
A Computational Framework for Comprehensive Genomic Profiling in Solid Cancers: The Analytical Performance of a High-Throughput Assay for Small and Copy Number Variants
 , , ,
, , ,  , , , ,
, , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
- research purpose;
- screening for clinical trials;
- drug development;
- tumor characterization of non-squamous non-small-cell lung cancer (NSCLC), prostate cancers, ovarian cancers and cholangiocarcinoma only in cases of acceptable additional cost;
- tumor characterization of colon cancer as an alternative option to PCR only in cases of acceptable additional cost;
- tumor characterization of all cancers for which agnostic drugs are available (i.e., pembrolizumab for high tumor mutational burden, TMB).
2. Materials and Methods
2.1. Samples and Orthogonal Assay
2.2. Library Set-Up
2.3. Sequencing
2.4. Bioinformatics Analysis
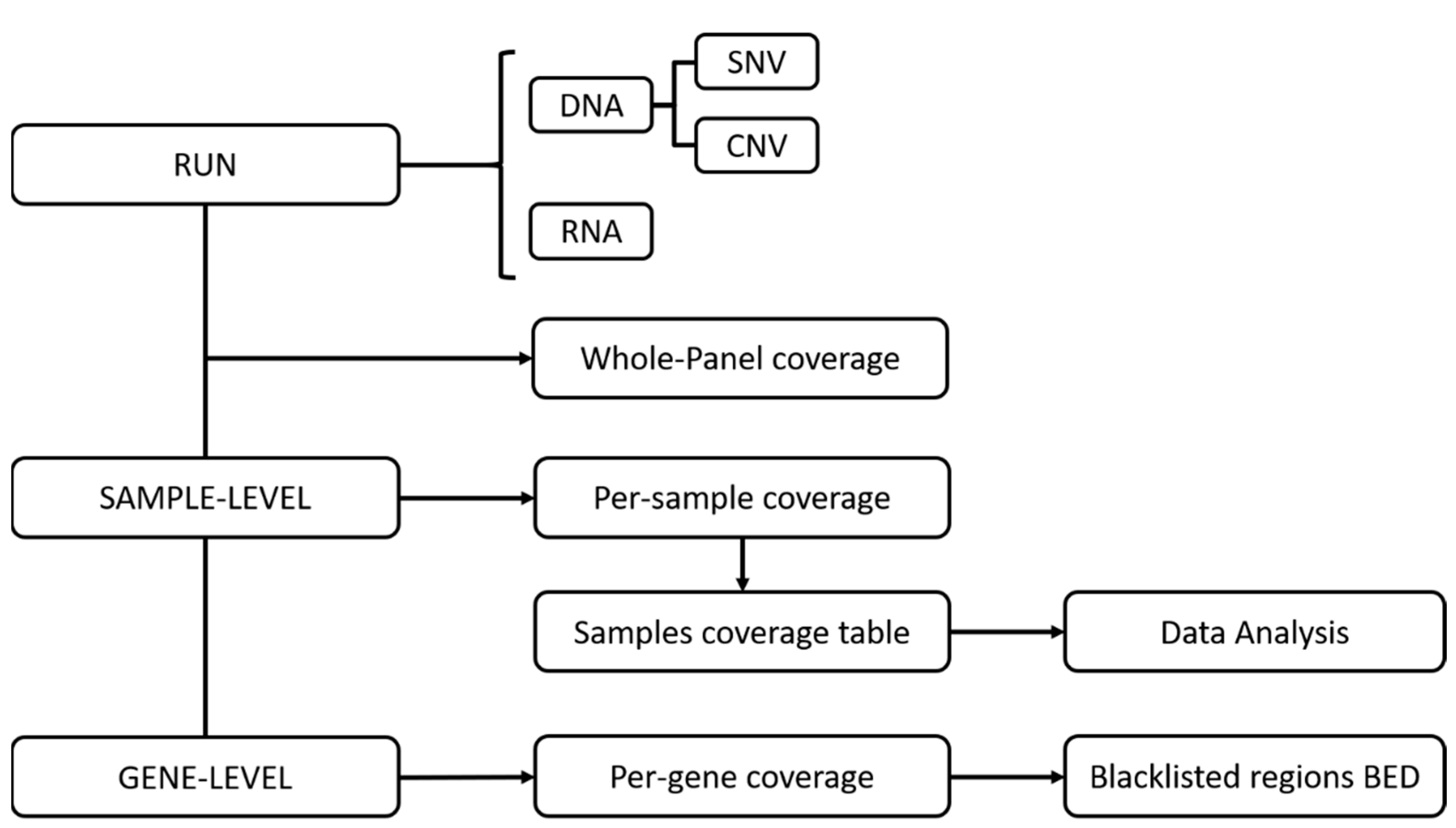
2.5. Coverage Analysis
2.6. Data Analysis
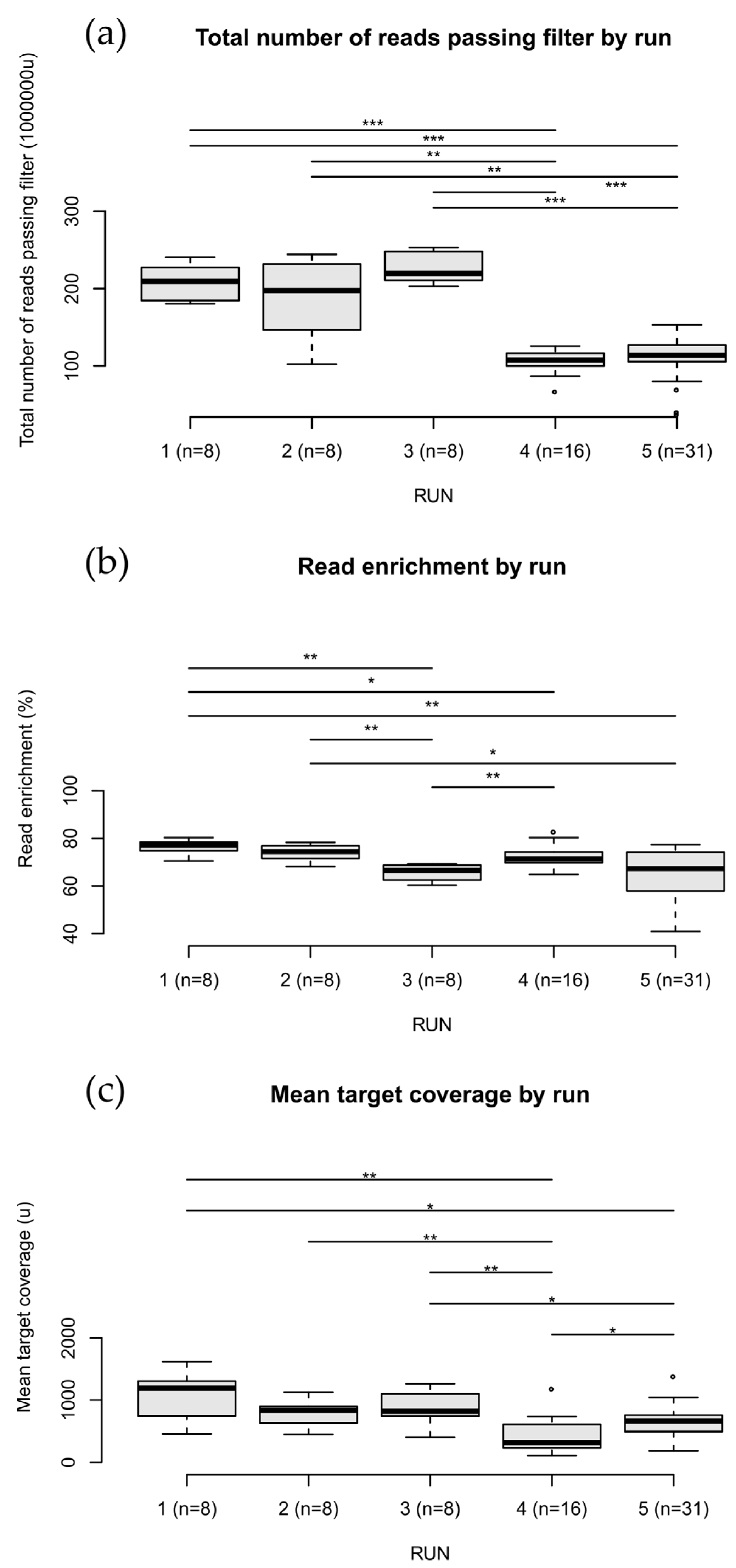
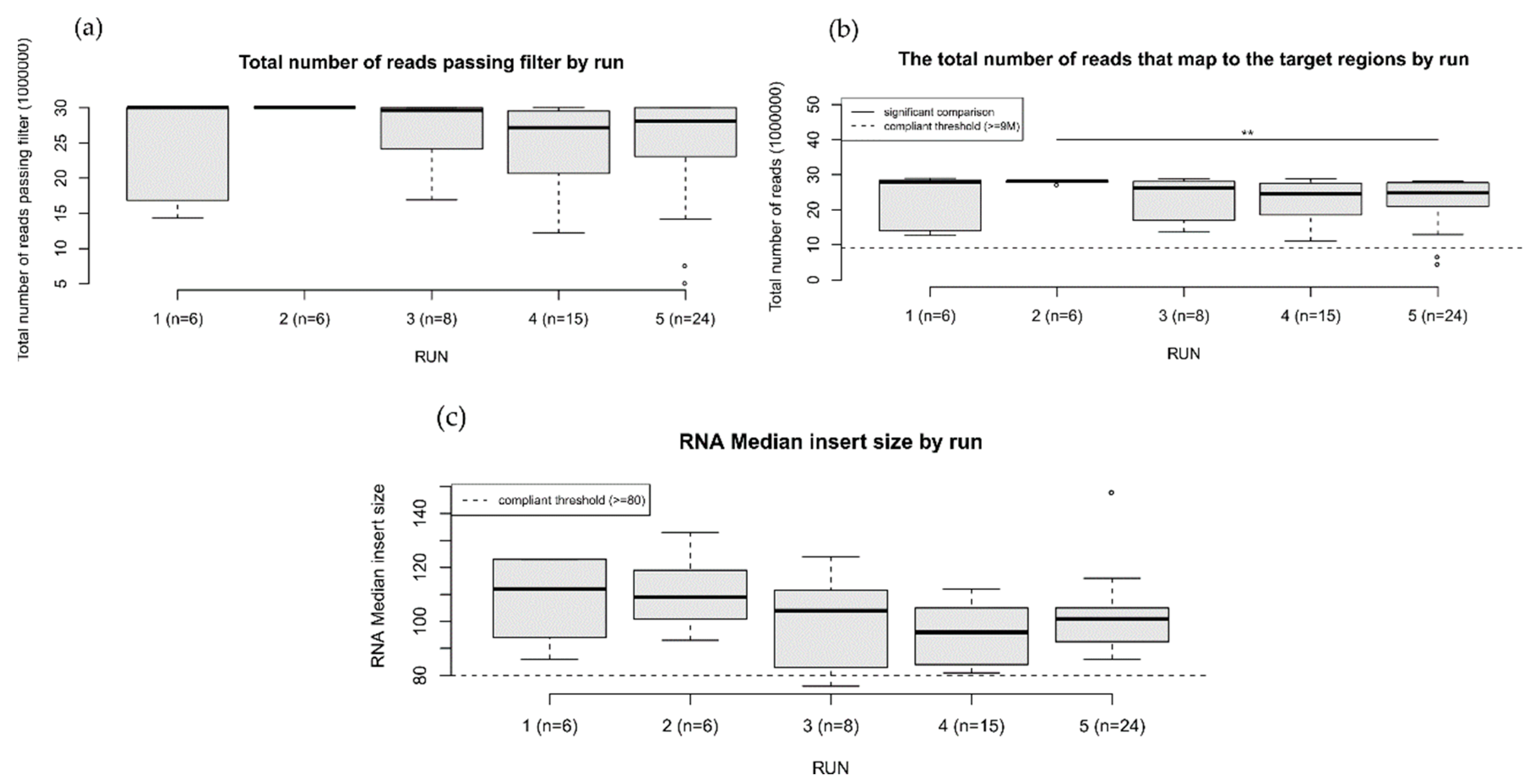
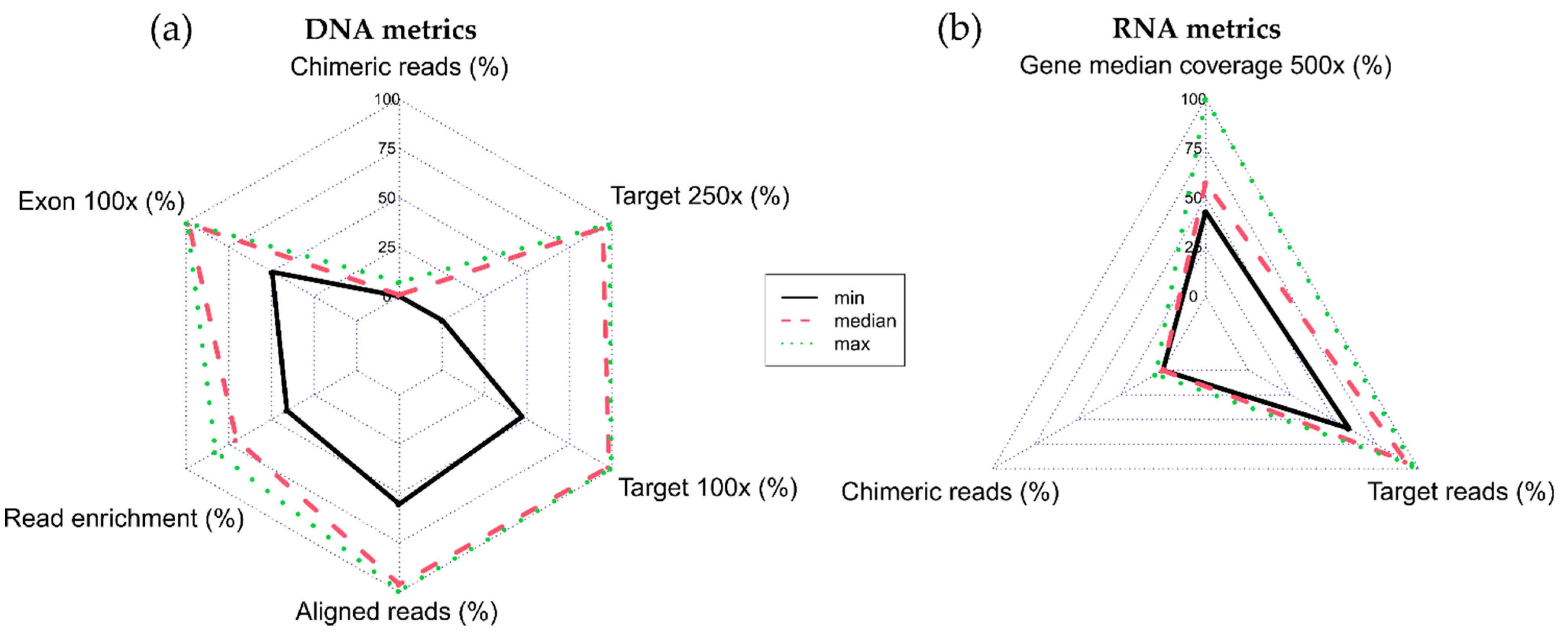
3. Results
3.1. Variant Results
3.2. Bioinformatics Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Conroy, J.M.; Pabla, S.; Glenn, S.T.; Seager, S.J.; Van Roey, E.; Gao, S.; Burgher, B.; Andreas, J.; Giamo, V.; Mallon, M.; et al. A scalable high-throughput targeted next-generation sequencing assay for comprehensive genomic profiling of solid tumors. PLoS ONE 2021, 16, e0260089. [Google Scholar] [CrossRef] [PubMed]
