


The Role of Cell-Free DNA in Cancer Treatment Decision Making


Abstract
:Simple Summary
Abstract
1. Introduction

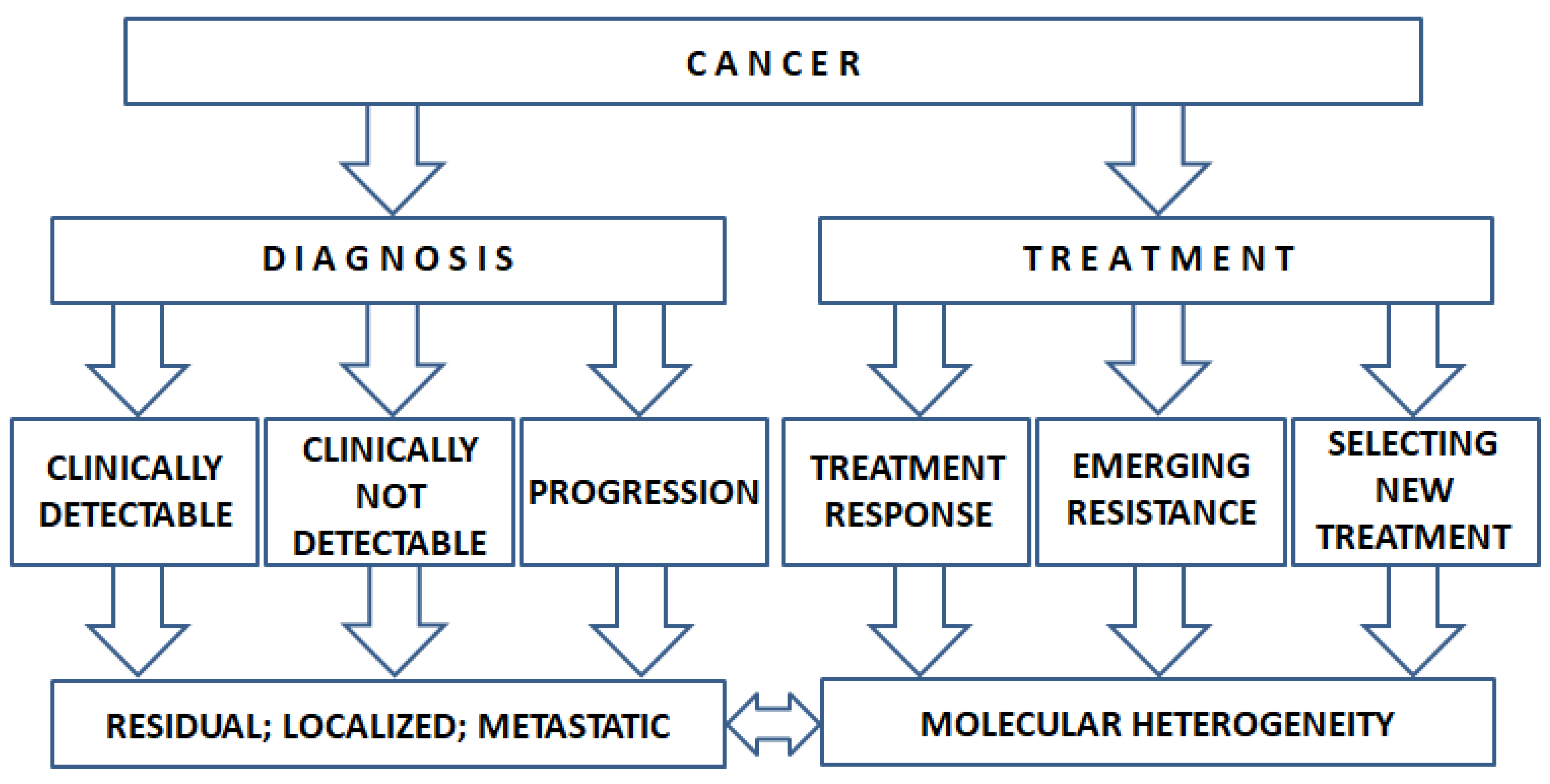{kind=link}
| Tumor Type | Screening | Early Stage | Monitoring for MRD | Metastatic Disease | To Prove Therapeutic Effect | Targeted Therapy Indicated Based on a ctDNA Test |
|---|---|---|---|---|---|---|
| Bladder [19,20,21] | Liquid biopsy in muscle-invasive bladder cancer (MIBC) and nonMIBC urinary-cfDNA for those, who are not taking flexible cystoscopy [22] | Yes | Yes | Yes | Yes | Ongoing trials [23]: deraazantinib, erdafitinib, futibatinib, infigratinib, lenvatinib, pemigatinib, regorafenib, regoratinib, |
| Breast [24,25,26,27,28] | ctDNA assays hold substantial potential as an early cancer screening test, but very early stage (asymptomatic) tumors are not likely to release enough ctDNA to be detectable in a typical blood draw of 10 mL [29] | No | Yes | Yes. Brain and meningeal metastasis from cerebrospinalfluid ctDNAs [30,31] | Yes | Alpelisib [32]; Ribociklib, Neratinib, abemaciklib Trials ongoing: PADA-1, SOLAR-1, MONALEESA-2,3,7; BELLE-2,3; PALOMA-3, POSEIDON, SUMMIT, BEECH, I-SPY2, MONARCH2, LOTUS, INSPIRE, Neo-ALTO, MONAL |
| CRC [33,34,35,36,37,38] | Yes | Yes | Yes | Visceral metastases were found to be associated with detectable ctDNA [39] | Yes | Trials ongoing [40]: CIRCULATE-Idea; GALAXY, ALTAIR, DYNAMIC, TRACC, MEDOCC-CrEATENRG-GI-005, VEGA, ACT-3, IMPROVE-IT2 |
| Esophagus [41,42,43] | Not good for screening [34] | No | Yes | Yes | No [44] | Afatinib, crizotinib, ABBV-321 trial: serclutamabtalirine [45] |
| Gastric cc [46,47] | Yes | Yes | Yes | Yes | Yes | – |
| Head and neck [48,49] | Not good for screening | No | Yes | Yes | After radiation therapy [50] | – |
| Liver/bileduct [33,51,52,53] | Yes Even from the bile fluid [54] | Yes | Yes | Yes | Yes | Trials ongoing in HCC [55]: Sorafenib, sunitinib, cedirabinib, linifanib, dovotinib, brivanib |
| Melanoma malignum [56,57] | No data | No data | Yes | Yes | Yes | Dabrafenib plus trametinib [58] |
| NSCLC [59,60,61,62,63] | Yes | Yes | Yes | Yes | Yes | Durvalumab, amivantamab [64] |
| Ovary [35,65,66] | Specificity and sensitivity is better than if CA-125and HE combined wihcfDNA [67] | Not yet | Not yet | Yes | Yes | – |
| Pancreas [68,69,70] | Not good for screening [71,72] | No | Yes | Yes | ctDNA and CA19- showed similar trends | – |
| Prostate cancer [73,74] | Yes | Yes | Yes | Yes | Yes [75] | – |
| SCLC [76,77] | Yes | Yes | Yes | May predict the progression of lung cancer patients earlier than imaging [78] | yes | – |
2. Liquid Biopsy
3. Cell-Free DNA (cfDNA)
4. The Circulating Tumor DNA (ctDNA)
5. The Applications of cfDNA and ctDNA in Clinical Oncology
6. Cell-Free DNA in Diagnosis
7. The cfDNA and ctDNA in Cancer Treatment
8. Combined Analysis of ctDNA with Other Parameters
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 5fC | 5-formylcytosine |
| ALK | anaplastic lymphoma kinase gene |
| BRAF | human gene that encodes a protein called B-Raf |
| CA-125 | cancer/ carcinoma antigen 125, or carbohydrate antigen 125 |
| CEA | carcinoembryonic antigen |
| cfDNA | cell-free DNA |
| cfRNA | cell-free RNA |
| CHIP | clonal hematopoiesis of indeterminate potential |
| CI | confidence interval |
| CpG | dinucleotide DNA where a cytosine (C) nucleotide is followed by a guanine (G) nucleotide |
| CRC | colorectal cancer |
| CRPC | castrate-resistant prostate cancer |
| CSC | cancer stem cell |
| CSF | cerebrospinal fluid |
| CTC | circulating tumor cell |
| ctDNA | circulating tumor-derived DNA |
| DELFI | DNA evaluation of fragments for early interception |
| DFS | disease-free survival |
| DNA | deoxyribonucleic acid |
| EC | endometrial cancer |
| EGFR | epidermal growth factor receptor |
| ER | estrogen receptor |
| ESMO | European Society for Medical Oncology |
| ET | end of treatment |
| EV | extracellular vesicle |
| FDA | Food and Drug Administration |
| FTV | functional tumor volume |
| HER2 | human epidermal growth factor receptor 2 |
| HR | hazard ratio |
| ICB | immune checkpoint blockade |
| KRAS | “Kirsten rat sarcoma virus” is a gene that provides instructions for making a protein called K-Ras |
| LB | liquid biopsy |
| mAb | monoclonal antibody |
| MRD | minimal residual disease |
| mRNA | messenger RNA |
| MSI | microsatellite instability |
| MT | middle of thetherapy |
| mtDNA | mitochondrial DNA |
| NAT | neoadjuvant therapy |
| NCCN | National Comprehensive Cancer Network |
| NCDB | National Cancer Database |
| NGS | next-generation sequencing |
| NSCLC | non-small-cell lung cancer |
| OC | ovarian cancer |
| OR | odds ratio |
| OS | overall survival |
| PCR | polymerase chain reaction |
| PFS | progression-free survival |
| PIK3CA | phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha |
| PSA | prostate-specific antigen |
| RFS | relapse-free survival |
| RNA | ribonucleic acid |
| RT | radiotherapy |
| SCLC | small cell lung cancer |
| TEP | tumor-educated platelets |
| TET | ten-eleven translocation enzyme |
| TKI | tyrosine kinase inhibitor |
| TMB | tumor mutational burden |
| TNBC | triple-negative breast cancer |
| TP53 | tumor protein p53 |
| VAF | variant allele frequency |
References
- Bankó, P.; Lee, S.Y.; Nagygyörgy, V.; Zrínyi, M.; Chae, C.H.; Cho, D.H.; Telekes, A. Technologies for circulating tumor cell separation from whole blood. J. Hematol. Oncol. 2019, 12, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salvi, S.; Gurioli, G.; De Giorgi, U.; Conteduca, V.; Tedaldi, G.; Calistri, D.; Casadio, V. Cell-free DNA as a diagnostic marker for cancer: Current insights. Oncotargets Ther. 2016, 9, 6549–6559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spisák, S.; Solymosi, N.; Ittzés, P.; Bodor, A.; Kondor, D.; Vattay, G.; Barták, B.K.; Sipos, F.; Galamb, O.; Tulassay, Z.; et al. Complete genes may pass from food to human blood. PLoS ONE 2013, 8, e69805. [Google Scholar] [CrossRef] [Green Version]
- Calabuig-Fariñas, S.; Jantus-Lewintre, E.; Herreros-Pomares, A.; Camps, C. Circulating tumor cells versus circulating tumor DNA in lung cancer-which one will win? Transl. Lung Cancer Res. 2016, 5, 466–482. [Google Scholar] [CrossRef] [Green Version]
- FDA Web Site. Available online: https://www.fda.gov/media/137482 (accessed on 21 April 2019).
- Bailey, C.; Black, J.R.M.; Reading, J.L.; Litchfield, K.; Turajlic, S.; McGranahan, N.; Jamal-Hanjani, M.; Swanton, C. Tracking Cancer Evolution through the Disease Course. Cancer Discov. 2021, 11, 916–932. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.nccn.org/professionals/physician_gls (accessed on 27 July 2021).
