
Mechanisms of Resistance in Gastroenteropancreatic Neuroendocrine Tumors


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
3. Resistance to Molecularly Targeted Therapies
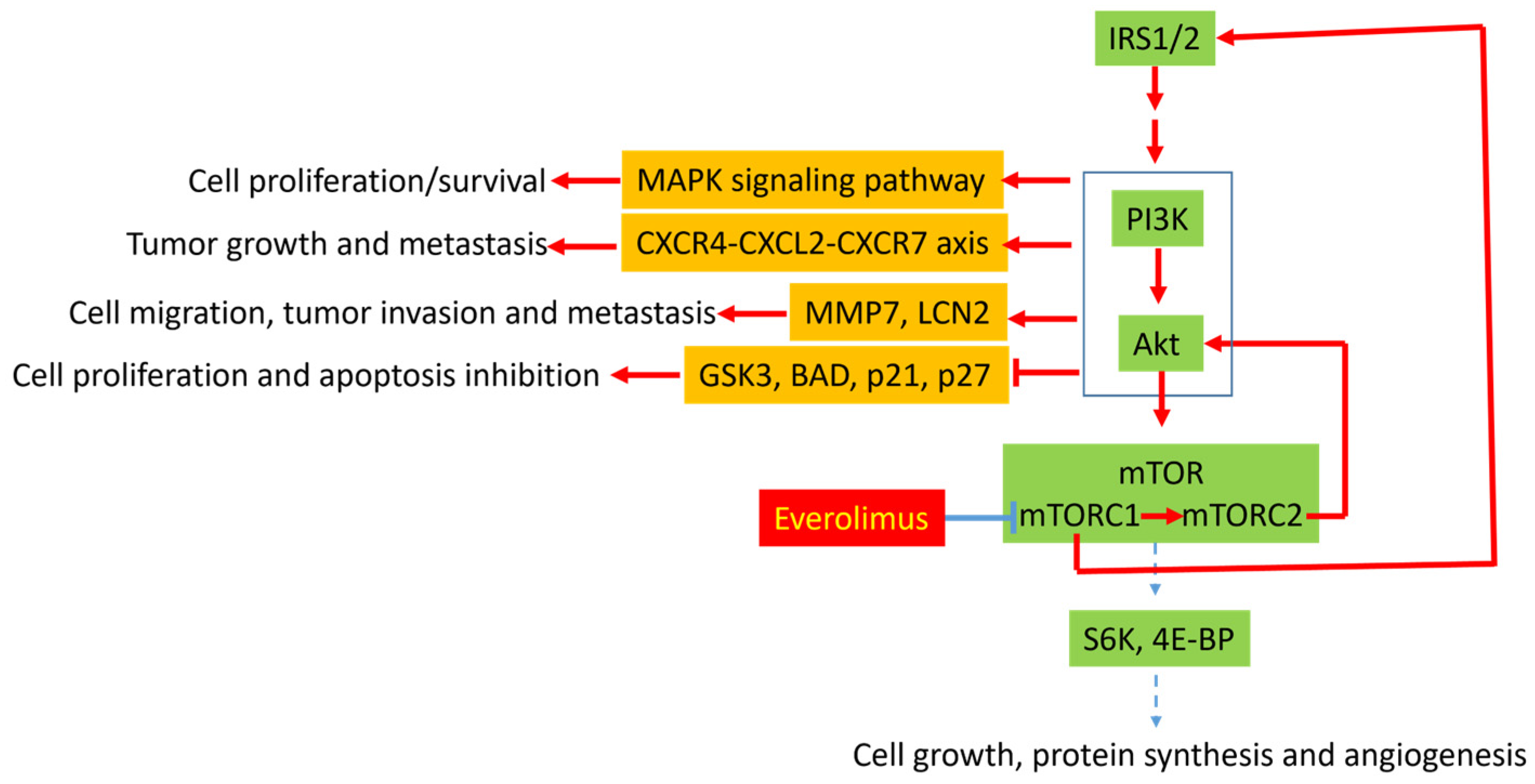
3.1. Molecular Mechanisms of Resistance to Everolimus
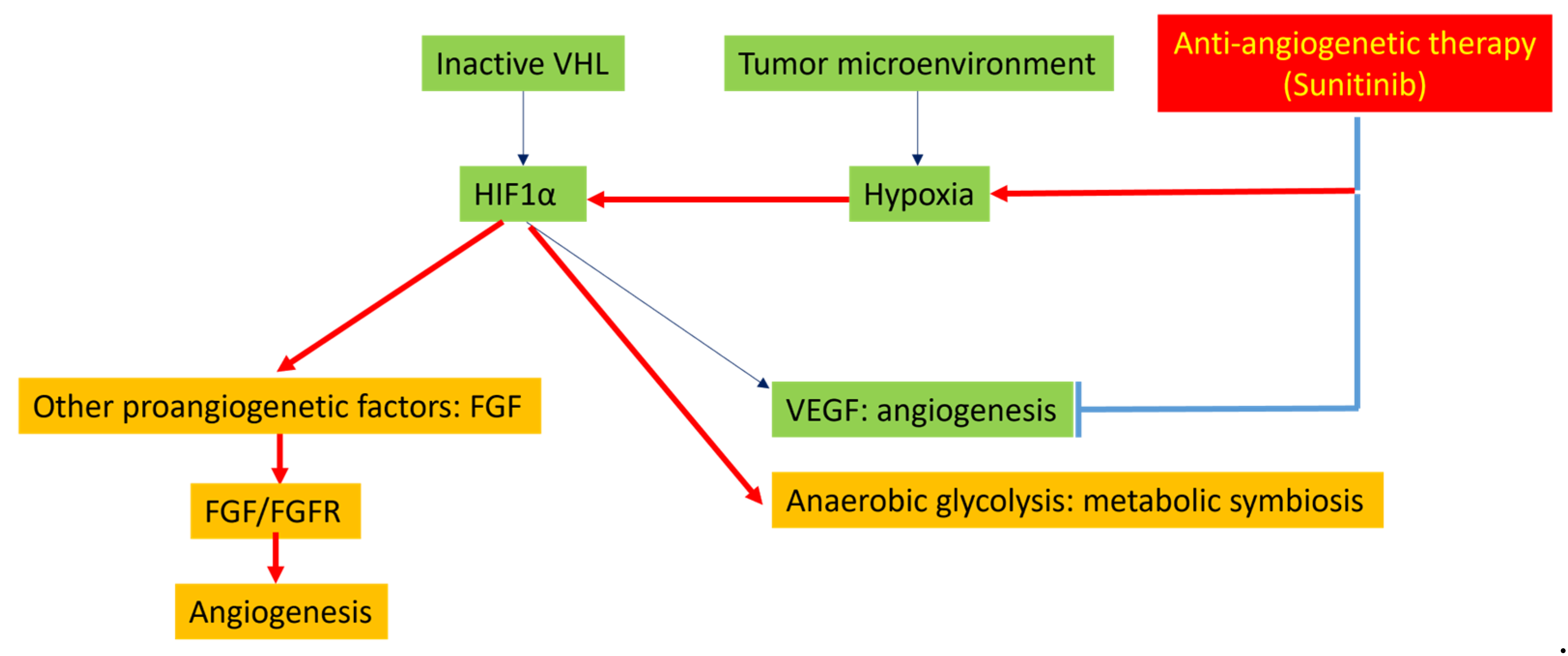
3.2. Molecular Mechanisms of Resistance to Multi-Kinase Inhibitors
3.3. Epigenetic/Genetic Dysregulation and Therapy Resistance
4. Resistance to SSTR-Targeted Therapy
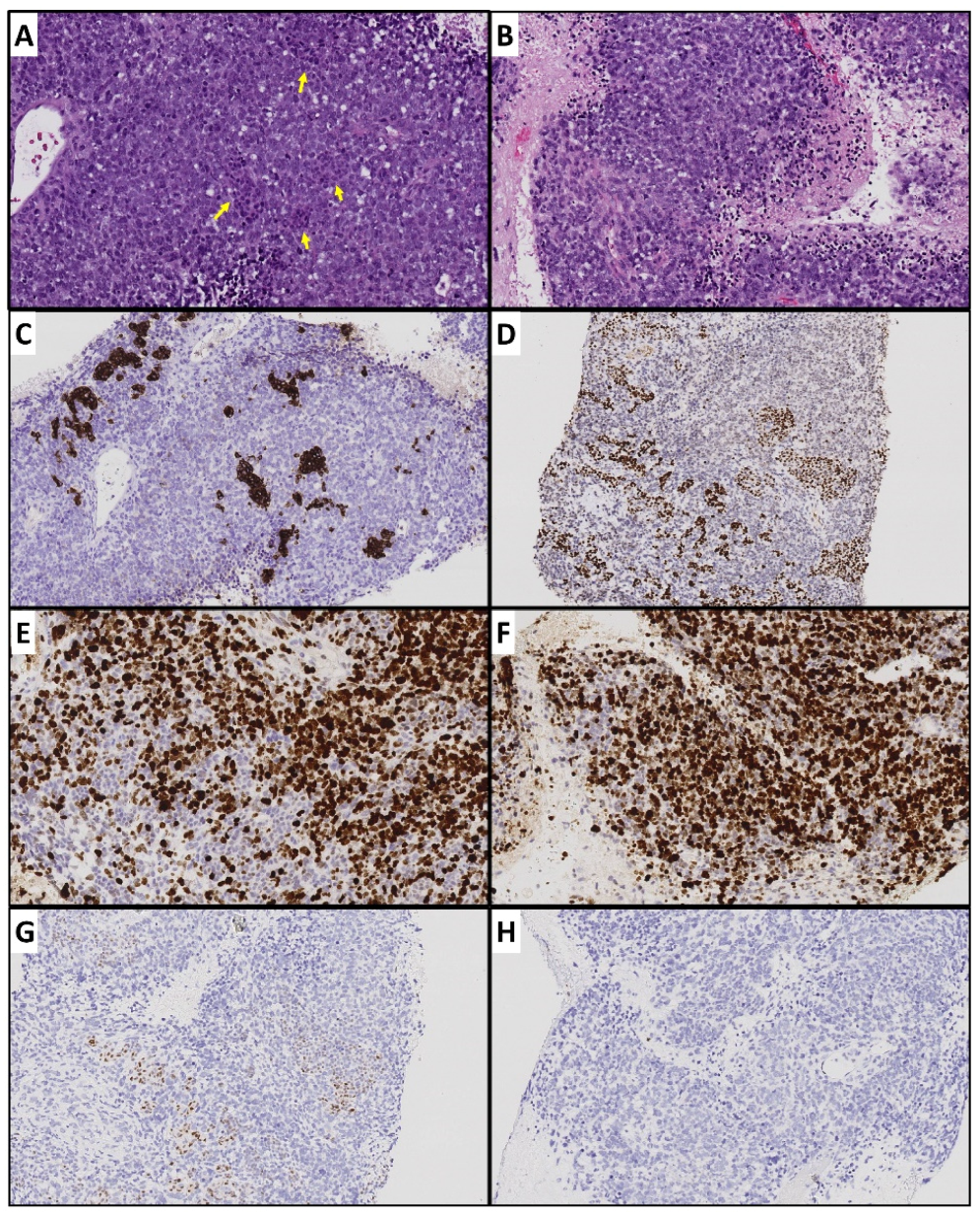
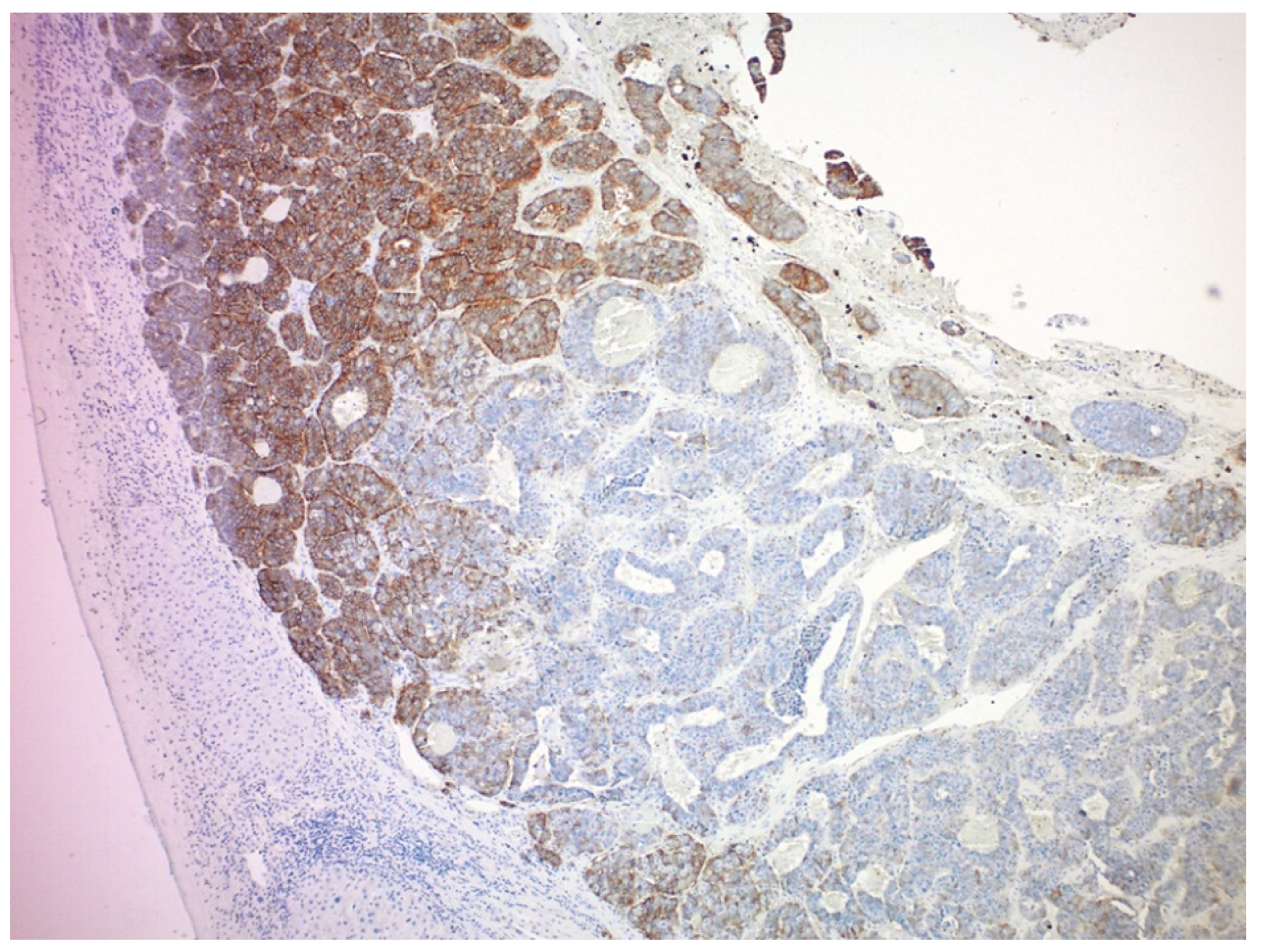
4.1. Tumor Heterogeneity
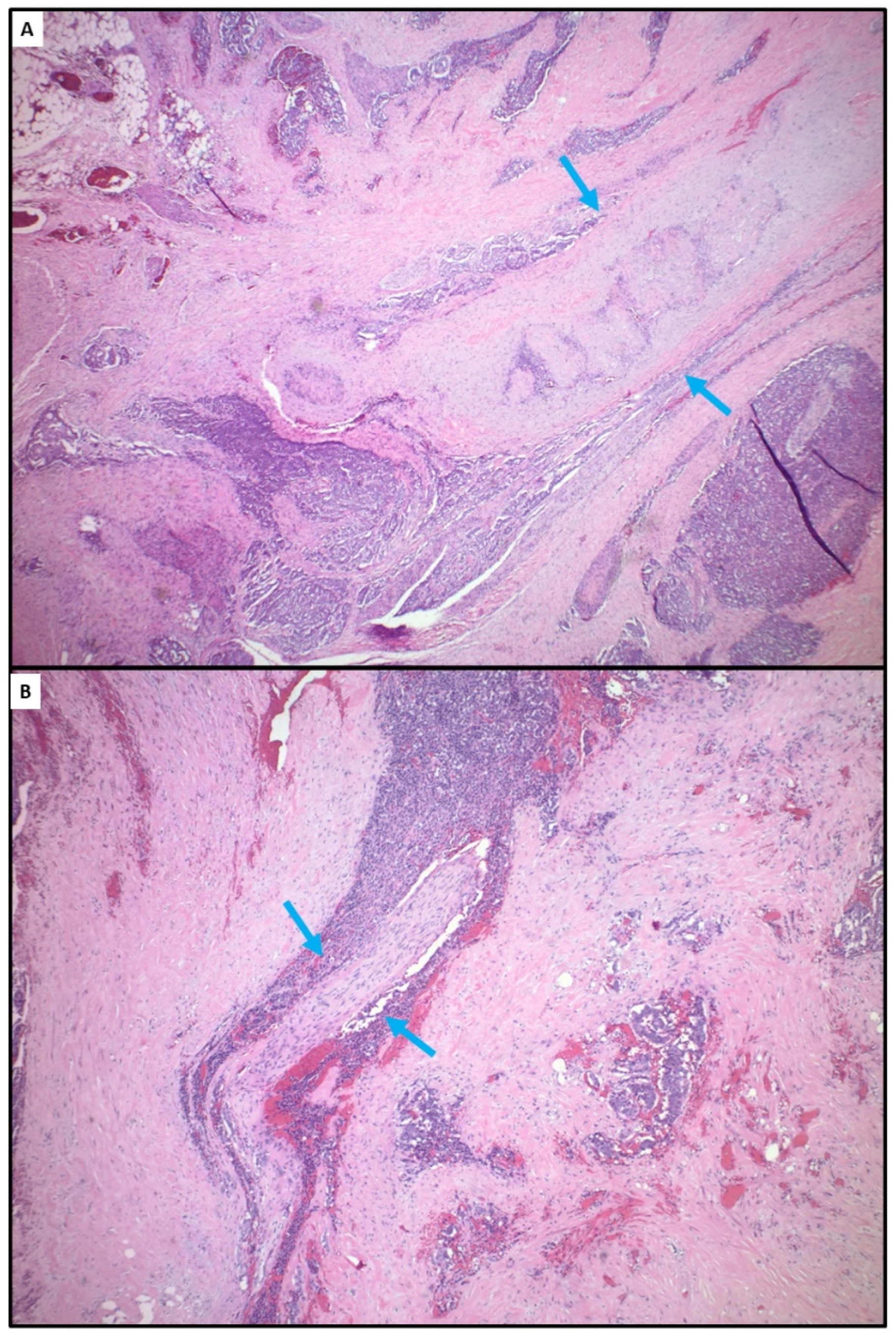
4.2. Tumor Hypoxia Related to Peritoneal Metastasis and Mesenteric Tumor Mass
5. Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Xu, Z.; Wang, L.; Dai, S.; Chen, M.; Li, F.; Sun, J.; Luo, F. Epidemiologic trends of and factors associated with overall survival for patients with gastroenteropancreatic neuroendocrine tumors in the United States. JAMA Netw. Open 2021, 4, e2124750. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekharan, C. Medical Management of gastroenteropancreatic neuroendocrine tumors. Surg. Oncol. Clin. N. Am. 2020, 29, 293–316. [Google Scholar] [CrossRef] [PubMed]
- Briest, F.; Grabowski, P. PI3K-AKT-mTOR-signaling and beyond: The complex network in gastroenteropancreatic neuroendocrine neoplasms. Theranostics 2014, 4, 336–365. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Shi, C.; Edil, B.H.; de Wilde, R.F.; Klimstra, D.S.; Maitra, A.; Schulick, R.D.; Tang, L.H.; Wolfgang, C.L.; Choti, M.A.; et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science 2011, 331, 1199–1203. [Google Scholar] [CrossRef]
- Vandamme, T.; Beyens, M.; Boons, G.; Schepers, A.; Kamp, K.; Biermann, K.; Pauwels, P.; De Herder, W.W.; Hofland, L.J.; Peeters, M.; et al. Hotspot DAXX, PTCH2 and CYFIP2 mutations in pancreatic neuroendocrine neoplasms. Endocr. Relat. Cancer 2019, 26, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ghayouri, M.; Boulware, D.; Nasir, A.; Strosberg, J.; Kvols, L.; Coppola, D. Activation of the serine/theronine protein kinase Akt in enteropancreatic neuroendocrine tumors. Anticancer Res. 2010, 30, 5063–5067. [Google Scholar] [PubMed]
- Beyens, M.; Vandamme, T.; Peeters, M.; Van Camp, G.; Op de Beeck, K. Resistance to targeted treatment of gastroenteropancreatic neuroendocrine tumors. Endocr. Relat. Cancer 2019, 26, R109–R130. [Google Scholar] [CrossRef]
- Yao, J.C.; Shah, M.H.; Ito, T.; Bohas, C.L.; Wolin, E.M.; Van Cutsem, E.; Hobday, T.J.; Okusaka, T.; Capdevila, J.; de Vries, E.G.; et al. Everolimus for advanced pancreatic neuroendocrine tumors. N. Engl. J. Med. 2011, 364, 514–523. [Google Scholar] [CrossRef]
