
Targeting Glioblastoma via Selective Alteration of Mitochondrial Redox State

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Cells and Treatment Protocol
2.3. Cell Proliferation and Viability Assays
2.4. Apoptosis Assay
2.5. Mitochondrial Superoxide Assay
2.6. Mitochondrial Membrane Potential
2.7. Succinate Assay
2.8. NAD+/NADH Quantification Assay
2.9. Total Glutathione Assay
2.10. Total Antioxidant Capacity (TAC) Assay
2.11. tNOX (ENOX2) Assay
2.12. TGF-β1 Assay
2.13. Animals and Treatment Protocol
2.14. Measurement of Hemoglobin, Hematocrit, and Thrombosis
2.15. In Vivo MRI Measurements
2.16. Statistical Analysis
3. Results
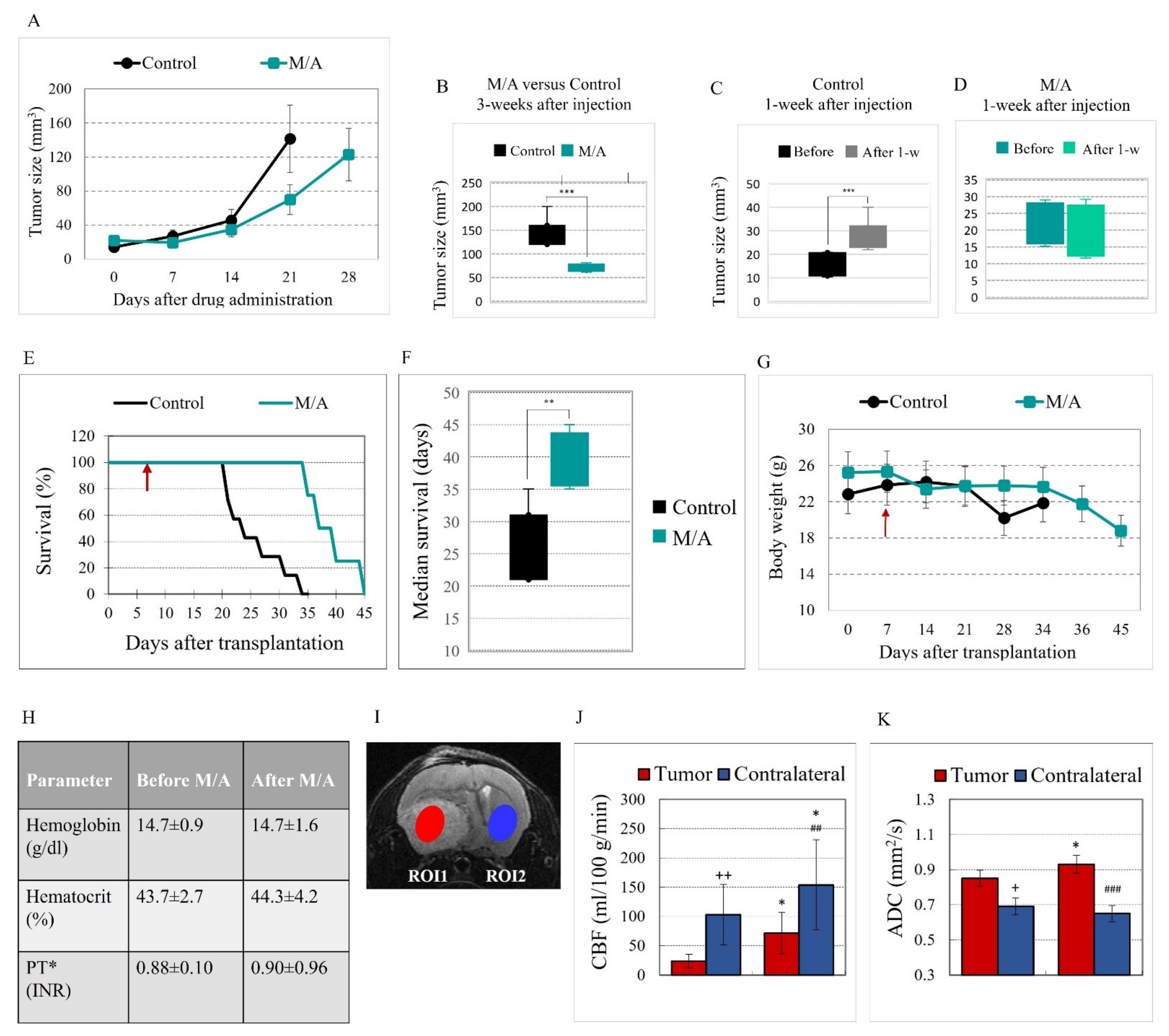
3.1. Effect of M/A on Tumor Growth and Survival of Glioblastoma-Bearing Mice
3.2. Effect of M/A on Brain Perfusion and Cell Density of Glioblastoma-Bearing Mice
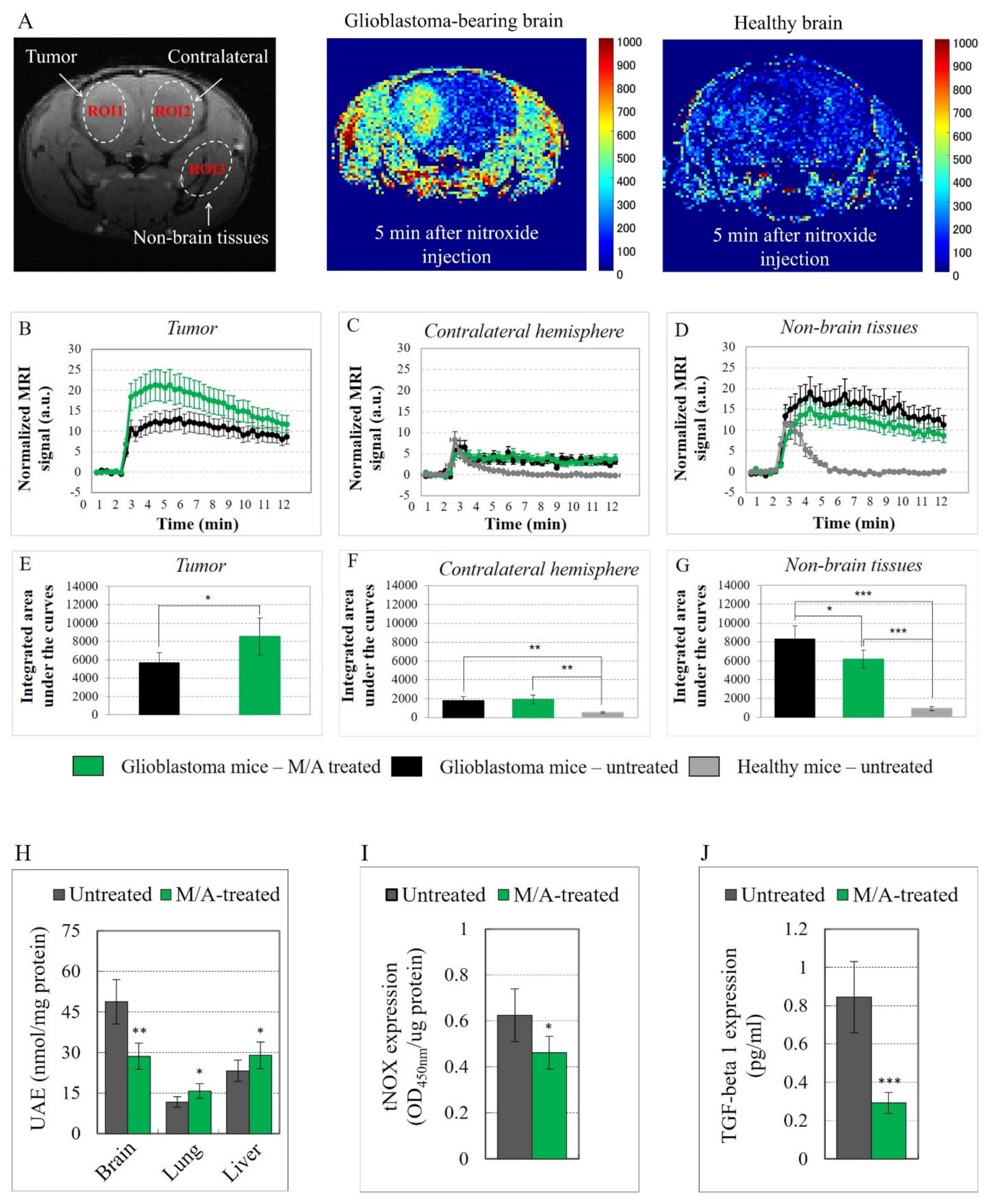
3.3. Effect of M/A on Tissue Redox State of Glioblastoma-Bearing Mice
3.4. Effect of M/A on Mitochondrial Functionality, tNOX Expression, and Prenylation
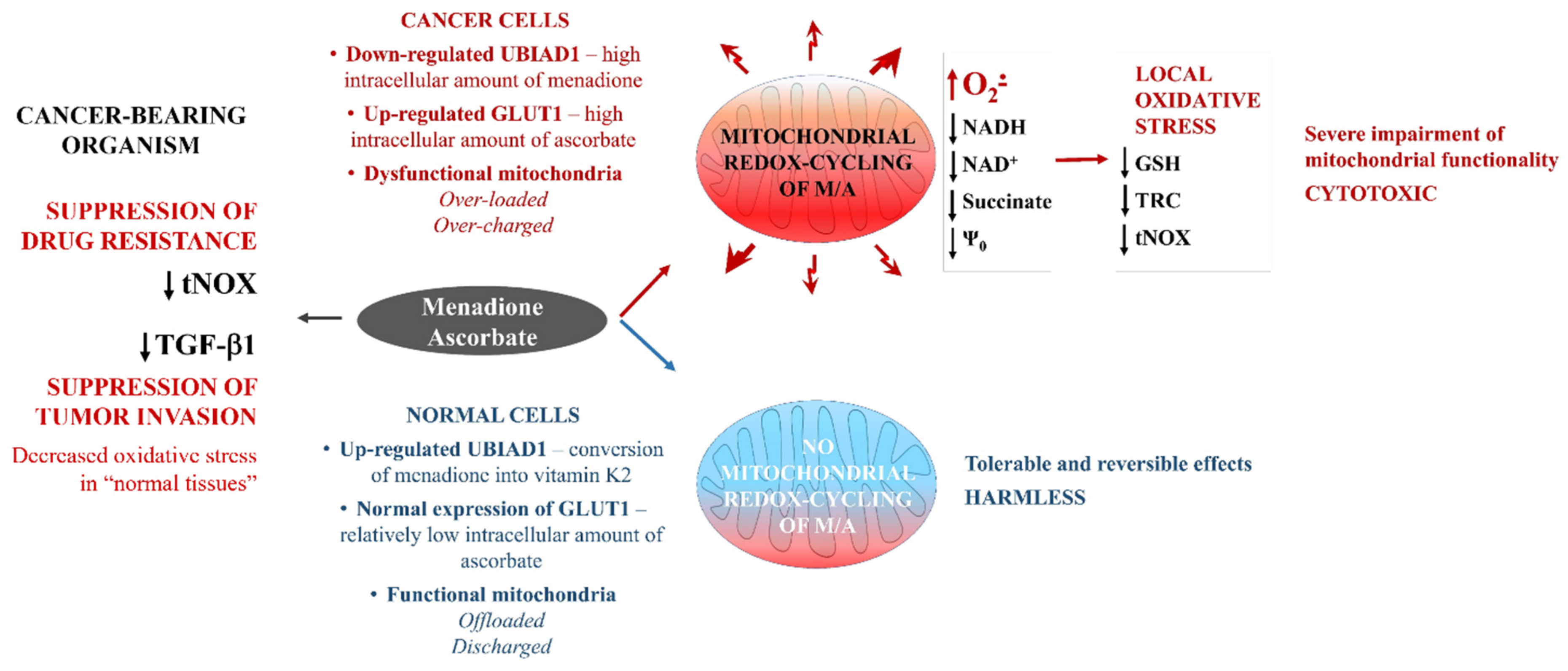
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bush, N.E.O.; Chang, S.M.; Berger, M.S. Current and Future Strategies for Treatment of Glioma. Neurosurg. Rev. 2017, 40, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Sampson, J.H.; Gunn, M.D.; Fecci, P.E.; Ashley, D.M. Brain Immunology and Immunotherapy in Brain Tumors. Nat. Rev. Cancer 2020, 20, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.; Shah, J. The Current and Future Aspects of Glioblastoma: Immunotherapy a New Hope? Eur. J. Neurosci. 2021, 54, 5120–5142. [Google Scholar] [CrossRef] [PubMed]
- Poon, M.T.C.; Sudlow, C.L.M.; Figueroa, J.D.; Brennan, P.M. Longer-Term (over 2 Years) Survival in Patients with Glioblastoma in Population-Based Studies Pre- and Post-2005: A Systematic Review and Meta-Analysis. Sci. Rep. 2020, 10, 11622. [Google Scholar] [CrossRef] [PubMed]
- Femandes, C.; Costa, A.; Osorio, L.; Lago, R.C.; Lonhares, P.; Carvalho, B.; Caeiro, C. Current Standards of Care in Glioblastoma Therapy. In Glioblastoma; Chapter 11; De Vleeschouwer, S., Ed.; Codon Publ.: Brisbane, Australia, 2017; ISBN -13: 978-0-9944381-2-6. [Google Scholar]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van Den Bert, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. European Organization for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups; National Cancer Institute of Canada Clinical Trials Group. Effects of Radiotherapy with Concomitant and Adjuvant Temozolomide versus Radiotherapy Alone on Survival in Glioblastoma in a Randomized Phase III Study: 5-Year Analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Zhang, J.; Stevens, M.F.; Bradshaw, T.D. Temozolomide: Mechanisms of Action, Repair and Resistance. Curr. Mol. Pharmacol. 2012, 5, 102–114. [Google Scholar] [CrossRef]
- Tatar, Z.; Thivat, E.; Planchat, E.; Gimbergues, P.; Gadea, E.; Abria, C.; Durando, X. Temozolomide and Unusual Indications: Review of Literature. Cancer Treat Rev. 2013, 39, 125–135. [Google Scholar] [CrossRef]
- Sengupta, S.; Marrian, J.; Frishman, C.; Sampath, P. Impact of Temozolomide on Immune Response during Malignant Glioma Chemotherapy. Clin. Dev. Immunol. 2012, 2012, 831090. [Google Scholar] [CrossRef] [Green Version]