- Pascual, J.; Attard, G.; Bidard, F.C.; Curigliano, G.; De Mattos-Arruda, L.; Diehn, M.; Italiano, A.; Lindberg, J.; Merker, J.D.; Montagut, C.; et al. ESMO recommendations on the use of circulating tumour DNA assays for patients with cancer: A report from the ESMO Precision Medicine Working Group. Ann. Oncol. 2022, 33, 750–768. [Google Scholar] [CrossRef] [PubMed]
- Kong, S.L.; Liu, X.; Tan, S.J.; Tai, J.A.; Phua, L.Y.; Poh, H.M.; Yeo, T.; Chua, Y.W.; Haw, Y.X.; Ling, W.H.; et al. Complementary Sequential Circulating Tumor Cell (CTC) and Cell-Free Tumor DNA (ctDNA) Profiling Reveals Metastatic Heterogeneity and Genomic Changes in Lung Cancer and Breast Cancer. Front. Oncol. 2021, 11, 698551. [Google Scholar] [CrossRef]
- Chedid, J.; Allam, S.; Chamseddine, N.; BouZerdan, M.; El Nakib, C.; Assi, H.I. Role of circulating tumor DNA and circulating tumor cells in breast cancer: History and updates. SAGE Open Med. 2022, 10, 20503121221077838. [Google Scholar] [CrossRef] [PubMed]
- Tivey, A.; Church, M.; Rothwell, D.; Dive, C.; Cook, N. Circulating tumour DNA—Looking beyond the blood. Nat. Rev. Clin. Oncol. 2022, 19, 600–612. [Google Scholar] [CrossRef]
- Sanz-Garcia, E.; Zhao, E.; Bratman, S.V.; Siu, L.L. Monitoring and adapting cancer treatment using circulating tumor DNA kinetics: Current research, opportunities, and challenges. Sci. Adv. 2022, 8, eabi8618. [Google Scholar] [CrossRef]
- Ignatiadis, M.; Sledge, G.W.; Jeffrey, S.S. Liquid biopsy enters the clinic—Implementation issues and future challenges. Nat. Rev. Clin. Oncol. 2021, 18, 297–312. [Google Scholar] [CrossRef] [PubMed]
- Deveson, I.W.; Gong, B.; Lai, K.; LoCoco, J.S.; Richmond, T.A.; Schageman, J.; Zhang, Z.; Novoradovskaya, N.; Willey, J.C.; Jones, W.; et al. Evaluating the analytical validity of circulating tumor DNA sequencing assays for precision oncology. Nat. Biotechnol. 2021, 39, 1115–1128. [Google Scholar] [CrossRef]
- Kilgour, E.; Rothwell, D.G.; Brady, G.; Dive, C. Liquid biopsy-based biomarkers of treatment response and resistance. Cancer Cell 2020, 37, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Andersson, D.; Kristiansson, H.; Kubista, M.; Ståhlberg, A. Ultrasensitive circulating tumor DNA analysis enables precision medicine: Experimental workflow considerations. Expert Rev. Mol. Diagn. 2021, 21, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Danesi, R.; Lo, Y.M.D.; Oellerich, M.; Beck, J.; Galbiati, S.; Re, M.D.; Lianidou, E.; Neumaier, M.; van Schaik, R.H.N. What do we need to obtain high quality circulating tumor DNA (ctDNA) for routine diagnostic test in oncology?—Considerations on pre-analytical aspects by the IFCC workgroup cfDNA. Clin. Chim. Acta 2021, 520, 168–171. [Google Scholar] [CrossRef] [PubMed]
- Hasenleithner, S.; Speicher, M.R. A clinician’s handbook for using ctDNA throughout the patient journey. Mol. Cancer 2022, 21, 81. [Google Scholar] [CrossRef]
- Liu, M.C.; Oxnard, G.R.; Klein, E.A.; Swanton, C.; Seiden, M.V.; CCGA Consortium. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann. Oncol. 2020, 31, 745–759. [Google Scholar] [CrossRef]
- Christensen, E.; Birkenkamp-Demtröder, K.; Sethi, H.; Shchegrova, S.; Salari, R.; Nordentoft, I.K.; Wu, H.-T.; Knudsen, M.; Lamy, P.; Lindskrog, S.V.; et al. Early Detection of Metastatic Relapse and Monitoring of Therapeutic Efficacy by Ultra-Deep Sequencing of Plasma Cell-Free DNA in Patients With Urothelial Bladder Carcinoma. J. Clin. Oncol. 2019, 37, 1547–1557. [Google Scholar] [CrossRef]
- Tan, M.P.; Attard, G.; Huddart, R.A. Circulating Tumour DNA in Muscle-Invasive Bladder Cancer. Int. J. Mol. Sci. 2018, 19, 2568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tse, R.T.; Zhao, H.; Wong, C.Y.; Cheng, C.; Kong, A.; Peng, Q.; Chiu, P.; Ng, C.-F.; Teoh, J. Urinary Cell-Free DNA in Bladder Cancer Detection. Diagnostics 2021, 11, 306. [Google Scholar] [CrossRef]
- Thomas, J.; Sonpavde, G. Molecularly Targeted Therapy towards Genetic Alterations in Advanced Bladder Cancer. Cancers 2022, 14, 1795. [Google Scholar] [CrossRef] [PubMed]
- Sant, M.; Bernat-Peguera, A.; Felip, E.; Margelí, M. Role of ctDNA in Breast Cancer. Cancers 2022, 14, 310. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Murillas, I.; Schiavon, G.; Weigelt, B.; Ng, C.; Hrebien, S.; Cutts, R.J.; Cheang, M.; Osin, P.; Nerurkar, A.; Kozarewa, I.; et al. Mutation Tracking in Circulating Tumor DNA Predicts Relapse in Early Breast Cancer. Sci. Transl. Med. 2015, 7, 302ra133. [Google Scholar] [CrossRef]
- Ivanova, E.; Ward, A.; Wiegmans, A.P.; Richard, D.J. Circulating Tumor Cells in Metastatic Breast Cancer: From Genome Instability to Metastasis. Front. Mol. Biosci. 2020, 7, 134. [Google Scholar] [CrossRef] [PubMed]
- Murtaza, M.; Dawson, S.J.; Pogrebniak, K.; Rueda, O.M.; Provenzano, E.; Grant, J.; Chin, S.-F.; Tsui, D.W.Y.; Marass, F.; Gale, D.; et al. Multifocal clonal evolution characterized using circulating tumour DNA in a case of metastatic breast cancer. Nat. Commun. 2015, 6, 8760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzpatrick, A.; Iravani, M.; Mills, A.; Childs, L.; Alaguthurai, T.; Clifford, A.; Garcia-Murillas, I.; Van Laere, S.; Dirix, L.; Harries, M.; et al. Assessing CSF ctDNA to Improve Diagnostic Accuracy and Therapeutic Monitoring in Breast Cancer Leptomeningeal Metastasis. Clin. Cancer Res. 2022, 28, 1180–1191. [Google Scholar] [CrossRef] [PubMed]
- Fiala, C.; Diamandis, E.P. Utility of circulating tumor DNA in cancer diagnostics with emphasis on early detection. BMC Med. 2018, 16, 166. [Google Scholar] [CrossRef] [Green Version]
- Boral, D.; Vishnoi, M.; Liu, H.N.; Yin, W.; Sprouse, M.L.; Scamardo, A.; Hong, D.S.; Tan, T.Z.; Thiery, J.P.; Chang, J.C.; et al. Molecular characterization of breast cancer CTCs associated with brain metastasis. Nat Commun. 2017, 8, 196. [Google Scholar] [CrossRef] [Green Version]
- Siravegna, G.; Geuna, E.; Mussolin, B.; Crisafulli, G.; Bartolini, A.; Galizia, D.; Casorzo, L.; Sarotto, I.; Scaltriti, M.; Sapino, A.; et al. Genotyping tumour DNA in cerebrospinal fluid and plasma of a HER2-positive breast cancer patient with brain metastases. ESMO Open 2017, 2, e000253. [Google Scholar] [CrossRef] [Green Version]
- Banys-Paluchowski, M.; Fehm, T.N.; Grimm-Glang, D.; Rody, A.; Krawczyk, N. Liquid Biopsy in Metastatic Breast Cancer: Current Role of Circulating Tumor Cells and Circulating Tumor DNA. Oncol. Res. Treat. 2022, 45, 4–11. [Google Scholar] [CrossRef]
- Molinari, C.; Chiadini, E. Mutational Analysis in Urinary Cell-Free DNA: KRAS in Colorectal Cancer. Methods Mol. Biol. 2021, 2292, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Gole, J.; Gore, A.; He, Q.; Lu, M.; Min, J.; Yuan, Z.; Yang, X.; Jiang, Y.; Zhang, T.; et al. Non-Invasive Early Detection of Cancer Four Years before Conventional Diagnosis Using a Blood Test. Nat. Commun. 2020, 11, 3475. [Google Scholar] [CrossRef]
- Yamada, T.; Matsuda, A.; Koizumi, M.; Shinji, S.; Takahashi, G.; Iwai, T.; Takeda, K.; Ueda, K.; Yokoyama, Y.; Hara, K.; et al. Liquid Biopsy for the Management of Patients with Colorectal Cancer. Digestion 2019, 99, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Phallen, J.; Sausen, M.; Adleff, V.; Leal, A.; Hruban, C.; White, J.; Anagnostou, V.; Fiksel, J.; Cristiano, S.; Papp, E.; et al. Direct detection of early-stage cancers using circulating tumor DNA. Sci. Transl. Med. 2017, 9, eaan2415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arisi, M.F.; Dotan, E.; Fernandez, S.V. Circulating Tumor DNA in Precision Oncology and Its Applications in Colorectal Cancer. Int. J. Mol. Sci. 2022, 23, 4441. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.; Siravegna, G.; Blaszkowsky, L.S.; Corti, G.; Crisafulli, G.; Ahronian, L.G.; Mussolin, B.; Kwak, E.L.; Buscarino, M.; Lazzari, L.; et al. Tumor Heterogeneity and Lesion-Specific Response to Targeted Therapy in Colorectal Cancer. Cancer Discov. 2016, 6, 147–153. [Google Scholar] [CrossRef] [Green Version]
- Hallermayr, A.; Steinke-Lange, V.; Vogelsang, H.; Rentsch, M.; de Wit, M.; Haberl, C.; Holinski-Feder, E.; Pickl, J.M.A. Clinical Validity of Circulating Tumor DNA as Prognostic and Predictive Marker for Personalized Colorectal Cancer Patient Management. Cancers 2022, 14, 851. [Google Scholar] [CrossRef]
- Naidoo, M.; Gibbs, P.; Tie, J. ctDNA and Adjuvant Therapy for Colorectal Cancer: Time to Re-Invent Our Treatment Paradigm. Cancers 2021, 13, 346. [Google Scholar] [CrossRef]
- Wallander, K.; Eisfeldt, J.; Lindblad, M.; Nilsson, D.; Billiau, K.; Foroughi, H.; Nordenskjöld, M.; Liedén, A.; Tham, E. Cell-free tumour DNA analysis detects copy number alterations in gastro-oesophageal cancer patients. PLoS ONE 2021, 16, e0245488. [Google Scholar] [CrossRef]
- Min, J.; Zhou, H.; Jiang, S.; Yu, H. A Review of Circulating Tumor DNA in the Diagnosis and Monitoring of Esophageal Cancer. Med. Sci. Monit. 2022, 28, e934106. [Google Scholar] [CrossRef]
- Kwak, E.L.; Ahronian, L.G.; Siravegna, G.; Mussolin, B.; Borger, D.R.; Godfrey, J.T.; Jessop, N.A.; Clark, J.W.; Blaszkowsky, L.S.; Ryan, D.P.; et al. Molecular Heterogeneity and Receptor Coamplification Drive Resistance to Targeted Therapy in MET-Amplified Esophagogastric Cancer. Cancer Discov. 2015, 5, 1271–1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maron, S.B.; Xu, J.; Janjigian, Y.Y. Targeting EGFR in Esophagogastric Cancer. Front. Oncol. 2020, 10, 553876. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.G.; Falls, H.D.; Mitten, M.J.; Oleksijew, A.; Vaidya, K.S.; Boghaert, E.R.; Gao, W.; Palma, J.P.; Cao, D.; Chia, P.L.; et al. Targeting Multiple EGFR-expressing Tumors with a Highly Potent Tumor-selective Antibody-Drug Conjugate. Mol. Cancer Ther. 2020, 19, 2117–2125. [Google Scholar] [CrossRef]
- Nakamura, Y.; Kawazoe, A.; Lordick, F.; Janjigian, Y.Y.; Shitara, K. Biomarker-targeted therapies for advanced-stage gastric and gastro-oesophageal junction cancers: An emerging paradigm. Nat. Rev. Clin. Oncol. 2021, 18, 473–487. [Google Scholar] [CrossRef] [PubMed]