- Pavel, M.E.; Singh, S.; Strosberg, J.R.; Bubuteishvili-Pacaud, L.; Degtyarev, E.; Neary, M.P.; Carnaghi, C.; Tomasek, J.; Wolin, E.; Raderer, M.; et al. Health-related quality of life for everolimus versus placebo in patients with advanced, non-functional, well-differentiated gastrointestinal or lung neuroendocrine tumours (RADIANT-4): A multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1411–1422. [Google Scholar] [CrossRef]
- Wang, X.; Yue, P.; Kim, Y.A.; Fu, H.; Khuri, F.R.; Sun, S.Y. Enhancing mammalian target of rapamycin (mTOR)-targeted cancer therapy by preventing mTOR/raptor inhibition-initiated, mTOR/rictor-independent Akt activation. Cancer Res. 2008, 68, 7409–7418. [Google Scholar] [CrossRef]
- Aristizabal Prada, E.T.; Spöttl, G.; Maurer, J.; Lauseker, M.; Koziolek, E.J.; Schrader, J.; Grossman, A.; Pacak, K.; Beuschlein, F.; Auernhammer, C.J.; et al. The role of GSK3 and its reversal with GSK3 antagonism in everolimus resistance. Endocr. Relat. Cancer 2018, 25, 893–908. [Google Scholar] [CrossRef] [PubMed]
- Tabernero, J.; Rojo, F.; Calvo, E.; Burris, H.; Judson, I.; Hazell, K.; Martinelli, E.; Ramon y Cajal, S.; Jones, S.; Vidal, L.; et al. Dose- and schedule-dependent inhibition of the mammalian target of rapamycin pathway with everolimus: A phase I tumor pharmacodynamic study in patients with advanced solid tumors. J. Clin. Oncol. 2008, 26, 1603–1610. [Google Scholar] [CrossRef] [PubMed]
- Miki, M.; Oono, T.; Fujimori, N.; Takaoka, T.; Kawabe, K.; Miyasaka, Y.; Ohtsuka, T.; Saito, D.; Nakamura, M.; Ohkawa, Y.; et al. CLEC3A, MMP7, and LCN2 as novel markers for predicting recurrence in resected G1 and G2 pancreatic neuroendocrine tumors. Cancer Med. 2019, 8, 3748–3760. [Google Scholar] [CrossRef] [PubMed]
- Djukom, C.; Porro, L.J.; Mrazek, A.; Townsend, C.M., Jr.; Hellmich, M.R.; Chao, C. Dual inhibition of PI3K and mTOR signaling pathways decreases human pancreatic neuroendocrine tumor metastatic progression. Pancreas 2014, 43, 88–92. [Google Scholar] [CrossRef]
- Curigliano, G.; Martin, M.; Jhaveri, K.; Beck, J.T.; Tortora, G.; Fazio, N.; Maur, M.; Hubner, R.A.; Lahner, H.; Donnet, V.; et al. Alpelisib in combination with everolimus ± exemestane in solid tumours: Phase Ib randomised, open-label, multicentre study. Eur. J. Cancer 2021, 151, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Hollebecque, A.; Houédé, N.; Cohen, E.E.; Massard, C.; Italiano, A.; Westwood, P.; Bumgardner, W.; Miller, J.; Brail, L.H.; Benhadji, K.A.; et al. A phase Ib trial of LY2584702 tosylate, a p70 S6 inhibitor, in combination with erlotinib or everolimus in patients with solid tumours. Eur. J. Cancer 2014, 50, 876–884. [Google Scholar] [CrossRef]
- De Luca, A.; Maiello, M.R.; D’Alessio, A.; Pergameno, M.; Normanno, N. The RAS/RAF/MEK/ERK and the PI3K/AKT signalling pathways: Role in cancer pathogenesis and implications for therapeutic approaches. Expert Opin. Targets 2012, 16 (Suppl. S2), S17–S27. [Google Scholar] [CrossRef]