- Mendez, J.S.; Govindan, A.; Leong, J.; Gao, F.; Huang, J.; Campian, J.L. Association between Treatment-Related Lymphopenia and Overall Survival in Elderly Patients with Newly Diagnosed Glioblastoma. J. Neurooncol. 2016, 127, 329–335. [Google Scholar] [CrossRef] [Green Version]
- Villano, J.L.; Letarte, N.; Yu, J.M.; Abdur, S. Hematologic Adverse Events Associated with Temozolomide. Cancer Chemother. Pharmacol. 2011, 69, 107–113. [Google Scholar] [CrossRef]
- Gilbar, P.J.; Pokharel, K.; Mangos, H.M. Temozolomide-Induced Aplastic Anemia: Case Report and Review of the Literature. J. Oncol. Pharm. Pract. 2020, 27, 1275–1280. [Google Scholar] [CrossRef]
- Alphandery, E. Glioblastoma Treatments: An Account of Recent Industrial Developments. Front. Pharmacol. 2018, 9, 879. [Google Scholar] [CrossRef] [Green Version]
- Auffinger, B.; Spencer, D.; Pytel, P.; Ahmed, A.U.; Lesniak, M. The Role of Glioma Stem Cells in Chemotherapy Resistance and Glioblastoma Multiforme Recurrence. Expert Rev. Neurother. 2015, 15, 741–752. [Google Scholar] [CrossRef] [Green Version]
- Bush, N.A.O.; Butowski, N. The Effect of Molecular Diagnostics on the Treatment of Glioma. Curr. Oncol. Rep. 2017, 19, 26. [Google Scholar] [CrossRef]
- Ghoneum, A.; Abdulfattah, A.Y.; Warren, B.O.; Shu, J.; Said, N. Redox Homeostasis and Metabolism in Cancer: A Complex Mechanism and Potential Targeted Therapeutics. Int. J. Mol. Sci. 2020, 21, 3100. [Google Scholar] [CrossRef]
- Cencioni, C.; Comunanza, V.; Middonti, E.; Valleriello, E.; Bussolino, F. The Role of Redox System in Metastasis Formation. Angiogenesis 2021, 24, 435–450. [Google Scholar] [CrossRef] [PubMed]
- Reczek, C.R.; Chandel, N.S. The Two Faces of Reactive Oxygen Species in Cancer. Annu. Rev. Cancer Biol. 2017, 1, 79–98. [Google Scholar] [CrossRef]
- Halliwell, B. Free Radicals and Antioxidants—Quo Vadis? Trends Pharmacol. Sci. 2011, 32, 125–130. [Google Scholar] [CrossRef]
- Friedman, J. Why Is the Nervous System Vulnerable to Oxidative Stress. In Oxidative Stress and Free Radical Damage in Neurology; Gadoth, N., Gobel, H.H., Eds.; Humana Press: New York, NY, USA, 2011; pp. 19–27. ISBN 978-1-60327-514-9. [Google Scholar]
- Wallace, D.C. Mitochondria in Cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in Cancer: Initiators, Amplifiers or an Achilles’ Heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.; Lu, P.; Beeraka, N.M.; Sukocheva, O.A.; Madhunapantula, S.V.; Liu, J.; Sinelnikov, M.Y.; Nikolenko, V.N.; Bulygin, K.V.; Mikhaleva, L.M.; et al. Mitochondrial Mutations and Mitoepigenetics: Focus on Regulation of Oxidative Stress-Induced Responses in Breast Cancers. Semin. Cancer Biol. 2020. [Google Scholar] [CrossRef]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting Cancer Cells by ROS-Mediated Mechanism: A Radical Therapeutic Approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef]
- Morry, J.; Ngamcherdtrakul, W.; Yantasee, W. Oxidative Stress in Cancer and Fibrosis: Opportunity for Therapeutic Intervention with Antioxidant Compounds, Enzymes, and Nanoparticles. Redox. Biol. 2017, 11, 240–253. [Google Scholar] [CrossRef]
- Cheng, H.-L.; Lee, Y.-H.; Yuan, T.-M.; Chen, S.-W.; Chuen, P.-J. Update on a Tumor-Associated NADH Oxidase in Gastric Cancer Cell Growth. World J. Gastroenter. 2016, 22, 2900–2905. [Google Scholar] [CrossRef]