- Leal, A.; van Grieken, N.C.T.; Palsgrove, D.N.; Phallen, J.; Medina, J.E.; Hruban, C.; Broeckaert, M.A.M.; Anagnostou, V.; Adleff, V.; Bruhm, D.C.; et al. White blood cell and cell-free DNA analyses for detection of residual disease in gastric cancer. Nat. Commun. 2020, 11, 525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akashi, K.; Sakai, T.; Fukuoka, O.; Saito, Y.; Yoshida, M.; Ando, M.; Ito, T.; Murakami, Y.; Yamasoba, T. Usefulness of circulating tumor DNA by targeting human papilloma virus-derived sequences as a biomarker in p16-positive oropharyngeal cancer. Sci. Rep. 2022, 12, 572. [Google Scholar] [CrossRef]
- Kong, L.; Birkeland, A.C. Liquid Biopsies in Head and Neck Cancer: Current State and Future Challenges. Cancers 2021, 13, 1874. [Google Scholar] [CrossRef]
- Muhanna, N.; Eu, D.; Chan, H.H.L.; Douglas, C.; Townson, J.L.; Di Grappa, M.A.; Mohamadi, R.M.; Kelley, S.O.; Bratman, S.V.; Irish, J.C. Cell-free DNA and circulating tumor cell kinetics in a pre-clinical head and neck Cancer model undergoing radiation therapy. BMC Cancer 2021, 21, 1075. [Google Scholar] [CrossRef]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014, 6, 224ra24. [Google Scholar] [CrossRef] [Green Version]
- Galun, D.; Mijac, D.; Filipovic, A.; Bogdanovic, A.; Zivanovic, M.; Masulovic, D. Precision Medicine for Hepatocellular Carcinoma: Clinical Perspective. J. Pers. Med. 2022, 12, 149. [Google Scholar] [CrossRef]
- Arechederra, M.; Rullán, M.; Amat, I.; Oyon, D.; Zabalza, L.; Elizalde, M.; Latasa, M.U.; Mercado, M.R.; Ruiz-Clavijo, D.; Saldaña, C.; et al. Next-generation sequencing of bile cell-free DNA for the early detection of patients with malignant biliary strictures. Gut 2022, 71, 1141–1151. [Google Scholar] [CrossRef] [PubMed]
- Han, J.Y.; Ahn, K.S.; Kim, T.S.; Kim, Y.H.; Cho, K.B.; Shin, D.W.; Baek, W.K.; Suh, S.I.; Jang, B.C.; Kang, K.J. Liquid Biopsy from Bile-Circulating Tumor DNA in Patients with Biliary Tract Cancer. Cancers 2021, 13, 4581. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Yang, X.R.; Chung, W.Y.; Dennison, A.R.; Zhou, J. Targeted therapy for hepatocellular carcinoma. Signal Transduct. Target. Ther. 2020, 5, 146. [Google Scholar] [CrossRef]
- Rowe, S.P.; Luber, B.; Makell, M.; Brothers, P.; Santmyer, J.; Schollenberger, M.D.; Quinn, H.; Edelstein, D.L.; Jones, F.S.; Bleich, K.B.; et al. From validity to clinical utility: The influence of circulating tumor DNA on melanoma patient management in a real-world setting. Mol. Oncol. 2018, 12, 1661–1672. [Google Scholar] [CrossRef] [Green Version]
- Gracie, L.; Pan, Y.; Atenafu, E.G.; Ward, D.G.; Teng, M.; Pallan, L.; Stevens, N.M.; Khoja, L. Circulating tumour DNA (ctDNA) in metastatic melanoma, a systematic review and meta-analysis. Eur. J. Cancer 2021, 158, 191–207. [Google Scholar] [CrossRef] [PubMed]
- Syeda, M.M.; Wiggins, J.M.; Corless, B.C.; Long, G.V.; Flaherty, K.T.; Schadendorf, D.; Nathan, P.D.; Robert, C.; Ribas, A.; Davies, M.A.; et al. Circulating tumour DNA in patients with advanced melanoma treated with dabrafenib or dabrafenib plus trametinib: A clinical validation study. Lancet Oncol. 2021, 22, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Nagasaka, M.; Uddin, M.H.; Al-Hallak, M.N.; Rahman, S.; Balasubramanian, S.; Sukari, A.; Azmi, A.S. Liquid biopsy for therapy monitoring in early-stage non-small cell lung cancer. Mol. Cancer 2021, 20, 82. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.M.; Kim, J.H.; Kim, S.K.; Kim, S.; Kwon, H.J.; Bae, J.S.; Lee, S.; Lee, H.S.; Choi, M.Y.; Jeon, B.H.; et al. Clinical Utility of Combined Circulating Tumor Cell and Circulating Tumor DNA Assays for Diagnosis of Primary Lung Cancer. Anticancer Res. 2020, 40, 3435–3444. [Google Scholar] [CrossRef]
- Kapeleris, J.; Ebrahimi Warkiani, M.; Kulasinghe, A.; Vela, I.; Kenny, L.; Ladwa, R.; O’Byrne, K.; Punyadeera, C. Clinical Applications of Circulating Tumour Cells and Circulating Tumour DNA in Non-Small Cell Lung Cancer—An Update. Front. Oncol. 2022, 12, 859152. [Google Scholar] [CrossRef] [PubMed]
- Guibert, N.; Hu, Y.; Feeney, N.; Kuang, Y.; Plagnol, V.; Jones, G.; Howarth, K.; Beeler, J.F.; Paweletz, C.P.; Oxnard, G.R. Amplicon-based next-generation sequencing of plasma cell-free DNA for detection of driver and resistance mutations in advanced non-small cell lung cancer. Ann. Oncol. 2018, 29, 1049–1055. [Google Scholar] [CrossRef]
- Cho, B.C.; Loong, H.H.F.; Tsai, C.M.; Teo, M.L.P.; Kim, H.R.; Lim, S.M.; Jain, S.; Olsen, S.; Park, K. Genomic Landscape of Non-Small Cell Lung Cancer (NSCLC) in East Asia Using Circulating Tumor DNA (ctDNA) in Clinical Practice. Curr. Oncol. 2022, 29, 2154–2164. [Google Scholar] [CrossRef] [PubMed]
- Parums, D.V. Editorial: Global Regulatory Initiatives Deliver Accelerated Approval of the First Bispecific Therapeutic Monoclonal Antibody for Advanced Non-Small Cell Lung Cancer (NSCLC). Med. Sci. Monit. 2021, 27, e934854. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.W.; Charkhchi, P.; Akbari, M.R. Potential clinical utility of liquid biopsies in ovarian cancer. Mol. Cancer 2022, 21, 114. [Google Scholar] [CrossRef]
- Asante, D.B.; Calapre, L.; Ziman, M.; Meniawy, T.M.; Gray, E.S. Liquid biopsy in ovarian cancer using circulating tumor DNA and cells: Ready for prime time? Cancer Lett. 2020, 468, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Shao, X.; He, Y.; Ji, M.; Chen, X.; Qi, J.; Shi, W.; Hao, T.; Ju, S. Quantitative analysis of cell-free DNA in ovarian cancer. Oncol. Lett. 2015, 10, 3478–3482. [Google Scholar] [CrossRef] [Green Version]
- Strijker, M.; Soer, E.C.; de Pastena, M.; Creemers, A.; Balduzzi, A.; Beagan, J.J.; Busch, O.R.; van Delden, O.M.; Halfwerk, H.; van Hooft, J.E.; et al. Circulating tumor DNA quantity is related to tumor volume and both predict survival in metastatic pancreatic ductal adenocarcinoma. Int. J. Cancer 2020, 146, 1445–1456. [Google Scholar] [CrossRef] [Green Version]
- Ying, L.; Sharma, A.; Chhoda, A.; Ruzgar, N.; Hasan, N.; Kwak, R.; Wolfgang, C.L.; Wang, T.H.; Kunstman, J.W.; Salem, R.R.; et al. Methylation-based Cell-free DNA Signature for Early Detection of Pancreatic Cancer. Pancreas 2021, 50, 1267–1273. [Google Scholar] [CrossRef]
- Bunduc, S.; Gede, N.; Váncsa, S.; Lillik, V.; Kiss, S.; Dembrovszky, F.; Eróss, B.; Szakács, Z.; Gheorghe, C.; Mikó, A.; et al. Prognostic role of cell-free DNA biomarkers in pancreatic adenocarcinoma: A systematic review and meta-analysis. Crit. Rev. Oncol. 2022, 169, 103548. [Google Scholar] [CrossRef]
- Huerta, M.; Roselló, S.; Sabater, L.; Ferrer, A.; Tarazona, N.; Roda, D.; Gambardella, V.; Alfaro-Cervelló, C.; Garcés-Albir, M.; Cervantes, A.; et al. Circulating Tumor DNA Detection by Digital-Droplet PCR in Pancreatic Ductal Adenocarcinoma: A Systematic Review. Cancers 2021, 13, 994. [Google Scholar] [CrossRef]
- Herreros-Villanueva, M.; Bujanda, L.; Ruiz-Rebollo, L.; Torremocha, R.; Ramos, R.; Martín, R.; Artigas, M.C. Circulating tumor DNA tracking in patients with pancreatic cancer using next-generation sequencing. Gastroenterol. Hepatol. 2022, 45, 637–644. [Google Scholar] [CrossRef]
- Bjerre, M.T.; Nørgaard, M.; Larsen, O.H.; Jensen, S.Ø.; Strand, S.H.; Østergren, P.; Fode, M.; Fredsøe, J.; Ulhøi, B.P.; Mortensen, M.M.; et al. Epigenetic Analysis of Circulating Tumor DNA in Localized and Metastatic Prostate Cancer: Evaluation of Clinical Biomarker Potential. Cells 2020, 9, 1362. [Google Scholar] [CrossRef] [PubMed]
- González-Billalabeitia, E.; Conteduca, V.; Wetterskog, D.; Jayaram, A.; Attard, G. Circulating tumor DNA in advanced prostate cancer: Transitioning from discovery to a clinically implemented test. Prostate Cancer Prostatic Dis. 2019, 22, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Pizzutilo, E.; Pedrani, M.; Amatu, A.; Ruggieri, L.; Lauricella, C.; Veronese, S.; Signorelli, D.; Cerea, G.; Giannetta, L.; Siena, S.; et al. Liquid Biopsy for Small Cell Lung Cancer either De Novo or Transformed: Systematic Review of Different Applications and Meta-Analysis. Cancers 2021, 13, 2265. [Google Scholar] [CrossRef] [PubMed]
- Devarakonda, S.; Sankararaman, S.; Herzog, B.H.; Gold, K.A.; Waqar, S.N.; Ward, J.P.; Raymond, V.M.; Lanman, R.B.; Chaudhuri, A.A.; Owonikoko, T.K.; et al. Circulating Tumor DNA Profiling in Small-Cell Lung Cancer Identifies Potentially Targetable Alterations. Clin. Cancer Res. 2019, 25, 6119–6126. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Huang, C.; Zhou, H.; Liu, D.; Chen, R.; Li, X.; Cheng, Y.; Gao, B.; Chen, J. Circulating tumor DNA predicts the outcome of chemotherapy in patients with lung cancer. Thorac. Cancer 2022, 13, 95–106. [Google Scholar] [CrossRef]
- Chaudhuri, A.A.; Chabon, J.J.; Lovejoy, A.F.; Newman, A.M.; Stehr, H.; Azad, T.D.; Khodadoust, M.S.; Esfahani, M.S.; Liu, C.L.; Zhou, L.; et al. Early Detection of Molecular Residual Disease in Localized Lung Cancer by Circulating Tumor DNA Profiling. Cancer Discov. 2017, 7, 1394–1403. [Google Scholar] [CrossRef] [Green Version]
- Poh, J.; Ngeow, K.C.; Pek, M.; Tan, K.H.; Lim, J.S.; Chen, H.; Ong, C.K.; Lim, J.Q.; Lim, S.T.; Lim, C.M.; et al. Analytical and clinical validation of an amplicon-based next generation sequencing assay for ultrasensitive detection of circulating tumor DNA. PLoS ONE 2022, 17, e0267389. [Google Scholar] [CrossRef]
- Heitzer, E.; Haque, I.S.; Roberts, C.E.S.; Speicher, M.R. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat. Rev. Genet. 2019, 20, 71–88. [Google Scholar] [CrossRef]
- Schwarzenbach, H.; Nishida, N.; Calin, G.A.; Pantel, K. Clinical relevance of circulating cell-free microRNAs in cancer. Nat. Rev. Clin. Oncol. 2014, 11, 145–156. [Google Scholar] [CrossRef]
- Fan, H.C.; Blumenfeld, Y.J.; Chitkara, U.; Hudgins, L.; Quake, S.R. Analysis of the size distributions of fetal and maternal cell-free DNA by paired-end sequencing. Clin. Chem. 2010, 56, 1279–1286. [Google Scholar] [CrossRef]
- Mandel, P.; Metais, P. Les acides nucléiques du plasma sanguin chez l’homme. CR Seances Soc. Biol. Fil. 1948, 142, 241–243. [Google Scholar]
- Tan, E.M.; Schur, P.H.; Carr, R.I.; Kunkel, H.G. Deoxybonucleic acid (DNA) and antibodies to DNA in the serum of patients with systemic lupus erythematosus. J. Clin. Investig. 1966, 45, 1732–1740. [Google Scholar] [CrossRef] [PubMed]
- Leon, S.A.; Shapiro, B.; Sklaroff, D.M.; Yaros, M.J. Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res. 1977, 37, 646–650. [Google Scholar] [PubMed]