- Carracedo, A.; Ma, L.; Teruya-Feldstein, J.; Rojo, F.; Salmena, L.; Alimonti, A.; Egia, A.; Sasaki, A.T.; Thomas, G.; Kozma, S.C.; et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J. Clin. Investig. 2008, 118, 3065–3074. [Google Scholar] [CrossRef]
- Valentino, J.D.; Li, J.; Zaytseva, Y.Y.; Mustain, W.C.; Elliott, V.A.; Kim, J.T.; Harris, J.W.; Campbell, K.; Weiss, H.; Wang, C.; et al. Cotargeting the PI3K and RAS pathways for the treatment of neuroendocrine tumors. Clin. Cancer Res. 2014, 20, 1212–1222. [Google Scholar] [CrossRef]
- Tolcher, A.W.; Bendell, J.C.; Papadopoulos, K.P.; Burris, H.A., III; Patnaik, A.; Jones, S.F.; Rasco, D.; Cox, D.S.; Durante, M.; Bellew, K.M.; et al. A phase IB trial of the oral MEK inhibitor trametinib (GSK1120212) in combination with everolimus in patients with advanced solid tumors. Ann. Oncol. 2015, 26, 58–64. [Google Scholar] [CrossRef]
- Höpfner, M.; Baradari, V.; Huether, A.; Schöfl, C.; Scherübl, H. The insulin-like growth factor receptor 1 is a promising target for novel treatment approaches in neuroendocrine gastrointestinal tumours. Endocr. Relat. Cancer 2006, 13, 135–149. [Google Scholar] [CrossRef]
- O’Reilly, K.E.; Rojo, F.; She, Q.B.; Solit, D.; Mills, G.B.; Smith, D.; Lane, H.; Hofmann, F.; Hicklin, D.J.; Ludwig, D.L.; et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006, 66, 1500–1508. [Google Scholar] [CrossRef] [PubMed]
- Strosberg, J.R.; Chan, J.A.; Ryan, D.P.; Meyerhardt, J.A.; Fuchs, C.S.; Abrams, T.; Regan, E.; Brady, R.; Weber, J.; Campos, T.; et al. A multi-institutional, phase II open-label study of ganitumab (AMG 479) in advanced carcinoid and pancreatic neuroendocrine tumors. Endocr. Relat. Cancer 2013, 20, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Dasari, A.; Phan, A.; Gupta, S.; Rashid, A.; Yeung, S.C.; Hess, K.; Chen, H.; Tarco, E.; Chen, H.; Wei, C.; et al. Phase I study of the anti-IGF1R antibody cixutumumab with everolimus and octreotide in advanced well-differentiated neuroendocrine tumors. Endocr. Relat. Cancer 2015, 22, 431–441. [Google Scholar] [CrossRef]
- Circelli, L.; Sciammarella, C.; Guadagno, E.; Tafuto, S.; del Basso de Caro, M.; Botti, G.; Pezzullo, L.; Aria, M.; Ramundo, V.; Tatangelo, F.; et al. CXCR4/CXCL12/CXCR7 axis is functional in neuroendocrine tumors and signals on mTOR. Oncotarget 2016, 7, 18865–18875. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, M.E.; Giaccone, G.; Liu, S.V.; Rajan, A.; Guha, U.; Halfdanarson, T.R.; Curtis, K.K.; Kunz, P.L.; Gabrail, N.; Hinson, J.M.; et al. Phase I, open-label, dose-escalation study of SNX-5422 plus everolimus in neuroendocrine tumors (NETs). Ann. Oncol. 2016, 27 (Suppl. S6), VI138. [Google Scholar] [CrossRef][Green Version]
- Pedersen, K.S.; Grierson, P.M.; Picus, J.; Lockhart, A.C.; Roth, B.J.; Liu, J.; Morton, A.; Chan, E.; Huffman, J.; Liang, C.; et al. Vorolanib (X-82), an oral anti-VEGFR/PDGFR/CSF1R tyrosine kinase inhibitor, with everolimus in solid tumors: Results of a phase I study. Investig. New Drugs 2021, 39, 1298–1305. [Google Scholar] [CrossRef]