- Jiang, Z.; Gorenstein, N.M.; Morré, D.M.; Morré, D.J. Molecular Cloning and Characterization of a Candidate Human Growth-Related and Time-Keeping Constitutive Cell Surface Hydroquinone (NADH) Oxidase. Biochemistry 2008, 47, 14028–14038. [Google Scholar] [CrossRef]
- Salazar-Ramiro, A.; Ramirez-Ortega, D.; de la Cruz, V.P.; Hernandez-Pedro, N.Y.; Gonzalez-Esquivel, D.F.; Sotelo, J.; Pineda, B. Role of Redox Status in Development of Glioblastoma. Front. Immunol. 2016, 7, 156. [Google Scholar] [CrossRef] [Green Version]
- Bakalova, R.; Zhelev, Z.; Aoki, I.; Saga, T. Tissue Redox Activity as a Hallmark of Carcinogenesis: From Early to Terminal Stages of Cancer. Clin. Cancer Res. 2013, 19, 2503–2517. [Google Scholar] [CrossRef] [Green Version]
- Duraj, T.; Garcia-Romero, N.; Carrion-Navarro, J.; Madurga, R.; de Mendivil, A.O.; Prat-Acin, R.; Garcia-Canamaque, L.; Ayuso-Sacido, A. Beyond the Warburg Effect: Oxidative and Glycolytic Phenotypes Coexist within the Metabolic Heterogeneity of Glioblastoma. Cells 2021, 10, 202. [Google Scholar] [CrossRef]
- Kim, J.; Han, J.; Jang, Y.; Kim, S.J.; Lee, M.J.; Ryu, M.J.; Kweon, G.R.; Heo, J.Y. High-Capacity Glycolytic and Mitochondrial Oxidative Metabolism Mediate the Growth Ability of Glioblastoma. Int. J. Oncol. 2015, 47, 1009–1016. [Google Scholar] [CrossRef] [Green Version]
- Gringuer, C.E.; Oliva, C.R. Bioenergetics Pathways and Therapeutic Resistance in Gliomas: Emerging Role of Mitochondria. Curr. Pharm. Des. 2011, 17, 2421–2427. [Google Scholar] [CrossRef]
- Duman, C.; Yaqubi, K.; Hoffmann, A.; Acikgöz, A.A.; Korshunov, A.; Bendszus, M.; Herold-Mende, C.; Liu, H.K.; Alfonso, J. Acyl-CoA-Binding Protein Drives Glioblastoma Tumorigenesis by Sustaining Fatty Acid Oxidation. Cell Metab. 2019, 30, 274–289.e5. [Google Scholar] [CrossRef]
- Sperry, J.; Condro, M.C.; Guo, L.; Braas, D.; Vanderveer-Harris, N.; Kim, K.K.O.; Pope, W.B.; Divakaruni, A.S.; Lai, A.; Christofk, H.; et al. Glioblastoma Utilizes Fatty Acids and Ketone Bodies for Growth Allowing Progression during Ketogenic Diet Therapy. iScience 2020, 23, 101453. [Google Scholar] [CrossRef]
- Bacic, G.; Pavicevic, A.; Peyrot, F. In Vivo Evaluation of Different Alterations of Redox Status by Studying Pharmacokinetics of Nitroxides Using Magnetic Resonance Techniques. Redox. Biol. 2016, 8, 226. [Google Scholar] [CrossRef] [Green Version]
- D’Autreaux, B.; Toledano, M.B. ROS as Signaling Molecules: Mechanisms That Generate Specificity in ROS Homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef]
- Zhelev, Z.; Aoki, I.; Gadjeva, V.; Nikolova, B.; Bakalova, R.; Saga, T. Tissue Redox Activity as a Sensing Platform for Imaging of Cancer Based on Nitroxide Redox Cycle. Eur. J. Cancer 2013, 49, 1467–1478. [Google Scholar] [CrossRef]
- Kroemer, G.; Pouyssegur, J. Tumor Cell Metabolism: Cancer’s Achilles’ Heel. Cancer Cell 2008, 13, 472–482. [Google Scholar] [CrossRef]
- Torrisi, F.; Vicario, N.; Spitale, F.M.; Cammarata, F.P.; Minafra, L.; Salvatorelli, L.; Russo, G.; Cuttone, G.; Valable, S.; Gulino, R.; et al. The Role of Hypoxia and SRC Tyrosine Kinase in Glioblastoma Invasiveness and Radioresistance. Cancers 2020, 12, 2860. [Google Scholar] [CrossRef]