- Froyen, G.; Geerdens, E.; Berden, S.; Cruys, B.; Maes, B. Diagnostic Validation of a Comprehensive Targeted Panel for Broad Mutational and Biomarker Analysis in Solid Tumors. Cancers 2022, 14, 2457. [Google Scholar] [CrossRef] [PubMed]
- Mosele, F.; Remon, J.; Mateo, J.; Westphalen, C.B.; Barlesi, F.; Lolkema, M.P.; Normanno, N.; Scarpa, A.; Robson, M.; Meric-Bernstam, F.; et al. Recommendations for the use of next-generation sequencing (NGS) for patients with metastatic cancers: A report from the ESMO Precision Medicine Working Group. Ann. Oncol. 2020, 31, 1491–1505. [Google Scholar] [CrossRef] [PubMed]
- Merkel, D. Docker: Lightweight linux containers for consistent development and deployment. Linux J. 2014, 2, 239. [Google Scholar]
- Lianne, Version 2.1. Fondazione Policlinico Universitario Agostino Gemelli IRCCS. 2022. Available online: https://github.com/lucianogiaco/lianne (accessed on 17 November 2022).
- Anaconda Software Distribution. Anaconda Documentation; Anaconda Inc.: Austin, TX, USA, 2020; Available online: https://docs.anaconda.com/ (accessed on 17 November 2022).
- Kurtzer, G.M.; Sochat, V.; Bauer, M.W. Singularity: Scientific containers for mobility of compute. PLoS ONE 2017, 12, e0177459. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.S.; Quinlan, A.R. Mosdepth: Quick coverage calculation for genomes and exomes. Bioinformatics 2018, 34, 867–868. Available online: https://github.com/brentp/mosdepth (accessed on 6 October 2022). [CrossRef] [PubMed] [Green Version]
- VarHound, Version 0.1.0-beta. Fondazione Policlinico Universitario Agostino Gemelli IRCCS. 2022. Available online: https://github.com/fernandoPalluzzi/VarHound (accessed on 17 November 2022).
