CEGRs/ALCDs Pathway
3.4. Inhibition of β-Catenin by PRI-724 Removes β-Catenin-TCF4-p300 Complexes from CEGRs/ALCDs and Represses Expression of CEGRs/ALCDs-Dependent Oncogenes
3.5. PRI-724-Mediated Inhibition of β-Catenin-TCF4-CEGRs/ALCDs axis Reduces Proliferation of Cancer Cells
3.6. Inhibition of β-Catenin in Hepatoblastoma Cells Removes β-Catenin-TCF4-p300 Complex from CEGR/ALCD of GPC3 and from the Promoter of AFP Gene and Represses Expression of GPC3 and AFP
3.7. Silencing CEGRs/ALCDs in HBL-Derived PDXs Inhibits Expression of GPC3 and AFP in Tumors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ranganathan, S.; Lopez-Terrada, D.; Alaggio, R. Hepatoblastoma and Pediatric Hepatocellular Carcinoma: An Update. Pediatr. Dev. Pathol. 2019, 23, 79–95. [Google Scholar] [CrossRef] [PubMed]
- Johnston, M.; Timchenko, N.A. Molecular Signatures of Aggressive Pediatric Liver Cancer. Arch. Stem Cell Ther. 2021, 2, 1. [Google Scholar] [PubMed]
- Haines, K.; Sarabia, S.; Alvarez, K.R.; Tomlinson, G.; Vasudevan, S.A.; Heczey, A.A.; Roy, A.; Finegold, M.J.; Parsons, D.W.; Plon, S.E.; et al. Characterization of pediatric hepatocellular carcinoma reveals genomic heterogeneity and diverse signaling pathway activation. Pediatr. Blood Cancer 2019, 66, e27745. [Google Scholar] [CrossRef] [PubMed]
- Sumazin, P.; Peters, T.L.; Sarabia, S.F.; Kim, H.R.; Urbicain, M.; Hollingsworth, E.F.; Alvarez, K.R.; Perez, C.R.; Pozza, A.; Panah, M.J.N.; et al. Hepatoblastomas with carcinoma features represent a biological spectrum of aggressive neoplasms in children and young adults. J. Hepatol. 2022, 77, 1026–1037. [Google Scholar] [CrossRef] [PubMed]
- Short, S.S.; Kastenberg, Z.J.; Wei, G.; Bondoc, A.; Dasgupta, R.; Tiao, G.M.; Watters, E.; Heaton, T.E.; Lotakis, D.; La Quaglia, M.P.; et al. Histologic type predicts disparate outcomes in pediatric hepatocellular neoplasms: A Pediatric Surgical Oncology Research Collaborative study. Cancer 2022, 128, 2786–2795. [Google Scholar] [CrossRef]
- Eichenmüller, M.; Trippel, F.; Kreuder, M.; Beck, A.; Schwarzmayr, T.; Häberle, B.; Cairo, S.; Leuschner, I.; von Schweinitz, D.; Strom, T.M.; et al. The genomic landscape of hepatoblastoma and their progenies with HCC-like features. J. Hepatol. 2014, 61, 1312–1320. [Google Scholar] [CrossRef]
- Cairo, S.; Armengol, C.; de Reynies, A.; Wei, Y.; Thomas, E.; Renard, C.; Goga, A.; Balakrishnan, A.; Semeraro, M.; Gresh, L.; et al. Hepatic stem-like phenotype and interplay of Wnt/beta-catenin and Myc signaling in aggressive childhood liver cancer. Cancer Cell 2008, 14, 471–484. [Google Scholar] [CrossRef]
- Min, Q.; Molina, L.; Li, J.; Michael, A.O.A.; Russell, J.O.; Preziosi, M.E.; Singh, S.; Poddar, M.; Matz-Soja, M.; Ranganathan, S.; et al. β-Catenin and Yes-Associated Protein 1 Cooperate in Hepatoblastoma Pathogenesis. Am. J. Pathol. 2019, 189, 1091–1104. [Google Scholar] [CrossRef]
- Zhang, W.; Meyfeldt, J.; Wang, H.; Kulkarni, S.; Lu, J.; Mandel, J.A.; Marburger, B.; Liu, Y.; Gorka, J.E.; Ranganathan, S.; et al. β-Catenin mutations as determinants of hepatoblastoma phenotypes in mice. J. Biol. Chem. 2019, 294, 17524–17542. [Google Scholar] [CrossRef]
- Krutsenko, Y.; Singhi, A.; Monga, S. β-Catenin Activation in Hepatocellular Cancer: Implications in Biology and Therapy. Cancers 2021, 13, 1830. [Google Scholar] [CrossRef]