- Jones, V.S.; Huang, R.-Y.; Chen, L.-P.; Chen, Z.-S.; Fu, L.; Huang, R.-P. Cytokines in Cancer Drug Resistance: Cues to New Therapeutic Strategies. Biochim. Biophys. Acta 2016, 1865, 255–265. [Google Scholar] [CrossRef] [Green Version]
- Berraondo, P.; Sanmamed, M.F.; Ochoa, M.C.; Etxeberria, I.; Aznar, M.A.; Perez-Gracia, J.L.; Rodriguez-Ruiz, M.E.; Ponz-Sarvise, M.; Castanon, E.; Melero, I. Cytokines in Clinical Cancer Immunotherapy. Brit. J. Cancer 2019, 120, 6–15. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Xu, Z.; Wang, Z.; Du, G.; Lun, L. TGF-Beta Signaling in Cancer Radiotherapy. Cytokine 2021, 148, 155709. [Google Scholar] [CrossRef]
- Magana-Maldonado, R.; Manoutcharian, K.; Hernandez-Pedro, N.Y.; Rangel-Lopez, E.; Perez-De la Cruz, V.; Rodriguez-Balderas, C.; Sotelo, J.; Pineda, B. Concomitant Treatment with Pertussis Toxin Plus Temozolomide Increases the Survival of Rats Bearing Intracerebral RG2 Glioma. J. Cancer Res. Clin. Oncol. 2014, 140, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Orozco-Morales, M.; Sanchez-Garcia, F.J.; Golan-Cancela, I.; Hernandez-Pedro, N.; Costoya, J.A.; de la Cruz, V.P.; Moreno-Jimenez, S.; Sotelo, J.; Pineda, B. RB Mutation and RAS Overexpression Induce Resistance to NK Cell-Mediated Cytotoxicity in Glioma Cells. Cancer Cell Int. 2015, 15, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, H.; Zhou, Y.; Wen, C.; Zhou, C.; Zhang, W.; Hu, X.; Wang, L.; You, C.; Shao, J. Curcumin Sensitizes Glioblastoma to Temozolomide by Simultaneously Generating ROS and Disrupting AKT/mTOR Signaling. Oncol. Rep. 2014, 32, 1610–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakalova, R.; Semkova, S.; Ivanova, D.; Zhelev, Z.; Miller, T.; Takeshima, T.; Shibata, S.; Lazarova, D.; Aoki, I.; Higashi, T. Selective Targeting of Cancerous Mitochondria and Suppression of Tumor Growth Using Redox-Active Treatment Adjuvant. Oxid. Med. Cell Longev. 2020, 2020, 6212935. [Google Scholar] [CrossRef] [PubMed]
- Vafai, S.B.; Mevers, E.; Higgins, K.W.; Fomina, Y.; Zhang, J.; Mandinova, A.; Newman, D.; Shaw, S.Y.; Clardy, J.; Mootha, V.K. Natural Product Screening Reveals Naphthoquinone Complex I Bypass Factors. PLoS ONE 2016, 11, e0162686. [Google Scholar] [CrossRef] [PubMed]
- Majamaa, K.; Rusanen, H.; Remes, A.; Hassinen, I.E. Metabolic interventions against complex I deficiency in MELAS syndrome. Mol. Cell Biochem. 1997, 174, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.S.; Teng, S.; Wilson, J.X.; Galati, G.; Khan, S.; O’Brien, P.J. Coenzyme Q Cytoprotective Metabolisms for Mitochondrial Complex I Cytopathies Involves NAD(P)H: Quinone Oxidoreductase (NQO1). Free Radic. Res. 2002, 36, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Eleff, S.; Kennaway, N.G.; Buist, N.R.; Darley-Usmar, V.M.; Capaldi, R.A.; Bank, W.J.; Chance, B. 31P NMR Study of Improvement in Oxidative Phosphorylation by Vitamins K3 and C in a Patient with a Defect in Electron Transport at Complex III in Skeletal Muscle. Proc. Natl. Acad. Sci. USA 1984, 81, 3529–3533. [Google Scholar] [CrossRef] [Green Version]
- U.S. Department of Health and Human Services (FDA) Home Page. National Institutes of Health (Office of Dietary Supplements). Dietary Supplements for Primary Mitochondrial Disorders. 2020. Available online: https://ods.od.nih.gov/factsheets/PrimaryMitochondrialDisorders-HealthProfessional/ (accessed on 5 January 2020).