- R Core Team. R: A language and environment for statistical computing; R Foundation for Statistical Computing: Vienna, Austria, 2022; Available online: https://www.R-project.org/ (accessed on 17 November 2022).
- Tso500 Validation Scripts. Fondazione Policlinico Universitario Agostino Gemelli IRCCS. 2022. Available online: https://github.com/lucianogiaco/TSO500-validation/tree/luciano (accessed on 17 November 2022).
- Hothorn, T.; Hornik, K.; van de Wiel, M.A.; Zeileis, A. A Lego system for conditional inference. Am. Stat. 2006, 60, 257–263. [Google Scholar] [CrossRef] [Green Version]
- Hothorn, T.; Hornik, K.; van de Wiel, M.A.; Zeileis, A. Implementing a class of permutation tests: The coin package. J. Stat. Softw. 2008, 28, 1–23. [Google Scholar] [CrossRef]
- Nakazawa, M. fmsb: Functions for Medical Statistics Book with Some Demographic Data. R Package Version 0.7.2. 2021. Available online: https://CRAN.R-project.org/package=fmsb (accessed on 17 November 2022).
- Pestinger, V.; Smith, M.; Sillo, T.; Findlay, J.M.; Laes, J.F.; Martin, G.; Middleton, G.; Taniere, P.; Beggs, A.D. Use of an Integrated Pan-Cancer Oncology Enrichment Next-Generation Sequencing Assay to Measure Tumour Mutational Burden and Detect Clinically Actionable Variants. Mol. Diagn. Ther. 2020, 24, 339–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, B.; Kang, J.; Kibukawa, M.; Arreaza, G.; Maguire, M.; Chen, L.; Qiu, P.; Lang, L.; Aurora-Garg, D.; Cristescu, R.; et al. Evaluation of the TruSight Oncology 500 Assay for Routine Clinical Testing of Tumor Mutational Burden and Clinical Utility for Predicting Response to Pembrolizumab. J. Mol. Diagn. 2022, 24, 600–608. [Google Scholar] [CrossRef] [PubMed]
- Sanghvi, R.V.; Buhay, C.J.; Powell, B.C.; Tsai, E.A.; Dorschner, M.O.; Hong, C.S.; Lebo, M.S.; Sasson, A.; Hanna, D.S.; McGee, S.; et al. Characterizing reduced coverage regions through comparison of exome and genome sequencing data across 10 centers. Genet. Med. 2018, 20, 855–866. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Shashikant, C.S.; Jensen, M.; Altman, N.S.; Girirajan, S. Novel metrics to measure coverage in whole exome sequencing datasets reveal local and global non-uniformity. Sci. Rep. 2017, 7, 885. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Overall | Run 1 | Run 2 | Run 3 | Run 4 | Run 5 | p | |