- Vasioukhin, V.; Anker, P.; Maurice, P.; Lyautey, J.; Lederrey, C.; Stroun, M. Point mutations of the N-ras gene in the blood plasma DNA of patients with myelodysplastic syndrome or acute myelogenous leukaemia. Br. J. Haematol. 1994, 86, 774–779. [Google Scholar] [CrossRef]
- Mouliere, F.; Thierry, A.R. The importance of examining the proportion of circulating DNA originating from tumor, microenviroment and normal cells in colorectal cancer patients. Expert Opin. Biol. Ther. 2012, 12 (Suppl. S1), S209–S215. [Google Scholar] [CrossRef]
- Fatouros, I.G.; Jamurtas, A.Z.; Nikolaidis, M.G.; Destouni, A.; Michailidis, Y.; Vrettou, C.; Douroudos, I.I.; Avloniti, A.; Chatzinikolaou, A.; Taxildaris, K.; et al. Time of sampling is crucial for measurement of cell-free plasma DNA following acute aseptic inflammation induced by exercise. Clin. Biochem. 2010, 43, 1368–1370. [Google Scholar] [CrossRef]
- Aucamp, J.; Bronkhorst, A.J.; Badenhorst, C.P.S.; Pretorius, P.J. The diverse origins of circulating cell-free DNA in the human body: A critical re-evaluation of the literature. Biol. Rev. 2018, 93, 1649–1683. [Google Scholar] [CrossRef]
- Fernando, M.R.; Chen, K.; Norton, S.; Krzyzanowski, G.; Bourne, D.; Hunsley, B.; Ryan, W.L.; Bassett, C. A new methodology to preserve the original proportion and integrity of cell-free fetal DNA in maternal plasma during sample processing and storage. Prenat. Diagn. 2010, 30, 418–424. [Google Scholar] [CrossRef]
- Madsen, A.T.; Hojbjerg, J.A.; Sorensen, B.S.; Winther-Larsen, A. Day-to-day and within-day biological variation of cell-free DNA. eBioMedicine 2019, 49, 284–290. [Google Scholar] [CrossRef] [Green Version]
- Salvi, S.; Martignano, F.; Molinari, C.; Gurioli, G.; Calistri, D.; De Giorgi, U.; Conteduca, V.; Casadio, V. The potential use of urine cell free DNA as a marker for cancer. Expert Rev. Mol. Diagn. 2016, 16, 1283–1290. [Google Scholar] [CrossRef]
- Rykova, E.Y.; Morozkin, E.S.; Ponomaryova, A.A.; Loseva, E.M.; Zaporozhchenko, I.A.; Cherdyntseva, N.V.; Vlassov, V.V.; Laktionov, P.P. Cell-free and cell-bound circulating nucleic acid complexes: Mechanisms of generation, concentration, and content. Expert Opin. Biol. Ther. 2012, 12 (Suppl. S1), S141–S153. [Google Scholar] [CrossRef] [PubMed]
- Korabecna, M.; Horinek, A.; Bila, N.; Opatrna, S. Circadian rhythmicity and clearance of cell-free DNA in human plasma. In Circulating Nucleic Acids in Plasma and Serum; Gahan, B.P., Ed.; Springer: Dordrecht, The Netherlands, 2011. [Google Scholar]
- Diehl, F.; Schmidt, K.; Choti, M.A.; Romans, K.; Goodman, S.; Li, M.; Thornton, K.; Agrawal, N.; Sokoll, L.; Szabo, S.A.; et al. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 2008, 14, 985–990. [Google Scholar] [CrossRef] [PubMed]
- Schwarzenbach, H.; Alix-Panabières, C.; Müller, I.; Letang, N.; Vendrell, J.P.; Rebillard, X.; Pantel, K. Cell-free tumor DNA in blood plasma as a marker for circulating tumor cells in prostate cancer. Clin. Cancer Res. 2009, 15, 1032–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peled, M.; Agassi, R.; Czeiger, D.; Ariad, S.; Riff, R.; Rosenthal, M.; Lazarev, I.; Novack, V.; Yarza, S.; Mizrakli, Y.; et al. Cell-free DNA concentration in patients with clinical or mammographic suspicion of breast cancer. Sci. Rep. 2020, 10, 14601. [Google Scholar] [CrossRef]
- Trigg, R.M.; Martinson, L.J.; Parpart-Li, S.; Shaw, J.A. Factors that influence quality and yield of circulating-free DNA: A systematic review of the methodology literature. Heliyon 2018, 4, e00699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.L.; Liang, Z.Y. Circulating free DNA in the era of precision oncology: Pre- and post-analytical concerns. Chronic Dis. Transl. Med. 2016, 2, 223–230. [Google Scholar] [CrossRef]
- Leest, P.V.; Boonstra, P.A.; Elst, A.T.; Kempen, L.C.V.; Tibbesma, M.; Koopmans, J.; Miedema, A.; Tamminga, M.; Groen, H.J.M.; Reyners, A.K.L.; et al. Comparison of Circulating Cell-Free DNA Extraction Methods for Downstream Analysis in Cancer Patients. Cancers 2020, 12, 1222. [Google Scholar] [CrossRef]
- Barták, B.K.; Kalmár, A.; Galamb, O.; Wichmann, B.; Nagy, Z.B.; Tulassay, Z.; Dank, M.; Igaz, P.; Molnár, B. Blood Collection and Cell-Free DNA Isolation Methods Influence the Sensitivity of Liquid Biopsy Analysis for Colorectal Cancer Detection. Pathol. Oncol. Res. 2019, 25, 915–923. [Google Scholar] [CrossRef]
- Tuboly, E.; Mcllroy, D.; Briggs, G.; Lott, N.; Balogh, Z.J. Clinical implications, and pathological associations of circulating mitochondrial DNA. Front. Biosci. 2017, 22, 1011–1022. [Google Scholar] [CrossRef] [Green Version]
- Mair, R.; Mouliere, F.; Smith, C.G.; Chandrananda, D.; Gale, D.; Marass, F.; Tsui, D.W.Y.; Massie, C.E.; Wright, A.J.; Watts, C.; et al. Measurement of Plasma Cell-free Mitochondrial Tumor DNA Improves Detection of Glioblastoma in Patient-derived Orthotopic Xenograft Models. Cancer Res. 2019, 79, 220–230. [Google Scholar] [CrossRef] [Green Version]
- Afrifa, J.; Zhao, T.; Yu, J. Circulating mitochondria DNA, a non-invasive cancer diagnostic biomarkercandidate. Mitochondrion 2019, 47, 238–243. [Google Scholar] [CrossRef] [PubMed]
- Burnham, P.; Kim, M.S.; Agbor-Enoh, S.; Luikart, H.; Valantine, H.A.; Khush, K.K.; De Vlaminck, I. Single-stranded DNA library preparation uncovers the origin and diversity of ultrashort cell-free DNA in plasma. Sci. Rep. 2016, 6, 27859. [Google Scholar] [CrossRef] [PubMed]
- Chiu, R.W.; Chan, L.Y.; Lam, N.Y.; Tsui, N.B.; Ng, E.K.; Rainer, T.H.; Lo, Y.M. Quantitative analysis of circulating mitochondrial DNA in plasma. Clin. Chem. 2003, 49, 719–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Zhou, K.; Guo, S.; Wang, Y.; Ji, X.; Yuan, Q.; Su, L.; Guo, X.; Gu, X.; Xing, J. NGS-based accurate and efficient detection of circulating cell-free mitochondrial DNA in cancer patients. Mol. Ther.-Nucleic Acids 2021, 23, 657–666. [Google Scholar] [CrossRef]
- Sundquist, K.; Sundquist, J.; Hedelius, A.; Memon, A.A. Diagnostic potential of circulating cell-free nuclear and mitochondrial DNA for several cancer types and nonmalignant diseases: A study on suspected cancer patients. Mol. Carcinog. 2020, 59, 1362–1370. [Google Scholar] [CrossRef]
- Deligezer, U.; Eralp, Y.; Akisik, E.E.; Akisik, E.Z.; Saip, P.; Topuz, E.; Dalay, N. Size distribution of circulating cell-free DNA in sera of breast cancer patients in the course of adjuvant chemotherapy. Clin. Chem. Lab. Med. 2008, 46, 311–317. [Google Scholar] [CrossRef]
- Anker, P.; Stroun, M.; Maurice, P.A. Spontaneous release of DNA by human blood lymphocytes as shown in an in vitro system. Cancer Res. 1975, 35, 2375–2382. [Google Scholar]
- Borenstein, S.; Ephrati-Elizur, E. Spontaneous release of DNA in sequential genetic order by Bacillus subtilis. J. Mol. Biol. 1969, 45, 137–152. [Google Scholar] [CrossRef]
- Bronkhorst, A.J.; Wentzel, J.F.; Aucamp, J.; van Dyk, E.; du Plessis, L.; Pretorius, P.J. Characterization of the cell-free DNA released by cultured cancer cells. Biochim. Biophys. Acta 2016, 1863, 157–165. [Google Scholar] [CrossRef] [Green Version]
- Aucamp, J.; Bronkhorst, A.J.; Peters, D.L.; Van Dyk, H.C.; Van der Westhuizen, F.H.; Pretorius, P.J. Kinetic analysis, size profiling, and bioenergetic association of DNA released by selected cell lines in vitro. Cell. Mol. Life Sci. 2017, 74, 2689–2707. [Google Scholar] [CrossRef]
- Wang, W.; Kong, P.; Ma, G.; Li, L.; Zhu, J.; Xia, T.; Xie, H.; Zhou, W.; Wang, S. Characterization of the release and biological significance of cell-free DNA from breast cancer cell lines. Oncotarget 2017, 8, 43180–43191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bronkhorst, A.J.; Wentzel, J.F.; Ungerer, V.; Peters, D.L.; Aucamp, J.; de Villiers, E.P.; Holdenrieder, S.; Pretorius, P.J. Sequence analysis of cell-free DNA derived from cultured human bone osteosarcoma (143B) cells. Tumor Biol. 2018, 40, 1010428318801190. [Google Scholar] [CrossRef] [PubMed]
- Thakur, B.K.; Zhang, H.; Becker, A.; Matei, I.; Huang, Y.; Costa-Silva, B.; Zheng, Y.; Hoshino, A.; Brazier, H.; Xiang, J.; et al. Double-stranded DNA in exosomes: A novel biomarker in cancer detection. Cell Res. 2014, 24, 766–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernando, M.R.; Jiang, C.; Krzyzanowski, G.D.; Ryan, W.L. New evidence that a large proportion of human blood plasma cell-free DNA is localized in exosomes. PLoS ONE 2017, 12, e0183915. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Zhang, F.; Du, M.; Zhang, P.; Fu, S.; Wang, L. Molecular characterization of cell-free eccDNAs in human plasma. Sci. Rep. 2017, 7, 10968. [Google Scholar] [CrossRef] [Green Version]
- Devereaux, K.A.; Souers, R.J.; Merker, J.D.; Lindeman, N.I.; Graham, R.P.; Hameed, M.R.; Vasalos, P.; Moncur, J.T.; Lockwood, C.M.; Xian, R.R. Clinical Testing for Tumor Cell-Free DNA. Arch. Pathol. Lab. Med. 2022. Epub ahead of print. [Google Scholar] [CrossRef]
- Swinkels, D.W.; Wiegerinck, E.; Steegers, E.A.; de Kok, J.B. Effects of blood-processing protocols on cell-free DNA quantification in plasma. Clin. Chem. 2003, 49, 525–526. [Google Scholar] [CrossRef] [Green Version]
- Alidousty, C.; Brandes, D.; Heydt, C.; Wagener, S.; Wittersheim, M.; Schäfer, S.C.; Holz, B.; Merkelbach-Bruse, S.; Büttner, R.; Fassunke, J.; et al. Comparison of Blood Collection Tubes from Three Different Manufacturers for the Collection of Cell-Free DNA for Liquid Biopsy Mutation Testing. J. Mol. Diagn. 2017, 19, 801–804. [Google Scholar] [CrossRef] [Green Version]
- El Messaoudi, S.; Rolet, F.; Mouliere, F.; Thierry, A.R. Circulating cell free DNA: Preanalytical considerations. Clin. Chim. Acta 2013, 424, 222–230. [Google Scholar] [CrossRef]
- Hindson, B.J.; Ness, K.D.; Masquelier, D.A.; Belgrader, P.; Heredia, N.J.; Makarewicz, A.J.; Bright, I.J.; Lucero, M.Y.; Hiddessen, A.L.; Legler, T.C.; et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal. Chem. 2011, 83, 8604–8610. [Google Scholar] [CrossRef]
- Alborelli, I.; Generali, D.; Jermann, P.; Cappelletti, M.R.; Ferrero, G.; Scaggiante, B.; Bortul, M.; Zanconati, F.; Nicolet, S.; Haegele, J.; et al. Cell-free DNA analysis in healthy individuals by next-generation sequencing: A proof of concept and technical validation study. Cell Death Dis. 2019, 10, 534. [Google Scholar] [CrossRef] [Green Version]