- Harvey, R.D.; Carthon, B.C.; Lewis, C.; Hossain, M.S.; Zhang, C.; Chen, Z.; Harris, W.B.; Alese, O.B.; Shaib, W.; Bilen, M.A.; et al. Phase 1 safety and pharmacodynamic study of lenalidomide combined with everolimus in patients with advanced solid malignancies with efficacy signal in adenoid cystic carcinoma. Br. J. Cancer 2020, 123, 1228–1234. [Google Scholar] [CrossRef]
- Chan, J.A.; Mayer, R.J.; Jackson, N.; Malinowski, P.; Regan, E.; Kulke, M.H. Phase I study of sorafenib in combination with everolimus (RAD001) in patients with advanced neuroendocrine tumors. Cancer Chemother. Pharm. 2013, 71, 1241–1246. [Google Scholar] [CrossRef]
- Pusceddu, S.; de Braud, F.; Concas, L.; Bregant, C.; Leuzzi, L.; Formisano, B.; Buzzoni, R. Rationale and protocol of the MetNET-1 trial, a prospective, single center, phase II study to evaluate the activity and safety of everolimus in combination with octreotide LAR and metformin in patients with advanced pancreatic neuroendocrine tumors. Tumori 2014, 100, e286–e289. [Google Scholar] [CrossRef]
- Raymond, E.; Dahan, L.; Raoul, J.L.; Bang, Y.J.; Borbath, I.; Lombard-Bohas, C.; Valle, J.; Metrakos, P.; Smith, D.; Vinik, A.; et al. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N. Engl. J. Med. 2011, 364, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Tijeras-Raballand, A.; Neuzillet, C.; Couvelard, A.; Serova, M.; de Gramont, A.; Hammel, P.; Raymond, E.; Faivre, S. Resistance to targeted therapies in pancreatic neuroendocrine tumors (PNETs): Molecular basis, preclinical data, and counteracting strategies. Target Oncol. 2012, 7, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Pinato, D.J.; Tan, T.M.; Toussi, S.T.; Ramachandran, R.; Martin, N.; Meeran, K.; Ngo, N.; Dina, R.; Sharma, R. An expression signature of the angiogenic response in gastrointestinal neuroendocrine tumours: Correlation with tumour phenotype and survival outcomes. Br. J. Cancer 2014, 110, 115–122. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cives, M.; Pelle, E.; Quaresmini, D.; Rizzo, F.M.; Tucci, M.; Silvestris, F. The Tumor Microenvironment in Neuroendocrine Tumors: Biology and Therapeutic Implications. Neuroendocrinology 2019, 109, 83–99. [Google Scholar] [CrossRef] [PubMed]
- Allen, E.; Walters, I.B.; Hanahan, D. Brivanib, a dual FGF/VEGF inhibitor, is active both first and second line against mouse pancreatic neuroendocrine tumors developing adaptive/evasive resistance to VEGF inhibition. Clin. Cancer Res. 2011, 17, 5299–5310. [Google Scholar] [CrossRef]
- Capdevila, J.; Fazio, N.; Lopez, C.; Teulé, A.; Valle, J.W.; Tafuto, S.; Custodio, A.; Reed, N.; Raderer, M.; Grande, E.; et al. Lenvatinib in Patients With Advanced Grade 1/2 Pancreatic and Gastrointestinal Neuroendocrine Tumors: Results of the Phase II TALENT Trial (GETNE1509). J. Clin. Oncol. 2021, 39, 2304–2312. [Google Scholar] [CrossRef]