|---|---|---|---|---|---|---|---|
| DNA (ng/ul) (QT) (CV if >3.5) | 34.58 ± 52.50 17.1 [3.11; 298] | 74.14 ± 86.11 24.45 [9.40; 242] | 16.448 ± 10.52 14.25 [5.4; 36.7] | 25.66 ± 27.13 17.75 [4.4; 88.7] | 44.792 ± 74.04 14.85 [3.11; 298] | 26.07 ± 33.28 17.1 [3.4; 184] | 0.598 |
| A 260/280 (u) (QL) (CV if >2) | 2.02 ± 0.219 1.97 [1.50; 3.06] | 2.058 ± 0.093 2.045 [1.93; 2.21] | 2.186 ± 0.145 2.195 [1.97; 2.36] | 2.107 ± 0.393 1.965 [1.9; 3.06] | 1.921 ± 0.141 1.9 [1.5; 2.12] | 1.997 ± 0.209 1.95 [1.58; 2.6] | 0.004 |
| A 260/230 (u) (QL) (CV if >2) | 0.884 ± 0.684 0.67 [0.1; 2.28] | 0.59 ± 0.448 0.38 [0.23; 1.49] | 0.303 ± 0.162 0.25 [0.18; 0.67] | 1.439 ± 0.909 2 [0.1; 2.28] | 1.183 ± 0.784 1.125 [0.26; 2.2] | 0.814 ± 0.539 0.75 [0.14; 2.12] | 0.005 |
| Delta Cq (u) (QL) (CV if <5) | 0.192 ± 1.541 0.3 [−4.7; 3.6] | 0.838 ± 0.851 0.45 [0.1; 2.5] | 0.437 ± 0.722 0.15 [−0.4; 1.7] | −0.185 ± 1.369 −0.5 [−1.6; 2] | −1.181 ± 1.818 −0.7 [−4.7; 1.4] | 0.769 ± 1.282 0.8 [−2.61; 3.6] | 0.001 |
| Quality Control post Fragmentation (bp) (QT) * | 235.1 ± 35.73 232 [173; 315] | 197.9 ± 17.59 207.5 [173; 216] | 197.1 ± 13.23 195.5 [181; 212] | 220.8 ± 25.49 219 [187; 265] | 226.8 ± 28.62 228.5 [174; 272] | 262.4 ± 26.21 261 [203; 315] | <0.001 |
| Pre-capture libraries metric (ng/ul) (QL) (CV if >20) | 47.43 ± 7.298 48.7 [26.4; 60] | 46.64 ± 3.613 47 [41.6; 52] | 49.85 ± 1.790 49.65 [47.7; 53] | 30.7 ± 3.001 30.45 [26.4; 35.6] | 49.71 ± 3.373 49.25 [44.4; 57] | 50.16 ± 5.089 50 [39.9; 60] | <0.001 |
| Enriched libraries metric (ng/ul) (QL) (CV if >3) | 16.7 ± 7.959 18.5 [1.53; 31.9] | 12.161 ± 4.368 13.95 [5.58; 16.4] | 17.49 ± 8.652 20.6 [3.4; 25.8] | 19.06 ± 1.694 18.6 [17; 21.7] | 10.125 ± 5.233 8.96 [3.34; 21.6] | 20.45 ± 8.173 23 [1.53; 31.9] | <0.001 |
| Overall | Run 1 | Run 2 | Run 3 | Run 4 | Run 5 | p | |
|---|---|---|---|---|---|---|---|
| RNA (ng/ul) (QT) (CV if >10.5) | 78.3 ± 63.40 66 [12.3; 312] | 70.33 ± 41.27 70.95 [23.7; 120] | 131.4 ± 105.13 89.7 [30.1; 312] | 46.1 ± 52.67 30.65 [12.3; 170] | 104.74 ± 58.11 86 [36; 235.8] | 61.23 ± 50.90 45.5 [13.1; 200.1] | 0.005 |