- Cohen, J.D.; Li, L.; Wang, Y.; Thoburn, C.; Afsari, B.; Danilova, L.; Douville, C.; Javed, A.A.; Wong, F.; Mattox, A.; et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 2018, 359, 926–930. [Google Scholar] [CrossRef] [PubMed]
- Shain, A.H.; Yeh, I.; Kovalyshyn, I.; Sriharan, A.; Talevich, E.; Gagnon, A.; Dummer, R.; North, J.; Pincus, L.; Ruben, B.; et al. The Genetic Evolution of Melanoma from Precursor Lesions. N. Engl. J. Med. 2015, 373, 1926–1936. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Ulrich, B.C.; Supplee, J.; Kuang, Y.; Lizotte, P.H.; Feeney, N.B.; Guibert, N.M.; Awad, M.M.; Wong, K.K.; Jänne, P.A.; et al. False-Positive Plasma Genotyping Due to Clonal Hematopoiesis. Clin. Cancer Res. 2018, 24, 4437–4443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef] [Green Version]
- Zill, O.A.; Banks, K.C.; Fairclough, S.R.; Mortimer, S.A.; Vowles, J.V.; Mokhtari, R.; Gandara, D.R.; Mack, P.C.; Odegaard, J.I.; Nagy, R.J.; et al. The Landscape of Actionable Genomic Alterations in Cell-Free Circulating Tumor DNA from 21,807 Advanced Cancer Patients. Clin. Cancer Res. 2018, 24, 3528–3538. [Google Scholar] [CrossRef] [Green Version]
- Lim, Z.F.; Ma, P.C. Emerging insights of tumor heterogeneity and drug resistance mechanisms in lung cancer targeted therapy. J. Hematol. Oncol. 2019, 12, 134. [Google Scholar] [CrossRef] [Green Version]
- Blakely, C.M.; Watkins, T.B.K.; Wu, W.; Gini, B.; Chabon, J.J.; McCoach, C.E.; McGranahan, N.; Wilson, G.A.; Birkbak, N.J.; Olivas, V.R.; et al. Evolution and clinical impact of co-occurring genetic alterations in advanced-stage EGFR-mutant lung cancers. Nat. Genet. 2017, 49, 1693–1704. [Google Scholar] [CrossRef]
- Aggarwal, C.; Thompson, J.C.; Black, T.A.; Katz, S.I.; Fan, R.; Yee, S.S.; Chien, A.L.; Evans, T.L.; Bauml, J.M.; Alley, E.W.; et al. Clinical Implications of Plasma-Based Genotyping with the Delivery of Personalized Therapy in Metastatic Non-Small Cell Lung Cancer. JAMA Oncol. 2019, 5, 173–180. [Google Scholar] [CrossRef]
- Murtaza, M.; Dawson, S.J.; Tsui, D.W.; Gale, D.; Forshew, T.; Piskorz, A.M.; Parkinson, C.; Chin, S.F.; Kingsbury, Z.; Wong, A.S.; et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature 2013, 497, 108–112. [Google Scholar] [CrossRef]
- Cisneros-Villanueva, M.; Hidalgo-Pérez, L.; Rios-Romero, M.; Cedro-Tanda, A.; Ruiz-Villavicencio, C.A.; Page, K.; Hastings, R.; Fernandez-Garcia, D.; Allsopp, R.; Fonseca-Montaño, M.A.; et al. Cell-free DNA analysis in current cancer clinical trials: A review. Br. J. Cancer 2022, 126, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Fazzari, M.J.; Greally, J.M. Introduction to epigenomics and epigenome-wide analysis. Methods Mol. Biol. 2010, 620, 243–265. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Samaranayake, M.; Pradhan, S. Epigenetic mechanisms in mammals. Cell. Mol. Life Sci. 2009, 66, 596–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kundu, S.; Peterson, C.L. Role of chromatin states in transcriptional memory. Biochim. Biophys. Acta 2009, 1790, 445–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jabbari, K.; Bernardi, G. Cytosine methylation and CpG, TpG (CpA) and TpA frequencies. Gene 2004, 333, 143–149. [Google Scholar] [CrossRef]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet. 2003, 33, 245–254. [Google Scholar] [CrossRef]
- Deaton, A.M.; Bird, A. CpG islands and the regulation of transcription. Genes Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef] [Green Version]
- Saxonov, S.; Berg, P.; Brutlag, D.L. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc. Natl. Acad. Sci. USA 2006, 103, 1412–1417. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J. Interplay between P53 and Epigenetic Pathways in Cancer. Ph.D. Thesis, University of Pennsylvania, Philadelphia, PA, USA, 2016. Available online: https://repository.upenn.edu/edissertations/2130 (accessed on 20 August 2022).
- Yagi, K.; Akagi, K.; Hayashi, H.; Nagae, G.; Tsuji, S.; Isagawa, T.; Midorikawa, Y.; Nishimura, Y.; Sakamoto, H.; Seto, Y.; et al. Three DNA methylation epigenotypes in human colorectal cancer. Clin. Cancer Res. 2010, 16, 21–33. [Google Scholar] [CrossRef] [Green Version]
- Baylin, S.B.; Esteller, M.; Rountree, M.R.; Bachman, K.E.; Schuebel, K.; Herman, J.G. Aberrant patterns of DNA methylation, chromatin formation and gene expression in cancer. Hum. Mol. Genet. 2001, 10, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Dor, Y.; Cedar, H. Principles of DNA methylation and their implication for biology and medicine. Lancet 2018, 392, 777–786. [Google Scholar] [CrossRef]
- Pfeifer, G.P.; Szabó, P.E. Gene body profiles of 5-hydroxymethylcytosine: Potential origin, function and use as a cancer biomarker. Epigenomics 2018, 10, 1029–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziller, M.J.; Gu, H.; Müller, F.; Donaghey, J.; Tsai, L.T.; Kohlbacher, O.; De Jager, P.L.; Rosen, E.D.; Bennett, D.A.; Bernstein, B.E.; et al. Charting adynamic DNA methylation landscape of the human genome. Nature 2013, 500, 477–481. [Google Scholar] [CrossRef] [Green Version]
- Diaz, L.A., Jr.; Bardelli, A. Liquid biopsies: Genotyping circulating tumor DNA. J. Clin. Oncol. 2014, 32, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Jammula, S.; Katz-Summercorn, A.C.; Li, X.; Linossi, C.; Smyth, E.; Killcoyne, S.; Biasci, D.; Subash, V.V.; Abbas, S.; Blasko, A.; et al. Identification of subtypes of Barett’s esophagus and esophageal adenocarcinoma based on DNA mathylation profiles and integration of transcriptome and genetic data. Gastroenterology 2020, 158, 1682–1697. [Google Scholar] [CrossRef] [PubMed]
- Nassiri, F.; Chakravarthy, A.; Feng, S.; Shen, S.Y.; Nejad, R.; Zuccato, J.A.; Voisin, M.R.; Patil, V.; Horbinski, C.; Aldape, K.; et al. Detection and discrimination of intracranial tumors using plasma cell-free DNA methylomes. Nat. Med. 2020, 26, 1044–1047. [Google Scholar] [CrossRef]
- Hirotsu, Y.; Otake, S.; Ohyama, H.; Amemiya, K.; Higuchi, R.; Oyama, T.; Mochizuki, H.; Goto, T.; Omata, M. Dual-molecular barcode sequencing detects rare variants in tumor and cell-free DNA in plasma. Sci. Rep. 2020, 10, 3391. [Google Scholar] [CrossRef] [Green Version]
- Keller, L.; Belloum, Y.; Wikman, H.; Pantel, K. Clinical relevance of blood-based ctDNA analysis: Mutation detection and beyond. Br. J. Cancer 2021, 124, 345–358. [Google Scholar] [CrossRef]
- Waarts, M.R.; Stonestrom, A.J.; Park, Y.C.; Levine, R.L. Targeting mutations in cancer. J. Clin. Investig. 2022, 132, e154943. [Google Scholar] [CrossRef]
- Bronkhorst, A.J.; Ungerer, V.; Holdenreider, S. The Emerging Role of Cell-Free DNA as a Molecular Marker for Cancer Management. Biomol. Detect. Quantif. 2019, 17, 100087. [Google Scholar] [CrossRef] [PubMed]
- Yao, W.; Mei, C.; Nan, X.; Hui, L. Evaluation and comparison of in vitro degradation kinetics of DNA in serum, urine, and saliva: A qualitative study. Gene 2016, 590, 142–148. [Google Scholar] [CrossRef]
- Underhill, H.R.; Kitzman, J.O.; Hellwig, S.; Welker, N.C.; Daza, R.; Baker, D.N.; Gligorich, K.M.; Rostomily, R.C.; Bronner, M.P.; Shendure, J. Fragment Length of Circulating Tumor DNA. PLoS Genet. 2016, 12, e1006162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez, C.; Snyder, M.W.; Tanos, R.; Shendure, J.; Thierry, A.R. New insights into structural features and optimal detection of circulating tumor DNA determined by single-strand DNA analysis. npj Genom. Med. 2018, 3, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jahr, S.; Hentze, H.; Englisch, S.; Hardt, D.; Fackelmayer, F.O.; Hesch, R.D.; Knippers, R. DNA fragments in the blood plasma of cancer patients: Quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001, 61, 1659–1665. [Google Scholar] [PubMed]
- Janku, F.; Huang, H.J.; Claes, B.; Falchook, G.S.; Fu, S.; Hong, D.; Ramzanali, N.M.; Nitti, G.; Cabrilo, G.; Tsimberidou, A.M.; et al. BRAF Mutation Testing in Cell-Free DNA from the Plasma of Patients with Advanced Cancers Using a Rapid, Automated Molecular Diagnostics System. Mol. Cancer Ther. 2016, 15, 1397–1404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janku, F.; Huang, H.J.; Fujii, T.; Shelton, D.N.; Madwani, K.; Fu, S.; Tsimberidou, A.M.; Piha-Paul, S.A.; Wheler, J.J.; Zinner, R.G.; et al. Multiplex KRASG12/G13 mutation testing of unamplified cell-free DNA from the plasma of patients with advanced cancers using droplet digital polymerase chain reaction. Ann. Oncol. 2017, 28, 642–650. [Google Scholar] [CrossRef]
- Yang, X.; Lay, F.; Han, H.; Jones, P.A. Targeting DNA methylation for epigenetic therapy. Trends Pharmacol. Sci. 2010, 31, 536–546. [Google Scholar] [CrossRef] [Green Version]
- Kelly, T.K.; De Carvalho, D.D.; Jones, P.A. Epigenetic modifications as therapeutic targets. Nat. Biotechnol. 2010, 28, 1069–1078. [Google Scholar] [CrossRef] [Green Version]
- Vrba, L.; Futscher, B.W. A suite of DNA methylation markers that can detect most common human cancers. Epigenetics 2018, 13, 61–72. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Wang, L. Cell-Free DNA Methylation Profiling Analysis-Technologies and Bioinformatics. Cancers 2019, 11, 1741. [Google Scholar] [CrossRef] [PubMed]
- Beck, S.; Rakyan, V.K. The methylome: Approaches for global DNA methylation profiling. Trends Genet. 2008, 24, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Warton, K.; Mahon, K.L.; Samimi, G. Methylated circulating tumor DNA in blood: Power in cancer prognosis and response. Endocr.-Relat. Cancer 2016, 23, R157–R171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Constantin, N.; Ibn Sina, A.A.; Korbie, D.; Trau, M. Opportunities for Early Cancer Detection: The Rise of ctDNA Methylation-Based Pan-Cancer Screening Technologies. Epigenomes 2022, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Lubotzky, A.; Zemmour, H.; Neiman, D.; Gotkine, M.; Loyfer, N.; Piyanzin, S.; Ochana, B.L.; Lehmann-Werman, R.; Cohen, D.; Moss, J.; et al. Liquid biopsy reveals collateral tissue damage in cancer. J. Clin. Investig. 2022, 7, e153559. [Google Scholar] [CrossRef] [PubMed]