- Bakalova, R.; Zhelev, Z.; Miller, T.; Aoki, I.; Higashi, T. Vitamin C versus Cancer: Ascorbic Acid Radical and Impairment of Mitochondrial Respiration? Oxid. Med. Cell Longev. 2020, 2020, 1504048. [Google Scholar] [CrossRef]
- Bakalova, R.; Zhelev, Z.; Miller, T.; Aoki, I.; Higashi, T. New Potential Biomarker for Stratification of Patients for Pharmacological Vitamin C in Adjuvant Settings of Cancer Therapy. Redox. Biol. 2020, 28, 101357. [Google Scholar] [CrossRef]
- De Cabo, R.; Siendones, E.; Minor, R.; Navas, R. Cyb5R3: A Key Player in Aerobic Metabolism and Aging? Aging 2009, 2, 63–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikiforova, A.B.; Saris, N.-E.L.; Kruglov, A.G. External Mitochondrial NADH-Dependent Reductase of Redox Cyclers: VDAC1 or Cyb5R3? Free Radic. Biol. Med. 2014, 74, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Ngo, B.; van Riper, J.; Cantley, L.C.; Yun, J. Targeting Cancer Vulnerabilities with High-Dose Vitamin C. Nat. Rev. Cancer 2019, 19, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Vita, M.F.; Nagachar, N.; Avramidis, D.; Delwar, Z.M.; Cruz, M.H.; Siden, A.; Paulsson, K.M.; Yakisich, J.S. Pankiller Effect of Prolonged Exposure to Menadione on Glioma Cells: Potentiation by Vitamin C. Investig. New Drugs 2011, 29, 1314–1320. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Agus, D.B.; Winfree, C.J.; Kiss, S.; Mack, W.; McTaggart, R.A.; Choudhri, T.F.; Kim, L.J.; Mocco, J.; Fox, W.D.; et al. Dehydroascorbic Acid, a Blood-Brain Barrier Transpotable form of Vitamin C.; Mediates Potent Cerebroprotection in Experimental Stroke. Proc. Natl. Acad. Sci. USA 2001, 98, 11720–11724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- May, J.M. Vitamin C Transport and Its Role in the Central Nervous System. Subcell. Biochem. 2012, 56, 85–103. [Google Scholar] [CrossRef] [Green Version]
- The Human Protein Atlas: ENOX2 Protein Expression Summary. Available online: http://www.proteinatlas.org/ENSG00000165675-ENOX2 (accessed on 2 November 2021).
- Castellano, A.; Bailo, M.; Cicone, F.; Carideo, L.; Quartuccio, N.; Mortini, P.; Falini, A.; Cascini, G.L.; Minniti, G. Advanced Imaging Techniques for Radiotherapy Planning of Gliomas. Cancers 2021, 13, 1063. [Google Scholar] [CrossRef]
- Song, J.; Kadaba, P.; Kravitz, A.; Hotmigo, A.; Friedman, J.; Belani, P.; Hadjipanayis, C.; Ellingston, B.M.; Nael, K. Multiparametric MRI for Early Identification of Therapeutic Response in Recurrent Glioblastoma Treated with Immune Checkpoint Inhibitors. Neurol. Oncol. 2020, 22, 1658–1666. [Google Scholar] [CrossRef]
- Dikalov, S.; Harrison, D.G. Methods for Detection of Mitochondrial and Cellular Oxygen Species. Antioxid. Redox. Signal. 2014, 20, 372–382. [Google Scholar] [CrossRef] [Green Version]
- Levraut, J.; Iwase, H.; Shao, Z.-H.; Vanden Hoek, T.L.; Schumacker, P.T. Cell Death during Ischemia: Relationship to Mitochondrial Depolarization and ROS Generation. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H549–H558. [Google Scholar] [CrossRef] [Green Version]
- Semkova, S.; Zhelev, Z.; Miller, T.; Sugaya, K.; Aoki, I.; Higashi, T.; Bakalova, R. Menadione/Ascorbate Induces Overproduction of Mitochondrial Superoxide and Impairs Mitochondrial Function in Cancer: Comparative study on Cancer and Normal Cells of the Same Origin. Anticancer Res. 2020, 40, 1963–1972. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.I.; Nakanishi, I.; Zhelev, Z.; Bakalova, R. Nitroxyl Radical as a Theranostic Contrast Agent in Magnetic Resonance Redox Imaging. Antioxid. Redox. Signal 2021. [Google Scholar] [CrossRef] [PubMed]
- Verrax, J.; Vanbever, S.; Stockis, J.; Taper, H.; Calderon, P.B. Role of Glycolysis Inhibition and Poly(ADP Ribose)Polymerase Activation in Necrotic-Like Death Caused by Ascorbate/Menadione-Induced Oxidative Stress in K562 Human Chronic Myelogenous Leukemia Cells. Int. J. Cancer 2007, 120, 1192–1197. [Google Scholar] [CrossRef] [PubMed]
- Beck, R.; Pedrosa, R.C.; Dejenas, N.; Glorieux, C.; Gallez, P.L.B.; Taper, H.; Eeckhoudt, S.; Knoops, L.; Calderon, P.B.; Verrax, J. Ascorbate/Menadione-Induced Oxidative stress Kills Cancer Cells That Express Normal or Mutated Forms of the Oncogenic Protein Bcr-Abl. An In Vitro and In Vivo Mechanistic Study. Investig. New Drugs 2011, 29, 891–900. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Santhosh, S.M.; Coppo, L.; Ogata, F.T.; Lu, J.; Holmgren, A. The Combination of Ascorbate and Menadione Causes Cancer Cell Death by Oxidative Stress and Replicative Stress. Free Radic. Biol. Med. 2019, 134, 350–358. [Google Scholar] [CrossRef]
- Greenwood, J.; Steinman, L.; Zamvil, S. Statin Therapy in Autoimmunity: From Protein Prenylation to Immunomodulation. Nat. Rev. Immunol. 2006, 6, 350–370. [Google Scholar] [CrossRef] [Green Version]
- Ryszawy, D.; Pudelek, M.; Catapano, J.; Ciarach, M.; Setkowicz, Z.; Konduracka, E.; Madeja, Z.; Czyz, J. High Doses of Sodium Ascorbate Interfere with the Expansion of Glioblastoma Multiform Cells In Vitro and In Vivo. Life Sci. 2019, 232, 116657. [Google Scholar] [CrossRef]
- Padayatty, S.; Sun, A.Y.; Chen, Q.; Epsey, M.G.; Drisko, J.; Levine, M. Vitamin C: Intravenous Use by Complementary and Alternative Medicine Practitioners and Adverse Effects. PLoS ONE 2010, 5, e11414. [Google Scholar] [CrossRef] [Green Version]