| RNA A260/280 (QL) (CV if >2) | 1.942 ± 0.101 1.96 [1.6; 2.2] | 2 ± 0.109 2 [1.9; 2.2] | 1.917 ± 0.098 1.95 [1.8; 2] | 1.976 ± 0.089 2 [1.76; 2.05] | 1.871 ± 0.066 1.9 [1.7; 1.97] | 1.968 ± 0.102 1.98 [1.6; 2.2] | <0.001 |
| RNA A260/230 (QL) (CV if >2) | 1.123 ± 0.635 1.36 [0.03; 1.95] | 1.105 ± 0.563 1.21 [0.09; 1.6] | 1.113 ± 0.609 1.1 [0.17; 1.75] | 0.89 ± 0.817 0.87 [0.03; 1.95] | 1.66 ± 0.314 1.7 [0.6; 1.9] | 0.871 ± 0.575 0.79 [0.16; 1.94] | 0.001 |
| DV200 (%) (QL) (CV if >20) | 59.2 ± 16.71 63.7 [2.6; 86.9] | 59.33 ± 15.49 64.7 [36.3; 73.2] | 60.32 ± 17.52 56.6 [41.7; 83.5] | 61.29 ± 18.73 68.1 [33.2; 82.5] | 65.25 ± 10.79 67.2 [42.6; 77.8] | 54.4 ± 18.92 55.95 [2.6; 86.9] | 0.455 |
| Pre-capture libraries metric (ng/ul) (QL) (CV if >20) | 48.84 ± 8.381 51 [25.7; 60] | 48.12 ± 2.778 47.4 [45.4; 53] | 45.93 ± 2.809 45.3 [43; 51] | 31.15 ± 3.787 30.85 [25.7; 36.4] | 52.05 ± 2.775 53 [46.1; 55] | 53.63 ± 4.780 54.5 [39.7; 60] | <0.001 |
| Enriched libraries metric (ng/ul) (QL) (CV if >3) | 7.033 ± 4.702 6.51 [0.80; 19.6] | 5.145 ± 3.821 5.755 [0.8; 9.03] | 9.685 ± 6.081 7.45 [3.8; 18.6] | 6.893 ± 6.023 5.615 [1.07; 17] | 6.981 ± 5.636 5.25 [1.12; 19.6] | 6.920 ± 3.372 6.925 [2.1; 13.8] | 0.635 |
| SNV 100X | SNV 250X | SNV 500X | CNV 100X | CNV 250X | CNV 500X | |
|---|---|---|---|---|---|---|
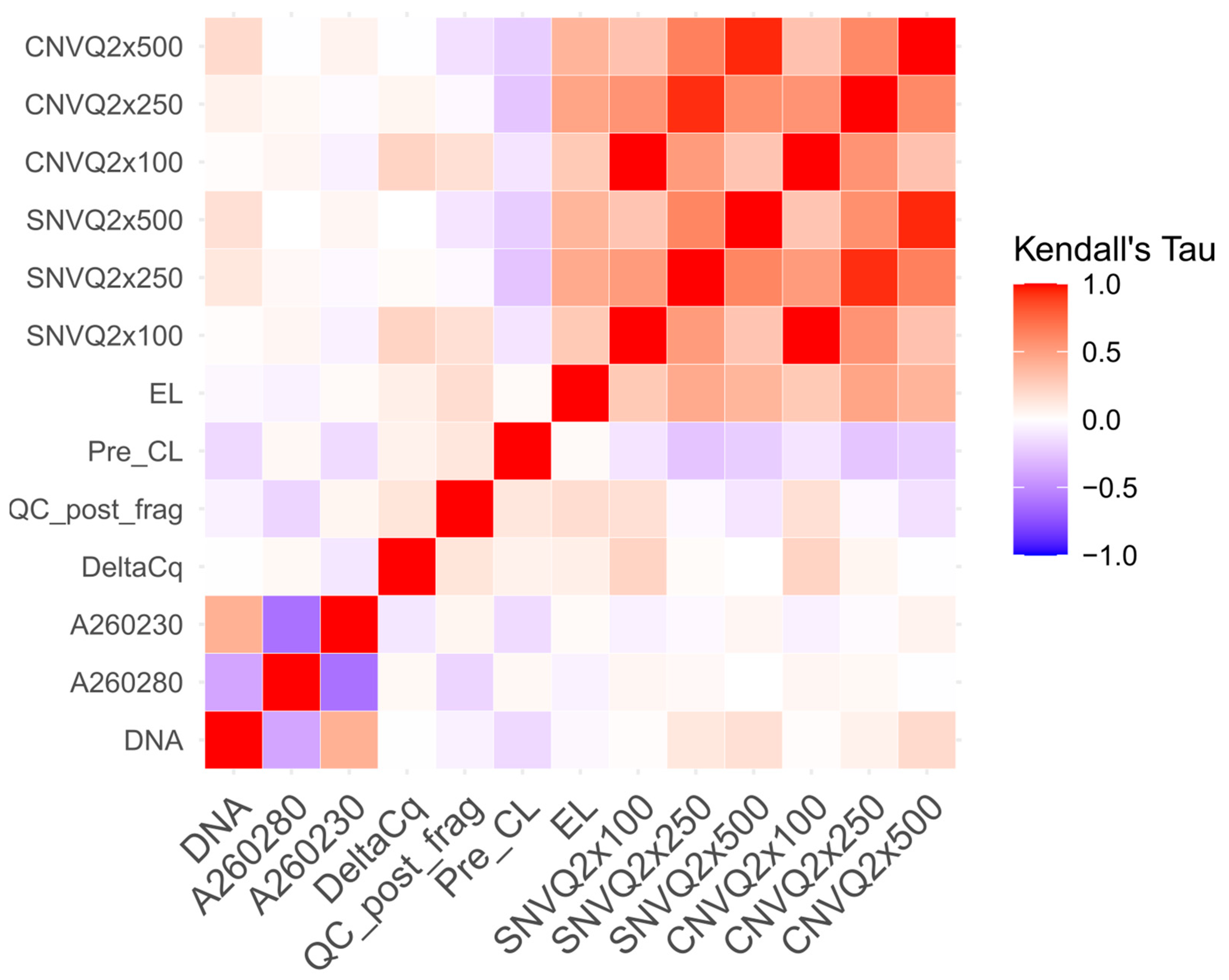
| DNA | 0.013 0.891 −0.222, 0.249 | 0.126 0.181 −0.076, 0.328 | 0.170 0.060 −0.009, 0.350 | 0.013 0.891 −0.222, 0.249 | 0.067 0.474 −0.138, 0.274 | 0.196 0.029 0.016, 0.375 |
| A 260/280 | 0.047 0.634 −0.158, 0.252 | 0.035 0.710 −0.145, 0.217 | 0 1 −0.180, 0.180 | 0.047 0.634 −0.158, 0.253 | 0.033 0.727 −0.149, 0.216 | −0.010 0.913 −0.190, 0.170 |
| A 260/230 | −0.058 0.549 −0.278, 0.161 | −0.027 0.774 −0.195, 0.141 | 0.051 0.578 −0.128, 0.229 | −0.058 0.550 −0.278, 0.161 | −0.019 0.836 −0.188, 0.149 | 0.062 0.489 −0.113, 0.238 |
| Delta Cq | 0.230 0.019 0.011, 0.449 | 0.022 0.812 −0.188, 0.234 | −0.001 0.991 −0.191, 0.189 | 0.230 0.019 0.012, 0.449 | 0.048 0.615 −0.166, 0.262 | −0.008 0.926 −0.196, 0.179 |
| Quality Control post Fragmentation | 0.170 0.081 −0.053, 0.393 | −0.031 0.737 −0.224, 0.161 | −0.114 0.201 −0.287, 0.058 | 0.170 0.081 −0.053, 0.393 | −0.028 0.762 −0.224, 0.167 | −0.136 0.130 −0.311, 0.037 |