- Allen, E.; Miéville, P.; Warren, C.M.; Saghafinia, S.; Li, L.; Peng, M.W.; Hanahan, D. Metabolic Symbiosis Enables Adaptive Resistance to Anti-angiogenic Therapy that Is Dependent on mTOR Signaling. Cell Rep. 2016, 15, 1144–1160. [Google Scholar] [CrossRef] [PubMed]
- Gotink, K.J.; Broxterman, H.J.; Labots, M.; de Haas, R.R.; Dekker, H.; Honeywell, R.J.; Rudek, M.A.; Beerepoot, L.V.; Musters, R.J.; Jansen, G.; et al. Lysosomal sequestration of sunitinib: A novel mechanism of drug resistance. Clin. Cancer Res. 2011, 17, 7337–7346. [Google Scholar] [CrossRef]
- Wiedmer, T.; Blank, A.; Pantasis, S.; Normand, L.; Bill, R.; Krebs, P.; Tschan, M.P.; Marinoni, I.; Perren, A. Autophagy Inhibition Improves Sunitinib Efficacy in Pancreatic Neuroendocrine Tumors via a Lysosome-dependent Mechanism. Mol. Cancer Ther. 2017, 16, 2502–2515. [Google Scholar] [CrossRef]
- Scarpa, A. The landscape of molecular alterations in pancreatic and small intestinal neuroendocrine tumours. Ann. D’endocrinologie 2019, 80, 153–158. [Google Scholar] [CrossRef]
- Karpathakis, A.; Dibra, H.; Pipinikas, C.; Feber, A.; Morris, T.; Francis, J.; Oukrif, D.; Mandair, D.; Pericleous, M.; Mohmaduvesh, M.; et al. Progressive epigenetic dysregulation in neuroendocrine tumour liver metastases. Endocr. Relat. Cancer 2017, 24, L21–L25. [Google Scholar] [CrossRef] [PubMed]
- Walter, D.; Harter, P.N.; Battke, F.; Winkelmann, R.; Schneider, M.; Holzer, K.; Koch, C.; Bojunga, J.; Zeuzem, S.; Hansmann, M.L.; et al. Genetic heterogeneity of primary lesion and metastasis in small intestine neuroendocrine tumors. Sci. Rep. 2018, 8, 3811. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.S.; Laddha, S.V.; Lewis, P.W.; Koletsky, M.S.; Robzyk, K.; Da Silva, E.; Torres, P.J.; Untch, B.R.; Li, J.; Bose, P.; et al. ATRX, DAXX or MEN1 mutant pancreatic neuroendocrine tumors are a distinct alpha-cell signature subgroup. Nat. Commun. 2018, 9, 4158. [Google Scholar] [CrossRef]
- Roy, S.; LaFramboise, W.A.; Liu, T.C.; Cao, D.; Luvison, A.; Miller, C.; Lyons, M.A.; O’Sullivan, R.J.; Zureikat, A.H.; Hogg, M.E.; et al. Loss of Chromatin-Remodeling Proteins and/or CDKN2A Associates With Metastasis of Pancreatic Neuroendocrine Tumors and Reduced Patient Survival Times. Gastroenterology 2018, 154, 2060–2063.e8. [Google Scholar] [CrossRef]
- Klieser, E.; Urbas, R.; Stättner, S.; Primavesi, F.; Jäger, T.; Dinnewitzer, A.; Mayr, C.; Kiesslich, T.; Holzmann, K.; Di Fazio, P.; et al. Comprehensive immunohistochemical analysis of histone deacetylases in pancreatic neuroendocrine tumors: HDAC5 as a predictor of poor clinical outcome. Hum. Pathol. 2017, 65, 41–52. [Google Scholar] [CrossRef]