- Laprovitera, N.; Riefolo, M.; Ambrosini, E.; Klec, C.; Pichler, M.; Ferracin, M. Cancer of Unknown Primary: Challenges and Progress in Clinical Management. Cancers 2021, 13, 451. [Google Scholar] [CrossRef]
- De Mattos-Arruda, L.; Weigelt, B.; Cortes, J.; Won, H.H.; Ng, C.K.Y.; Nuciforo, P.; Bidard, F.C.; Aura, C.; Saura, C.; Peg, V.; et al. Capturing intra-tumor genetic heterogeneity by de novo mutation profiling of circulating cell-free tumor DNA: A proof-of-principle. Ann. Oncol. 2014, 25, 1729–1735. [Google Scholar] [CrossRef]
- Jamal-Hanjani, M.; Wilson, G.A.; Horswell, S.; Mitter, R.; Sakarya, O.; Constantin, T.; Salari, R.; Kirkizlar, E.; Sigurjonsson, S.; Pelham, R.; et al. Detection of ubiquitous and heterogeneous mutations in cell-free DNA from patients with early-stage non-small-cell lung cancer. Ann. Oncol. 2016, 27, 862–867. [Google Scholar] [CrossRef]
- Chae, Y.K.; Davis, A.A.; Carneiro, B.A.; Chandra, S.; Mohindra, N.; Kalyan, A.; Kaplan, J.; Matsangou, M.; Pai, S.; Costa, R.; et al. Concordance between genomic alterations assessed by next-generation sequencing in tumor tissue or circulating cell-free DNA. Oncotarget 2016, 7, 65364–65373. [Google Scholar] [CrossRef] [Green Version]
- Kuderer, N.M.; Burton, K.A.; Blau, S.; Rose, A.L.; Parker, S.; Lyman, G.H.; Blau, C.A. Comparison of 2 Commercially Available Next-Generation Sequencing Platforms in Oncology. JAMA Oncol. 2017, 3, 996–998. [Google Scholar] [CrossRef]
- Zill, O.A.; Greene, C.; Sebisanovic, D.; Siew, L.M.; Leng, J.; Vu, M.; Hendifar, A.E.; Wang, Z.; Atreya, C.E.; Kelley, R.K.; et al. Cell-Free DNA Next-Generation Sequencing in Pancreatobiliary Carcinomas. Cancer Discov. 2015, 5, 1040–1048. [Google Scholar] [CrossRef] [PubMed]
- Adalsteinsson, V.A.; Ha, G.; Freeman, S.S.; Choudhury, A.D.; Stover, D.G.; Parsons, H.A.; Gydush, G.; Reed, S.C.; Rotem, D.; Rhoades, J.; et al. Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nat. Commun. 2017, 8, 1324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strickler, J.H.; Loree, J.M.; Ahronian, L.G.; Parikh, A.R.; Niedzwiecki, D.; Pereira, A.A.L.; McKinney, M.; Korn, W.M.; Atreya, C.E.; Banks, K.C.; et al. Genomic Landscape of Cell-Free DNA in Patients with Colorectal Cancer. Cancer Discov. 2018, 8, 164–173. [Google Scholar] [CrossRef] [Green Version]
- Martinelli, E.; Martini, G.; Famiglietti, V.; Troiani, T.; Napolitano, S.; Pietrantonio, F.; Ciardiello, D.; Terminiello, M.; Borrelli, C.; Vitiello, P.P.; et al. Cetuximab Rechallenge Plus Avelumab in Pretreated Patients With RAS Wild-type Metastatic Colorectal Cancer: The Phase 2 Single-Arm Clinical CAVE Trial. JAMA Oncol. 2021, 7, 1529–1535. [Google Scholar] [CrossRef]
- Lanman, R.B.; Mortimer, S.A.; Zill, O.A.; Sebisanovic, D.; Lopez, R.; Blau, S.; Collisson, E.A.; Divers, S.G.; Hoon, D.S.; Kopetz, E.S.; et al. Analytical and Clinical Validation of a Digital Sequencing Panel for Quantitative, Highly Accurate Evaluation of Cell-Free Circulating Tumor DNA. PLoS ONE 2015, 10, e0140712. [Google Scholar] [CrossRef]
- Garraway, L.A.; Jänne, P.A. Circumventing cancer drug resistance in the era of personalized medicine. Cancer Discov. 2012, 2, 214–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrock, A.B.; Pavlick, D.; Klempner, S.J.; Chung, J.H.; Forcier, B.; Welsh, A.; Young, L.; Leyland-Jones, B.; Bordoni, R.; Carvajal, R.D.; et al. Hybrid Capture-Based Genomic Profiling of Circulating Tumor DNA from Patients with Advanced Cancers of the Gastrointestinal Tract or Anus. Clin. Cancer Res. 2018, 24, 1881–1890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parikh, A.R.; Leshchiner, I.; Elagina, L.; Goyal, L.; Levovitz, C.; Siravegna, G.; Livitz, D.; Rhrissorrakrai, K.; Martin, E.E.; Van Seventer, E.E.; et al. Liquid versus tissue biopsy for detecting acquired resistance and tumor heterogeneity in gastrointestinal cancers. Nat. Med. 2019, 25, 1415–1421. [Google Scholar] [CrossRef]
- Radovich, M.; Jiang, G.; Hancock, B.A.; Chitambar, C.; Nanda, R.; Falkson, C.; Lynce, F.C.; Gallagher, C.; Isaacs, C.; Blaya, M.; et al. Association of Circulating Tumor DNA and Circulating Tumor Cells After Neoadjuvant Chemotherapy With Disease Recurrence in Patients With Triple-Negative Breast Cancer: Preplanned Secondary Analysis of the BRE12-158 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1410–1415. [Google Scholar] [CrossRef]
- Ortolan, E.; Appierto, V.; Silvestri, M.; Miceli, R.; Veneroni, S.; Folli, S.; Pruneri, G.; Vingiani, A.; Belfiore, A.; Cappelletti, V.; et al. Blood-based genomics of triple-negative breast cancer progression in patients treated with neoadjuvant chemotherapy. ESMO Open 2021, 6, 100086. [Google Scholar] [CrossRef]
- Fang, X.; Yu, S.; Jiang, Y.; Xiang, Y.; Lu, K. Circulating tumor DNA detection in MRD assessment and diagnosis and treatment of non-small cell lung cancer. Front. Oncol. 2022, 12, 1027664. [Google Scholar] [CrossRef] [PubMed]
- Dawson, S.J.; Tsui, D.W.; Murtaza, M.; Biggs, H.; Rueda, O.M.; Chin, S.F.; Dunning, M.J.; Gale, D.; Forshew, T.; Mahler-Araujo, B.; et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N. Engl. J. Med. 2013, 368, 1199–1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamkovich, S.N.; Cherepanova, A.V.; Kolesnikova, E.V.; Rykova, E.Y.; Pyshnyi, D.V.; Vlassov, V.V.; Laktionov, P.P. Circulating DNA and DNase activity in human Blood. Ann. N. Y. Acad. Sci. 2006, 1075, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Diehl, F.; Li, M.; Dressman, D.; He, Y.; Shen, D.; Szabo, S.; Diaz, L.A., Jr.; Goodman, S.N.; David, K.A.; Juhl, H.; et al. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc. Natl. Acad. Sci. USA 2005, 102, 16368–16373. [Google Scholar] [CrossRef] [Green Version]
- Reckamp, K.L.; Melnikova, V.O.; Karlovich, C.; Sequist, L.V.; Camidge, D.R.; Wakelee, H.; Perol, M.; Oxnard, G.R.; Kosco, K.; Croucher, P.; et al. A Highly Sensitive and Quantitative Test Platform for Detection of NSCLC EGFR Mutations in Urine and Plasma. J. Thorac. Oncol. 2016, 11, 1690–1700. [Google Scholar] [CrossRef] [Green Version]
- Thierry, A.R.; El Messaoudi, S.; Gahan, P.B.; Anker, P.; Stroun, M. Origins, structures, and functions of circulating DNA in oncology. Cancer Metastasis Rev. 2016, 35, 347–376. [Google Scholar] [CrossRef] [Green Version]
- Chelobanov, B.P.; Laktionov, P.P.; Vlasov, V.V. Proteins involved in binding and cellular uptake of nucleic acids. Biochemistry 2006, 71, 583–596. [Google Scholar] [CrossRef]
- Kustanovich, A.; Schwartz, R.; Peretz, T.; Grinshpun, A. Life and death of circulating cell-free DNA. Cancer Biol. Ther. 2019, 20, 1057–1067. [Google Scholar] [CrossRef] [Green Version]
- Cristiano, S.; Leal, A.; Phallen, J.; Fiksel, J.; Adleff, V.; Bruhm, D.C.; Jensen, S.Ø.; Medina, J.E.; Hruban, C.; White, J.R.; et al. Genome-wide cell-free DNA fragmentation in patients with cancer. Nature 2019, 570, 385–389. [Google Scholar] [CrossRef]
- Klein, E.A.; Richards, D.; Cohn, A.; Tummala, M.; Lapham, R.; Cosgrove, D.; Chung, G.; Clement, J.; Gao, J.; Hunkapiller, N.; et al. Clinical validation of a targeted methylation-based multi-cancer early detection test using an independent validation set. Ann. Oncol. 2021, 32, 1167–1177. [Google Scholar] [CrossRef]
- Kim, Y.J.; Jeon, H.; Jeon, S.; Lee, S.H.; Kim, C.; Ahn, J.H.; Um, H.; Woo, Y.J.; Jeong, S.H.; Kim, Y.; et al. A method for early diagnosis of lung cancer from tumor originated DNA fragments using plasma cfDNA methylome and fragmentome profiles. Mol. Cell. Probes 2022, 66, 101873. [Google Scholar] [CrossRef] [PubMed]
- Mathios, D.; Johansen, J.S.; Cristiano, S.; Medina, J.E.; Phallen, J.; Larsen, K.R.; Bruhm, D.C.; Niknafs, N.; Ferreira, L.; Adleff, V.; et al. Detection and characterization of lung cancer using cell-free DNA fragmentomes. Nat. Commun. 2021, 12, 5060. [Google Scholar] [CrossRef] [PubMed]
- Pellini, B.; Chaudhuri, A. Circulating Tumor DNA Minimal Residual Disease Detection of Non-Small-Cell Lung Cancer Treated with Curative Intent. J. Clin. Oncol. 2022, 40, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Chidharla, A.; Rapaport, E.; Agarwal, K.; Linares, B.; Chakrabarti, S.; Kasi, A. Circulating tumor DNA (ctDNA) as a minimal residual disease (MRD) assessment and recurrence risk in patients undergoing curative intent resection with or without adjuvant chemotherapy in colorectal cancer: A systematic review and meta-analysis. J. Clin. Oncol. 2022, 40, 3623. [Google Scholar] [CrossRef]
- Bent, A.; Raghavan, S.; Dasari, A.; Kopetz, S. The Future of ctDNA-Defined Minimal Residual Disease: Personalizing Adjuvant Therapy in Colorectal Cancer. Clin. Color. Cancer 2022, 21, 89–95. [Google Scholar] [CrossRef]
- Tie, J.; Wang, Y.; Tomasetti, C.; Li, L.; Springer, S.; Kinde, I.; Silliman, N.; Tacey, M.; Wong, H.L.; Christie, M.; et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci. Transl. Med. 2016, 8, 346ra92. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.; Zhang, L.; Chen, Y.; Qing, C. Circulating cell-free DNA and circulating tumor cells, the “liquid biopsies” in ovarian cancer. J. Ovarian Res. 2017, 10, 75. [Google Scholar] [CrossRef] [Green Version]
- Casas-Arozamena, C.; Díaz, E.; Moiola, C.P.; Alonso-Alconada, L.; Ferreirós, A.; Abalo, A.; Gil, C.L.; Oltra, S.S.; de Santiago, J.; Cabrera, S.; et al. Genomic Profiling of Uterine Aspirates and cfDNA as an Integrative Liquid Biopsy Strategy in Endometrial Cancer. J. Clin. Med. 2020, 9, 585. [Google Scholar] [CrossRef] [Green Version]
- Goering, W.; Kloth, M.; Schulz, W.A. DNA methylation changes in prostate cancer. Methods Mol. Biol. 2012, 863, 47–66. [Google Scholar] [CrossRef]
- Sobhani, N.; Sirico, M.; Generali, D.; Zanconati, F.; Scaggiante, B. Circulating cell-free nucleic acids as prognostic and therapy predictive tools for metastatic castrate-resistant prostate cancer. World J. Clin. Oncol. 2020, 11, 450–463. [Google Scholar] [CrossRef]
- Tulpule, V.; Morrison, G.J.; Falcone, M.; Quinn, D.I.; Goldkorn, A. Integration of Liquid Biopsies in Clinical Management of Metastatic Prostate Cancer. Curr. Oncol. Rep. 2022, 24, 1287–1298. [Google Scholar] [CrossRef]