| Pre-capture libraries metric | −0.120 0.221 −0.323, 0.083 | −0.257 0.007 −0.420, −0.094 | −0.216 0.018 −0.374, −0.058 | −0.120 0.222 −0.323, 0.083 | −0.250 0.008 −0.419, −0.080 | −0.216 0.017 −0.373, −0.059 |
| Enriched libraries metric | 0.283 0.003 0.032, 0.534 | 0.450 <0.001 0.280, 0.620 | 0.391 <0.001 0.245, 0.536 | 0.283 0.003 0.032, 0.534 | 0.474 <0.001 0.314, 0.633 | 0.392 <0.001 0.238, 0.545 |
| 5X | 10X | 50X | |
|---|---|---|---|
| RNA | 0.141 0.144 −0.047; 0.329 | 0.175 0.062 −0.207; 0.559 | 0.188 0.036 0.039; 0.336 |
| RNA A260/280 | −0.306 0.002 −0.473; −0.138 | −0.361 <0.001 −0.531; −0.192 | −0.344 <0.001 −0.507; −0.181 |
| RNA A260/230 | 0.265 0.006 0.087; 0.445 | 0.252 0.008 0.073; 0.430 | 0.218 0.016 0.050; 0.386 |
| DV200 | 0.249 0.010 0.082; 0.415 | 0.202 0.032 0.039; 0.366 | 0.109 0.226 −0.058; 0.275 |
| Pre-capture libraries metric | 0.105 0.284 −0.080; 0.290 | 0.132 0.165 −0.041; 0.306 | 0.093 0.303 −0.084; 0.272 |
| Enriched libraries metric | 0.356 <0.001 0.184; 0.528 | 0.279 0.003 0.102; 0.456 | 0.172 0.056 −0.015; 0.358 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giacò, L.; Palluzzi, F.; Guido, D.; Nero, C.; Giacomini, F.; Duranti, S.; Bria, E.; Tortora, G.; Cenci, T.; Martini, M.; et al. A Computational Framework for Comprehensive Genomic Profiling in Solid Cancers: The Analytical Performance of a High-Throughput Assay for Small and Copy Number Variants. Cancers 2022, 14, 6152. https://doi.org/10.3390/cancers14246152
Giacò L, Palluzzi F, Guido D, Nero C, Giacomini F, Duranti S, Bria E, Tortora G, Cenci T, Martini M, et al. A Computational Framework for Comprehensive Genomic Profiling in Solid Cancers: The Analytical Performance of a High-Throughput Assay for Small and Copy Number Variants. Cancers. 2022; 14(24):6152. https://doi.org/10.3390/cancers14246152
Chicago/Turabian StyleGiacò, Luciano, Fernando Palluzzi, Davide Guido, Camilla Nero, Flavia Giacomini, Simona Duranti, Emilio Bria, Giampaolo Tortora, Tonia Cenci, Maurizio Martini, and et al. 2022. "A Computational Framework for Comprehensive Genomic Profiling in Solid Cancers: The Analytical Performance of a High-Throughput Assay for Small and Copy Number Variants" Cancers 14, no. 24: 6152. https://doi.org/10.3390/cancers14246152