- Tang, L.H.; Untch, B.R.; Reidy, D.L.; O’Reilly, E.; Dhall, D.; Jih, L.; Basturk, O.; Allen, P.J.; Klimstra, D.S. Well-Differentiated Neuroendocrine Tumors with a Morphologically Apparent High-Grade Component: A Pathway Distinct from Poorly Differentiated Neuroendocrine Carcinomas. Clin. Cancer Res. 2016, 22, 1011–1017. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.R.; LaBauve, E.; Pomo, J.M.; Chiu, V.K.; Hanson, J.A.; Gullapalli, R.R. Site-Specific Genomic Alterations in a Well-Differentiated Pancreatic Neuroendocrine Tumor With High-Grade Progression. Pancreas 2018, 47, 502–510. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.L.; Yang, K.C.; Shen, Y.; Zhao, E.Y.; Loree, J.M.; Kennecke, H.F.; Kalloger, S.E.; Karasinska, J.M.; Lim, H.J.; Mungall, A.J.; et al. Molecular characterization of metastatic pancreatic neuroendocrine tumors (PNETs) using whole-genome and transcriptome sequencing. Cold Spring Harb. Mol. Case Stud. 2018, 4, a002329. [Google Scholar] [CrossRef]
- Allen, A.; Qin, A.; Raj, N.; Wang, J.; Uddin, S.; Yao, Z.; Tang, L.; Meyers, P.A.; Taylor, B.S.; Berger, M.F.; et al. Rare BRAF mutations in pancreatic neuroendocrine tumors may predict response to RAF and MEK inhibition. PLoS ONE 2019, 14, e0217399. [Google Scholar] [CrossRef]
- Gomes-Porras, M.; Cárdenas-Salas, J.; Álvarez-Escolá, C. Somatostatin Analogs in Clinical Practice: A Review. Int. J. Mol. Sci. 2020, 21, 1682. [Google Scholar] [CrossRef]
- Hennrich, U.; Kopka, K. Lutathera®: The First FDA- and EMA-Approved Radiopharmaceutical for Peptide Receptor Radionuclide Therapy. Pharmaceuticals 2019, 12, 114. [Google Scholar] [CrossRef] [PubMed]
- Rinke, A.; Müller, H.H.; Schade-Brittinger, C.; Klose, K.J.; Barth, P.; Wied, M.; Mayer, C.; Aminossadati, B.; Pape, U.F.; Bläker, M.; et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: A report from the PROMID Study Group. J. Clin. Oncol. 2009, 27, 4656–4663. [Google Scholar] [CrossRef] [PubMed]
- Lamberti, G.; Faggiano, A.; Brighi, N.; Tafuto, S.; Ibrahim, T.; Brizzi, M.P.; Pusceddu, S.; Albertelli, M.; Massironi, S.; Panzuto, F.; et al. Nonconventional Doses of Somatostatin Analogs in Patients With Progressing Well-Differentiated Neuroendocrine Tumor. J. Clin. Endocrinol. Metab. 2020, 105, dgz035. [Google Scholar] [CrossRef]
- Park, S.; Parihar, A.S.; Bodei, L.; Hope, T.A.; Mallak, N.; Millo, C.; Prasad, K.; Wilson, D.; Zukotynski, K.; Mittra, E. Somatostatin Receptor Imaging and Theranostics: Current Practice and Future Prospects. J. Nucl. Med. 2021, 62, 1323–1329. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.R.; Li, T.; Ter-Minassian, M.; Yang, J.; Chan, J.A.; Brais, L.K.; Masugi, Y.; Thiaglingam, A.; Brooks, N.; Nishihara, R.; et al. Association Between Somatostatin Receptor Expression and Clinical Outcomes in Neuroendocrine Tumors. Pancreas 2016, 45, 1386–1393. [Google Scholar] [CrossRef]