- Shohdy, K.S.; Villamar, D.M.; Cao, Y.; Trieu, J.; Price, K.S.; Nagy, R.; Tagawa, S.T.; Molina, A.M.; Sternberg, C.N.; Nanus, D.M.; et al. Serial ctDNA analysis predicts clinical progression in patients with advanced urothelial carcinoma. Br. J. Cancer 2022, 126, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Wu, X.; Ling, Y.; Liu, X.; Zheng, X.; Ye, W.; Gong, Y. The prognostic impact of variant allele frequency (VAF) in TP53 mutant patiens with MDS: A systematic review and meta-analyísis. Eur. J. Haematol. 2020, 105, 524–539. [Google Scholar] [CrossRef]
- Siravegna, G.; Mussolin, B.; Buscarino, M.; Corti, G.; Cassingena, A.; Crisafulli, G.; Ponzetti, A.; Cremolini, C.; Amatu, A.; Lauricella, C.; et al. Clonal evolution, and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat. Med. 2015, 21, 795–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sausen, M.; Phallen, J.; Adleff, V.; Jones, S.; Leary, R.J.; Barrett, M.T.; Anagnostou, V.; Parpart-Li, S.; Murphy, D.; Kay Li, Q.; et al. Clinical implications of genomic alterations in the tumour and circulation of pancreatic cancer patients. Nat. Commun. 2015, 6, 7686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henriksen, T.V.; Tarazona, N.; Frydendahl, A.; Reinert, T.; Gimeno-Valiente, F.; Carbonell-Asins, J.A.; Sharma, S.; Renner, D.; Hafez, D.; Roda, D.; et al. Circulating Tumor DNA in Stage III Colorectal Cancer, beyond Minimal Residual Disease Detection, toward Assessment of Adjuvant Therapy Efficacy and Clinical Behavior of Recurrences. Clin. Cancer Res. 2022, 28, 507–517. [Google Scholar] [CrossRef]
- Morais, M.; Pinto, D.M.; Machado, J.C.; Carneiro, S. ctDNA on liquid biopsy for predicting response and prognosis in locally advanced rectal cancer: A systematic review. Eur. J. Surg. Oncol. 2022, 48, 218–227. [Google Scholar] [CrossRef]
- Kasi, P.M.; Fehringer, G.; Taniguchi, H.; Starling, N.; Nakamura, Y.; Kotani, D.; Powles, T.; Li, B.T.; Pusztai, L.; Aushev, V.N.; et al. Impact of Circulating Tumor DNA-Based Detection of Molecular Residual Disease on the Conduct and Design of Clinical Trials for Solid Tumors. JCO Precis. Oncol. 2022, 6, e2100181. [Google Scholar] [CrossRef]
- Powles, T.; Assaf, Z.J.; Davarpanah, N.; Banchereau, R.; Szabados, B.E.; Yuen, K.C.; Grivas, P.; Hussain, M.; Oudard, S.; Gschwend, J.E.; et al. ctDNA guiding adjuvant immunitherapy in urothelial carcinoma. Nature 2021, 595, 432–437. [Google Scholar] [CrossRef]
- Ziogas, D.E.; Kyrochristos, I.D.; Lykoudis, E.G.; Roukos, D.H. Early solid tumor diagnosis through next-generation sequencing of cell-free DNA. Biomark. Med. 2018, 12, 1197–1201. [Google Scholar] [CrossRef]
- Husain, H.; Melnikova, V.O.; Kosco, K.; Woodward, B.; More, S.; Pingle, S.C.; Weihe, E.; Park, B.H.; Tewari, M.; Erlander, M.G.; et al. Monitoring Daily Dynamics of Early Tumor Response to Targeted Therapy by Detecting Circulating Tumor DNA in Urine. Clin. Cancer Res. 2017, 23, 4716–4723. [Google Scholar] [CrossRef] [PubMed]
- Goyal, L.; Saha, S.K.; Liu, L.Y.; Siravegna, G.; Leshchiner, I.; Ahronian, L.G.; Lennerz, J.K.; Vu, P.; Deshpande, V.; Kambadakone, A.; et al. Polyclonal Secondary FGFR2 Mutations Drive Acquired Resistance to FGFR Inhibition in Patients with FGFR2 Fusion-Positive Cholangiocarcinoma. Cancer Discov. 2017, 7, 252–263. [Google Scholar] [CrossRef] [Green Version]
- Diaz, L.A., Jr.; Williams, R.T.; Wu, J.; Kinde, I.; Hecht, J.R.; Berlin, J.; Allen, B.; Bozic, I.; Reiter, J.G.; Nowak, M.A.; et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 2012, 486, 537–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crowley, E.; Di Nicolantonio, F.; Loupakis, F.; Bardelli, A. Liquid biopsy: Monitoring cancer-genetics in the blood. Nat. Rev. Clin. Oncol. 2013, 10, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.C.; Jiang, P.; Zheng, Y.W.; Liao, G.J.; Sun, H.; Wong, J.; Siu, S.S.N.; Chan, W.C.; Chan, S.L.; Chan, A.T.; et al. Cancer genome scanning in plasma: Detection of tumor-associated copy number aberrations, single-nucleotide variants, and tumoral heterogeneity by massively parallel sequencing. Clin. Chem. 2013, 59, 211–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandarlapaty, S.; Chen, D.; He, W.; Sung, P.; Samoila, A.; You, D.; Bhatt, T.; Patel, P.; Voi, M.; Gnant, M.; et al. Prevalence of ESR1 Mutations in Cell-Free DNA and Outcomes in Metastatic Breast Cancer: A Secondary Analysis of the BOLERO-2 Clinical Trial. JAMA Oncol. 2016, 2, 1310–1315. [Google Scholar] [CrossRef] [Green Version]
- Turner, N.C.; Kingston, B.; Kilburn, L.S.; Kernaghan, S.; Wardley, A.M.; Macpherson, I.R.; Baird, R.D.; Roylance, R.; Stephens, P.; Oikonomidou, O.; et al. Circulating tumour DNA analysis to direct therapy in advanced breast cancer (plasmaMATCH): A multicentre, multicohort, phase 2a, platform trial. Lancet Oncol. 2020, 21, 1296–1308. [Google Scholar] [CrossRef]
- Parsons, H.A.; Beaver, J.A.; Cimino-Mathews, A.; Ali, S.M.; Axilbund, J.; Chu, D.; Connolly, R.M.; Cochran, R.L.; Croessmann, S.; Clark, T.A.; et al. Individualized Molecular Analyses Guide Efforts (IMAGE): A Prospective Study of Molecular Profiling of Tissue and Blood in Metastatic Triple-Negative Breast Cancer. Clin. Cancer Res. 2017, 23, 379–386. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Gampenrieder, S.P.; Frantal, S.; Rinnerthaler, G.; Singer, C.F.; Egle, D.; Pfeiler, G.; Bartsch, R.; Wette, V.; Pichler, A.; et al. Persistence of ctDNA in Patients with Breast Cancer During Neoadjuvant Treatment Is a Significant Predictor of Poor Tumor Response. Clin. Cancer Res. 2022, 28, 697–707. [Google Scholar] [CrossRef]
- Papakonstantinou, A.; Gonzalez, N.S.; Pimentel, I.; Suñol, A.; Zamora, E.; Ortiz, C.; Espinosa-Bravo, M.; Peg, V.; Vivancos, A.; Saura, C.; et al. Prognostic value of ctDNA detection in patients with early breast cancer undergoing neoadjuvant therapy: A systematic review and meta-analysis. Cancer Treat. Rev. 2022, 104, 102362. [Google Scholar] [CrossRef]
- Tissot, C.; Toffart, A.C.; Villar, S.; Souquet, P.J.; Merle, P.; Moro-Sibilot, D.; Pérol, M.; Zavadil, J.; Brambilla, C.; Olivier, M.; et al. Circulating free DNA concentration is an independent prognostic biomarker in lung cancer. Eur. Respir. J. 2015, 46, 1773–1780. [Google Scholar] [CrossRef] [PubMed]
- Chabon, J.J.; Simmons, A.D.; Lovejoy, A.F.; Esfahani, M.S.; Newman, A.M.; Haringsma, H.J.; Kurtz, D.M.; Stehr, H.; Scherer, F.; Karlovich, C.A.; et al. Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients. Nat. Commun. 2016, 7, 11815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oxnard, G.R.; Thress, K.S.; Alden, R.S.; Lawrance, R.; Paweletz, C.P.; Cantarini, M.; Yang, J.C.; Barrett, J.C.; Jänne, P.A. Association Between Plasma Genotyping and Outcomes of Treatment with Osimertinib (AZD9291) in Advanced Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2016, 34, 3375–3382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remon, J.; Steuer, C.E.; Ramalingam, S.S.; Felip, E. Osimertinib and other third-generation EGFR TKI in EGFR-mutant NSCLC patients. Ann. Oncol. 2018, 29 (Suppl. 1), i20–i27. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Jung, H.Y.; Kim, K.; Kim, M.; Hong, S. Rational Computational Design of Fourth-Generation EGFR Inhibitors to Combat Drug-Resistant Non-Small Cell Lung Cancer. Int. J. Mol. Sci. 2020, 21, 9323. [Google Scholar] [CrossRef]
- O’Sullivan, H.; d’Arienzo, P.D.; Yousaf, N.; Cui, W.; Popat, S. Inadequacy of PCR genotyping in advanced non-small cell lung cancer: EGFR L747_A755delinsSS exon 19 deletion is not detected by the real-time PCR Idylla™ EGFR mutation test but is detected by ctDNA next generation sequencing and responds to osimertinib. Eur. J. Cancer 2022, 166, 38–40. [Google Scholar] [CrossRef]
- Gandara, D.R.; Paul, S.M.; Kowanetz, M.; Schleifman, E.; Zou, W.; Li, Y.; Rittmeyer, A.; Fehrenbacher, L.; Otto, G.; Malboeuf, C.; et al. Blood-based tumor mutational burden as a predictor of clinical benefit in non-small-cell lung cancer patients treated with atezolizumab. Nat. Med. 2018, 24, 1441–1448. [Google Scholar] [CrossRef]
- Stadler, J.C.; Belloum, Y.; Deitert, B.; Sementsov, M.; Heidrich, I.; Gebhardt, C.; Keller, L.; Pantel, K. Current and Future Clinical Applications of ctDNA in Immuno-Oncology. Cancer Res. 2021, 82, 349–358. [Google Scholar] [CrossRef]
- Lee, J.H.; Long, G.V.; Menzies, A.M.; Lo, S.; Guminski, A.; Whitbourne, K.; Peranec, M.; Scolyer, R.; Kefford, R.F.; Rizos, H.; et al. Association between circulating tumor DNA and pseudoprogression in patients with metastatic melanoma treated with anti-programmed cell death 1 antibodies. JAMA Oncol. 2018, 4, 717–721. [Google Scholar] [CrossRef] [Green Version]
- McGrail, D.J.; Pilié, P.G.; Rashid, N.U.; Voorwerk, L.; Slagter, M.; Kok, M.; Jonasch, E.; Khasraw, M.; Heimberger, A.B.; Lim, B.; et al. High tumor mutation burden fails to predict immune checkpoint blockade response across all cancer types. Ann. Oncol. 2021, 32, 661–672. [Google Scholar] [CrossRef]
- Zheng, M. Tumor mutation burden for predicting immune checkpoint blockade response: The more, the better. J. Immunother. Cancer 2022, 10, e003087. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Luo, J.; Wu, S.; Si, H.; Gao, C.; Xu, W.; Abdullah, S.E.; Higgs, B.W.; Dennis, P.A.; van der Heijden, M.S.; et al. Prognostic and predictive impact of circulating tumor DNA in patients with advanced cancers treated with immune checkpoint blockade. Cancer Discov. 2020, 10, 1842–1853. [Google Scholar] [CrossRef] [PubMed]
- Drusbosky, L.; Bilen, M.A.; Azzi, G.; Barata, P.C.; Boland, P.M.; Bryce, A.H.; Chae, Y.K.; Force, J.M.; Gutierrez, M.; Kasi, P.M.; et al. Blood-based tumor mutational burden from circulating tumor DNA (ctDNA) across advanced solid malignancies using a commercially available liquid biopsy assay. J. Clin. Oncol. 2021, 39, 3040. [Google Scholar] [CrossRef]
- Chae, Y.K.; Davis, A.A.; Agte, S.; Pan, A.; Simon, N.I.; Iams, W.T.; Cruz, M.R.; Tamragouri, K.; Rhee, K.; Mohindra, N.; et al. Clinical Implications of Circulating Tumor DNA Tumor Mutational Burden (ctDNA TMB) in Non-Small Cell Lung Cancer. Oncologist 2019, 24, 820–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heitzer, E.; van den Broek, D.; Denis, M.G.; Hofman, P.; Hubank, M.; Mouliere, F.; Paz-Ares, L.; Schuuring, E.; Sültmann, H.; Vainer, G.; et al. Recommendations for a practical implementation of circulating tumor DNA mutation testing in metastatic non-small-cell lung cancer. ESMO Open 2022, 7, 100399. [Google Scholar] [CrossRef]