- Carr, A.C.; Cook, J. Intravenous Vitamin C for Cancer Therapy—Identifying the Current Gaps in Our Knowledge. Front. Physiol. 2018, 9, 1182. [Google Scholar] [CrossRef]
- Kim, K.; Bae, O.-N.; Koh, S.-H.; Kang, S.; Lim, K.-M.; Noh, J.-Y.; Shin, S.; Kim, I.; Chung, J.-H. High-Dose Vitamin C Injection to Cancer Patients May Promote Thrombosis through Procoagulant Activation of Erythrocytes. Toxicol. Sci. 2015, 147, 350–359. [Google Scholar] [CrossRef] [Green Version]
- Grasso, C.; Fabre, M.-S.; Collis, S.V.; Castro, M.L.; Field, C.S.; Schleich, N.; McConnell, M.J.; Herst, P.M. Pharmacological Doses of Daily Ascorbate Protect Tumors from Radiation Damage after a Single Dose of Radiation in an Intracranial Mouse Glioma model. Front. Oncol. 2014, 4, 356. [Google Scholar] [CrossRef] [Green Version]
- Taper, H.S.; Roberfroid, M. Non-Toxic Sensitization of Cancer Chemotherapy by Combined Vitamin C and K3 Pre-Treatment in a Mouse Tumor Resistant to Oncovin. Anticancer Res. 1992, 12, 1651–1654. [Google Scholar]
- Taper, H.S.; Keyeus, A.; Roberfroid, M. Potentiation of Radiotherapy by Nontoxic Pre-Treatment with Combined Vitamins C and K3 in Mice Bearing Solid Transplantable Tumor. Anticancer Res. 1996, 16, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Calderon, P.; Cadrobbi, J.; Marques, C.; Hong-Ngoc, N.; Jamison, J.M.; Summers, J.L.; Taper, H.S. Potential Therapeutic Application of the Association of Vitamins C and K3 in Cancer Treatment. Curr. Med. Chem. 2002, 9, 2271–2285. [Google Scholar] [CrossRef]
- Taper, H.S.; Jamison, J.M.; Gilloteaux, J.; Summers, J.L.; Calderon, P.B. Inhibition of the Development of Metastases by Dietary Vitamin C: K3 Combination. Life Sci. 2004, 75, 955–967. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.F.; Yang, C.M.; Su, C.M.; Liao, J.W.; Hu, M.L. Inhibitory Effect of Vitamin C in Combination with Vitamin K3 on Tumor Growth and Metastasis of Lewis Lung Carcinoma Xenografted in C57Bl/6 Mice. Nutr. Cancer 2011, 63, 1036–1043. [Google Scholar] [CrossRef]
- Du, J.; Gullen, J.J.; Buettner, G.R. Ascorbic Acid: Chemistry, Biology and the Treatment of Cancer. Biochim. Biophys. Acta 2012, 1826, 443–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Criddle, D.N.; Gillies, S.; Baumgartner-Wilson, H.K.; Jaffar, M.; Chinje, E.C.; Passmore, S.; Chvanov, M.; Barrow, S.; Gerasimenko, O.V.; Tepikin, A.V.; et al. Menadione-Induced ROS Generation via Redox Cycling Promotes Apoptosis of Muruine Pancreatic Acinar Cells. J. Biol. Chem. 2006, 281, 40485–40492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tielens, A.G.M.; Rotte, C.; van Hellemond, J.J.; Martin, W. Mitochondria as We Don’t Know them. Trends Biol. Sci. 2002, 27, 564–572. [Google Scholar] [CrossRef]
- Dubouchaud, H.; Walter, L.; Rigoulet, M.; Batandier, C. Mitochondrial NADH Redox Potential Impacts the ROS Production of Reverse Electron Transfer through Complex I. J. Bioenerg. Biomembr. 2018, 50, 367–377. [Google Scholar] [CrossRef]
- Nakagawa, K.; Hirota, Y.; Sawada, N.; Yuge, N.; Watanabe, M.; Uchino, Y.; Okuda, N.; Shimomura, Y.; Suhara, Y.; Okano, T. Identification of UBIAD1 as a Novel Human Menaquinone-4 Biosynthetic Enzyme. Nature 2010, 468, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, K.; Fujiwara, K.; Nishimura, A.; Murakami, C.; Kawamoto, K.; Ichinose, C.; Kunitou, Y.; Suhara, Y.; Okano, T.; Hasegawa, H. UBIAD1 Plays an Essential Role in the Survival of Pancreatic Acinar Cells. Int. J. Mol. Sci. 2019, 20, 1971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Human Protein Atlas: UBIAD1 Protein Expression Summary. Available online: http://www.proteinatlas.org/ENSG00000120942-UBUAD1 (accessed on 2 November 2021).
- Li, H.; Qu, L.; Zhang, H.; Liu, J.; Zhang, X. A Comprehensive Transcriptomic Landscape of Cholangiosarcoma Based on Bioinformatics Analysis from Large Cohort of Patients. Sci. Rep. 2021, 11, 13713. [Google Scholar] [CrossRef] [PubMed]
- Park, S.B.; Chung, C.K.; Gonzalez, E.; Yoo, C. Causal Inference Network of Genes Relates with Bone Metastasis of Breast Cancer and Osteoblasts Using Causal Bayesian Networks. J. Bone Metab. 2018, 25, 251–266. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Wei, X.; Wu, S.; Wang, B.; Hong, L. Down-Regulation of TERE1/UBIAD1 Activated Ras-MAPK Signalling and Induced Cell Proliferation. Cell Biol. Int. Rep. 2010, 17, e00005. [Google Scholar] [CrossRef] [Green Version]
- Mao, H.; LeBrun, D.G.; Yang, J.; Zhu, V.F.; Li, M. Deregulated Signaling Pathways in Glioblastoma Multiform: Mechanisms and Therapeutic Targets. Cancer Investig. 2012, 30, 48–56. [Google Scholar] [CrossRef] [Green Version]
- Frederiks, W.J.; Sepulveda, J.; Lai, P.; Tomaszewski, J.E.; Lin, M.-F.; McGarvey, T.; Rauscher, F.J.; Malkovicz, S.B. The Tumor Suppressor TERE1 (UBIAD1) Prenyltransferase Regulates the Elevated Cholesterol Phenotype in Castration Resistant Prostate Cancer by Controlling a Program of Ligand Dependent SXR Target Genes. Oncotarget 2013, 4, 1075–1092. [Google Scholar] [CrossRef]
- Liu, S.; Guo, W.; Han, X.; Dai, W.; Diao, Z.; Liu, W. Role of UBIAD1 in Intracellular Cholesterol Metabolism and Vascular Cell Calcification. PLoS ONE 2016, 11, e0149639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, T.L.; Emily, E.L.; Cimmino, L. Reprogramming the Epigemome with Vitamin C. Front. Cell Dev. Biol. 2019, 7, 128. [Google Scholar] [CrossRef]