- Ortega, C.; Wong, R.; Schaefferkoetter, J.; Veit-Haibach, P.; Myrehaug, S.; Juergens, R.; Laidley, D.; Anconina, R.; Liu, A.; Metser, U. Quantitative 68Ga-DOTATATE PET/CT Parameters for the Prediction of Therapy Response in Patients with Progressive Metastatic Neuroendocrine Tumors Treated with 177Lu-DOTATATE. J. Nucl. Med. 2021, 62, 1406–1414. [Google Scholar] [CrossRef] [PubMed]
- Feijtel, D.; Doeswijk, G.N.; Verkaik, N.S.; Haeck, J.C.; Chicco, D.; Angotti, C.; Konijnenberg, M.W.; de Jong, M.; Nonnekens, J. Inter and intra-tumor somatostatin receptor 2 heterogeneity influences peptide receptor radionuclide therapy response. Theranostics 2021, 11, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Charoenpitakchai, M.; Liu, E.; Zhao, Z.; Koyama, T.; Huh, W.J.; Berlin, J.; Hande, K.; Walker, R.; Shi, C. In liver metastases from small intestinal neuroendocrine tumors, SSTR2A expression is heterogeneous. Virchows Arch. 2017, 470, 545–552. [Google Scholar] [CrossRef]
- Shaheen, S.; Moradi, F.; Gamino, G.; Kunz, P.L. Patient Selection and Toxicities of PRRT for Metastatic Neuroendocrine Tumors and Research Opportunities. Curr. Treat. Options Oncol. 2020, 21, 25. [Google Scholar] [CrossRef]
- Strosberg, J.; El-Haddad, G.; Wolin, E.; Hendifar, A.; Yao, J.; Chasen, B.; Mittra, E.; Kunz, P.L.; Kulke, M.H.; Jacene, H.; et al. Phase 3 Trial of 177Lu-Dotatate for Midgut Neuroendocrine Tumors. N. Engl. J. Med. 2017, 376, 125–135. [Google Scholar] [CrossRef]
- Bodei, L.; Schöder, H.; Baum, R.P.; Herrmann, K.; Strosberg, J.; Caplin, M.; Öberg, K.; Modlin, I.M. Molecular profiling of neuroendocrine tumours to predict response and toxicity to peptide receptor radionuclide therapy. Lancet Oncol. 2020, 21, e431–e443. [Google Scholar] [CrossRef] [PubMed]
- Graf, F.; Fahrer, J.; Maus, S.; Morgenstern, A.; Bruchertseifer, F.; Venkatachalam, S.; Fottner, C.; Weber, M.M.; Huelsenbeck, J.; Schreckenberger, M.; et al. DNA double strand breaks as predictor of efficacy of the alpha-particle emitter Ac-225 and the electron emitter Lu-177 for somatostatin receptor targeted radiotherapy. PLoS ONE 2014, 9, e88239. [Google Scholar] [CrossRef]
- Shi, C.; Gonzalez, R.S.; Zhao, Z.; Koyama, T.; Cornish, T.C.; Hande, K.R.; Walker, R.; Sandler, M.; Berlin, J.; Liu, E.H. Liver metastases of small intestine neuroendocrine tumors: Ki-67 heterogeneity and World Health Organization grade discordance with primary tumors. Am. J. Clin. Pathol. 2015, 143, 398–404. [Google Scholar] [CrossRef] [PubMed]
- Assi, H.A.; Hornbacker, K.; Shaheen, S.; Wittenberg, T.; Silberman, R.; Kunz, P.L. Rapid Progression After 177Lu-DOTATATE in Patients With Neuroendocrine Tumors. Pancreas 2021, 50, 890–894. [Google Scholar] [CrossRef] [PubMed]
- Chicago Consensus Working Group. The Chicago Consensus on peritoneal surface malignancies: Management of neuroendocrine tumors. Cancer 2020, 126, 2561–2565. [Google Scholar] [CrossRef]