- Selvaggi, F.; Catalano, T.; Cotellese, R.; Aceto, G.M. Targeting Wnt/β-Catenin Pathways in Primary Liver Tumours: From Microenvironment Signaling to Therapeutic Agents. Cancers 2022, 14, 1912. [Google Scholar] [CrossRef] [PubMed]
- Apte, U.; Zeng, G.; Thompson, M.D.; Müller, P.; Micsenyi, A.; Cieply, B.; Kaestner, K.H.; Monga, S.P.S. β-Catenin is critical for early postnatal liver growth. Am. J. Physiol. Liver Physiol. 2007, 292, G1578–G1585. [Google Scholar] [CrossRef]
- Apte, U.; Singh, S.; Zeng, G.; Cieply, B.; Virji, M.A.; Wu, T.; Monga, S.P. Beta-Catenin Activation Promotes Liver Regeneration after Acetaminophen-Induced Injury. Am. J. Pathol. 2009, 175, 1056–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saggi, H.; Maitra, D.; Jiang, A.; Zhang, R.; Wang, P.; Cornuet, P.; Singh, S.; Locker, J.; Ma, X.; Dailey, H.; et al. Loss of hepatocyte β-catenin protects mice from experimental porphyria-associated liver injury. J. Hepatol. 2019, 70, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xu, H.; Cui, G.; Liang, B.; Chen, X.; Ko, S.; Affo, S.; Song, X.; Liao, Y.; Feng, J.; et al. β-Catenin Sustains and Is Required for YES-associated Protein Oncogenic Activity in Cholangiocarcinoma. Gastroenterology 2022, 163, 481–494. [Google Scholar] [CrossRef]
- Valanejad, L.; Lewis, K.; Wright, M.; Jiang, Y.; D’Souza, A.; Karns, R.; Sheridan, R.; Gupta, A.; Bove, K.; Witte, D.; et al. FXR-Gankyrin axis is involved in development of pediatric liver cancer. Carcinogenesis 2017, 38, 738–747. [Google Scholar] [CrossRef] [Green Version]
- Valanejad, L.; Cast, A.; Wright, M.; Bissig, K.-D.; Karns, R.; Weirauch, M.T.; Timchenko, N. PARP1 activation increases expression of modified tumor suppressors and pathways underlying development of aggressive hepatoblastoma. Commun. Biol. 2018, 1, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, M.E., II; Rivas, M.P.; Nicolle, D.; Gorse, A.; Gulati, R.; Kumbaji, M.; Weirauch, M.T.; Bondoc, A.; Cairo, S.; Geller, J.; et al. Olaparib Inhibits Tumor Growth of Hepatoblastoma in Patient Derived Xenograft Models. Hepatology 2021, 74, 2201–2215. [Google Scholar] [CrossRef] [PubMed]
- Rivas, M.; Johnston, M.E., II; Gulati, R.; Kumbaji, M.; Aguiar, T.F.M.; Timchenko, L.; Krepischi, A.; Shin, S.; Bondoc, A.; Tiao, G.; et al. HDAC1-Dependent Repression of Markers of Hepatocytes and P21 is Involved in Development of Pediatric Liver Cancer. Cell. Mol. Gastroenterol. Hepatol. 2021, 12, 1669–1682. [Google Scholar] [CrossRef]
- Gulati, R.; Johnston, M.; Rivas, M.; Cast, A.; Kumbaji, M.; Hanlon, M.A.; Lee, S.; Zhou, P.; Lake, C.; Schepers, E.; et al. β-catenin cancer–enhancing genomic regions axis is involved in the development of fibrolamellar hepatocellular carcinoma. Hepatol. Commun. 2022, 6, 2950–2963. [Google Scholar] [CrossRef]
- Zhou, S.; O’Gorman, M.R.G.; Yang, F.; Andresen, K.; Wang, L. Glypican 3 as a Serum Marker for Hepatoblastoma. Sci. Rep. 2017, 7, srep45932. [Google Scholar] [CrossRef]
- Zhou, P.; Gao, S.; Hu, B. Exploration of Potential Biomarkers and Immune Landscape for Hepatoblastoma: Evidence from Machine Learning Algorithm. Evid.-Based Complement. Altern. Med. 2022, 2022, 2417134. [Google Scholar] [CrossRef]