- Corcoran, R.B.; André, T.; Atreya, C.E.; Schellens, J.H.M.; Yoshino, T.; Bendell, J.C.; Hollebecque, A.; McRee, A.J.; Siena, S.; Middleton, G.; et al. Combined BRAF, EGFR, and MEK Inhibition in Patients with BRAFV600E-Mutant Colorectal Cancer. Cancer Discov. 2018, 8, 428–443. [Google Scholar] [CrossRef] [Green Version]
- Pinheiro, M.; Peixoto, A.; Rocha, P.; Veiga, I.; Pinto, C.; Santos, C.; Pinto, P.; Guerra, J.; Escudeiro, C.; Barbosa, A.; et al. KRAS and NRAS mutational analysis in plasma ctDNA from patients with metastatic colorectal cancer by real-time PCR and digital PCR. Int. J. Color. Dis. 2022, 37, 895–905. [Google Scholar] [CrossRef]
- Chakrabarti, S.; Kasi, A.K.; Parikh, A.R.; Mahipal, A. The Evolving Paradigm of Circulating Tumor DNA (ctDNA)—Guided Minimal Residual Disease (MRD) Assessment in Colorectal Cancer (CRC). Cancers 2022, 14, 3078. [Google Scholar] [CrossRef]
- Dall’Olio, F.G.; Marabelle, A.; Caramella, C.; Garcia, C.; Aldea, M.; Chaput, N.; Robert, C.; Besse, B. Tumour burden and efficacy of immune-checkpoint inhibitors. Nat. Rev. Clin. Oncol. 2022, 19, 75–90. [Google Scholar] [CrossRef]
- Bhang, H.E.; Ruddy, D.A.; Krishnamurthy Radhakrishna, V.; Caushi, J.X.; Zhao, R.; Hims, M.M.; Singh, A.P.; Kao, I.; Rakiec, D.; Shaw, P.; et al. Studying clonal dynamics in response to cancer therapy using high-complexity barcoding. Nat. Med. 2015, 21, 440–448. [Google Scholar] [CrossRef]
- Hazar-Rethinam, M.; Kleyman, M.; Han, G.C.; Liu, D.; Ahronian, L.G.; Shahzade, H.A.; Chen, L.; Parikh, A.R.; Allen, J.N.; Clark, J.W.; et al. Convergent Therapeutic Strategies to Overcome the Heterogeneity of Acquired Resistance in BRAFV600E Colorectal Cancer. Cancer Discov. 2018, 8, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piotrowska, Z.; Niederst, M.J.; Karlovich, C.A.; Wakelee, H.A.; Neal, J.W.; Mino-Kenudson, M.; Fulton, L.; Hata, A.N.; Lockerman, E.L.; Kalsy, A.; et al. Heterogeneity Underlies the Emergence of EGFRT790 Wild-Type Clones Following Treatment of T790M-Positive Cancers with a Third-Generation EGFR Inhibitor. Cancer Discov. 2015, 5, 713–722. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Hu, S.; Zhang, L.; Xin, J.; Sun, C.; Wang, L.; Ding, K.; Wang, B. Tumor circulome in the liquid biopsies for cancer diagnosis and prognosis. Theranostics 2020, 10, 4544–4556. [Google Scholar] [CrossRef] [PubMed]
- Amelio, I.; Bertolo, R.; Bove, P.; Buonomo, O.C.; Candi, E.; Chiocchi, M.; Cipriani, C.; Di Daniele, N.; Ganini, C.; Juhl, H.; et al. Liquid biopsies and cancer omics. Cell Death Discov. 2020, 6, 131. [Google Scholar] [CrossRef] [PubMed]
- Ascencio Gonzáles, C.D. Liquid Biopsy perspectives theranostic and personalized oncology. Ann. Cytol. Pathol. 2020, 5, 073–077. [Google Scholar] [CrossRef]
- Best, M.G.; Sol, N.; Kooi, I.; Tannous, J.; Westerman, B.A.; Rustenburg, F.; Schellen, P.; Verschueren, H.; Post, E.; Koster, J.; et al. RNA-seq of tumor-educated platelts enables blood-based pan-cancer, multiclass, and molecular pathway cancer diagnostic. Cancer Cell 2015, 28, 666–676. [Google Scholar] [CrossRef] [Green Version]
- Lazzari, C.; Gregorc, V.; Cangi, M.G.; Giovannetti, E.; Bulotta, A.; Santarpia, M. Combined exosomal RNA and circulating tumor DNA for epidermal growth factor mutation detection in non-small cell lung cancer. J. Thorac. Dis. 2018, 10, S4023–S4027. [Google Scholar] [CrossRef]
- Qu, F.; Gu, Y.; Wang, Q.; He, M.; Zhou, F.; Sun, J.; Wang, G.; Peng, Y. Metabolomic profiling to evaluate the efficacy of proxalutamide, a novel androgen receptor antagonist, in prostate cancer cells. Investig. New Drugs 2020, 38, 1292–1302. [Google Scholar] [CrossRef]
- Fidler, I.J. Biological behavior of malignant melanoma cells correlated to their survival in vivo. Cancer Res. 1975, 35, 218–224. [Google Scholar]
- Costa-Silva, B.; Aiello, N.M.; Ocean, A.J.; Singh, S.; Zhang, H.; Thakur, B.K.; Becker, A.; Hoshino, A.; Mark, M.T.; Molina, H.; et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat. Cell Biol. 2015, 17, 816–826. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, A.; Costa-Silva, B.; Shen, T.L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastases. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Lai, W.; Fang, D.; Fang, Q. Protein biomarkers in breast cancer-derived extracellular vesicles for use in liquid biopsies. Am. J. Physiol. Cell Physiol. 2021, 321, C779–C797. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tao, R. 1265P Combined detection of tumor markers and ctDNA in cerebrospinal fluid: A good strategy in diagnoses of leptomeningeal metastases. Ann. Oncol. 2021, 32, S990. [Google Scholar] [CrossRef]
- Isaksson, S.; George, A.M.; Jönsson, M.; Cirenajwis, H.; Jönsson, P.; Bendahl, P.O.; Brunnström, H.; Staaf, J.; Saal, L.H.; Planck, M. Pre-operative plasma cell-free circulating tumor DNA and serum protein tumor markers as predictors of lung adenocarcinoma recurrence. Acta Oncol. 2019, 58, 1079–1086. [Google Scholar] [CrossRef] [Green Version]
- Magbanua, M.J.M.; Li, W.; Wolf, D.M.; Yau, C.; Hirst, G.L.; Swigart, L.B.; Newitt, D.C.; Gibbs, J.; Delson, A.L.; Kalashnikova, E.; et al. Circulating tumor DNA and magnetic resonance imaging to predict neoadjuvant chemotherapy response and recurrence risk. NPJ Breast Cancer 2021, 7, 32. [Google Scholar] [CrossRef]
- Metzenmacher, M.; Hegedüs, B.; Forster, J.; Schramm, A.; Horn, P.A.; Klein, C.A.; Bielefeld, N.; Ploenes, T.; Aigner, C.; Theegarten, D.; et al. Combined multimodal ctDNA analysis and radiological imaging for tumor surveilance in non-small cell lung cancer. Transl. Oncol. 2021, 15, 101279. [Google Scholar] [CrossRef]
- Fakih, M.; Sandhu, J.; Wang, C.; Kim, J.; Chen, Y.J.; Lai, L.; Melstrom, K.; Kaiser, A. Evaluation of Comparative Surveillance Strategies of Circulating Tumor DNA, Imaging, and Carcinoembryonic Antigen Levels in Patients with Resected Colorectal Cancer. JAMA Netw. Open 2022, 5, e221093. [Google Scholar] [CrossRef]
- Wong, S.Q.; Raleigh, J.M.; Callahan, J.; Vergara, I.A.; Ftouni, S.; Hatzimihalis, A.; Colebatch, A.J.; Li, J.; Semple, T.; Doig, K.; et al. Cirulating Tumor DNA Analysis and Functional Imaging Provide Complementary Approaches for Comprehensive Disease Monitoring in Metastatic Melanoma. JCO Precis. Oncol. 2017, 1, 1–14. [Google Scholar]
- Batth, I.S.; Mitra, A.; Manier, S.; Ghobrial, I.M.; Menter, D.; Kopetz, S.; Li, S. Circulating tumor markers: Harmonizing the yin and yang of CTCs and ctDNA for precision medicine. Ann. Oncol. 2017, 28, 468–477. [Google Scholar] [CrossRef]
- van Rensburg, H.J.J.; Spiliopoulou, P.; Siu, L.L. Circulating Biomarkers for Therapeuting Monitoring of Anti-cancer Agents. Oncologist 2022, 27, 352–362. [Google Scholar] [CrossRef]
- Bedard, P.L.; Hansen, A.R.; Ratain, M.J.; Siu, L.L. Tumour heterogeneity in the clinic. Nature 2013, 501, 355–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Y.; Mei, W.; Ma, K.; Zeng, C. Circulating Tumor DNA and Minimal Residual Disease (MRD) in Solid Tumors: Current Horizons and Future Perspectives. Front. Oncol. 2021, 11, 763790. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J.; Crown, J. Use of Circulating Tumour DNA (ctDNA) for Measurement of Therapy Predictive Biomarkers in Patients with Cancer. J. Pers. Med. 2022, 12, 99. [Google Scholar] [CrossRef] [PubMed]
- De Mattos-Arruda, L.; Mayor, R.; Ng, C.K.Y.; Weigelt, B.; Martínez-Ricarte, F.; Torrejon, D.; Oliveira, M.; Arias, A.; Raventos, C.; Tang, J.; et al. Cerebrospinal fluid-derived circulating tumor DNA better represents the genomic alterations of brain tumors than plasma. Nat. Commun. 2015, 6, 8839. [Google Scholar] [CrossRef] [Green Version]
- Dang, D.K.; Park, B.H. Circulating tumor DNA: Current challenges for clinical utility. J. Clin. Investig. 2022, 132, e154941. [Google Scholar] [CrossRef]
| Aspect | ctDNA | CTC |
|---|---|---|
| Procedure | Minimally invasive | Minimally invasive |
| Sample collection | Prospective | Prospective |
| Intratumoral heterogeneity | Adequately represent, but false-negative and false-positive errors could occur more than 15% [5] May identify resistance mechanisms, discordant clinical history, and intertumor/intratumor heterogeneity | Adequately represent, but small and fragile sample population; detection increased with increasing stage |
| Phenotype [6] | Study the methylome may provide information about phenotype | Structural evaluation of the tumor might be possible |
| Tumor burden | Sensitive indicator | Still obscure how adequately represent tumor burden |
| Resistance | The emergence of resistance could be addressed | The emergence of resistance could be addressed |
| Recommended application NCCN and ESMO [7,8] | Should not be used to diagnose! Analysis DNA methylation changes, copy, and mutations can be used to identify EGFR, ALK, and other oncogenic biomarkers that would not otherwise be identified in patientswith metastatic cancer In ER+ breast cancer PIK3CA mutations to identify candidates for alpelisib plus fulvestrant In CRC, post-surgical ctDNA is a marker for an elevated risk of recurrence in stage I–III colon cancer | Should not be used to diagnose! May be considered at progression of NSCLC instead of tissue biopsy to detect whether patients have lung cancer; however, if plasma testing is negative, then tissue biopsy is recommended |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Telekes, A.; Horváth, A. The Role of Cell-Free DNA in Cancer Treatment Decision Making. Cancers 2022, 14, 6115. https://doi.org/10.3390/cancers14246115
Telekes A, Horváth A. The Role of Cell-Free DNA in Cancer Treatment Decision Making. Cancers. 2022; 14(24):6115. https://doi.org/10.3390/cancers14246115
Chicago/Turabian StyleTelekes, András, and Anna Horváth. 2022. "The Role of Cell-Free DNA in Cancer Treatment Decision Making" Cancers 14, no. 24: 6115. https://doi.org/10.3390/cancers14246115
APA StyleTelekes, A., & Horváth, A. (2022). The Role of Cell-Free DNA in Cancer Treatment Decision Making. Cancers, 14(24), 6115. https://doi.org/10.3390/cancers14246115