- Weller, M.; Stupp, R.; Reifenberger, G.; Brandes, A.A.; van den Bent, M.J.; Wick, W.; Hegi, M.E. MGMT Promoter Methylation in Malignant Gliomas: Ready for Personalized Medicine? Nat. Rev. Neurol. 2010, 6, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Fresnais, M.; Turcan, S.; Theile, D.; Ungermann, J.; Abou Zeed, Y.; Lindner, J.R.; Breitkopf, M.; Burhenne, J.; Haefeli, W.E.; Longuespee, R. Approaching Sites of Action of Temozolomide for Pharmacological and Clinical Studies in Glioblastoma. Biomedicines 2022, 10, 1. [Google Scholar] [CrossRef]
- Hsieh, P.-F.; Chueh, P.J.; Liu, P.-F.; Liao, J.-W.; Hsieh, M.-K. Immune Response Evoked by Tumor-Associated NADH Oxidase (tNOX) Confers Potential Inhibitory Effect on Lung Carcinoma in a Mouse Model. Am. J. Cancer Res. 2019, 9, 740–751. [Google Scholar] [PubMed]
- Yang, L.; Pang, Y.; Moses, H.L. TGB-b and Immune Cells: An Important Regulatory Axis in the Tumor Microenvironment and Progression. Trends Immunol. 2010, 31, 220–227. [Google Scholar] [CrossRef] [Green Version]
- Travis, M.A.; Sheppard, D. TGF-b Activation and Function in Immunity. Annu. Rev. Immunol. 2014, 32, 51–82. [Google Scholar] [CrossRef] [Green Version]
- Park, B.V.; Freeman, Z.T.; Ghasemzadeh, A.; Chattergoon, M.A.; Rutebenberwa, A.; Steigner, J.; Winter, M.E.; Huynh, T.V.; Sebald, S.M.; Lee, S.-J.; et al. TGF-b1-Mediated Smad3 Enhances PD-1 Expression on Antigen-Specific T Cells in Cancer. Cancer Discov. 2016, 6, 1366–1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, D.G.; Murphy, M.P.; Frezza, C.; Prag, H.A.; Chouchani, E.T.; O’Neill, L.A.; Mills, E.L. Coupling Krebs Cycle Metabolites to Signaling in Immunity and Cancer. Nat. Metab. 2019, 1, 16–33. [Google Scholar] [CrossRef]
- Hostetler, B.; Weston, N.; Kim, C.; Morre, D.M.; Morre, D.J. Cancer Site-Specific Isoforms of ENOX2 (tNOX): A Cancer-Specific Cell Surface Oxidase. Clin. Proteom. 2009, 5, 46–51. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.Y.; Cheng, H.L.; Lee, Y.H.; Yuan, T.M.; Chen, S.W.; Lin, Y.Y.; Chueh, P.J. Tumor-Associated NADH Oxidase (tNOX)-NAD+-Sirtuin 1 Axis Contributes to Oxaliplatin-Induced Apoptosis of Gastric Cancer Cells. Oncotarget 2017, 8, 15338–15348. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.Y.; Islam, A.; Yuan, T.M.; Chen, S.W.; Liu, P.F.; Chuen, P.J. Regulation of tNOX Expression through the ROS-p53-POU3F2 Axis Contributes to Cellular Responses against Oxaliplatin in Human Colon Cancer Cells. J. Exp. Clin. Cancer Res. 2018, 37, 161. [Google Scholar] [CrossRef]
- Lee, Y.H.; Chen, H.Y.; Su, L.J.; Chueh, P.J. Sirtuin 1 (SIRT1) Deacetylase Activity and NAD+/NADH Ratio Are Imperative for Capsaicin-Mediated Programmed Cell Death. J. Agric. Food Chem. 2015, 63, 7361–7367. [Google Scholar] [CrossRef]
- Islam, A.; Hsieh, P.-F.; Chou, J.-C.; Liao, J.-W.; Hsieh, M.-K.; Chuen, P.J. Capsaicin Exerts Therapeutic Effects by Targeting tNOX-SIRT1 Axis and Augmenting ROS-Dependent Cytotoxic Autophagy in Melanoma Cancer Cells. Am. J. Cancer Res. 2021, 11, 4199–4219. [Google Scholar] [PubMed]
- Su, Y.C.; Lin, Y.H.; Zeng, Z.M.; Shao, K.N.; Chueh, P.J. Chemotherapeutic Agents Enhance Cell Migration and Epithelial-to-Mesenchymal Transition through Transient Up-Regulation of tNOX (ENOX2) Protein. Biochim. Biophys. Acta 2012, 1820, 1744–1752. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.C.; Hsieh, P.F.; Hsieh, M.K.; Zeng, Z.M.; Cheng, H.L.; Liao, J.W.; Chueh, P.J. Capsaicin-Mediated tNOX (ENOX2) Up-Regulation Enhances Cell Proliferation and Migration In Vitro and In Vivo. J. Agric. Food Chem. 2012, 60, 2758–2765. [Google Scholar] [CrossRef] [PubMed]
- Gunderson, A.J.; Yamazaki, T.; McCarty, K.; Fox, N.; Phillips, M.; Alice, A.; Blair, T.; Whiteford, M.; O’Brien, D.; Ahmad, R.; et al. TGF-b Suppresses CD8+ T Cell Expression of CXCR3 and Tumor Trafficking. Nat. Commun. 2020, 11, 1749. [Google Scholar] [CrossRef] [Green Version]
- Neganova, M.; Liu, J.; Aleksandrova, Y.; Kluchkov, S.; Fan, R. Therapeutic Influence on Important Targets Associated with Chronic Inflammation and Oxidative Stress in Cancer Treatment. Cancers 2021, 13, 6062. [Google Scholar] [CrossRef]
- Lan, T.; Chen, L.; Wei, X. Inflammatory Cytokines in Cancer: Comprehensive Understanding and Clinical Progress in Gene Therapy. Cells 2021, 10, 100. [Google Scholar] [CrossRef] [PubMed]
- Kundu, J.K.; Surh, Y.-J. Emerging Avenues Linking Inflammation and Cancer. Free Radic. Biol. Med. 2012, 52, 2013–2037. [Google Scholar] [CrossRef]
- Gregg, D.; de Carvalho, D.D.; Kavacic, H. Integrins and Coagulation: A Role for ROS/Redox Signaling? Antioxid. Redox. Signal 2004, 6, 757–764. [Google Scholar] [CrossRef]
- Malenica, I.; Adam, J.; Corgnac, S.; Mezquita, L.; Auclin, E.; Damei, I.; Grynszpan, L.; Gros, G.; de Montpreville, V.; Planchard, D.; et al. Integrin-AV-Mediated Activation of TGF-b Regulates Anti-Tumor CD8 T Cell Immunity and Response to PD-1 Blockade. Nat. Commun. 2021, 12, 5209. [Google Scholar] [CrossRef]