- Koumarianou, A.; Alexandraki, K.I.; Wallin, G.; Kaltsas, G.; Daskalakis, K. Pathogenesis and Clinical Management of Mesenteric Fibrosis in Small Intestinal Neuroendocrine Neoplasms: A Systematic Review. J. Clin. Med. 2020, 9, 1777. [Google Scholar] [CrossRef]
- Merola, E.; Prasad, V.; Pascher, A.; Pape, U.F.; Arsenic, R.; Denecke, T.; Fehrenbach, U.; Wiedenmann, B.; Pavel, M.E. Peritoneal Carcinomatosis in Gastro-Entero-Pancreatic Neuroendocrine Neoplasms: Clinical Impact and Effectiveness of the Available Therapeutic Options. Neuroendocrinology 2020, 110, 517–524. [Google Scholar] [CrossRef]
- Wilson, R.B. Hypoxia, cytokines and stromal recruitment: Parallels between pathophysiology of encapsulating peritoneal sclerosis, endometriosis and peritoneal metastasis. Pleura Peritoneum 2018, 3, 20180103. [Google Scholar] [CrossRef]
- Kassis, A.I. Therapeutic radionuclides: Biophysical and radiobiologic principles. Semin. Nucl. Med. 2008, 38, 358–366. [Google Scholar] [CrossRef]
- Blažević, A.; Brabander, T.; Zandee, W.T.; Hofland, J.; Franssen, G.; van Velthuysen, M.F.; Feelders, R.A.; De Herder, W.W. Evolution of the Mesenteric Mass in Small Intestinal Neuroendocrine Tumours. Cancers 2021, 13, 443. [Google Scholar] [CrossRef]
- Xu, J. Current treatments and future potential of surufatinib in neuroendocrine tumors (NETs). Adv. Med. Oncol. 2021, 13, 17588359211042689. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, R.L.; Weissbach, J.; Kleilein, J.; Bell, J.; Hüttelmaier, S.; Viol, F.; Clauditz, T.; Grabowski, P.; Laumen, H.; Rosendahl, J.; et al. Targeting HDACs in Pancreatic Neuroendocrine Tumor Models. Cells 2021, 10, 1408. [Google Scholar] [CrossRef] [PubMed]
- Pollard, J.H.; Menda, Y.; Zamba, K.D.; Madsen, M.; O’Dorisio, M.S.; O’Dorisio, T.; Bushnell, D. Potential for Increasing Uptake of Radiolabeled 68Ga-DOTATOC and 123I-MIBG in Patients with Midgut Neuroendocrine Tumors Using a Histone Deacetylase Inhibitor Vorinostat. Cancer Biother. Radiopharm. 2021, 36, 632–641. [Google Scholar] [CrossRef] [PubMed]
- Si, Y.; Kim, S.; Ou, J.; Lu, Y.; Ernst, P.; Chen, K.; Whitt, J.; Carter, A.M.; Markert, J.M.; Bibb, J.A.; et al. Anti-SSTR2 antibody-drug conjugate for neuroendocrine tumor therapy. Cancer Gene 2021, 28, 799–812. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, C.; Morse, M.A. Mechanisms of Resistance in Gastroenteropancreatic Neuroendocrine Tumors. Cancers 2022, 14, 6114. https://doi.org/10.3390/cancers14246114
Shi C, Morse MA. Mechanisms of Resistance in Gastroenteropancreatic Neuroendocrine Tumors. Cancers. 2022; 14(24):6114. https://doi.org/10.3390/cancers14246114
Chicago/Turabian StyleShi, Chanjuan, and Michael A. Morse. 2022. "Mechanisms of Resistance in Gastroenteropancreatic Neuroendocrine Tumors" Cancers 14, no. 24: 6114. https://doi.org/10.3390/cancers14246114
APA StyleShi, C., & Morse, M. A. (2022). Mechanisms of Resistance in Gastroenteropancreatic Neuroendocrine Tumors. Cancers, 14(24), 6114. https://doi.org/10.3390/cancers14246114