- Zhang, S.; Mo, Q.; Wang, X. Oncological role of HMGA2 (Review). Int. J. Oncol. 2019, 55, 775–788. [Google Scholar] [CrossRef] [PubMed]
- Cavard, C.; Terris, B.; Grimber, G.; Christa, L.; Audard, V.; Radenen-Bussiere, B.; Simon, M.-T.; Renard, C.-A.; Buendia, M.-A.; Perret, C. Overexpression of regenerating islet-derived 1 alpha and 3 alpha genes in human primary liver tumors with β-catenin mutations. Oncogene 2005, 25, 599–608. [Google Scholar] [CrossRef] [Green Version]
- Kimura, K.; Ikoma, A.; Shibakawa, M.; Shimoda, S.; Harada, K.; Saio, M.; Imamura, J.; Osawa, Y.; Kimura, M.; Nishikawa, K.; et al. Safety, Tolerability, and Preliminary Efficacy of the Anti-Fibrotic Small Molecule PRI-724, a CBP/β-Catenin Inhibitor, in Patients with Hepatitis C Virus-related Cirrhosis: A Single-Center, Open-Label, Dose Escalation Phase 1 Trial. eBioMedicine 2017, 23, 79–87. [Google Scholar] [CrossRef]
- Bottomly, D.; Kyler, S.L.; McWeeney, S.K.; Yochum, G.S. Identification of beta-catenin binding regions in colon cancer cells using ChIP-Seq. Nucleic Acids Res. 2010, 38, 5735–5745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, K.; Kanto, T.; Shimoda, S.; Harada, K.; Kimura, M.; Nishikawa, K.; Imamura, J.; Ogawa, E.; Saio, M.; Ikura, Y.; et al. Safety, tolerability, and anti-fibrotic efficacy of the CBP/β-catenin inhibitor PRI-724 in patients with hepatitis C and B virus-induced liver cirrhosis: An investigator-initiated, open-label, non-randomised, multicentre, phase 1/2a study. Ebiomedicine 2022, 80, 104069. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Dai, X.; Dai, J.; Ding, C.; Zhang, Z.; Lin, Z.; Hu, J.; Lu, M.; Wang, Z.; Qi, Y.; et al. AFP promotes HCC progression by suppressing the HuR-mediated Fas/FADD apoptotic pathway. Cell Death Dis. 2020, 11, 822. [Google Scholar] [CrossRef]
- Wang, S.; Chen, N.; Chen, Y.; Sun, L.; Li, L.; Liu, H. Elevated GPC3 level promotes cell proliferation in liver cancer. Oncol. Lett. 2018, 16, 970–976. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Wang, M.; Zheng, C.; Zhong, Q.; Shi, Y.; Han, X. Diagnostic value of serum glypican-3 alone and in combination with AFP as an aid in the diagnosis of liver cancer. Clin. Biochem. 2020, 79, 54–60. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gulati, R.; Hanlon, M.A.; Lutz, M.; Quitmeyer, T.; Geller, J.; Tiao, G.; Timchenko, L.; Timchenko, N. Phosphorylation-Mediated Activation of β-Catenin-TCF4-CEGRs/ALCDs Pathway Is an Essential Event in Development of Aggressive Hepatoblastoma. Cancers 2022, 14, 6062. https://doi.org/10.3390/cancers14246062
Gulati R, Hanlon MA, Lutz M, Quitmeyer T, Geller J, Tiao G, Timchenko L, Timchenko N. Phosphorylation-Mediated Activation of β-Catenin-TCF4-CEGRs/ALCDs Pathway Is an Essential Event in Development of Aggressive Hepatoblastoma. Cancers. 2022; 14(24):6062. https://doi.org/10.3390/cancers14246062
Chicago/Turabian StyleGulati, Ruhi, Margaret A. Hanlon, Maggie Lutz, Tyler Quitmeyer, James Geller, Gregory Tiao, Lubov Timchenko, and Nikolai Timchenko. 2022. "Phosphorylation-Mediated Activation of β-Catenin-TCF4-CEGRs/ALCDs Pathway Is an Essential Event in Development of Aggressive Hepatoblastoma" Cancers 14, no. 24: 6062. https://doi.org/10.3390/cancers14246062