- De Bleser, P.J.; Xu, G.; Rombouts, K.; Rogiers, V.; Geerts, A. Glutathione Levels Discriminate between Oxidative Stress and Transforming Growth Factor-B Signaling in Activated Rat Hepatic Stellate Cells. J. Biol. Chem. 1999, 274, 33881–33887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahmani, A.; Selisle, J.-S. TGF-B in T Cell Biology: Implications for Cancer Immunotherapy. Cancers 2018, 10, 194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marin-Acevedo, J.A.; Kimbrough, E.O.; Lou, Y. Next Generation of Immune Checkpoint Inhibitors and Beyond. J. Hematol. Oncol. 2021, 14, 45. [Google Scholar] [CrossRef] [PubMed]
- Khasraw, M.; Reardon, D.A.; Weller, M.; Sampson, J.H. PD-1 Inhibitors: Do They Have a Future in the Treatment of Glioblastoma? Clin. Cancer Res. 2020, 26, 5287–5296. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Yan, W. Succinate in the Cancer-Immune Cycle. Cancer Lett. 2017, 390, 45–47. [Google Scholar] [CrossRef]
- Martinez-Reyes, I.; Chandel, N.S. Mitochondrial TCA Cycle Metabolites Control Physiology and Disease. Nat. Commun. 2020, 11, 102. [Google Scholar] [CrossRef] [Green Version]
- Checker, R.; Sharma, D.; Sandur, S.K.; Khan, N.M.; Patwardhan, R.S.; Kohli, V.; Sainis, K.B. Vitamin K3 Suppressed Inflammatory and Immune Responses in a Redox-Dependent Manner. Free Radic. Res. 2011, 45, 975–985. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Hou, Y.; He, H.; Chen, Y.; Zhou, R.; Wang, X.; Gong, T.; Jiang, W. Synthetic Vitamin K Analogs Inhibit Inflammation by Targeting the NLRP3 Inflammasome. Cell Mol. Immunol. 2021, 18, 2422–2430. [Google Scholar] [CrossRef]
- Zitvogel, L.; Pietrocola, F.; Kroemer, G. Nutrition, Inflammation, and Cancer. Nat. Immunol. 2017, 18, 843–850. [Google Scholar] [CrossRef]
- Eilaghi, A.; Yeung, T.; d’Esterre, C.; Bauman, G.; Yartsev, S.; Easaw, J.; Fainardi, E.; Lee, T.-Y.; Frayne, R. Quantitative Perfusion and Permeability Biomarkers in Brain Cancer from Tomographic CT and MR Images. Biomark. Cancer 2016, 8 (Suppl. 2), 47–59. [Google Scholar] [CrossRef]
- Van Dijken, B.R.J.; Van Laar, P.J.; Smits, M.; Dankbaar, J.W.; Enting, R.H.; van der Hoom, A. Perfusion MRI in Treatment Evaluation of Glioblastomas: Clinical Relevance of Current and Future Techniques. J. Magn. Reson. Imaging 2019, 49, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Lah, T.T.; Novak, M.; Breznik, B. Brain Malignancies: Glioblastoma and Brain Metastasis. Semin. Cancer Biol. 2020, 60, 262–273. [Google Scholar] [CrossRef] [PubMed]
- Couto, M.; Coelho-Santos, V.; Santos, L.; Fontes-Ribeiro, C.; Silva, A.P.; Gomes, C.M.F. The Interplay between Glioblastoma and Microglial Cells Leads to Endothelial Cell Monolayer Dysfunction via the Interleukin-6-Induced JAK2/STAT3 Pathway. J. Cell Physiol. 2019, 234, 19750–19760. [Google Scholar] [CrossRef] [PubMed]
- Sugahara, T.; Korogi, Y.; Kochi, M.; Ikushima, I.; Shigematu, Y.; Hirai, T.; Okuda, T.; Liang, L.; Ge, Y.; Komohara, Y.; et al. Usefulness of Diffusion-Weighted MRI with Echo-Planar Technique in the Evaluation of Cellularity in Glioma. Magn. Reson. Imaging 1999, 9, 53–60. [Google Scholar] [CrossRef]
- Tourell, M.C.; Shokoohmand, A.; Landgraf, M.; Holzapfel, N.P.; Poh, P.S.P.; Loessner, D.; Momot, K.I. The Distribution of the Apparent Diffusion Coefficient as an Indicator of the Response to Chemotherapeutics in Ovarian Tumor Xenografts. Sci. Rep. 2017, 7, 42905. [Google Scholar] [CrossRef] [Green Version]
- Surov, A.; Meyer, H.J.; Wienke, A. Correlation between Apparent Diffusion Coefficient (ADC) and Cellularity Is Different in Several Tumors: A Meta-Analysis. Oncotarget 2017, 8, 59492–59499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fliedner, F.P.; Engel, T.B.; El-Ali, H.H.; Hansen, A.E. Diffusion Weighted Magnetic Resonance Imaging (DW-MRI) as a Non-Invasive, Tissue Cellularity Marker to Monitor Cancer Treatment Response. BMC Cancer 2020, 20, 134. [Google Scholar] [CrossRef] [Green Version]
- Surov, A.; Meyer, H.J.; Wienke, A. Apparent Diffusion Coefficient for Distinguishing between Malignant and Benign Lesions in the Head and Neck Region: A Systematic Review and Meta-Analysis. Front. Oncol. 2019, 9, 1362. [Google Scholar] [CrossRef]
- Gilloteaux, J.; Jamison, J.M.; Arnold, D.; Taper, H.S.; Summers, J.L. Ultrastructural Aspects of Autoschizis: A New Cancer Cell Death Induced by the Synergistic Action of Ascorbate/Menadione on Human Bladder Carcinoma Cells. Ultrastruct. Pathol. 2001, 25, 183–192. [Google Scholar] [CrossRef]
- Nguyen, C.; Pandey, S. Exploiting Mitochondrial Vulnerabilities to Trigger Apoptosis Selectively in Cancer Cells. Cancers 2019, 11, 916. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sumiyoshi, A.; Shibata, S.; Zhelev, Z.; Miller, T.; Lazarova, D.; Aoki, I.; Obata, T.; Higashi, T.; Bakalova, R. Targeting Glioblastoma via Selective Alteration of Mitochondrial Redox State. Cancers 2022, 14, 485. https://doi.org/10.3390/cancers14030485
Sumiyoshi A, Shibata S, Zhelev Z, Miller T, Lazarova D, Aoki I, Obata T, Higashi T, Bakalova R. Targeting Glioblastoma via Selective Alteration of Mitochondrial Redox State. Cancers. 2022; 14(3):485. https://doi.org/10.3390/cancers14030485
Chicago/Turabian StyleSumiyoshi, Akira, Sayaka Shibata, Zhivko Zhelev, Thomas Miller, Dessislava Lazarova, Ichio Aoki, Takayuki Obata, Tatsuya Higashi, and Rumiana Bakalova. 2022. "Targeting Glioblastoma via Selective Alteration of Mitochondrial Redox State" Cancers 14, no. 3: 485. https://doi.org/10.3390/cancers14030485